

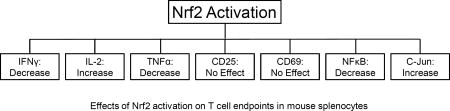
Abstract
We previously demonstrated that activation of the transcription factor, nuclear factor erythroid 2-related factor 2 (Nrf2) promotes CD4+ Th2 differentiation. In the current study, we assessed the role of Nrf2 in early events following T cell activation. The Nrf2 activators, tBHQ (tert-butylhydroquinone) and CDDO-Im (the imidazolide derivative of the triterpenoid CDDO), were used in conjunction with splenocytes derived from wild-type and Nrf2-null mice to distinguish between Nrf2-specific and off-target effects. CDDO-Im inhibited early IFNγ and TNFα production in a largely Nrf2-dependent manner. In contrast, tBHQ and CDDO-Im had little effect on expression of CD25 or CD69. Furthermore, tBHQ inhibited GM-CSF and IL-2 production in both wild-type and Nrf2-null T cells, suggesting this effect is Nrf2-independent. Conversely, CDDO-Im caused a concentration-dependent increase in IL-2 secretion in wild-type, but not Nrf2-null, splenocytes, suggesting that Nrf2 promotes IL-2 production. Interestingly, both compounds inhibit NFκB DNA binding, where the suppression by tBHQ is Nrf2-independent and CDDO-Im is Nrf2-dependent. Surprisingly, as compared to wild-type splenocytes, Nrf2-null splenocytes showed lower nuclear accumulation of c-Jun, a member of the AP-1 family of transcription factors, which have been shown to drive multiple immune genes, including IL-2. Both Nrf2 activators caused a Nrf2-dependent trend towards increased nuclear accumulation of c-Jun. These data suggest that modulation of cytokine secretion by tBHQ likely involves multiple pathways, including AP-1, NFκB, and Nrf2. Overall, the data suggest that Nrf2 activation inhibits secretion of the Th1 cytokine IFNγ, and increases early production of IL-2, which has been shown to promote Th2 differentiation, and may support the later occurrence of Th2 polarization.
Keywords: Nrf2, IL-2, IFNγ, T cell, CDDO-Im, tBHQ, NFκB
Graphical abstract
1 INTRODUCTION
Nrf2 (Nuclear factor erythroid 2-related factor 2) is a transcription factor that is activated by various types of cellular stress, including oxidative stress and electrophilic insult [1,2]. In the absence of cell stress, Nrf2 is bound to its repressor protein, Keap1 (Kelch-like ECH-associated protein 1), in the cytosol and is subsequently ubiquitinated and degraded by the proteasome [3,4]. Upon introduction of cell stress, Nrf2 is no longer degraded and translocates to the nucleus where it induces transcription of its target genes [5]. Nrf2 regulates a battery of target genes that perform a variety of functions, including detoxification and metabolism of reactive compounds, transport of xenobiotics, antioxidant functions, and upregulation of the proteasomal subunits [6,7].
Nrf2 is activated by numerous compounds, including heavy metals, phenols, reactive oxygen species, and many others [7]. In addition, there are numerous activators that are commonly used experimentally to activate Nrf2, which include 1[2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole (CDDO-Im), tert-butylhydroquinone (tBHQ), butylated hydroxyanisole and sulforaphane. CDDO-Im is one of the most potent known activators of Nrf2, active at nanomolar concentrations, but it also activates other pathways at micromolar concentrations [8,9]. Although both compounds activate Nrf2 by modification of key cysteine residues on Keap1, there are distinct potency differences, as 0.1–1 µM tBHQ was used vs. 0.001–0.1 µM CDDO-Im in our experiments. The regulation of the Nrf2 pathway by CDDO-Im is also dependent on cell type, as is likely true for tBHQ as well [8]. At the concentrations of CDDO-Im used in the current study (nM range), no decrease in viability was observed. In addition to CDDO-Im, tBHQ was also used in these studies as we have determined previously that it is a potent activator of Nrf2 in T cells and does not cause cytotoxicity at the concentrations used in our studies [10]. Interestingly, tBHQ is also a commonly used food preservative found in a variety of foods, including cereals, crackers, and fast food items, among others [11].
In addition to its functions in detoxification and the antioxidant response, Nrf2 also has anti-inflammatory activity, which has been reported in numerous inflammatory models. For instance, Nrf2 appears to play a protective role in experimental autoimmune encephalitis, experimental sepsis from cecal ligation and puncture or lipopolysaccharide administration [12], traumatic brain injury [13], endotoxemia-induced vascular inflammation [14] and experimental autoimmune hepatitis [15]. In addition, Nrf2 seems to be particularly important in lung inflammation. Nrf2-null mice have increased susceptibility to ovalbumin-induced asthma [16], hyperoxia-induced acute lung injury [17], pulmonary fibrosis [18], allergic airway inflammation [19] and Staphylococcus aureus pneumonia [20]. It is also notable that aged female Nrf2-null mice develop an autoimmune disease that resembles systemic lupus erythematosus in humans, which suggests an endogenous role for Nrf2 in immune regulation [21,22,23].
T cell activation occurs upon ligation of the T cell receptor and a costimulatory receptor by their respective agonists. Experimentally, T cells can be activated by treatment with monoclonal antibodies directed against the T cell receptor (CD3) and a costimulatory receptor, such as CD28. Activation of T cells by anti-CD3/anti-CD28 triggers a signaling cascade that ultimately results in a number of cellular events, such as activation of NFκB and AP-1, as well as other transcription factors, and induction of early cytokines, such as IL-2,GM-CSF, TNFα, and IFNγ [24]. In addition, a number of cell surface proteins are also induced, including CD25, which is the high-affinity IL-2 receptor, and CD69, which is a C-type lectin receptor. Although the exact function of CD69 is not fully known, both CD25 and CD69 are highly expressed after T cell stimulation and serve as markers of activation.
Our previous studies have demonstrated that activation of Nrf2 skews CD4+ T cells toward Th2 differentiation, whereas absence of Nrf2 skews CD4+ T cells toward Th1 differentiation [10]. However, little is known about the role of Nrf2 in the early events following T cell activation, which can influence polarization. Whereas our previous studies necessarily focused on events occurring 4 to 6 days after T cell activation (which is the time needed for CD4+ T cell differentiation), the purpose of the present studies is to investigate a potential role for Nrf2 in T cell activation within 24 h after CD3/CD28 ligation. It has been previously reported that the early events, such as cytokine release, post T cell stimulation are integral in determination of CD4+ T cell effector function and polarization.
2 MATERIALS AND METHODS
Materials
CDDO-Im was synthesized as previously described at >95% purity [25,26]. tBHQ, and all other reagents were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise noted.
Nrf2-null mice
Nrf2-null mice on a mixed C57BL/6 and AKR background were generated as described previously and received from Dr. Jefferson Chan [1]. The mice were subsequently back-crossed 8 generations onto the C57BL/6 background and are 99% congenic (analysis performed by Jackson Laboratories, Bar Harbor, ME). Female mice were used for the current studies. Age-matched wild-type female C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Mice were given food and water ad libitum. All animal studies were conducted in accordance with the Guide for the Care and Use of Animals as adopted by the National Institutes of Health, and were approved by the Institutional Animal Care and Use Committee (IACUC) at Michigan State University.
Cell culture
Splenocyte isolation: Single-cell suspensions from spleens were washed, filtered, counted and cell density adjusted to 5 × 106 c/ml, unless otherwise noted. Cells were cultured in DMEM (with L-glutamine, sodium bicarbonate and D-glucose) supplemented with 100 units penicillin/ml, 100 units streptomycin/ml, 50 µM 2-mercaptoethanol, and 10% fetal bovine serum (FBS). For most studies, cells were treated with vehicle, tBHQ, or CDDO-Im at the indicated concentrations for 30 min prior to T cell activation. 24 h post T cell receptor/coreceptor ligation, supernatants and cells were harvested and analyzed as noted in the figure legends. T cells were activated with purified hamster anti-mouse CD3ε (500A2, 1.5 µg/ml), purified hamster anti-mouse CD28 (37.51, 1.5 µg/ml), and an F(ab’)2 fragment specific for anti-Syrian hamster IgG that was used to cross-link CD3 and CD28 to enhance activation. Anti-CD3 and anti-CD28 were purchased from E-Biosciences (San Diego, CA), and the F(ab’)2 cross-linker was purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
Cytokine protein quantification: ELISA
IFNγ, IL-2, GM-CSF, and TNFα protein was quantified by sandwich ELISAs using commercially-available kits following the manufacturer’s protocol (For GM-CSF, TNFα, and IFNγ: Biolegend, San Diego, CA; for IL-2: E-Bioscience, San Diego, CA).
Measurement of CD25 and CD69 expression by flow cytometry
Freshly-isolated splenocytes were washed and resuspended in FACS buffer (PBS, 1% FBS). The cells were then incubated with anti-CD4/FITC, anti-CD25/APC, and/or anti-CD69/PE-Cy7 for 30 min at 4° C, after which the cells were washed and resuspended in FACS buffer. The fluorescence was then detected and quantified with a BD Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). The data were analyzed using CFlow software (BD Accuri, San Jose, CA). The CD4 antibody was purchased from E-Bioscience (San Diego, CA) and the CD25 and CD69 antibodies were purchased from Biolegend, (San Diego, CA).
ELISA-based DNA binding assay
Four hours after activation by anti-CD3/anti-CD28, nuclear protein was extracted from 1×107 wild-type and Nrf2-null splenocytes, using a commercially available kit (Active Motif, Carlsbad, CA). After extraction, nuclear protein was quantified via Bradford assay (Bio-Rad, Hercules, CA). Five micrograms of nuclear protein was used to quantify NFκB DNA binding using a commercially available ELISA-based DNA binding assay (Active Motif). Assays were performed per the manufacturer’s protocol.
Nuclear protein isolation
Wild-type and Nrf2-null mouse splenocytes were collected and resuspended at 5×106 c/mL. 3 h after activation by anti-CD3/anti-CD28, cells were lysed using a solution containing 10mM HEPES, 100mM KCl, 1.5mM MgCl2, 0.1 mM EGTA, 0.5mM DTT, 0.5% NP-40 substitute, and 1x Halt protease inhibitor cocktail (Thermo Scientific, Waltham, MA). Cell lysates were then pelleted by centrifugation. The resulting nuclear pellets were then resuspended in nuclear extraction buffer containing 10mM HEPES, 420mM NaCl, 1.5 mM MgCl2, 0.1mM EGTA, 0.5mM DTT, 5% glycerol, and 1x Halt protease inhibitor cocktail. Samples were incubated on ice for 1 h, vortexing every 10 min. After 1 h samples were spun down and nuclear protein was quantified. Nuclear protein was quantified via Bradford assay (Bio-Rad, Hercules, CA).
Western analysis
After protein quantification, samples were diluted in Laemmli Sample Buffer (Bio-Rad) containing 2.5% 2-mercaptoethanol. Samples were subjected to SDS-PAGE and subsequently transferred to a PVDF membrane using 10µg of each sample. Membranes were blocked with 5% BSA (Bovine Serum Albumin) or NFDM (nonfat dry milk) in PBS containing 0.05% Tween 20 (PBST). Histone H3 (7074S) and c-Jun (Cell Signaling) primary antibodies were diluted 1:1000 in PBST containing 5% NFDM and 5% BSA, respectively. The H3 primary antibody was purchased from Cell Signaling (Danvers, MA). c-Jun antibodies were purchased from Santa-Cruz Biotechnology (Santa Cruz, CA) and Cell signaling. Anti-rabbit IgG HRP-linked secondary antibody and was diluted 1:2000 in 0.05% PBST containing 2% of the blocking protein and was purchased form Cell Signaling (Danvers, MA). All blots were developed via Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL) per the manufacturer’s protocol. The bands were visualized by the LI-COR Odyssey FC infrared imaging system (Lincoln, NE).
Statistical Analysis
The mean ± standard error was determined for each treatment group in the individual experiments. Homogeneous data were evaluated by two-way parametric analysis of variance. When significant differences were observed, the Holm-Sidak post-hoc test was used to compare treatment groups to the vehicle (VEH) control using SigmaPlot 12.3 software (Systat Software, Inc., Chicago, IL).
3 RESULTS
3.1 Activation of Nrf2 by CDDO-Im inhibits early IFNγ production by activated T cells
We previously demonstrated that activation of Nrf2 by tBHQ skews CD4+ T cells toward Th2 differentiation, whereas absence of Nrf2 results in Th1-skewing [10]. CD4+ T cell differentiation is a process that generally occurs several days after T cell activation and little is known about the role of Nrf2 on early T cell activation. Thus, the purpose of the present studies was to determine the role of Nrf2 in early events following T cell activation. The current studies demonstrate that CDDO-Im markedly inhibits IFNγ secretion in anti-CD3/anti-CD28-activated T cells from wild-type mice (Fig. 1), similar to the effects of tBHQ on IFNγ reported in our previous studies [10]. Also similar to previous observations, activated T cells derived from Nrf2-null mice produced significantly more IFNγ than those derived from wild-type mice at early time-points. The highest concentration of CDDO-Im caused a modest, though statistically significant, inhibition of IFNγ secretion. Collectively, the current studies suggest that activation of Nrf2 inhibits early IFNγ production following T cell stimulation.
Fig. 1.

The Nrf2 activator, CDDO-Im, inhibits IFNγ secretion in activated mouse T cells. Splenocytes from Nrf2-null and wild-type mice were isolated from whole spleens and were either left untreated (BKG) or pretreated with either vehicle (VEH, 0.01% DMSO) or CDDO-Im for 30 min prior to activation with the T cell activator, anti-CD3/anti-CD28. After 24 h, the cell supernatants were collected and IFNγ production was quantified by sandwich ELISA. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.2 The synthetic food preservative tBHQ inhibits IL-2 secretion independently of Nrf2 while the triterpenoid CDDO-Im promotes IL-2 secretion in a Nrf2-dependent manner
Because activation of Nrf2 by tBHQ and CDDO-Im inhibited early IFNγ production by activated T cells, we investigated the effects of both Nrf2 activators on IL-2, which is also produced at early time-points after T cell activation. Similar to IFNγ, tBHQ significantly inhibited IL-2 secretion by wild-type anti-CD3/anti-CD28-activated T cells (Fig. 2a). The decrease in IL-2 production by tBHQ was also observed in Nrf2-null T cells however, suggesting that the effect is independent of Nrf2. In contrast, wild-type, but not Nrf2-null, T cells treated with CDDO-Im showed a concentration-dependent increase in IL-2 secretion at 24 h (Fig. 2b). Overall, the data demonstrate that activation of Nrf2 promotes IL-2 production.
Fig. 2.

The Nrf2 activators, CDDO-Im and tBHQ have differential effects on IL-2 secretion. Freshly isolated splenocytes from Nrf2-null and wild-type mice were either left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% ethanol for tBHQ or 0.01% DMSO for CDDO-Im), tBHQ, or CDDO-Im for 30 min prior to activation with anti-CD3/anti-CD28. After 24 h, the cell supernatants from A.) tBHQ treated splenocytes or B.) CDDO-Im treated splenocytes was collected and IL-2 was quantified by sandwich ELISA a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.3 tBHQ inhibits and CDDO-Im promotes GM-CSF secretion in a Nrf2-independent manner
Although not known for driving the polarization of CD4 T cells toward a Th2 phenotype, the early cytokine GM-CSF has been shown to be highly proinflammatory. Therefore, we investigated the effect of the Nrf2-activators CDDO-Im and tBHQ in the regulation of GM-CSF, as Nrf2 has been shown to be important in regulating inflammation. Treatment of wild-type and Nrf2-null splenocytes with tBHQ resulted in a Nrf2-independent suppression of GM-CSF, as both genotypes appear to be equally suppressed (Fig. 3a). Conversely, treatment of splenocytes with the synthetic triterpenoid CDDO-Im, resulted in a modest Nrf2-independent increase in the secretion of GM-CSF (Fig. 3b). These data further demonstrate the differential effects of tBHQ and CDDO-Im on immune endpoints.
Fig. 3.

The Nrf2 activators, CDDO-Im and tBHQ have differential effects on GM-CSF secretion. Freshly isolated splenocytes from Nrf2-null and wild-type mice were either left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% ethanol for tBHQ or 0.01% DMSO for CDDO-Im), tBHQ, or CDDO-Im for 30 min prior to activation with anti-CD3/anti-CD28. After 24 h, the cell supernatants from A.) tBHQ-treated splenocytes or B.) CDDO-Im treated splenocytes were collected and GM-CSF was quantified by sandwich ELISA. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.4 CDDO-Im induces TNFα secretion independently of Nrf2, whereas tBHQ inhibits TNFα secretion in a Nrf2-dependent manner
To further characterize the role of Nrf2 in early cytokine production, we assessed the effects of tBHQ and CDDO-Im on TNFα induction. Similar to the effects upon GM-CSF, tBHQ and CDDO-Im had differential effects on TNFα production. Treatment of wild-type and Nrf2-null splenocytes with CDDO-Im resulted in a modest Nrf2-independent induction of TNFα secretion (Fig. 4b). In contrast, treatment of splenocytes with tBHQ resulted in a Nrf2-dependent suppression of TNFα secretion (Fig. 4a). Furthermore, wild-type splenocytes produced significantly more TNFα than Nrf2-null cells, which is consistent with what has been previously reported [27]. Overall, these studies demonstrate that Nrf2 inhibits TNFα secretion.
Fig. 4.

The Nrf2 activators, CDDO-Im and tBHQ have differential effects on TNFα secretion. Freshly isolated splenocytes from Nrf2-null and wild-type mice were either left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% ethanol for tBHQ or 0.01% DMSO for CDDO-Im), tBHQ, or CDDO-Im for 30 min prior to activation with anti-CD3/anti-CD28. After 24 h, the cell supernatants from A.) tBHQ-treated splenocytes or B.) CDDO-Im treated splenocytes were collected and TNFα was quantified by sandwich ELISA. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.5 The Nrf2 activators tBHQ and CDDO-Im have little effect on the induction of CD25 and CD69 in activated mouse splenocytes
The differential effects of Nrf2 on early cytokine production by activated T cells led us to investigate the role of Nrf2 in other early events associated with T cell activation. In addition to increased cytokine production, T cells rapidly upregulate expression of the cell surface proteins CD25 and CD69 upon activation. In contrast to cytokine production, tBHQ and CDDO-Im had no effect on expression of CD25 or CD69 (Figs. 5–6). Collectively, the data demonstrate that Nrf2 plays a negligible role in the regulation of expression of the early T cell activation markers CD25 and CD69.
Fig. 5.

The Nrf2 activator tBHQ has little effect on CD25/CD69 expression in activated T cells. Freshly isolated splenocytes from wild-type or Nrf2-null mice were left untreated (BKG) or pretreated with either vehicle (VEH, 0.01% ethanol) or tBHQ (0.1–1µM) for 30 min prior to activation with anti-CD3/anti-CD28. A.) 24 h after activation, cells were collected, labeled with anti-CD4/FITC, anti-CD69/PECy7 and anti-CD25/APC and analyzed by flow cytometry. CD25/CD69 fluorescence was quantified in the gated CD4+ T cell population. Six representative dot plots are shown. B.) Graphical representation of CD25 expression. C.) Graphical representation of CD69 expression. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
Fig. 6.

The Nrf2 activator CDDO-Im has little effect on CD25/CD69 expression in activated T cells. Freshly isolated splenocytes from wild-type or Nrf2-null mice were left untreated (BKG) or pretreated with either vehicle (VEH, 0.01% DMSO) or CDDO-Im (0.001–0.1 µM) for 30 min prior to activation with anti-CD3/anti-CD28. A.) 24 h after activation, cells were collected, labeled with anti-CD4/FITC, anti-CD69/PECy7 and anti-CD25/APC and analyzed by flow cytometry. CD25/CD69 fluorescence was quantified in the gated CD4+ T cell population. Six representative dot plots are shown. B.) Graphical representation of CD25 expression by flow cytometry. C.) Graphical representation of CD69 expression by flow cytometry. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.6 Inhibition of NFκB DNA binding by CDDO-Im and tBHQ is Nrf2-dependent and -independent, respectively
Previous studies have shown cross-talk between the Nrf2 and the NFκB signaling pathways, whereby activation of NFκB is inhibited by Nrf2 through suppression of its activating kinase, inhibitor of kappa B kinase. In Jurkat T cells, we have previously shown that the Nrf2 activator tBHQ suppresses NFκB luciferase activity. Accordingly, we assessed the effects of Nrf2 activation by tBHQ and CDDO-Im in the DNA binding activity of p65, a major component of the NFκB signaling complex. tBHQ inhibited NFκB p65 DNA binding activity in both wild-type and Nrf2-null primary splenocytes, suggesting the effect is Nrf2-independent (Fig. 7a). The Nrf2-independent suppression of NFκB by tBHQ was consistent with its effect on IL-2 and GM-CSF secretion. In contrast, CDDO-Im inhibited p65 DNA binding in wild-type, but not Nrf2-null, splenocytes, suggesting that the effect of CDDO-Im is Nrf2-dependent. Taken together, these data suggest that although both compounds are Nrf2 activators, they are likely exerting their effects by different mechanisms (Fig 7b).
Fig 7.

Suppression of p65 DNA binding by tBHQ is Nrf2-independent whereas suppression by CDDO-Im is Nrf2-dependent. Freshly isolated splenocytes isolated from wild-type and Nrf2-null mice were either left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% ethanol for tBHQ or 0.01% DMSO for CDDO-Im), tBHQ, or CDDO-Im for 30 min prior to activation with anti-CD3/anti-CD28. After 4 h, nuclear proteins were extracted from either A.) tBHQ-treated splenocytes or B.) CDDO-Im treated splenocytes and p65 DNA binding was quantified by a commercially-available kit. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
3.7 The Nrf2 activators tBHQ and CDDO-Im increase c-jun nuclear accumulation in a Nrf2-dependent manner
It has been previously shown that Nrf2 can partner with and potentiate AP-1 signaling in some models, which has potential implications in immune endpoints, as AP-1 is known to drive many immune functions, including the transcription of IL-2 [28,29]. We, therefore, analyzed the nuclear accumulation of c-jun, an AP-1 family member, in splenocytes that were treated with either tBHQ or CDDO-Im by PAGE and Western blot analysis. The resulting blots showed that activated splenocytes derived from Nrf2-null mice had markedly less nuclear accumulation of c-jun as compared to wild-type (Fig. 8, Fig. 9). Furthermore, treatment of splenocytes with tBHQ resulted in a trend towards a Nrf2-dependent increase in c-jun nuclear accumulation (Fig. 8). Similarly, treatment of wild-type and Nrf2-null splenocytes with CDDO-Im also resulted in a trend towards a Nrf2-dependent increase in nuclear accumulation of c-jun (Fig. 9). Taken together, these data suggest that Nrf2 activity promotes c-jun nuclear accumulation in activated T cells, as loss of Nrf2 reduces nuclear c-jun accumulation and activation of Nrf2 results in a trend towards increased nuclear c-jun.
Fig. 8.

tBHQ increases nuclear accumulation of c-jun in wild-type, but not Nrf2-null, primary mouse splenocytes. Wild-type and Nrf2-null mouse splenocytes were left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% ethanol) or tBHQ (1µM) for 30 min prior to activation with anti-CD3/anti-CD28. After 4 h nuclear protein was then isolated and expression of c-jun and the housekeeper histone H3 was probed by western analysis. (A) Representative western blot. Wild-type and Nrf2-null samples are from the same blot, and have been separated for clarity. (B) Densitometry of the resulting western blots was calculated as a ratio of the c-jun band to the H3 band, and then normalized for each replicate as a percent of the wild-type vehicle value. Results are depicted as averages of the normalized values from four experiments. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
Fig. 9.

CDDO-Im increases nuclear accumulation of c-jun in wild-type, but not Nrf2-null, primary mouse splenocytes. Wild-type and Nrf2-null mouse splenocytes were left untreated (BKG) or were pretreated with vehicle (VEH, 0.01% DMSO) or CDDO-Im (0.1µM) for 30 min prior to activation with anti-CD3/anti-CD28. After 4 h nuclear protein was then isolated and expression of c-jun and the housekeeper histone H3 was probed by western analysis. (A) Representative western blot. Wild-type and Nrf2-null samples are from the same blot, and have been separated for clarity. (B) Densitometry of the resulting western blots was calculated as a ratio of the c-jun band to the H3 band, and then normalized for each replicate as a percent of the wild-type vehicle value. Results are depicted as averages of the normalized values from four experiments. a = p<0.05 versus wild-type VEH, b = p<0.05 versus Nrf2-null VEH, c = p<0.05 between the wild-type and Nrf2-null genotypes.
4 DISCUSSION
Our previous studies demonstrated that activation of Nrf2 results in skewing of CD4+ T cells toward Th2 differentiation [10]. However, these studies did not address the role of Nrf2 in early T cell activation. Thus, the purpose of the present studies was to determine the role of Nrf2 in early events following T cell activation. The data demonstrate that Nrf2 inhibits early IFNγ production, but has no effect on activation-induced CD25 or CD69 expression. Treatment of wild-type and Nrf2-null splenocytes with tBHQ decreased IL-2 production in both genotypes, suggesting the effect of the synthetic food preservative tBHQ on IL-2 secretion is independent of Nrf2. Interestingly, the triterpenoid, CDDO-Im, caused a concentration-dependent increase in IL-2 secretion in wild-type, but not Nrf2-null, splenocytes. Thus, the differential effects of CDDO-Im and tBHQ upon the secretion of IL-2 reflect differences in mechanism of action. Indeed, tBHQ and CDDO-Im had differential effects on the induction of all the early cytokines we analyzed, with the exception of IFNγ, which was decreased by both Nrf2 activators. The differential effects of tBHQ and CDDO-Im on cytokine production underscores the capacity of these Nrf2 activators for off-target effects in activated T cells. In general, tBHQ decreased early cytokine production, whereas CDDO-Im tended to increase cytokine production with the exception of IFNγ, which was decreased.
Overall, the data suggest that rather than simply turning T cell activation on or off, Nrf2 plays a more complex role in modulating T cell function. Nrf2 has no effect on the expression of CD25 or CD69, inhibits secretion of the Th1 cytokines, IFNγ and TNFα, and promotes early production of IL-2, which has been shown to promote Th2 differentiation [30,31]. In addition, other studies have implicated Nrf2 in the promotion of Th2 differentiation in both mouse and human models [10,32]. For example, the Nrf2 activator dimethyl fumarate, which is used to treat multiple sclerosis, has been shown to promote Th2 cytokine production [33,34]. Taken together, the findings of the current study suggest that the effect of Nrf2 activation on the early events following T cell activation support the later occurrence of Th2 polarization.
The transcription factor AP-1 plays a critical role in driving transcription of many immune endpoints, including the proinflammatory cytokines IFNγ, TNFα, and GM-CSF. Interestingly, nuclear accumulation of c-jun was decreased in Nrf2-null splenocytes as compared to wild-type splenocytes, and treatment of with both tBHQ and CDDO-Im resulted in a Nrf2-dependent trend towards an increase in the nuclear accumulation of c-jun, a key member of the AP-1 family of proteins. Although it seems logical that an increase in c-Jun nuclear accumulation should result in a subsequent increase in AP-1 signaling, this may not be the case. It has been previously shown that some AP-1 family members, such as Fra-1, can partner with activated Nrf2 and serve to inhibit Nrf2 signaling through the ARE, after being bound by Nrf2 [5]. Knowing this, it is plausible that a similar mechanism may be functioning on AP-1 binding sites, resulting in the suppression of target gene transcription.
Similar to AP-1, NFκB is a transcription factor that drives gene transcription through the IL-2 promoter. Previous studies have shown cross-talk between the Nrf2 and NFκB signaling pathways, which results in a Nrf2-dependent suppression of NFκB activation, by inhibition of inhibitor of kappa B kinase [10]. Therefore, the present studies investigated the effects of the Nrf2 activators CDDO-Im and tBHQ upon NFκB DNA binding. The results demonstrate a Nrf2-independent suppression of NFκB DNA binding by tBHQ. This finding was unexpected, as the literature has shown activation of Nrf2 to suppress NFκB function. Given the electrophilic properties of tBHQ, it likely has a complex mechanism of action. In addition to activating Nrf2, it is possible that tBHQ directly interacts with NFκB and thereby suppresses it, as has been shown with a similar antioxidant, BHA [35]. In contrast to tBHQ, CDDO-Im-induced suppression of NFκB is Nrf2-dependent. These findings coupled with the Nrf2-dependent increase in the secretion of IL-2 are interesting, as NFκB is an important positive regulator of the IL-2 promoter. According to sequence analysis by the ECR Browser (www.ecrbrowser.dcode.org), the promoter region of the mouse IL-2 gene contains at least one Nrf2 binding site. It is possible that the increase in IL-2 secretion may be due to Nrf2 directly binding to the IL-2 promoter. The partial inhibition of NFkB by CDDO-Im may not be a robust enough suppression to negatively impact IL-2 induction. Taken together, the data show that the mechanism of action of CDDO-Im and tBHQ is complex and operates through multiple signaling pathways, including NFκB, c-jun, and Nrf2.
The increase in IL-2 production by Nrf2 may appear to conflict with the notion that Nrf2 plays a protective inhibitory role within the immune system, preventing inappropriate induction of pro-inflammatory immune responses. However, unlike IFNγ and IL-17, IL-2 does not necessarily play a pro-inflammatory role in most immune responses. Whereas IFNγ, for example, is most notably associated with Th1 responses, which are often pro-inflammatory, IL-2 plays a complex role in T cell activity. IL-2 is a T cell growth factor that contributes to T cell proliferation following activation and is key to the induction of an effective T cell response [36]. However, IL-2 is also vital for the differentiation and function of Treg cells. Treg cells are CD4+ T cells that function to control and/or suppress the proliferation and function of effector T cells, which are responsible for launching and maintaining immune responses. Treg cells, therefore, play a protective regulatory role within the immune system to maintain peripheral tolerance and prevent the development of autoimmunity. Thus, promotion of IL-2 production by Nrf2 is consistent with a protective, anti-inflammatory role. Furthermore, the literature shows that an early increase in IL-2 production promotes the development of Th2 cells. Taken together, the effects of Nrf2 on early cytokine production further support a role for Nrf2 in Th2 polarization [30,37].
Overall, this study shows Nrf2 has differential effects on the early events of T cell activation. Whereas Nrf2 inhibits early TNFα and IFNγ production, it promotes IL-2 production, and has no effect on induction of CD25 or CD69. Importantly, this is the first paper to report that activation of Nrf2 increases early IL-2 production by activated T cells. Furthermore, the Nrf2-mediated inhibition of the Th1 cytokine, IFNγ (which suppresses Th2 differentiation), coupled with enhanced early production of IL-2, which is known to promote Th2 differentiation, suggest that the effects of Nrf2 on Th2 differentiation may occur earlier during polarization than previously thought. Collectively, these studies suggest an important and novel role for Nrf2 in regulating T cell activation, differentiation and cytokine production.
Acknowledgments
ACKNOWLEDGMENTS AND DISCLOSURES
We thank Dr. Jefferson Chan for generously providing the Nrf2-null mice. We also thank Dr. Bryan Copple and his laboratory for lending materials and instrumentation used in these studies. These studies were supported by National Institute of Health grants: GM092715 (to J.W.Z), ES007255 (to A.E.T) R01 ES024966 and R00 ES018885 (to C.E.R.).
ABBREVIATIONS
- IFNγ
interferon γ
- IL-2
interleukin-2
- Keap1
Kelch ECH associating protein 1
- NFAT
nuclear factor of activated T cells
- Nrf2
nuclear factor erythroid 2-related factor 2
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- tBHQ
tert-butylhydroquinone
- CDDO-Im
1[2-Cyano-3,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole
- TCR
T cell receptor
- Treg
regulatory T cell
- DMSO
Dimethylsulfoxide
- GM-CSF
Granulocyte-macrophage colony-stimulating factor
- TNFα
Tumor necrosis factor-alpha
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
K.L. has patent interests in synthetic triterpenoids. The other authors have no conflict of interest to disclose.
References
- 1.Chan K, Lu R, Chang JC, Kan YW. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci U S A. 1996;93:13943–13948. doi: 10.1073/pnas.93.24.13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 5.Venugopal R, Jaiswal AK. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci U S A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y-i. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochemical and Biophysical Research Communications. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 7.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 8.Liby KT, Sporn MB. Synthetic Oleanane Triterpenoids: Multifunctional Drugs with a Broad Range of Applications for Prevention and Treatment of Chronic Disease. Pharmacological Reviews. 2012;64:972–1003. doi: 10.1124/pr.111.004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dinkova-Kostova AT, Liby KT, Stephenson KK, Holtzclaw WD, Gao XQ, Suh N, Williarrli C, Risingsong R, Honda T, Gribble GW, Sporn MB, Talalay P. Extremely potent triterpenoid inducers of the phase 2 response: Correlations of protection against oxidant and inflammatory stress. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4584–4589. doi: 10.1073/pnas.0500815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rockwell CE, Zhang M, Fields PE, Klaassen CD. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol. 2012;188:1630–1637. doi: 10.4049/jimmunol.1101712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shahidi F. Antioxidants in food and food antioxidants. Nahrung. 2000;44:158–163. doi: 10.1002/1521-3803(20000501)44:3<158::AID-FOOD158>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 12.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saykally JN, Rachmany L, Hatic H, Shaer A, Rubovitch V, Pick CG, Citron BA. The nuclear factor erythroid 2-like 2 activator, tert-butylhydroquinone, improves cognitive performance in mice after mild traumatic brain injury. Neuroscience. 2012;223:305–314. doi: 10.1016/j.neuroscience.2012.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Choi YK, Lee KS, Cho DH, Baek YY, Lee DK, Ha KS, Choe J, Won MH, Jeoung D, Lee H, Kwon YG, Kim YM. Functional dissection of Nrf2-dependent phase II genes in vascular inflammation and endotoxic injury using Keap1 siRNA. Free Radic Biol Med. 2012;53:629–640. doi: 10.1016/j.freeradbiomed.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 15.Osburn WO, Yates MS, Dolan PD, Chen S, Liby KT, Sporn MB, Taguchi K, Yamamoto M, Kensler TW. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104:218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho HY, Reddy SP, Kleeberger SR. Nrf2 defends the lung from oxidative stress. Antioxid Redox Signal. 2006;8:76–87. doi: 10.1089/ars.2006.8.76. [DOI] [PubMed] [Google Scholar]
- 18.Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, Yamamoto M, Hizawa N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res. 2010;11:31. doi: 10.1186/1465-9921-11-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YJ, Takizawa H, Azuma A, Kohyama T, Yamauchi Y, Takahashi S, Yamamoto M, Kawada T, Kudoh S, Sugawara I. Nrf2 is closely related to allergic airway inflammatory responses induced by low-dose diesel exhaust particles in mice. Clin Immunol. 2010;137:234–241. doi: 10.1016/j.clim.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 20.Athale J, Ulrich A, Chou Macgarvey N, Bartz RR, Welty-Wolf KE, Suliman HB, Piantadosi CA. Nrf2 promotes alveolar mitochondrial biogenesis and resolution of lung injury in Staphylococcus aureus pneumonia in mice. Free Radic Biol Med. 2012;53:1584–1594. doi: 10.1016/j.freeradbiomed.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Stein TD, Johnson JA. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol Genomics. 2004;18:261–272. doi: 10.1152/physiolgenomics.00209.2003. [DOI] [PubMed] [Google Scholar]
- 22.Ma Q, Battelli L, Hubbs AF. Multiorgan autoimmune inflammation, enhanced lymphoproliferation, and impaired homeostasis of reactive oxygen species in mice lacking the antioxidant-activated transcription factor Nrf2. Am J Pathol. 2006;168:1960–1974. doi: 10.2353/ajpath.2006.051113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int. 2001;60:1343–1353. doi: 10.1046/j.1523-1755.2001.00939.x. [DOI] [PubMed] [Google Scholar]
- 24.Cantrell D. T cell antigen receptor signal transduction pathways. Annu Rev Immunol. 1996;14:259–274. doi: 10.1146/annurev.immunol.14.1.259. [DOI] [PubMed] [Google Scholar]
- 25.Liby K, Royce DB, Williams CR, Risingsong R, Yore MM, Honda T, Gribble GW, Dmitrovsky E, Sporn TA, Sporn MB. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Research. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 26.Honda T, Honda Y, Favaloro FG, Gribble GW, Suh N, Place AE, Rendi MH, Sporn MB. A novel dicyanotriterpenoid, 2-cyano-3,12-dioxooleana-1,9(11)-dien-28-onitrile, active at picomolar concentrations for inhibition of nitric oxide production. Bioorganic & Medicinal Chemistry Letters. 2002;12:1027–1030. doi: 10.1016/s0960-894x(02)00105-1. [DOI] [PubMed] [Google Scholar]
- 27.Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol. 2008;181:680–689. doi: 10.4049/jimmunol.181.1.680. [DOI] [PubMed] [Google Scholar]
- 28.Yang HP, Magilnick N, Lee C, Kalmaz D, Ou XP, Chan JY, Lu SC. Nrf1 and Nrf2 regulate rat glutamate-cysteine ligase catalytic subunit transcription indirectly via NF-kappa B and AP-1. Molecular and Cellular Biology. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cantrell DA. T cell antigen receptor signal transduction pathways. Cancer Surv. 1996;27:165–175. [PubMed] [Google Scholar]
- 30.Cote-Sierra J, Foucras G, Guo LY, Chiodetti L, Young HA, Hu-Li J, Zhu JF, Paul WE. Interleukin 2 plays a central role in Th2 differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3880–3885. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao W, Schones DE, Oh J, Cui YZ, Cui KR, Roh TY, Zhao K, Leonard WJ. Priming for T helper type 2 differentiation by interleukin 2-mediated induction of interleukin 4 receptor alpha-chain expression. Nature Immunology. 2008;9:1288–1296. doi: 10.1038/ni.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seumois G, Chavez L, Gerasimova A, Lienhard M, Omran N, Kalinke L, Vedanayagam M, Ganesan AP, Chawla A, Djukanovic R, Ansel KM, Peters B, Rao A, Vijayanand P. Epigenomic analysis of primary human T cells reveals enhancers associated with TH2 memory cell differentiation and asthma susceptibility. Nat Immunol. 2014;15:777–788. doi: 10.1038/ni.2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Bruck W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 34.de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw-Kraan T, Ottenhoff TH, Nibbering PH. Selective stimulation of T helper 2 cytokine responses by the anti-psoriasis agent monomethylfumarate. Eur J Immunol. 1996;26:2067–2074. doi: 10.1002/eji.1830260916. [DOI] [PubMed] [Google Scholar]
- 35.Schulze-Osthoff K, Beyaert R, Vandevoorde V, Haegeman G, Fiers W. Depletion of the mitochondrial electron transport abrogates the cytotoxic and gene-inductive effects of TNF. EMBO J. 1993;12:3095–3104. doi: 10.1002/j.1460-2075.1993.tb05978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657–679. doi: 10.1146/annurev.immunol.24.021605.090727. [DOI] [PubMed] [Google Scholar]
- 37.Le Gros G, Ben-Sasson SZ, Seder R, Finkelman FD, Paul WE. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells (Reprinted from The Journal of Experimental Medicine, vol 172, pg 921–929, 1990) Journal of Immunology. 2008;181:2943–2951. doi: 10.1084/jem.172.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]