

Abstract
Women with preeclampsia (PE) produce agonistic autoantibodies to the Angiotensin II type 1 receptor (AT1-AA), which stimulate reactive oxygen species (ROS), inflammatory factors, and hypertensive mechanisms (Endothelin and sFlt-1) in rodent models of PE. The placental ischemic reduced uterine perfusion pressure (RUPP) rat model of PE exhibits many of these features. In this study we examined the maternal outcomes of AT1-AA inhibition (‘n7AAc’) in RUPP rats. Blood pressure was higher in RUPP rats vs. normal pregnant (NP) rats (123±2 vs. 99± 2 mmHg, p< 0.05), which was reduced in RUPP+‘n7AAc’ (105±3 vs.123±2 mmHg, p<0.05 vs RUPP). Uterine artery resistant index was increased in RUPP vs. NP rats (0.71± 0.02 vs. 0.49± 0.02, p<0.05) and normalized in RUPP + ‘n7AAc’ rats (0.55± 0.03). Antiangiogenic factor sFlt1 was elevated in RUPP vs. NP rats (176±37 vs. 77±15 pg/ml, p<0.05) but normalized in RUPP+‘n7AAc’ (86±9, p=0.05 vs. RUPP). Plasma nitrate and nitrite was decreased (14±1 vs 20±1 μMNO3, p<0.05) and isoprostanes were elevated (20,117± 6304 vs. 2809±1375pg/ml, p<0.05) in RUPP vs. NP rats; and normalized in RUPP+‘n7AAc rats; (18±2 μMNO3; 4311±1 pg/ml). Pre-proendothelin-1 expression increased 4 fold in RUPP vs. NP rats which was prevented with ‘n7AAc’ Importantly placental cytolytic natural killer cells were elevated in RUPP vs. NP rats (8±2 vs. 2±2 % gated, p<0.05), which was prevented in RUPP+‘n7AAc’ total (3±1 % gated, p<0.05) In conclusion, AT1-AA inhibition prevents the rise in maternal blood pressure and several pathophysiological factors associated with PE in RUPP rats and could be a potential therapy for PE.
Keywords: AT1-AA, ANGII, renal, pregnancy, preeclampsia, natural killer cells
Introduction
Preeclampsia (PE) is a multi-systemic disorder characterized by new onset hypertension usually occurring in the third trimester of pregnancy.1 Preeclampsia effects ~ 5-10% of all births in the USA each year and is the leading cause of preterm births, morbidity, and mortality for both the mother and fetus during pregnancy.1–4 The origins of PE are unknown; however, it is widely believed that the placenta is the culprit of the disease. During PE several factors are released that play a role in the disease, such as soluble fms-like tyrosine kinase-1 (sFlt-1), inflammatory cytokines, oxidative stress molecules, cytolytic natural killer cells (NK), and the agonistic angiotensin II type 1 receptor autoantibody (AT1-AA).1–3,5–7 As of today there is no cure for PE except for the delivery of the fetal placenta unit. To better understand the etiology, pathology, and efficacy of possible treatments for PE, we use the reduced uterine perfusion pressure (RUPP) rat model.8 Similar to women with PE, RUPP rats have increased sFlt-1, inflammatory cytokines, cytolytic NK cells, ROS and AT1-AAs.3,8,9
AT1-AAs bind with high affinity to a 7 amino acid sequence on the second extracellular loop causing activation of the angiotensin II type 1 (AT1) receptor.6,7,10–15 AT1-AA binding of the receptor has been shown to increase intracellular calcium levels and stimulate activation of intracellular MAP/ERK kinase and TNFα pathways similar to ANG II.6,7,10–15 Both human and animal AT1-AAs isolated from PE patients or RUPP rats reproduce the PE-like phenotype when given to pregnant rodents. These models of AT1-AA induced hypertension during pregnancy have reduced glomerular filtration rate (GFR), increased blood pressure, oxidative stress, renal artery resistant index, reduced pup weight, and several circulating factors associated with PE, such as sFlt-1, sEng, inflammatory cytokines, endothelin-1 (ET-1), and endothelial microparticles.10,16–24
Landmark studies by the Xia laboratory demonstrated that AT1-AA titer in PE women is proportional to sFlt-1 and the severity of preeclampsia.25 Furthermore studies by Walther et al show that AT1-AAs are detected within 2nd trimester of a human pregnancy is associated with abnormal uterine perfusion pregnancies, suggesting that AT1-AA’s appear early in the diagnosis of preeclampsia and may play an critical role in the pathogenesis of preeclampsia and intrauterine growth restriction.26 Additional studies by the Xia laboratory showed that hypertension and elevated sFlt-1 l in pregnant mice, mediated by AT1-AAs from PE women, was blunted by ARB treatment, suggesting that the increase in sFlt-1 is downstream of AT1 receptor activation.27 Our previous studies have shown that AT1-AA induce hypertension in pregnant rats occurs with increased ET-1,sFlt-1 and oxidative stress10,16,18,20. Administration of an ET-A receptor antagonist or an AT1 receptor blocker (ARB) reduced blood pressure and these stimulated factors supporting the idea that AT1-AAs cause hypertension during pregnancy through At1 r mediated ET-1, oxidative stress and sFlt-1 upregulation16,18,20,42. However we cannot use losartan during pregnancy due to fetal toxicity, therefore the utilization of an inhibitory peptide to prevent the AT1-AA from activating the AT1 receptor could not only attenuate the increase in vasoactive factors and oxidative stress but also prevent the rise in maternal blood pressure and thereby could be advantageous to improve outcomes in response to placental ischemia during pregnancy.
Therapies targeted specifically against the AT1-AA to keep it from binding to the 2nd extracellular loop of the AT1 receptor have previously been performed by the laboratory of Yang Xia. Xia laboratory administered a linear seven amino acid sequence peptide corresponding to the binding site on the AT1 receptor to pregnant mice receiving the human AT1-AA. These studies showed that the peptide was able to improve trophoblast invasion, calcium mobilization, intrauterine growth restriction, proteinuria, renal pathology, and hypertension.14,16,28,29 In-vitro studies from our lab show that a similar linear seven amino acid sequence significantly improves renal afferent arteriolar vasoconstriction.16 However none of these studies address the role of the naturally occurring AT1-AA produced in response to placental ischemia. In this study, we examined the effects of a newly constructed, modified AT1-AA inhibitory peptide, chronically administered to RUPP rat model of PE during pregnancy. It is important to note that this modified peptide is different from the linear peptide used in previous publications.14,16,28,29 This newly modified peptide contains the 7AA sequence along with protein capping of the N and C terminus of the peptide. Protein capping is a process commonly used to increase peptide half-life and to protect exogenous peptides from protein lysis and degradation when used in the whole animal. Based on previous studies, we hypothesized that this modified peptide will bind to circulating AT1-AAs, to inhibit AT1-AAs from binding to the AT1 receptor thereby decreasing sflt-1, ET-1, and ROS; thus improving blood pressure and renal and vascular function in RUPP rats.
Methods
The techniques and data that support the findings of this study are available from the corresponding author upon reasonable request.
All animal protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Mississippi Medical Center. Pregnant Sprague Dawley rats purchased from Envigo (Indianapolis, IN) were used in this study. Rats were housed in a temperature-controlled room (75˚F) with a 12 hour light and dark cycle each day and maintained on a normal diet with free access to food and water. All experiments performed were in accordance with the National Institutes of Health guidelines for use and care of animals.
Measurement of MAP and renal function in RUPP rats
Pregnant SD rats were randomly dived into 4 groups: Normal pregnant (NP) rats (n= 23), NP + AT1-AA inhibitory peptide (‘n7AAc’) (n=3), reduced uterine perfusion pressure (RUPP) rats (n=32), and RUPP + ‘n7AAc’ (n=24). On day 14 of pregnancy, the RUPP surgery was performed, with one group of RUPP rats receiving a mini-osmotic pump. RUPP surgery consisted of placing constrictive silver clips on the aorta (0.203 mm clips) superior to the iliac bifurcation and on the ovarian vessels (0.100 mm clips). The mini-pump implanted IP delivered the modified capped AT1-AA inhibitory peptide (‘n7AAc’) (Thermo Fisher Scientific, Waltham, MA,) at a dose of 144μg/day through day 19 of gestation. The modified AT1-AA inhibitory peptide is different from the linear peptide sequence, used in previous studies6,16,18,22,25,27,28 by a process referred to as protein capping (Thermo Fisher Scientific, Waltham, MA). The advantages of capping the peptide is for stabilization of the peptide in-vivo, protection against peptidases, and to more accurately mimic natural peptides in the circulation. Mini-pumps were also inserted into NP rats on day 14 of pregnancy. This dose was based on previous studies performed in our laboratory.16,23,30 Note that we only measured the blood pressure, pup weight, litter size, and placental size for NP +‘n7AAc’ rats. We did not perform further analysis on these rats because the blood pressure and maternal outcomes of these rats were similar to NP values and did not appear to alter the physiological state of a normal pregnancy in rats. Furthermore the AT1-AA is not detectable in NP rats thus further molecular analysis was not performed.18 On day 18 of gestation, using isoflurane anesthesia, catheters were inserted into the carotid artery and jugular vein to measure the blood pressure and infusion of FITC-sinistrin to determine glomerular filtration rate (GFR). On day 19 of gestation, GFR and conscious blood pressure (MAP) were determined. On day 19, rats were anesthetized and all of the hair on the upper back below the ears were shaved and removed to reduce interference or auto-fluorescence of sinistrin. For determination of GFR, a miniaturized device (NIC-Kidney, Mannheim Pharma & Diagnostics, Mannheim, Germany) composed of 2-light-emmiting diodes that can transcutaneously excite and measure the clearance of FITC-sinistrin was used and baseline fluorescence was collected for 10-15 minutes, followed by a bolus injection of FITC-sinistrin (3mg/100 g body weight in 0.2mL of 0.9% irrigation saline). Continuous fluorescence was measured for 2hrs and clearance curves analyzed using the MPD Lab Ver 1.0RC3 software. The half-life (t1/2) for the clearance of FITC-sinistrin is determined 45 min post-injection using a one-compartment model. The t1/2 value is converted to GFR (mL/min/100 g body weight) using the following semi-empirical equation developed and validated by the manufacturer: GFR = 31.26 [mL/100g body weight]/t1/2 [min].31 MAP was measured after placing the rats in individual restraining cages.23,30 Briefly, arterial pressure was monitored with a pressure transducer (Cobe III transducer CDX Sema) and recorded continuously for 45 min after a 30-min stabilization period. Subsequently, blood and urine samples were collected. Pup weights were obtained and placentas and kidneys were harvested, weighed, and stored in -80˚C for further use.23,30 Plasma creatinine was measured using the LabAsay Creatinine kit (Jaffe method; Wako Pure Chemical Industries, Ltd., Osaka, Japan) according to the manufactures instructions. Standards ranged from 10 -1.25 mg/dl. This colorimetric assay determined the amount of plasma creatinine, which was expressed as mg/dL.
Effect of ‘n7AAc’ on uterine artery resistance index (UARI) in RUPP rats
UARIs measurements were obtained on gestational day 18 via Doppler sonography, using the Vevo 770 unit (Visual Sonics) with a 30-Hz transducer (model no. 710B) as described previously.32,42 Briefly, 2- 4 images and measurements of Doppler velocimetry were taken on both the left and right uterine arteries. The peak systolic velocity (PSV) and end-diastolic flow velocity (EDV) measurements were taken from the Doppler images and used to calculate UARI, using this equation UARI = (PSV − EDV)/PSV.32
Effect of ‘n7AAc’ on placental PPET-1 expression in RUPP rats
Placental total RNA was extracted using the RNeasy ® Protect Mini kit (Qiagen, Hilden, Germany). Real-time PCR was used to determine levels of placental preproendothelin-1 (PPET-1). cDNA was synthesized from 1μg of RNA with Bio-Rad Iscript cDNA reverse transcriptase, and real-time PCR was performed using the Bio-Rad Sybre Green Supermix (Bio-Rad, Hercules, CA). Placental PPE expression is measured as the fold difference from RUPP vs. NP and RUPP+’n7AAc’ vs. NP.16,32
The effect of ‘n7AAc’ on circulating factors: nitrate and nitrite, isoprostanes, and sFlt-1
Plasma nitrate and nitrite was measured using the Nitrate/Nitrite Colorimetric Assay Kit (Cayman Chemical, Ann Harbor, MI) according to the manufactures instructions and reagents. Plasma isoprostanes levels were measured using the 8-Isoprostane EIA kit (Cayman Chemical, Ann Harbor, MI) according to the manufactures instructions.23 The intra and inter-assay variation in concentration (CV) for this kit were determined at multiple points on the standard curve, with CVs lower than 20%. Plasma soluble vascular endothelial growth factor receptor 1 (sFlt-1) content was determined using commercial Quantikine ELISA kits (R&D Systems, Minneapolis, MN) according the manufacturer's instructions. The inter- and intra-assay variability for the concentration was determined by the kit to be 8.4% and 7.2%, respectively.
Effect of placenta natural killer (NK) cells measured by flow cytometry
At the time of harvest, kidneys and placentas will be collected. Lymphocytes were isolated from tissues via centrifugation on a cushion of Ficoll-Hypaque (Lymphoprep, Accurate Chemical & Scientific Corp., Westbury, NY). For flow cytometric analysis, 1 × 106 cells were incubated for 30 minutes at 4 °C with antibodies against rat Anti-Natural Killer Cell Activation Structures (ANK61) or rat Anti-Natural Killer Cell antibody (ANK44) (AbCam, Cambridge, MA). ANK61 binds to the killer cell activation structure that is expressed on all NK cells, while ANK44 is only expressed on stimulated, cytotoxic NK cells.33 After washing, cells were labeled with secondary Fluorescein isothiocyanate (FITC; AbCam) antibody for 30 minutes at 4 °C. As a negative control for each individual rat, cells were treated exactly as described above except they were incubated with isotype controls antibodies conjugated to FITC alone. Subsequently, cells were washed, fixed, and resuspended in 500 μL of Rosswell Park Memorial Institute medium (RPMI) and analyzed for single staining on a Gallios flow cytometer (Beckman Coulter, Brea, CA). Lymphocytes were gated in the forward and side scatter plot. Cells that stained as ANK61+ will were designated as NK cells. Cells that stain as ANK44+ were designated as activated NK cells. The percent of positive stained cells above the negative control was collected for individual rats and the mean values for each experimental group were taken.
Statistical Analysis
All data are presented as mean ± SEM. Data were analyzed by one-way ANOVA with Bonferroni post hoc analysis or by student t-test comparing each group to each other as needed. All statistical analysis was performed with Graphpad Prism 6 software (GraphPad Software, La Jolla, CA). P < 0.05 was considered statistically significant.
Results
The effect of ‘n7AAc’ on MAP, fetal growth, UARI, and renal function in RUPP rats
Mean arterial pressure (MAP), a major diagnostic factor of PE, was elevated in RUPP rats (n=32,123± 2 mmHg) compared to NP rats (n=23, 99± 2 mmHg, p< 0.05) and was normalized to NP levels with RUPP+‘n7AAc’ (n=24, 105± 3 mmHg, p<0.05 vs RUPP; Figure 1) MAP was similar to NP rats in NP+‘n7AAc’ rats (100± 7 mmHg), suggesting that the inhibitor does not affect the blood pressure in NP rats. These data indicate that inhibiting AT1-AA activation of the AT1R decreases blood pressure in response to placental ischemia suggesting that the AT1-AA is a major contributor to hypertension in the RUPP rats.
Figure 1.
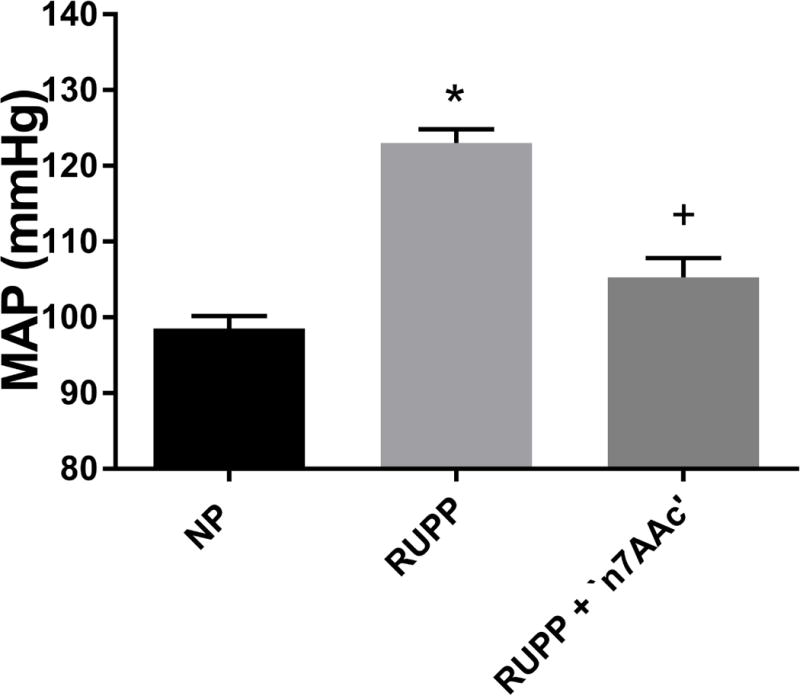
MAP was elevated in RUPP vs. normal pregnant rats (NP), and returned to baseline levels in RUPP + ‘n7AAc’. Statistical changes is MAP was derived using one-way ANOVA with Bonferroni post hoc analysis, *p<0.05 vs NP and +p<0.05 vs. RUPP.
Next we examined markers of fetal growth restriction, such as litter size, pup weight, and placental weight. The litter size was lower in RUPP rats (n=32, 6± 0.7) compared to NP rats (n=23, 13±0.4, p<0.05; Figure 2A) and was unchanged in RUPP+‘n7AAc’ rats (n= 24, 6± 0.9, ns vs. RUPP; Figure 2A). The average pup was smaller for RUPP (1.98± 0.04g) and RUPP+‘n7AAc’ (2.13± 0.05 g) rats in comparison to NP (2.49± 0.08 g) rats (p < 0.05) (Figure 2B). Placental weight was decreased for RUPPs (0.54± 0.02 g) and RUPP+‘n7AAc’ (0.53± 0.02 g) rats in comparison to NP (0.69± 0.05 g) rats (p < 0.05) (Figure 2C). No difference was observed in the litter size, average pup, and placental weight between RUPP and RUPP+‘n7AAc’ rats (Figure 2A–2C). In addition the litter size, pup weight, and placental weight was 15 ±1, 2.04g ±0.08, and 0.51g± 0.01 respectively for NP + ‘n7AAc’ rats which was not different than NP rats. Although AT1-AA inhibition did not prevent fetal growth restriction in RUPP rats, there was no further reduction of fetal growth restriction with AT1-AA inhibition.
Figure 2.
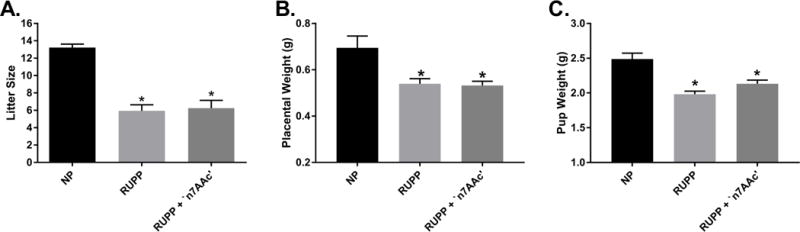
A) The number of pups was decreased for RUPP and RUPP + ‘n7AAc’ vs NP rats. B) Placental weight was decreased for RUPP and RUPP + ‘n7AAc’ vs NP rats. C) Pup weight was decreased for RUPP and RUPP + ‘n7AAc’ vs NP rats. Although, there was no difference between RUPP and RUPP + ‘n7AAc’ with number of pups, placental weight, and pup weight, there was no further decrease with RUPP + ‘n7AAc’. Statistical changes was determined using one-way ANOVA with Bonferroni post hoc analysis, *p<0.05 vs NP.
The UARI, which is a measurement of uterine artery vascular resistance, was increased in RUPP (n=9) vs NP (n=11) rats (0.71± 0.02 vs. 0.49± 0.02, p<0.05) (Figure 3). UARI decreased in RUPP+‘n7AAc (n=8) vs. RUPP rats (0.55± 0.03 vs. 0.71± 0.02, p<0.05), and was similar to NP values (Figure 3).
Figure 3.
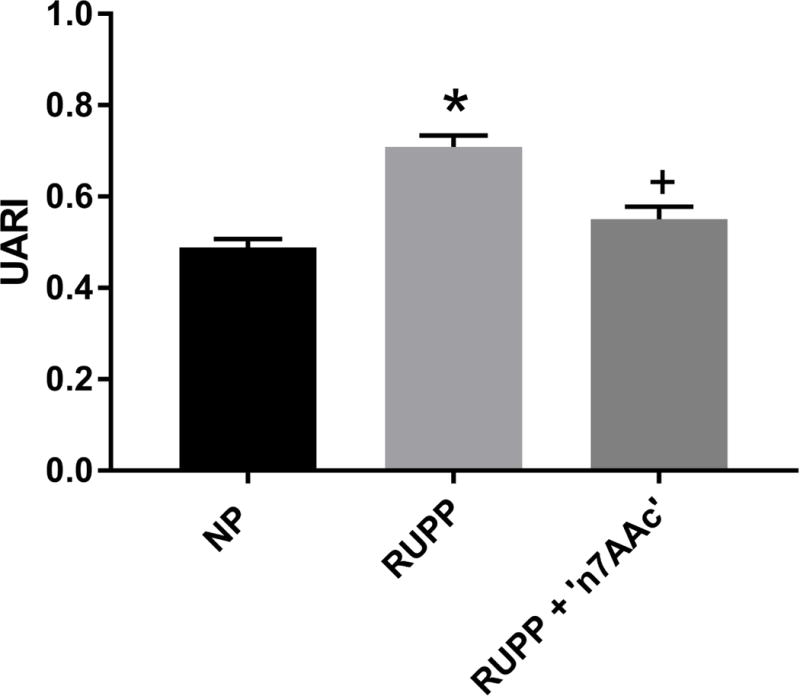
Uterine artery resistant index was increased in RUPP vs. NP rats and restored to NP levels in RUPP + ‘n7AAc’ rats. Statistical changes was determined using one-way ANOVA with Bonferroni post hoc analysis, *p<0.05 vs NP and +p<0.05 vs. RUPP.
There was a tendency for a decrease in renal function (GFR) for RUPP (n=6, 3.1± 0.2 ml/min) vs. NP rats (n=9, 3.4± 0.2 ml/min, ns) (Figure 4A). GFR was normalized to NP levels in RUPP+‘n7AAc’ (n=8, 3.4± 0.3 ns v; Figure 4A). Plasma creatinine was more than doubled in RUPP rats (n=9), (0.86± 0.32 mg/dL) compared to NP rats (n=7) 0.30± 0.02 mg/dL, p=0.14). The same trend was also observed comparing RUPP to RUPP+‘n7AAc’ rats (n=7) (0.86± 0.32 vs. 0.38± 0.05 mg/dL, p=0.21).). (Figure 4B). These data taken together suggest that RUPP decreases renal function, which can be prevented in pregnant rats by AT1-AA inhibition.
Figure 4.
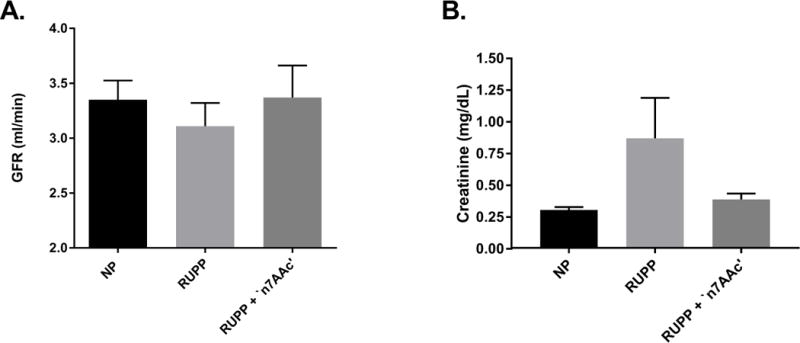
A) GFR had a tendency to decrease with RUPP vs. NP rats. B) Plasma creatinine was increased with RUPP vs. NP rats. Both were normalized in RUPP + ‘n7AAc’ to that of NP levels.
The effect of ‘n7AAc’ on circulating factors: sFlt-1, nitrate and nitrite, isoprostanes, and placental ET-1 in RUPP rats
Plasma sFlt-1, a potent anti-angiogenic factor that causes vasoconstriction and is strongly associated with PE, levels were elevated in RUPP (n=4) vs. NP (n=4) rats (176± 37 vs. 77± 15 ρg/ml, p<0.05) (Figure 5A). Importantly, the rise in sFlt-1 was prevented with ‘n7AAc’ as there was no change between NP and RUPP + ‘n7AAc’ rats (n=4) (77± 15 vs. 86± 9 ρg/ml, ns) (Figure 5A). Plasma levels of nitrate and nitrite, metabolites of nitric oxide (NO), were decreased in RUPP (n=5) rats compared to NP (n=4) rats (14.01± 1.47 vs. 19.98 ± 1.02 μM of NO3, p< 0.05) (Figure 5B). Plasma nitrate and nitrite levels were returned to NP levels in RUPP+‘n7AAc’ (n=8) (18.27 ± 1.56 μM of NO3) (Figure 5B), suggesting that NO bioavailability is maintained in RUPP rats treated with AT1-AA inhibition. There was a 7 fold increase in circulating isoprostane levels, a reactive oxygen species and marker of oxidative stress which can cause vasoconstriction, in RUPP (n=9) rats vs. NP (n=4) rats (20,117± 6,304 vs. 2,809± 1,375 pg/ml, p< 0.05) and aa 4.5 fold increase in RUPP vs. RUPP + ‘n7AAc’ rats (n=8) (4,311± 2,249 vs. 2,809± 1,375pg/ml, p< 0.05) (Figure 5C). No difference in NP and RUPP + ‘n7AAc’ rats levels, suggesting that AT1-AA inhibition normalized RUPP isoprostanes to that of NP values. (Figure 5C). RUPP (n=7) rats displayed a 4 fold increase in placental pre-pro-endothelin, precursor for endothelin 1 a potent vasoconstrictor, message vs. NP (n=6) rats (4.2± 0.20 vs. 1.0± 0.33, p< 0.05) (Figure 5D). Placental pre-pro-endothelin expression was also elevated in RUPP vs. RUPP + ‘n7AAc’ (n=4) rats (4.2± 0.20 vs. 2.3± 0.39, p< 0.05) (Figure 5D). These data suggest that AT1-AA inhibition prevents vasoconstriction and hypertension, by decreasing sFlt-1, oxidative stress, ET-1, and increasing NO bioavailability in RUPP rats.
Figure 5.
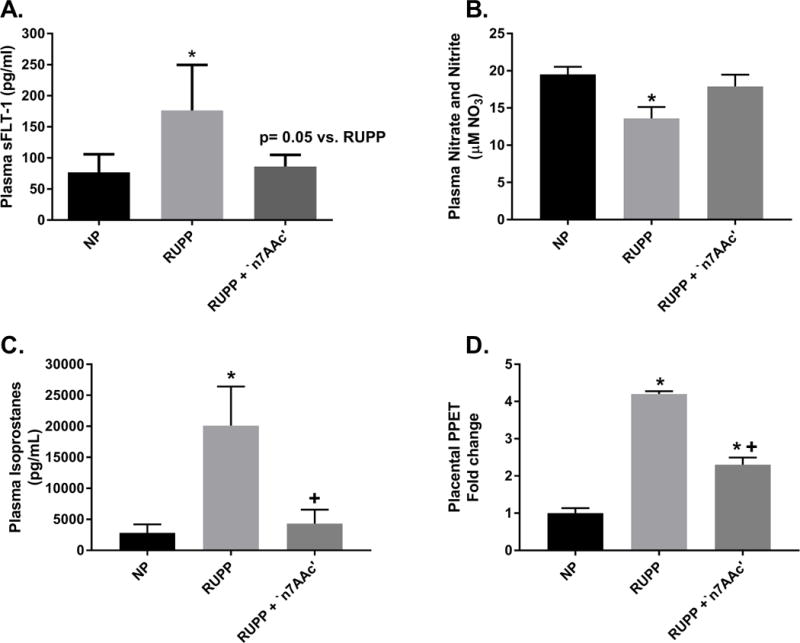
A) Plasma sFlt-1 is increased in RUPP vs. NP rats and returned to NP levels in RUPP + ‘n7AAc’ rats. B) Plasma nitrate and nitrite (metabolites of NO) was decreased in RUPP vs. NP rats and restored to NP levels in RUPP + ‘n7AAc’ rats. C) Plasma circulating isoprostanes showed a 7-fold increase in RUPP vs. NP rats. Isoprostanes significantly and greatly decreased in RUPP + ‘n7AAc’ vs. RUPP rats to NP levels. D) Placental Pre-proendothelin (precursor to endothelin 1) message expression was increased 4 fold in RUPPs vs. NP rats. Placental PPE expression was decreased, but not to NP levels, in RUPP + ‘n7AAc’ vs. RUPP rats. Statistical changes was determined using one-way ANOVA with Bonferroni post hoc analysis, *p<0.05 vs NP and +p<0.05 vs. RUPP.
The effect of n’7AAc’ on placental NK cells in RUPP rats
NK cells which are elevated in PE, were measure by flow cytometry in the placenta. Total placental NK cells in RUPP rats (n=11; 18.6 ± 6.6% gated) are elevated over NP (n=9; 7.5 ± 2.3% gated) and RUPP + ‘n7AAc’ (n=6; 6.4 ± 3.2 %gated) rats (Figure 6A). Placental cytolytic NK cells were significantly increased in RUPP (n=11) rats vs. NP (n=9) rats (8.3± 1.7 vs. 2.1± 1.6 % gated, p< 0.05) (Figure 6B) and were decreased in RUPP + ‘n7AAc’ (n=6) rats (8.3± 1.7 vs. 2.8± 0.9% gated, p< 0.05, RUPP vs. RUPP + ‘n7AAc’) (Figure 6B). Thus AT1-AA blockade prevents placental NK cell activation in RUPP rats.
Figure 6.
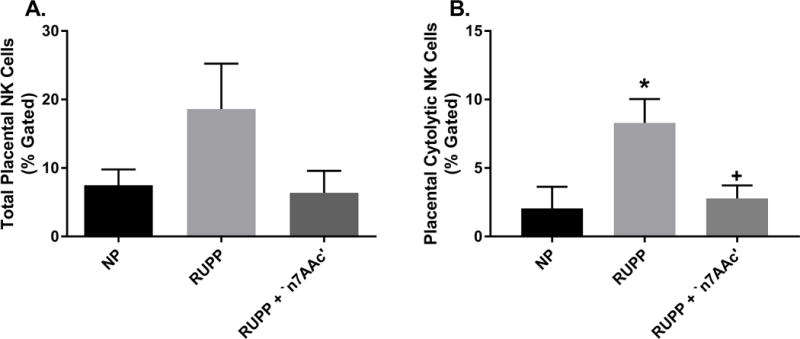
A) Total placental NK cells was elevated in RUPPs vs. NP rats and returned to NP levels in RUPP + ‘n7AAc’ (ns). B) Cytolytic NK cells was significantly increased in RUPPs vs. NP rats. RUPP + ‘n7AAc’ rats displayed a decrease in activated NK cells in comparison to RUPP rats. Activated NK cells were at similar levels in RUPP + ‘n7AAc’ and NP rats. Statistical changes was determined using one-way ANOVA with Bonferroni post hoc analysis, *p<0.05 vs NP and +p<0.05 vs. RUPP.
Conclusion
The major finding of this study is that epitope binding of the AT1-AA in placental ischemic rats prevents the pathophysiological symptoms associated with PE in women. Inhibition of AT1-AA binding to the AT1 receptor normalized the increase in blood pressure, in which we believe was achieved by suppressing ET-1, plasma sFlt-1 and isoprostanes. It also improved systemic NO bioavailability and reduced cytolytic NK cells. Moreover, renal function was improved, due to reduced oxidative stress, ET-1, sFlt-1 and improved NO bioavailability, which contributed to the improved blood pressure response to placental ischemia. This study supports findings that a major culprit responsible for the etiology of preeclampsia are AT1-AAs. Therefore this study, in conjunction with studies performed by the Xia laboratory, support the idea that inhibiting the AT1-AA from activating the AT1R could be a potential therapeutic option for PE.
AT1-AAs were first discovered in women with PE by Wallukat et al in 1999, when they isolated immunoglobulins from the serum of preeclamptic women. They showed that these antibodies had the same affinity and ability to activate the AT1 receptor as ANG II.6 In these initial studies, AT1-AA activation of the AT1 receptor on cultured neonatal rat cardiomyocytes was inhibited with the addition of clinically used AT1 receptor blockers (ARBs) to the media.6 However the use of ARBs and other clinical pharmaceuticals used to inhibit the endogenous Renin-Angiotensin-Aldosterone System (RAAS) during pregnancy are deleterious to the fetus. Therefore, new modalities to block the actions of endogenously produced AT1-AA without interfering with the RAAS could improve the clinical management of PE.
An important new finding in this study is the decrease in placental natural killer cells. We have recently shown that RUPP rats have increased cytolytic NK cells which are mediators of oxidative stress and secrete inflammatory cytokines.9 In the case of autoimmunity, cytolytic NK cells are targeted toward normal tissues thereby causing inflammation and tissue damage.34 Therefore we hypothesize that the increase in cytolytic NK cells in RUPP rats contributes to the pathology of PE. In fact we recently depleted placental NK cells in RUPP rats and found that this significantly improved blood pressure and inflammation, however this technique may not be possible for treatment of the pregnant mom.9 Thus the utilization of the method presented in this study to block AT1-AA lead to a reduction in cytolytic NK cells, could be efficacious to the treatment of PE.
In addition to studying NK cells, we previously examined the effects of an agent used clinically to suppress B lymphocytes known as Rutiximab. Administration of Rituximab to RUPP rats depleted B cell and also reduced the AT1-AA in RUPP rats.35 Although administration of Rutiximab depleted B cells, significantly reduced AT1-AA and blood pressure in RUPP rats, the effects of B cell depletion could have detrimental effects in the fetus. In fact in the drug information pamphlet accompanying Rituximab warns that B cell depleting agents cross the placental barrier and are present in the newborns of treated women and macaques while pregnant. Given the rapid maturation of the immune system that occurs in newborns B cell depletion could have serious long term negative effects on children. Therefore other therapeutic strategies are needed to combat the detrimental effects caused by the AT1-AA during pregnancy, while leaving B cell function intact. Thus although standard techniques of immune suppression could be advantageous to use during PE, designing a new therapeutic which allows for normal B cell function, normal NK cell function without interrupting the RAAS would greatly advance the treatment of this disease.
In-vivo studies in our lab and others have shown that AT1-AA infusion into rats increases the blood pressure, oxidative stress, the vasoconstrictor endothelin-1, and other circulating factors associated with preeclampsia, such as sFlt-1, sEng, and endothelial microparticles8,10,16,18,20. ET-1 and sFlt-1 increase vasoconstriction of blood vessels and decrease vasodilators, such as NO. In this study we demonstrate that the AT1-AA inhibiting peptide improved endothelial function as demonstrated by reduced UARI, ET-1 sFlt-1 and improved NO! Reduced NO is a marker of endothelial dysfunction which contributes to hypertension. Plasma nitrate and nitrite, metabolites of NO, were decreased in RUPP rats and returned to normal pregnant levels in RUPP rats treated n’7AAc’. There was a striking 7-fold increase in circulating isoprostanes in RUPP rats that returned to NP levels after AT1-AA inhibition. These data suggest the overall importance of AT1-AA blockade to restore circulating factors to a more normal pregnant milieu, thus indicating one pathway to improve maternal symptoms and thus allow PE pregnancies to continue, which could then improve fetal outcomes as well.
In RUPP + ‘n7AAc’ there was a significant decrease in circulating sFlt-1. SFlt-1 is a splice variant of the VEGF receptor Flt1 lacking the transmembrane and cytoplasmic domains. This molecule is produced by the placenta and functions as a vascular endothelial growth factor (VEGF) antagonist.36–38 VEGF is important for angiogenesis, which is the formation of new vessels, and is needed during trophoblast invasion for placental formation and endothelial cell function.37 Under physiological conditions VEGF functions through the NO pathway and helps to maintain vascular tone and normalize blood pressure. Early studies by Maynard et al, showed that both plasma levels and placental mRNA expression of sFlt-1 was elevated in in preeclamptic patients vs. normal pregnant patients. Furthermore the same study showed that severity of preeclampsia was correlated with sFlt-1 levels.37 Xia et al, also verified that severity of PE was correlated with sFlt-1 along with a strong correlation between sFlt-1 and AT1-AAs.29 Our data from this study confirms the important role that AT1-AAs play to stimulate sFlt-1 and suggests that a reduction in sFlt-1 via ‘n7AAc’ may serve as a mechanism to improve NO production, oxidative stress, endothelial function, and thus hypertension in RUPP rats.
One mechanism important for either AT1-AA or sFlt-1 induced hypertension is the upregulation of ET-1.36–39 ET-1 is a potent vasoconstrictor made primarily by endothelial cells40. ET-1 regulates salt and water homeostasis via it effects on the RAAS and vascular tone.40 ET-1 opposes the actions of potent vasodilators, such as NO, prostacyclin, prostaglandins, and natriuretic peptides.40 The overall actions of ET-1 is to increase vascular tone and cause hypertension. ET-1 elevation is prevalent and part of the pathology of many disease including preeclampsia via its activation of the ET-1 A receptor.16,40,41 ET-1 is derived from its initial product pre-proendothelin (PPE), which is cleaved into proendothlin-1, then cleaved to big endothelin, and lastly cleaved to its biological active form of endothelin.40 A number of experimental studies with animal models of preeclampsia, including the RUPP model, have elevated renal, placental, and vascular mRNA expression of pre-proendothelin.4,41 Importantly we have shown that AT1-AA induced hypertension is associated with increased production of PPET-1 in the kidney, vasculature, and placenta. AT1-AA induced hypertension can be blocked with an ET-1 A receptor antagonist; however this is not recommended for controlling blood pressure during pregnancy. This data highlights the importance of inhibiting endogenous AT1-AA mediated stimulation of ET-1 as yet another mechanism to reduce blood pressure in response to placental ischemia.
The decrease in UARI with AT1-AA inhibition treated RUPP rats, suggest a decrease in placental vasoconstriction and placental ischemia in these rats, despite the RUPP surgery. This decrease maybe due to the increase NO bioavailability and decrease in vasoactive agents, such as ROS, endothelin 1, and sFlt-1. We believe that the inhibition of AT1-AA from binding to the AT1 receptor will directly and indirectly decreases vasoconstriction. Directly, AT1-AAs are known to increase vascular tone when placed on renal afferent arterioles and to increase renal vascular resistance when administered in vivo.16,23 Furthermore blockade of the AT1 receptor activation, via administration of an angiotensin II converting enzyme inhibitor and/or angiotensin II receptor blocker, blunted the increase in AT1-AA vasoconstriction and increased vascular resistance.16 Indirectly, the decrease in vascular resistance in the uterine arterioles maybe facilitated by the mechanisms mentioned above. We have shown in earlier studies that tempol (an antioxidant) treated rats with AT1-AA infusion improved both blood pressure and the renal artery resistance index (RARI), while the endothelin 1 receptor blocker improved the blood pressure and the UARI in RUPP rats.4,42 Thus suggesting that reducing oxidative stress and ET-1 are mechanism of improved renal and uterine endothelial function.
Although there were no changes or improvements with pup weight, placental weight, or litter size in RUPP rats treated with AT1-AA inhibition, it is important to note that we did not see a further decrease in pup weight, placental weight, or litter size with treatment. This suggest that the AT1-AA inhibition is not toxic to the pups. Moreover, it is important to remember that the invasive RUPP procedure, is induced by manually placing surgical clips on blood vessels to reduce flow and nutrients to the growing fetus. Thus it’s possible that without mechanical restraint of blood vessels, AT1-AA inhibition might be able to improve fetal outcomes in addition to those observed in the mother.
Perspectives
In summary AT1-AA inhibition prevents maternal blood pressure, secretion of anti-angiogenic and vasoconstrictive factors, renal function, systemic NO bioavailability, and oxidative stress in response to placental ischemia. Improvement in these factors contributed to improved blood pressure and renal function in the moms. Moreover, we hypothesize that AT1-AA inhibition during pregnancy could lead to short term improvements for mom and the baby during pregnancy which could result in long term improvements for both individual’s health risks of developing cardiovascular-renal disease and stroke later in life. Due to complications caused by angiotensin-converting-enzyme inhibitors, angiotensin II receptor blockers, or B cell depletion during pregnancy, specific AT1-AA inhibition may be an option to consider for better management of PE.
Novelty and Significance.
What is New?
This study tested the efficacy of administering an improved targeted peptide against the AT1-AA in placental ischemic rats during pregnancy.
The modified peptide improved hypertension and did not further cause any harm to the fetus.
What is Relevant?
AT1-AA inhibition improve many factors associated with the pathology of PE such as ROS, antiangiogenic factors, endothelin, and NO bioavailability.
This study emphasizes the importance of drug discovery for AT1-AA inhibition to improve pregnancy outcomes in hypertensive patients with PE.
Summary
Understanding the role of AT1-AAs in pathophysiology of preeclampsia is important as we search for new therapies for preeclamptic pregnancies.
Acknowledgments
Special Thanks to Dr. Ed Cable and Ferring Pharmaceuticals for assistance with developing the AT1-AA inhibition peptide. Thanks to Dr. Frank T. Spradley in the department of Surgery at University of Mississippi Medical for assistance in initial renal clearance experiments.
Funding Sources
This work was supported by NIH grants HL78147 and HL51971 and HD067541 awarded to BL. Funding supported by P20GM104357 and DK109133-01 awarded to J.M.W. DC is supported by NIH grant HL130456 and the American Heart Association 16SDG27520000.
Footnotes
Conflict of Interest/Disclosure Statement
There are no disclosures or conflicts of interest.
References
- 1.LaMarca B, Amaral LM, Harmon AC, Cornelius DC, Faulkner JL, Cunningham MW., Jr Placental Ischemia and Resultant Phenotype in Animal Models of Preeclampsia. Current hypertension reports. 2016 Apr;18(5):38. doi: 10.1007/s11906-016-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LaMarca B, Cornelius DC, Harmon AC, et al. Identifying immune mechanisms mediating the hypertension during preeclampsia. American journal of physiology Regulatory, integrative and comparative physiology. 2016 Jul 01;311(1):R1–9. doi: 10.1152/ajpregu.00052.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amaral LM, Cunningham MW, Jr, Cornelius DC, LaMarca B. Preeclampsia: long-term consequences for vascular health. Vascular health and risk management. 2015;11:403–415. doi: 10.2147/VHRM.S64798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MC, Best KE, Pearce MS, Waugh J, Robson SC, Bell R. Cardiovascular disease risk in women with pre-eclampsia: systematic review and meta-analysis. European journal of epidemiology. 2013 Jan;28(1):1–19. doi: 10.1007/s10654-013-9762-6. [DOI] [PubMed] [Google Scholar]
- 5.Shah DA, Khalil RA. Bioactive factors in uteroplacental and systemic circulation link placental ischemia to generalized vascular dysfunction in hypertensive pregnancy and preeclampsia. Biochemical pharmacology. 2015 Jun 15;95(4):211–226. doi: 10.1016/j.bcp.2015.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallukat G, Homuth V, Fischer T, et al. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. The Journal of clinical investigation. 1999 Apr;103(7):945–952. doi: 10.1172/JCI4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herse F, LaMarca B. Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. American journal of reproductive immunology. 2013 Apr;69(4):413–418. doi: 10.1111/aji.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li J, LaMarca B, Reckelhoff JF. A model of preeclampsia in rats: the reduced uterine perfusion pressure (RUPP) model. American journal of physiology Heart and circulatory physiology. 2012 Jul;303(1):H1–8. doi: 10.1152/ajpheart.00117.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elfarra J, Amaral LM, McCalmon M, et al. Natural Killer Cells Mediate Pathophysiology in Response to Reduced Uterine Perfusion Pressure. Clinical science. 2017 Oct;131(23):2753–2762. doi: 10.1042/CS20171118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parrish MR, Wallace K, Tam Tam KB, et al. Hypertension in response to AT1-AA: role of reactive oxygen species in pregnancy-induced hypertension. American journal of hypertension. 2011 Jul;24(7):835–840. doi: 10.1038/ajh.2011.62. [DOI] [PubMed] [Google Scholar]
- 11.Chai W, Zhang W, Jin Z, Feng Y, Kuang Y, Zhi J. Angiotensin II type I receptor agonistic autoantibody-induced apoptosis in neonatal rat cardiomyocytes is dependent on the generation of tumor necrosis factor-alpha. Acta biochimica et biophysica Sinica. 2012 Dec;44(12):984–990. doi: 10.1093/abbs/gms087. [DOI] [PubMed] [Google Scholar]
- 12.Jin Z, Wang J, Zhang W, Zhang G, Jiao X, Zhi J. Changes in cardiac structure and function in rats immunized by angiotensin type 1 receptor peptides. Acta biochimica et biophysica Sinica. 2011 Dec;43(12):970–976. doi: 10.1093/abbs/gmr096. [DOI] [PubMed] [Google Scholar]
- 13.Yang S, Zhong Q, Qiu Z, et al. Angiotensin II receptor type 1 autoantibodies promote endothelial microparticles formation through activating p38 MAPK pathway. Journal of hypertension. 2014 Apr;32(4):762–770. doi: 10.1097/HJH.0000000000000083. [DOI] [PubMed] [Google Scholar]
- 14.Thway TM, Shlykov SG, Day MC, et al. Antibodies from preeclamptic patients stimulate increased intracellular Ca2+ mobilization through angiotensin receptor activation. Circulation. 2004 Sep 21;110(12):1612–1619. doi: 10.1161/01.CIR.0000142855.68398.3A. [DOI] [PubMed] [Google Scholar]
- 15.Dechend R, Homuth V, Wallukat G, et al. AT(1) receptor agonistic antibodies from preeclamptic patients cause vascular cells to express tissue factor. Circulation. 2000 May 23;101(20):2382–2387. doi: 10.1161/01.cir.101.20.2382. [DOI] [PubMed] [Google Scholar]
- 16.Brewer J, Liu R, Lu Y, et al. Endothelin-1, oxidative stress, and endogenous angiotensin II: mechanisms of angiotensin II type I receptor autoantibody-enhanced renal and blood pressure response during pregnancy. Hypertension. 2013 Nov;62(5):886–892. doi: 10.1161/HYPERTENSIONAHA.113.01648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhillion P, Wallace K, Herse F, et al. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. American journal of physiology Regulatory, integrative and comparative physiology. 2012 Aug 15;303(4):R353–358. doi: 10.1152/ajpregu.00051.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LaMarca B, Parrish M, Ray LF, et al. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension. 2009 Oct;54(4):905–909. doi: 10.1161/HYPERTENSIONAHA.109.137935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenzel K, Rajakumar A, Haase H, et al. Angiotensin II type 1 receptor antibodies and increased angiotensin II sensitivity in pregnant rats. Hypertension. 2011 Jul;58(1):77–84. doi: 10.1161/HYPERTENSIONAHA.111.171348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parrish MR, Murphy SR, Rutland S, et al. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. American journal of hypertension. 2010 Aug;23(8):911–916. doi: 10.1038/ajh.2010.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Novotny SR, Wallace K, Heath J, et al. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. American journal of physiology Regulatory, integrative and comparative physiology. 2012 May 15;302(10):R1197–1201. doi: 10.1152/ajpregu.00623.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou CC, Zhang Y, Irani RA, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nature medicine. 2008 Aug;14(8):855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cunningham MW, Jr, Williams JM, Amaral L, et al. Agonistic Autoantibodies to the Angiotensin II Type 1 Receptor Enhance Angiotensin II-Induced Renal Vascular Sensitivity and Reduce Renal Function During Pregnancy. Hypertension. 2016 Nov;68(5):1308–1313. doi: 10.1161/HYPERTENSIONAHA.116.07971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Warrington J, F F, LaMarca BB, Dechend R, Wallukat G, Roman R, Drummond H, Granger J, Ryan M. Agonistic Autoantibodies to Angiotensin II Type I Receptor Contributes Partly to Placental Ischemia-induced Cerebrovascular Abnormalities. AHA Conference: HBPR. 2015:127. (Abstract) [Google Scholar]
- 25.Siddiqui AH, Irani RA, Blackwell SC, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibody is highly prevalent in preeclampsia: correlation with disease severity. Hypertension. 2010 Feb;55(2):386–393. doi: 10.1161/HYPERTENSIONAHA.109.140061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walther T, Wallukat G, Jank A, et al. Angiotensin II type 1 receptor agonistic antibodies reflect fundamental alterations in the uteroplacental vasculature. Hypertension. 2005 Dec;46(6):1275–1279. doi: 10.1161/01.HYP.0000190040.66563.04. [DOI] [PubMed] [Google Scholar]
- 27.Zhou CC, Ahmad S, Mi T, et al. Autoantibody from women with preeclampsia induces soluble Fms-like tyrosine kinase-1 production via angiotensin type 1 receptor and calcineurin/nuclear factor of activated T-cells signaling. Hypertension. 2008 Apr;51(4):1010–1019. doi: 10.1161/HYPERTENSIONAHA.107.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia Y, Wen H, Bobst S, Day MC, Kellems RE. Maternal autoantibodies from preeclamptic patients activate angiotensin receptors on human trophoblast cells. Journal of the Society for Gynecologic Investigation. 2003 Feb;10(2):82–93. doi: 10.1016/s1071-5576(02)00259-9. [DOI] [PubMed] [Google Scholar]
- 29.Xia Y, Kellems RE. Angiotensin receptor agonistic autoantibodies and hypertension: preeclampsia and beyond. Circulation research. 2013 Jun 21;113(1):78–87. doi: 10.1161/CIRCRESAHA.113.300752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu F, Bytautiene E, Tamayo E, et al. Gender-specific effect of overexpression of sFlt-1 in pregnant mice on fetal programming of blood pressure in the offspring later in life. American journal of obstetrics and gynecology. 2007 Oct;197(4):418 e411–415. doi: 10.1016/j.ajog.2007.06.064. [DOI] [PubMed] [Google Scholar]
- 31.Schock-Kusch D, Sadick M, Henninger N, et al. Transcutaneous measurement of glomerular filtration rate using FITC-sinistrin in rats. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2009 Oct;24(10):2997–3001. doi: 10.1093/ndt/gfp225. [DOI] [PubMed] [Google Scholar]
- 32.Santiago-Font JA, Amaral LM, Faulkner J, et al. Serelaxin improves the pathophysiology of placental ischemia in the reduced uterine perfusion pressure rat model of preeclampsia. American journal of physiology Regulatory, integrative and comparative physiology. 2016 Dec 01;311(6):R1158–R1163. doi: 10.1152/ajpregu.00192.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giezeman-Smits KM, Jonges LE, Chambers WH, et al. Novel monoclonal antibodies against membrane structures that are preferentially expressed on IL-2-activated rat NK cells. J Leukoc Biol. 1998 Feb;63(2):209–215. doi: 10.1002/jlb.63.2.209. [DOI] [PubMed] [Google Scholar]
- 34.Small HY, Cornelius DC, Guzik TJ, Delles C. Natural killer cells in placentation and cancer: Implications for hypertension during pregnancy. Placenta. 2017 Aug;56:59–64. doi: 10.1016/j.placenta.2017.03.003. [DOI] [PubMed] [Google Scholar]
- 35.LaMarca B, Wallace K, Herse F, et al. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension. 2011 Apr;57(4):865–871. doi: 10.1161/HYPERTENSIONAHA.110.167569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamarca B. Endothelial dysfunction. An important mediator in the pathophysiology of hypertension during pre-eclampsia. Minerva ginecologica. 2012 Aug;64(4):309–320. [PMC free article] [PubMed] [Google Scholar]
- 37.Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. The Journal of clinical investigation. 2003 Mar;111(5):649–658. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy SR, LaMarca B, Cockrell K, Arany M, Granger JP. L-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. American journal of physiology Regulatory, integrative and comparative physiology. 2012 Jan 15;302(2):R259–263. doi: 10.1152/ajpregu.00319.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murphy SR, LaMarca BB, Cockrell K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension. 2010 Feb;55(2):394–398. doi: 10.1161/HYPERTENSIONAHA.109.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Agapitov AV, Haynes WG. Role of endothelin in cardiovascular disease. Journal of the renin-angiotensin-aldosterone system : JRAAS. 2002 Mar;3(1):1–15. doi: 10.3317/jraas.2002.001. [DOI] [PubMed] [Google Scholar]
- 41.Bakrania B, Duncan J, Warrington JP, Granger JP. The Endothelin Type A Receptor as a Potential Therapeutic Target in Preeclampsia. International journal of molecular sciences. 2017 Feb 28;18(3) doi: 10.3390/ijms18030522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tam Tam KB, George E, Cockrell K, et al. Endothelin type A receptor antagonist attenuates placental ischemia-induced hypertension and uterine vascular resistance. American journal of obstetrics and gynecology. 2011 Apr;204(4):330 e331–334. doi: 10.1016/j.ajog.2011.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]