

Abstract
There is growing evidence that environmental toxicants can affect various physiological processes by altering DNA methylation patterns. However, very little is known about the impact of toxicant-induced DNA methylation changes on gene expression patterns. The objective of this study was to determine the genome-wide changes in DNA methylation concomitant with altered gene expression patterns in response to 3, 3’, 4, 4’, 5-pentachlorobiphenyl (PCB126) exposure. We used PCB126 as a model environmental chemical because the mechanism of action is well-characterized, involving activation of aryl hydrocarbon receptor, a ligand-activated transcription factor. Adult zebrafish were exposed to 10 nM PCB126 for 24 h (water-borne exposure) and brain and liver tissues were sampled at 7 days post-exposure in order to capture both primary and secondary changes in DNA methylation and gene expression. We used enhanced Reduced Representation Bisulfite Sequencing and RNAseq to quantify DNA methylation and gene expression, respectively. Enhanced reduced representation bisulfite sequencing analysis revealed 573 and 481 differentially methylated regions in the liver and brain, respectively. Most of the differentially methylated regions are located more than 10 kilobases upstream of transcriptional start sites of the nearest neighboring genes. Gene Ontology analysis of these genes showed that they belong to diverse physiological pathways including development, metabolic processes and regeneration. RNAseq results revealed differential expression of genes related to xenobiotic metabolism, oxidative stress and energy metabolism in response to polychlorinated biphenyl exposure. There was very little correlation between differentially methylated regions and differentially expressed genes suggesting that the relationship between methylation and gene expression is dynamic and complex, involving multiple layers of regulation.
Keywords: enhanced reduced representation bisulfite sequencing (eRRBS), RNAseq, PCBs, liver, brain
Introduction
One of the extensively studied epigenetic effects of environmental chemicals is altered DNA methylation, a covalent modification of cytosine nucleotides in a CG dinucleotide context and an important mechanism of gene regulation. Toxicant effects on the DNA methylation process demonstrated so far include alterations in the expression of genes encoding DNA methyltransferases (DNMTs), the availability of DNA methylation substrates and the degree of CpG methylation in different regions of the genome. The majority of studies to date have focused on gene or locus-specific changes in DNA methylation. With the advent of high throughput bisulfite sequencing methods such as reduced representation bisulfite sequencing (RRBS) and whole genome bisulfite sequencing (WGBS), it has become possible to measure genome-wide changes in DNA methylation at base pair (bp) resolution [1, 2]. These methods have enabled identification of differentially methylated regions (DMRs) in diverse genomic locations including promoters, CpG islands, gene bodies and intergenic regions. As DNA methylation is one of the key mechanisms for regulating gene expression, concomitant analysis of the transcriptome and DNA methylome have been conducted to demonstrate the role of DNA methylation in transcriptional regulation. Recent studies have demonstrated very little correlation between these two processes [3–5]. Similar studies investigating the impact of environmental chemicals on DNA methylation and gene expression are lacking except for one study on the effect of vinclozolin on DNA methylation and gene expression [6, 7].
Persistent organic pollutants such as polychlorinated biphenyls (PCBs) are ubiquitously distributed in the environment and exert a wide range of toxic effects. The most toxic PCBs are non-ortho substituted congeners (dioxin-like PCBs) such as 3, 3’, 4, 4’, 5-pentachlorobiphenyl (PCB126). There is accumulating evidence that exposure to PCBs can cause a number of metabolic, behavioral and neurodevelopmental defects. The mode of action of dioxin-like PCBs is well understood; it involves activation of aryl hydrocarbon receptor (AHR), a ligand-activated transcription factor [8]. There is extensive knowledge of target genes regulated by the AHR [9], but there is very limited information on whether the effects of AHR ligands on these genes involve changes in DNA methylation. Recent studies have demonstrated an association between PCB exposure and altered DNA methylation in humans [10–14]. In addition, exposure to dioxin-like PCBs has been shown to cause changes in DNMT gene expression and gene-specific DNA methylation [15–20]. Very few studies have determined genome-wide changes in DNA methylation in response to AHR agonists [21]. The objective of this study is to identify genome-wide changes in DNA methylation concomitant with gene expression patterns in the liver and brain tissues of zebrafish exposed to PCB126. We hypothesized that DNA methylation changes play a role in altered gene expression in response to PCB126 exposure.
Zebrafish is a well-established toxicology model system and has been used to study the developmental toxicity of dioxin-like PCBs [22], as well as the latent effects of developmental exposure [23, 24]. The DNA methylomes of different life history stages of developing zebrafish, as well as adult tissues have been sequenced [25–28]. We have previously shown that developmental exposure to the potent AHR ligand, 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD or dioxin) altered the expression of DNMTs and differentially methylated the CpG islands in the promoter regions of some AHR target genes in zebrafish [29]. However, the relationship between genome-wide changes in DNA methylation and altered patterns of gene expression in response to any AHR ligand is unknown, in any model system. Our results demonstrate that PCB126 exposure alters DNA methylation across the genome with the majority of the sites localizing to the distal promoter regions in both liver and brain tissues. Transcriptional profiling suggests that there is very little concordance between these DNA methylation changes and gene expression patterns.
Materials and Methods
Experimental Animals
Adult TL (Tupfel/Long fin mutations) strain of zebrafish was used in this study. The zebrafish were maintained in a 10 l tank (density of 2 fish per litre) at 28°C system water with a 14-h light, 10-h dark cycle. The fish were fed twice daily; morning feeding with freshly hatched brine shrimp (Artemia salina) and afternoon feeding with GEMMA Micro 300 microencapsulated diet (Skretting USA, Tooele, Utah). The procedures used in this study were approved by the Animal Care and Use Committee of the Woods Hole Oceanographic Institution.
PCB126 Exposure
Adult male and female zebrafish (6 months old) were exposed to either 10 nM PCB126 or solvent carrier (0.01% DMSO) in system water (475 mg/l Instant Ocean, 79 mg/l sodium bicarbonate and 53 mg/l calcium sulfate; pH 7.2) for 24 h. We chose a 24 h exposure period in order to ensure AHR activation in both liver and brain tissues. Exposures were carried out in 2 gallon capacity glass aquarium tanks in 5 l of water at density of 1 fish per 1.25 l of water. This concentration of PCB126 was chosen because it does not elicit any overt morphological defects, but induces gene expression changes. This concentration is also ten times lower than the concentration used in a previous study where zebrafish juveniles (∼2 months old) were exposed to PCB126 by a similar route of exposure [30]. We chose 6 month old fish (standard length 23.5 ± 3.5 mm) for this experiment for practical reasons of dissecting liver and brain tissues. Zebrafish are considered as adults starting from 3 months of age to 2 years [31].
Each treatment had 4 biological replicates (2 males and 2 females). At the end of the exposure, fish were transferred to clean water with constant aeration and heating and maintained for 7 days. Fish were not fed during the 24 h exposure period. During the 7 days post-exposure, the husbandry conditions were the same as described in the previous section. At 7 days post-exposure, fish were euthanized with MS-222 (150 mg/l) buffered with sodium bicarbonate (pH 7.2) prior to tissue sampling. We chose this experimental design in order to capture both primary and secondary changes in DNA methylation and gene expression. Liver and brain tissues were dissected and snap frozen in liquid nitrogen and stored at –80°C until nucleic acids were isolated.
Isolation of Total RNA and Genomic DNA
Simultaneous isolation of genomic DNA and total RNA was performed using the ZR-Duet™ DNA/RNA MiniPrep kit (Zymo Research, CA). RNA was treated with DNase during the isolation process. DNA and RNA were quantified using the Nanodrop Spectrophotometer. The quality of DNA and RNA was checked using the Agilent 4200 and 2200 TapeStation systems, respectively. The DNA and RNA integrity numbers of all samples were between 9 and 10.
Quantitative Real-Time PCR
Complementary DNA was synthesized from 1 μg total RNA using the iScript cDNA Synthesis Kit (Bio-Rad, CA). Quantitative PCR was performed with iQ SYBR Green Supermix in a MyiQ Single-Color Real-Time PCR Detection System (Bio-Rad, CA). Real-time PCR primers used for amplifying β-actin were 5’-CAACAGAGAGAAGATGACACAGATCA-3’ (Forward) and 5’-GTCACACCATCACCAGAGTCCATCAC-3’ (Reverse). These primers amplify both β-actin paralogs (actb1 and actb2). Cyp1a forward and reverse primers were 5’-GCATTACGATACGTTCGATAAGGAC-3’ and 5’-GCTCCGAATAGGTCATTGACGAT-3’, respectively.
The PCR conditions used were 95°C for 3 min (1 cycle) and 95°C for 15 s/65°C for 1 min (40 cycles). At the end of each PCR run, a melt curve analysis was performed to ensure that only a single product was amplified. Three technical replicates were used for each sample. A no-template control was included on each plate to ensure the absence of background contamination. We did not observe any significant differences in β-actin levels between DMSO and PCB126 both in qPCR and in our RNAseq data. Relative expression was normalized to that of β-actin (2–ΔCt; where ΔCt = [Ct(cyp1a)–Ct(β-actin)]). One-way ANOVA was used to determine the effect of PCB126 on cyp1a induction (GraphPad Prism version 5.3). A probability level of P < 0.05 was considered statistically significant.
Enhanced Reduced Representation Bisulfite Sequencing and Data Analysis
Enhanced reduced representation bisulfite sequencing (eRRBS) library preparation and sequencing was conducted by ZymoResearch. Briefly, libraries were prepared from 200–500 ng of genomic DNA digested with 60 units of TaqαI and 30 units of MspI (New England Biolabs, MA) sequentially and then extracted with DNA Clean and Concentrator™-5 kit (ZymoResearch, CA). Fragments were ligated to pre-annealed adapters containing 5’-methylcytosine instead of cytosine according to Illumina’s specified guidelines. Adaptor-ligated fragments of 150–250 bp and 250–350 bp in size were recovered from a 2.5% NuSieve 1 : 1 agarose gel (Zymoclean™ Gel DNA Recovery Kit). The fragments were then bisulfite-treated using the EZ DNA Methylation-Lightning™ Kit. Preparative-scale PCR was performed and the resulting products were purified and 50 bp paired end (PE) sequencing was performed on an Illumina HiSeq2500 platform. Sequence reads from eRRBS libraries were identified using standard Illumina base-calling software.
Raw reads were pre-processed using TrimGalore and aligned to the zebrafish genome (GRCz10) using the Bismark alignment software [32] with maximum 2 mismatches, and only uniquely aligned reads were retained for subsequent analysis. Index files were generated using the bismark_genome_preparation command and the entire reference genome. The –non_directional parameter was applied while running Bismark. All other parameters were set to default. Filled-in nucleotides were trimmed off when doing methylation calling. The methylation level of each sampled cytosine was estimated as the number of reads reporting a C, divided by the total number of reads reporting a C or T.
Differentially methylated CpG regions were analysed using methylKit [33]. Sex of the fish was used as a co-variate in the analysis. Briefly, the numbers of methylated and unmethylated CpG sites were counted for each region and descriptive statistics such as read coverage and percentage methylation were calculated for each sample. Samples were filtered based on read coverage and any reads with <10x coverage were discarded. We further filtered the reads by only selecting the CpG sites covered in all the samples. To determine differential methylation, we used the tiling window approach with a 300 bp window length and 300 bp step-size. The logistic regression method was used to calculate P-values. We empirically determined that the 300 bp window length was appropriate in our case as the majority of the tiles had approximately the same number of CpG sites and the DMRs were not fragmented into multiple small regions. The SLIM method [34] was used to calculate q-values. We used a q-value of less than 0.01 and percent methylation difference larger than 25% as the statistical cutoff for determining DMRs. We used the Genomation package [35] to classify the DMRs into different genomic regions. Genomic coordinates of zebrafish refseq genes, CpG islands and repeat sequences were downloaded from the UCSC genome browser. CpG shores were defined as 2000 bp flanking regions on upstream and downstream of a given CpG island.
DMR Gene Ontology Enrichment Analysis
We used Genomic Regions Enrichment of Annotations Tool (GREAT) to associate DMRs with genes [36]. GREAT predicts gene functions of cis regulatory elements by assigning each gene a regulatory domain. To use GREAT, we converted the genomic coordinates of DMRs from GRCz10 version to Zv9 version of the genome using the UCSC genome browser liftOver utility (https://genome.ucsc.edu/cgi-bin/hgLiftOver). We used default parameters with a basal domain that extends 5 kb upstream and 1 kb downstream of the TSS and conducted gene ontology (GO) (biological process and molecular function) enrichment analysis.
RNA Sequencing and Data Analysis
Unstranded RNAseq library preparation using the Illumina TruSeq total RNA library prep kit and 50 bp single-ends sequencing on the HiSeq2000 platform were performed at the Tufts University Core Facility. Raw data files were assessed for quality using FastQC [37] and pre-processed as described previously [38]. Pre-processed reads were mapped to the genome. We used Ensembl version 84 (GRCz10) of the zebrafish genome and annotations (gtf) in this analysis [39]. HTSeq-count was used to obtain the number of reads mapped to annotated regions of the genome [40]. Statistical analysis was conducted using edgeR, a Bioconductor package [41], using sex as an independent variable. Only genes with false discovery rate (FDR) of less than 5% were considered to be differentially expressed. Raw data has been deposited into Gene Expression Omnibus (accession number GSE104221).
GO analysis of differentially expressed genes (DEGs) was done using gProfiler [42]. Zebrafish ensembl IDs were used as input and Bonferroni correction for multiple testing (P-value <0.05) was used while determining the fold enrichment. To understand the relationship between GO terms, Directed Acyclic Graphs of significantly enriched GO terms were drawn using GOView (webgestalt.org/GOView).
Comparing DNA Methylation Changes with Gene Expression
To determine the overlap between DMRs and gene expression, we compared the gene annotation of DMRs obtained from GREAT analysis with the DEGs determined by RNA-seq. For the set of overlapping genes, we determined the Spearman’s correlation coefficient between difference in methylation of the DMR and the altered expression of the target gene.
Results
Cyp1a Gene Expression
To confirm the activation of AHR by PCB126, we quantified cyp1a gene expression using quantitative real-time PCR (Fig. 1). We observed 48-fold and 672-fold increase in cyp1a expression in liver and brain, respectively. The higher fold increase in cyp1a expression in the brain compared to liver is partly due to a lower basal expression.
Figure 1:
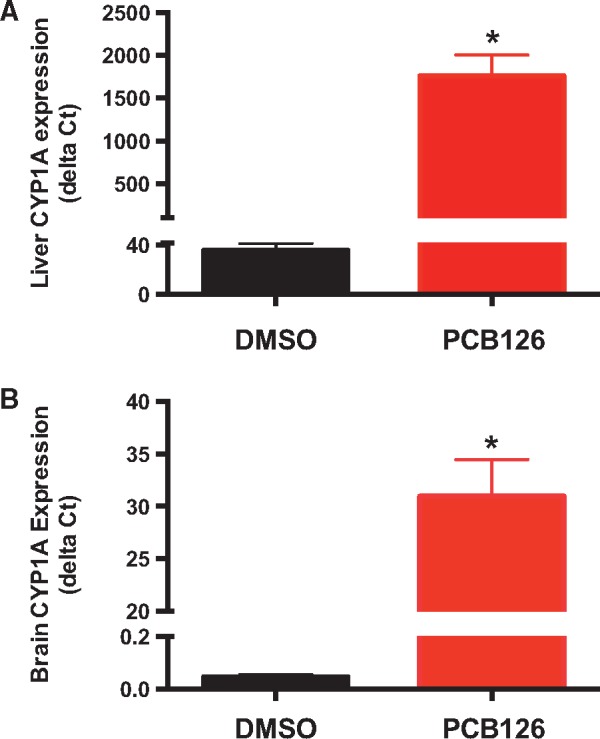
PCB126-induced cyp1a gene expression in the liver (A) and brain (B). Cyp1a expression relative to the reference gene was calculated using the delta Ct method. β-Actin was used as a reference gene. * Represents significant difference from DMSO control (One-way ANOVA; P < 0.01)
DNA Methylation Profiling
We used enhanced RRBS (eRRBS) to identify DMRs in response to PCB126 exposure. eRRBS is a modified version of RRBS that includes fragments of MspI (C^CG_G) and Taqα1 (A^CG_T) restriction enzyme digest and captures CpG sites beyond the CpG islands [43]. RRBS and eRRBS have been previously used to profile baseline DNA methylation in adult zebrafish tissues [28, 44] and in response to toxicants [45].
Using eRRBS, we sequenced an average of 34 million and 31.66 million paired-ends reads from liver and brain samples, respectively. The bisulfite conversion efficiency was 98–99%. The mapping efficiency of these reads to the bisulfite converted zebrafish genome was between 36% and 38%, which is comparable to previously published studies in zebrafish [28, 44]. On average, 257 million cytosines were sequenced. Of these 28.5 and 15.2 million were methylated and unmethylated, respectively, in a CpG context. In both liver and brain tissues, we sequenced approximately 1.25 million unique CpGs per sample with 10x coverage. A detailed summary of the number of CpGs and their coverage is provided in the Supplementary Material (eRRBS_summary_statistics.xlsx).
Using a 300-bp tiling window size, we observed 125 008 unique tiles that were represented in all eight liver samples. The number of unique 300 bp tiles that were shared among all eight brain samples was 103 673. Based on the analysis of all the samples, the genome wide methylation level in the liver and brain was found to be 66.87% and 63.12%, respectively. Methylation level was low in the promoter regions and CpG islands (Fig. 2A and B). Annotation of these tiles revealed that the majority of the CpGs are located in the distal promoter regions in both tissues (Fig. 2C). Only 8% of the features are associated with CpG islands and shores (Fig. 2C).
Figure 2:
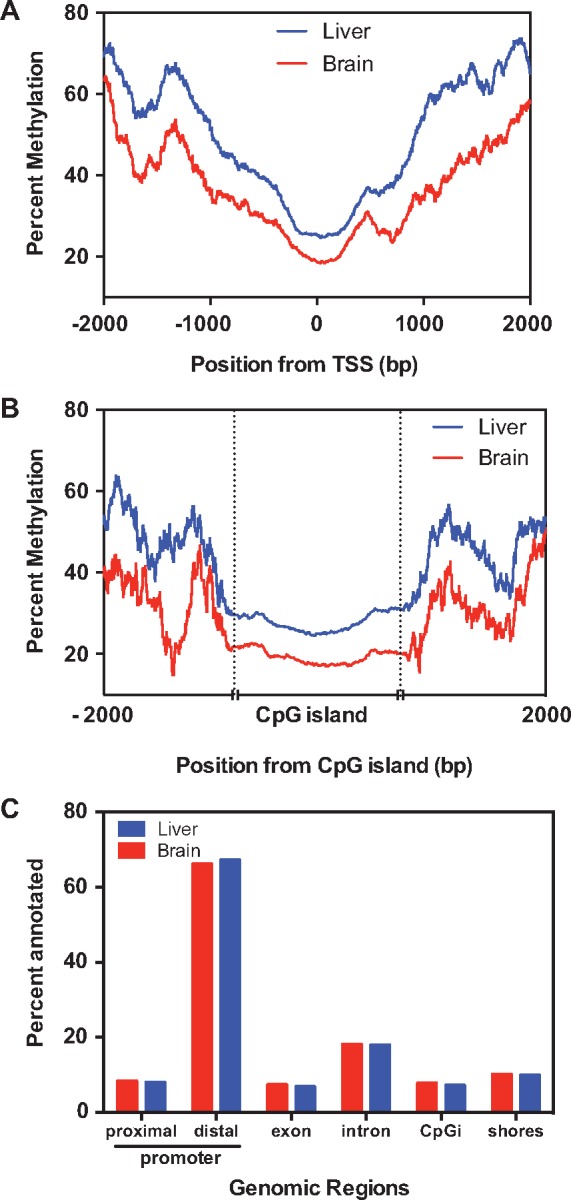
Tissue-specific DNA methylation profiles and their genomic location. Percentage of methylation levels in the proximal promoter regions (A) and CpG islands (B) in the liver and brain are plotted. (C) Percentage of 300 bp tiles overlapping with different genomic regions. These plots are based on the tiles represented in all samples. Proximal (up to 5 kb upstream) and distal promoter (>5 kb) regions are classified based on the distance from the transcriptional start site
PCB126-Induced Changes in DNA Methylation in the Liver
Differential methylation analysis revealed a total of 573 DMRs in the liver in response to PCB126 exposure (Fig. 3A), out of which 327 were hypomethylated and 246 were hypermethylated. Only 59 hypomethylated DMRs (15% of total) and 47 hypermethylated DMRs (19% of total) showed a percent methylation difference larger than or equal to 40%. DMRs were distributed throughout the genome with no discernable patterns in their chromosomal location. The complete list of liver DMRs is provided in the Supplementary Material (Liver_DMRs.xlsx).
Figure 3:

PCB126-induced tissue-specific changes in DNA methylation. Volcano plots showing DMRs in response to PCB126 exposure in the liver (A) and brain (B). Percent methylation difference (x-axis) between PCB126 and Control are plotted against q-value (y-axis). Red vertical lines represent the 25% methylation difference and the blue horizontal line represents a q-value of 0.05, which are used as a statistical cutoff in differential methylation analysis. Each green and red spot represents a statistically significant hypo and hypermethylated region, respectively
The hypermethylated DMRs were enriched in GO biological process terms such as otic placode development, positive regulation of Wnt receptor signaling pathway and integrin-mediated signaling pathway (Fig. 4A). The enriched GO molecular function terms include dopamine beta-monoxygenase activity, inositol 1, 4, 5-trisphosphate 3-kinase (IP3 3-kinase/IP3K) and GABA receptor activity (Fig. 4B). The complete list of all GO terms, their fold enrichment, the list of genes associated with each GO term and the DMR distance from the transcriptional start site are provided in the Supplementary Material (Liver_DMR_GO_analysis.xlsx).
Figure 4:

GO term analysis of DMRs in the liver (A, C) and brain (B, D). Top panel represents GO biological process terms and the bottom panel contains the GO molecular function terms. GO term analysis was performed on DMRs using GREAT. Only statistically significant GO terms are shown. The fold enrichment of GO terms and the genes associated with each term are provided in the Supplementary Material
GO analysis of hypomethylated DMRs revealed enrichment of biological process terms such as negative regulation of apoptosis, secretion, regulation of collateral sprouting and thyroid gland development (Fig. 4A). The hypomethylated DMRs were enriched in GO molecular function terms sugar-phosphatase activity, transferase activity, transferring sulfur-containing groups, thyroxine 5’-deiodinase activity, cGMP binding, intracellular ligand-gated ion channel activity and heparan sulfate sulfotransferase activity (Fig. 4B).
PCB126-Induced Changes in DNA Methylation in the Brain
In the brain, a total of 481 DMRs were observed with 294 hypomethylated and 187 hypermethylated regions (Fig. 3B). Only a small proportion of these (19% of hypomethylated and 14% of hypermethylated) showed a percent methylation difference larger than 40%. Similar to the liver DMRs, the DMRs in the brain were distributed randomly along chromosomes. The complete list of brain DMRs is provided in the Supplementary Material (Brain_DMRs.xlsx).
Hypermethylated DMRs in the brain showed significant enrichment of two GO molecular function terms (cyclin-dependent protein kinase 5 activator activity and protein kinase activator activity) and three biological process terms (piwi-interacting RNA (piRNA) metabolic process, regulation of receptor activity and monocarboxylic acid biosynthetic process) (Fig. 4C). On the other hand, the hypomethylated DMRs were enriched in GO molecular function terms such as acetylglucosaminyltransferase activity, GABA receptor activity and transcription regulatory region DNA binding (Fig. 4D). The complete list of all GO terms, their fold enrichment, the list of genes associated with each GO term and the DMR distance from the transcriptional start site are provided in the Supplementary Material (Brain_DMR_GO_analysis.xlsx).
PCB126-Induced Transcriptional Changes
We obtained an average of 32.3 and 30.6 million raw reads from liver and brain samples, respectively. Of these, 83% (liver) and 88% (brain) of the reads were mapped uniquely to the zebrafish genome. Multi-dimensional scaling indicated two liver samples [one control (male) and one PCB126 treated (female)] to be outliers and hence these were not included in the differential expression analysis. These two samples had only 68% and 74% of the reads uniquely mapped to the zebrafish genome. The summary of the raw data is presented in the Supplemental Material (RNAseq_Summary.xlsx).
There were a total of 585 and 1715 DEGs in the liver and brain, respectively (Fig. 5). Among the 585 DEGs in the liver, 300 were upregulated and 285 were downregulated. All DEGs in the liver were altered by a fold change of 2 or higher even with a FDR of 10% as a statistical cutoff (Supplementary Material – Liver_DEGs.xlsx). The upregulated genes were enriched in GO terms such as response to xenobiotic stimulus (biological process), and acrosin binding (molecular function). Many of the upregulated genes are known AHR target genes (Table 1). Cell cycle and glutathione metabolism are the two KEGG pathways enriched among the upregulated genes. The downregulated genes in the liver represent pathways associated with sterol, ion, oxygen and heme transport activity. The GO terms and the associated genes for both up and downregulated genes in the liver are shown in Table 2.
Figure 5:

Tissue-specific gene expression changes. MA plots showing the DEGs in the liver (A) and brain (B). Horizontal blue lines represent fold change of ± 2. Each red spot represent a statistically differentially expressed gene
Table 1:
Representative xenobiotic responsive genes differentially expressed in liver and brain in response to PCB126 exposure
| Gene name | Gene symbol | Liver |
Brain |
||
|---|---|---|---|---|---|
| logFC | FDR | logFC | FDR | ||
| AHR repressor a | ahrra | 10.677 | 7.21E–46 | 3.850 | 1.12E–02 |
| AHR repressor b | ahrrb | 7.500 | 4.89E–04 | 2.669 | 3.68E–02 |
| AHR interacting protein | aip | 0.553 | 3.90E–02 | ||
| AHR nuclear translocator-like 1a | arntl1a | –0.694 | 2.71E–02 | ||
| AHR nuclear translocator-like 1b | arntl1b | –0.792 | 3.38E–02 | ||
| AHR nuclear translocator-like 2 | arntl2 | –1.758 | 2.75E–03 | –1.033 | 2.53E–02 |
| Cytochrome P4501a | cyp1a | 5.420 | 3.40E–20 | 8.571 | 8.60E–85 |
| Cytochrome P4501b1 | cyp1b1 | 7.750 | 1.35E–16 | 1.834 | 3.11E–03 |
| Cytochrome P450, family 1, subfamily C, polypeptide 1 | cyp1c1 | 5.433 | 6.30E–17 | 4.745 | 7.26E–06 |
| Cytochrome P450, family 1, subfamily C, polypeptide 2 | cyp1c2 | 2.887 | 2.44E–06 | 3.058 | 6.81E–03 |
| Cytochrome P450, family 2, subfamily AA, polypeptide 12 | cyp2aa12 | 1.443 | 3.88E–02 | ||
| Cytochrome P450, family 2, subfamily AA, polypeptide 4 | cyp2aa4 | 2.662 | 2.04E–02 | ||
| Cytochrome P450, family 2, subfamily K, polypeptide 22 | cyp2k22 | –5.654 | 2.50E–02 | ||
| Cytochrome P450, family 2, subfamily K, polypeptide 31 | cyp2k31 | –5.169 | 3.10E–07 | ||
| Cytochrome P450, family 2, subfamily K, polypeptide 6 | cyp2k6 | 8.607 | 1.52E–02 | ||
| Cytochrome P450, family 2, subfamily X, polypeptide 10.2 | cyp2x10.2 | 3.495 | 2.32E–10 | ||
| Cytochrome P450, family 2, subfamily AD, polypeptide 3 | cyp2ad3 | 1.055 | 1.79E–02 | ||
| Cytochrome P450, family 2, subfamily P, polypeptide 6 | cyp2p6 | 1.005 | 6.51E–03 | ||
| Cytochrome P450, family 2, subfamily V, polypeptide 1 | cyp2v1 | 1.348 | 9.56E–03 | ||
| Cytochrome P450, family 26, subfamily b, polypeptide 1 | cyp26b1 | 0.985 | 9.56E–03 | ||
| Glutathione S-transferase theta 1b | gstt1b | 1.349 | 4.30E–02 | ||
| Microsomal glutathione S-transferase 3b | mgst3b | 2.050 | 7.38E–04 | 0.762 | 1.18E–02 |
| Glutathione S-transferase theta 1a | gstt1a | 3.983 | 3.73E–02 | ||
| Glutathione S-transferase mu, tandem duplicate 1 | gstm.1 | 0.619 | 3.15E–02 | ||
| Glutathione S-transferase rho | gstr | 0.768 | 3.90E–02 | ||
| Glutathione S-transferase pi 1 | gstp1 | 0.883 | 3.43E–02 | ||
| Glutamate-cysteine ligase, modifier subunit | gclm | 1.779 | 1.52E–03 | ||
| Glutathione reductase | gsr | 1.498 | 3.79E–03 | ||
| Glucose-6-phosphate dehydrogenase | g6pd | 1.610 | 1.96E–02 | ||
| NAD(P)H dehydrogenase, quinone 1 | nqo1 | 1.951 | 3.42E–02 | ||
| Kelch-like ECH-associated protein 1b | keap1b | 1.308 | 4.11E–02 | ||
| UDP glucuronosyltransferase 1 family, polypeptide B5 | ugt1b5 | 1.633 | 2.20E–02 | ||
| UDP glucuronosyltransferase 1 family, polypeptide B1 | ugt1b1 | 1.667 | 6.72E–03 | ||
| UDP glucuronosyltransferase 1 family, polypeptide B2 | ugt1b2 | –2.217 | 3.98E–02 | ||
| UDP glucuronosyltransferase 1 family, polypeptide B7 | ugt1b7 | –2.779 | 8.30E–05 | ||
| UDP glucuronosyltransferase 5 family, polypeptide C2 | ugt5c2 | –2.099 | 1.03E–02 | ||
| UDP glucuronosyltransferase 1 family, polypeptide A6 | ugt1a6 | 4.251 | 3.13E–02 | ||
| Phosphogluconate dehydrogenase | pgd | 1.856 | 4.99E–03 | ||
Table 2:
GO and KEGG pathway analysis of DEGs in the liver (A) Upregulated genes and (B) downregulated genes in the liver were analysed separately for enrichment of GO biological process and molecular function terms and KEGG pathways
| A | ||
|---|---|---|
| Term ID | Name | Adjusted P-value |
| GO: 0009410 | Response to xenobiotic stimulus | 0.002 |
| GO: 0032190 | Acrosin binding | 0.000152 |
| KEGG | ||
| KEGG: 04110 | Cell cycle | 0.0000199 |
| B | ||
| Term ID | Name | Adjusted P-value |
| Biological process | ||
| GO: 0015669 | Gas transport | 0.0000129 |
| GO: 0006820 | Anion transport | 0.000632 |
| GO: 0010873 | Positive regulation of cholesterol esterification | 0.00185 |
| GO: 0033700 | Phospholipid efflux | 0.00185 |
| GO: 0065005 | Protein-lipid complex assembly | 0.00185 |
| GO: 0055088 | Lipid homeostasis | 0.00365 |
| GO: 0006695 | Cholesterol biosynthetic process | 0.0141 |
| Molecular function | ||
| GO: 0015248 | Sterol transporter activity | 0.00123 |
| GO: 0060228 | Phosphatidylcholine-sterol-O-acyltransferase activator activity | 0.00185 |
| GO: 0005344 | Oxygen transporter activity | 0.00365 |
| GO: 0019825 | Oxygen binding | 0.00365 |
| GO: 0020037 | Heme binding | 0.0376 |
| GO: 0015485 | Cholesterol binding | 0.0417 |
| GO: 0005215 | Transporter activity | 0.0463 |
| KEGG | ||
| KEGG: 00910 | Nitrogen metabolism | 0.0386 |
In the brain, PCB126 exposure resulted in the differential expression of 1715 genes. Among these 899 genes were upregulated and 816 genes were downregulated (Fig. 5; Supplementary Material – Brain_DEGs.xlsx). Similar to the responses in the liver, genes associated with xenobiotic metabolism were upregulated in the brain (Table 1). The GO terms (molecular function) enriched among the upregulated genes include acrosin binding, actin binding and ribosomal constituents. The upregulated KEGG pathway terms include metabolism of xenobiotics by cytochrome P450 and ribosome. The downregulated genes in the brain were enriched in GO terms (molecular function) such as nucleoside-triphosphate activity, structural constituent of cytoskeleton and tubulin binding. KEGG terms represented include dorso-ventral axis formation, FoxO signaling pathway, ErbB signaling pathway and mRNA surveillance pathway. The GO terms and the associated genes for both up and downregulated genes in the brain are shown in Table 3.
Table 3:
GO and KEGG pathway analysis of DEGs in the brain (A) Upregulated genes and (B) downregulated genes in the brain were analysed separately for enrichment of GO biological process and molecular funtion terms and KEGG pathways
| A | ||
|---|---|---|
| Term ID | GO term | Adjusted P-value |
| Biological process | ||
| GO: 0006412 | Translation | 0.0000253 |
| GO: 0009410 | Response to xenobiotic stimulus | 0.0000854 |
| GO: 0003012 | Muscle system process | 0.000151 |
| GO: 0060326 | Cell chemotaxis | 0.00487 |
| GO: 0045861 | Negative regulation of proteolysis | 0.0158 |
| GO: 0043902 | Positive regulation of multi-organism process | 0.0161 |
| GO: 0009605 | Response to external stimulus | 0.024 |
| Molecular function | ||
| GO: 0003735 | Structural constituent of ribosome | 9.07E–13 |
| GO: 0003779 | Actin binding | 0.0000803 |
| GO: 0032190 | Acrosin binding | 0.0299 |
| KEGG | ||
| KEGG: 03010 | Ribosome | 1.66E–11 |
| KEGG: 00980 | Metabolism of xenobiotics by cytochrome P450 | 0.000786 |
| B | ||
| Term ID | GO term | Adjusted P-value |
| Biological process | ||
| GO: 0016043 | Cellular component organization | 2.28E–15 |
| GO: 0060627 | Regulation of vesicle-mediated transport | 0.00154 |
| GO: 0043087 | Regulation of GTPase activity | 0.0023 |
| GO: 0006836 | Neurotransmitter transport | 0.00343 |
| GO: 0035556 | Intracellular signal transduction | 0.00659 |
| Molecular function | ||
| GO: 0000902 | Cell morphogenesis | 0.0188 |
| GO: 0017111 | Nucleoside-triphosphatase activity | 0.00641 |
| GO: 0015631 | Tubulin binding | 6.44E–11 |
| GO: 0008047 | Enzyme activator activity | 0.00309 |
| GO: 0005200 | Structural constituent of cytoskeleton | 0.00375 |
| GO: 0098772 | Molecular function regulator | 0.0042 |
| KEGG | ||
| KEGG: 04320 | Dorso-ventral axis formation | 0.0043 |
| KEGG: 04068 | FoxO signaling pathway | 0.0147 |
| KEGG: 03015 | mRNA surveillance pathway | 0.0167 |
| KEGG: 04012 | ErbB signaling pathway | 0.0368 |
Relationship between DMRs and Altered Gene Expression
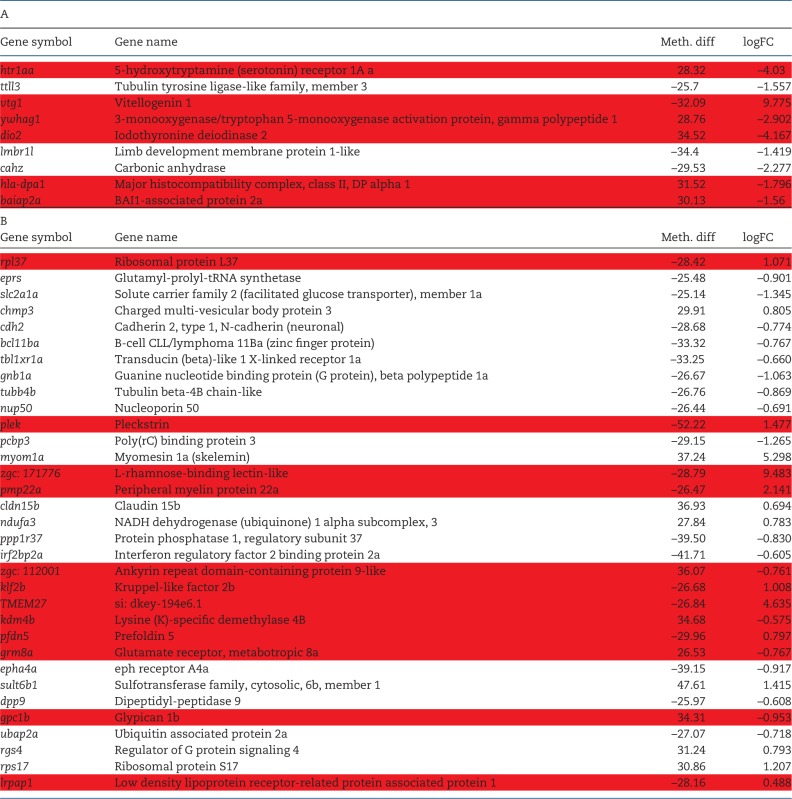
There was very little correlation between DMRs and gene expression patterns. In the liver, only nine genes were both differentially methylated and differentially expressed. Of these, six genes showed the expected inverse relationship between DNA methylation and gene expression. In the brain, 33 genes showed both altered methylation and altered gene expression and only 12 of these were inversely related (Table 4). For both liver and brain, there was not a significant correlation between the degree of altered DNA methylation and the degree of altered gene expression (Spearman r2 = 0.2719 for liver and 0.0078 for brain).
Table 4:
Relationship between DMRs and DEGs (A) List of genes in the liver (A) and brain (B) that are both differentially methylated and differentially expressed. Spearman correlation coefficient was not statistically significant (r2 = 0.2719 for liver and 0.0078 for brain). There is an inverse relationship between DNA methylation (Meth. diff) and gene expression (logFC) for the genes highlighted in red
 |
Discussion
AHR is a ligand activated transcription factor known to regulate a wide variety of target genes upon PCB126 exposure. The results indicate that PCB126 exposure altered DNA methylation patterns mostly in the distal promoter regions (>10 kb). Transcriptomic profiling revealed classical responses to dioxin-like PCBs including upregulation of xenobiotic metabolism genes and downregulation of genes associated with growth and energy metabolism. However, the genes associated with DMRs represent diverse physiological pathways in both tissues. We observed very little overlap between DEGs and the genes associated with DMRs, suggesting distinct effects of PCB126 on DNA methylation and gene expression. We attempted to capture both the primary and secondary changes in DNA methylation and gene expression by sampling at 7 days post-exposure. As DNA methylation and transcription are dynamic processes, our results represent a snapshot of the changes. In order to capture the relationship between these processes multiple time points need to be analysed.
The effects of dioxin and dioxin-like PCBs on DNA methylation have previously been demonstrated but most of these studies have used gene or locus-specific methods to measure DNA methylation [15–18, 20, 29]. Using eRRBS we show that there are strikingly high levels (>60%) of DNA methylation in the distal promoter regions in zebrafish compared to other species [46]. Our results demonstrate differential methylation in response to PCB126 exposure at multiple loci distributed throughout the genome in both tissues. We could not detect any sex-specific differences, possibly due to small sample size (n = 2) and lack of statistical power.
Differential Methylation in the Liver
In the liver, DMRs are associated with a variety of genes including transcription factors, receptors, transporters, cell adhesion molecules, endocrine and metabolic enzymes. Most of the genes play an important role in hepatic development and regeneration in response to injury. For instance, we observed PCB126-induced hypermethylation of sox9a and sox9b genes, which encode transcription factors critical for normal development. In mammals, SOX9 is responsible for maintaining homeostasis in the liver and the regenerative capacity is dependent on SOX9-positive hepatic progenitor cells [47]. Similarly, in the zebrafish regeneration model, ablation of hepatocytes causes transdifferentiation of biliary cells into hepatocytes and this process is blocked in sox9b mutants [48], suggesting an important role in liver regeneration. Previous studies have shown downregulation of sox9b expression with AHR agonists in zebrafish embryos and in regenerating tissues (caudal fin) [49–52]. However, we did not observe differential expression of sox9b in either the liver or brain, suggesting tissue-specific differences in its regulation. As SOX9 genes are master regulators of cell fate and tissue morphogenesis, it is possible that their expression is regulated by multiple mechanisms including DNA methylation.
One such mechanism of sox9b regulation is by non-coding RNA. A recent study has demonstrated that a long intervening non-coding RNA (lincRNA; si: ch1073–384e4.1) regulates the expression of sox9b in zebrafish embryos [53]. We also observed significant upregulation of this lincRNA in response to PCB126 exposure (4.5 log FC; FDR 6.03E–12), suggesting multiple modes of action of AHR agonists in altering sox9b expression in zebrafish. Garcia et al. [53] also demonstrated that this lincRNA expression is AHR-dependent and is required for sox9b expression during normal development.
Other hypermethylated genes that are critical for liver growth and regeneration include hepatocyte growth factor (hgfa) and fibroblast growth factor 8a (fgf8a). Hgfa is an acute phase protein induced in the liver in response to injury and is required for liver regeneration [54, 55]. Fgf8a is a member of the FGF family of growth factors and a morphogen [56]. Given the role of these genes in repair and regeneration, it is possible that hypermethylation (and therefore downregulated expression) of these genes exacerbate injury by reduced regeneration.
Another important enriched pathway among the hypermethylated DMRs includes thyroid hormone signaling. We observed hypermethylation of paired box 8 (pax8) and iodothyronine deiodinase genes (dio2 and dio3a). Pax8 is a transcription factor involved in thyroid differentiation and its altered expression by either mutations or epigenetic modifications disrupts thyroid morphogenesis leading to a variety of phenotypes ranging from thyroid agenesis, ectopia and hypoplasia [57]. Dio2 catalyzes the conversion of prohormone thyroxine (T4) to the bioactive thyroid hormone (T3), and dio3a converts T4 and T3 into inactive metabolites. As liver has an important role in thyroid hormone metabolism and hypothyroidism is linked to liver diseases [58], it is possible that altered methylation of these genes could affect downstream signaling. It is very well documented that dioxins and dioxin-like PCBs cause hypothyroidism by altering thyroid hormone levels [59–62]. Concomitant with hypermethylation of dio2, we observed a decrease in its expression, suggesting a potential mechanism of action of PCB126 in causing hypothyroidism. In addition, we also observed hypermethylation of genes that encode sulfotransferases (gal3st1, hs3st2, hs3st4, hs6st1a, ndst3, nfs1, sult6b1), key players in the metabolism of xenobiotics, drugs and endogenous hormones. Hydroxylated PCBs, the metabolic byproducts of PCBs, disrupt endocrine signaling by inhibiting sulfotransferases [63]. Indeed there is an association between increased body burden of hydroxylated PCBs and altered thyroid hormone status in humans [64]. It remains to be determined if altered methylation of these genes is a potential mechanism of action of dioxin-like PCBs. Our RNAseq data do not reveal differential expression of these genes.
We also observed several hypomethylated DMRs in the liver and enrichment analysis revealed these DMRs to be associated with transcription factors belonging to the homeobox family (dmbx1b, pax2a, pbx2, pbx4, lhx9), nuclear receptor superfamily (nr2f2) and others (gata5). We did not find any studies demonstrating the involvement of these transcription factors in liver metabolism or their altered DNA methylation or gene expression in response to dioxin-like PCB exposure. However, these transcription factors are important players in early cellular differentiation suggesting that altered methylation could affect tissue differentiation and regeneration capacity in response to injury.
Differential Methylation in the Brain
In contrast to the liver, hypermethylated DMRs in the brain did not show significant enrichment of many GO terms. Among the enriched GO terms, one that is particularly interesting is piRNA metabolic process. piRNAs are 23–32 nucleotide single stranded RNA molecules derived from the intergenic regions or 3’ untranslated regions of mRNAs. They are involved in transposable element (TE) repression [65] and preserving genomic integrity [66, 67]. piRNAs were originally discovered in the germline, but their presence and function in somatic cells including neurons has been reported [68, 69]. Defects in piRNA production result in TE de-repression and cell death. We observed hypomethylation of HEN methyltransferase (henmt1) and tudor domain containing protein 1 (tdrd1) genes, key players in piRNA biogenesis. Henmt1 adds a 2’-O-methyl group at the 3’-end of piRNAs and protects the 3’-end of piRNAs from degradation. In zebrafish, loss of henmt1 has been shown to reduce piRNA content in oocytes and cause female sterility. Tdrd1 acts as a molecular scaffold for piwi proteins and is required for proper functioning of the piRNA pathway. If hypomethylation of these genes could lead to increased gene expression and subsequently to an increase in piRNA production, it is possible that this is in response to an increase in TE re-activation and retrotransposition in the brain resulting from PCB126 exposure. We observed significant induction of tdrd1 expression in the brain (logFC 4.03; FDR 0.01) suggesting increased piRNA biosynthesis to suppress TE activation. Previous studies have demonstrated that xenobiotics can cause TE activation [70, 71].
In the brain, many of the hypomethylated DMRs are associated with genes related to the Wnt/β – catenin signaling pathway, crucial for the formation of the blood-brain barrier (BBB) [72]. BBB is a semi-permeable barrier that restricts the movement of ions and molecules between the blood and brain, and its disruption can lead to inflammation. We observed hypomethylation of a Wnt ligand (Wnt4a), a Wnt signaling antagonist (dkk1a) and associated genes (ctnnbip1, ascl1a). In addition, we observed hypomethylation of cell adhesion genes [cadherins (cdh2, cdh6) and protocadherins (pcdh10b, pcdh15a, pcdh17, pcdh18b, pcdh1a, pcdh2g12)], which are modulated by Wnt/β – catenin signaling. Cell adhesion genes are critical for the development and maintenance of proper vascular networks such as the BBB [73]. It is not yet clear how hypomethylation of these gene regulatory regions can alter their expression. We did not observe differential expression of these genes in our RNAseq data, but other cell adhesion genes (cdh2, cdh7, cdh17, cdh18a, pcdh2g5) were differentially expressed. Previous research in murine models has shown that endothelial cells and astrocytes are targets of AHR-dependent TCDD toxicity [74].
It remains to be determined how DNA methylation is regulated by AHR ligands. We have previously demonstrated that TCDD exposure alters DNMT gene expression patterns in developing embryos. Furthermore, we have shown in vitro that XREs in the DNMT proximal promoter regions influence DNMT expression [29]. However, further studies are necessary to determine the mechanism of action of AHR activation in altering genome-wide DNA methylation patterns. We hypothesize that AHR activation alters the accessibility of DNA to DNMTs by altering the chromatin state.
PCB126 Induced Transcriptional Responses
We observed classical responses to PCB126 exposure in the liver and brain, where genes associated with xenobiotic metabolism were upregulated. Almost all of these are AHR target genes and have been shown previously to be altered by dioxins and dioxin-like PCBs in a variety of animals including zebrafish (Table 1). In addition to cytochrome P450s, several oxidative stress response (OSR) genes were upregulated in both tissues. Glutathione (GSH) is critical for intracellular redox homeostasis and protects against oxidative damage caused by reactive oxygen species. It is well established that dioxin and dioxin-like PCBs contribute to oxidative stress [75]. In this study, upregulation of OSR genes suggest that PCB126-exposed fish are trying to maintain cellular redox balance.
PCB126 exposure downregulated the expression of several genes encoding lipid-binding and transport proteins in the liver including apolipoproteins (apoa1a, apoa4b.2, apoba, apoea, apoeb, apol6), fatty acid binding proteins (fabp1b.1, fabp2) and solute carriers. AHR-mediated disruption of hepatic lipid metabolism has been well documented and altered expression of transport proteins is the hallmark of dioxin-induced hepatic inflammation, non-alcoholic fatty liver disease and steatosis [76]. Our results agree with these findings that exposure to dioxin-like PCBs alters lipid metabolism by altering the expression of lipid transport proteins and suggest that the effects of PCBs on lipid homeostasis are conserved among vertebrates. We also observed downregulation of genes encoding heme-binding proteins (cyp2k31, ido1, cyp2k22, hbba1, hbaa1, hbba1, hbaa2 and PXDN) in the liver. Heme is critical for oxygen transport, energy metabolism and drug biotransformation, and previous studies have shown that exposure to AHR agonists’ increases cellular heme levels to compensate for the increased demand (e.g. cyp450 induction). Downregulation of these genes suggests reduced demand for heme or dysregulation of the compensatory mechanism potentially leading to metabolic disruption.
In the brain, we observed a relatively large number of DEGs compared to the liver suggesting that the brain is sensitive to exposure to environmental chemicals, particularly the neurotoxic effects of dioxin-like PCBs. We recently reported similar findings where developmental exposure to low levels of PCB126 altered the expression of thousands of genes in the adult brain [77]. The differences in the sensitivity of tissues to PCB exposure at the molecular level are yet to be determined. In addition to the induction of xenobiotic response genes, we observed upregulation of a large number of ribosomal genes. This is in contrast to our previous observation that developmental exposure to PCB126 caused downregulation of ribosomal genes in the adult zebrafish brain [38], sugggesting that the effects on gene expression vary depending on the duration and dose of exposure as well as the life stage at which the exposure has occurred. Ribosome biogenesis and protein synthesis are essential for cellular differentiation and proliferation and any alterations to these processes can severely impair cell growth. It has been shown that xenobiotics, nutrient depletion and hypoxia can affect ribosome biogenesis leading to ribosomal stress, a condition where unassembled ribosomal proteins are accumulated [78]. Some ribosomal genes are shown to induce the p53 pathway in response to ribosomal stress. Further studies are necessary to understand the functional significance of the upregulation of ribosomal genes in PCB-induced toxicity.
In addition to the potential effects on ribosome biogenesis, PCB126 exposure downregulated genes associated with neurotransmitter transport, cytoskeletal network and genes involved in FoxO, ErbB and mRNA surveillance pathways. FoxO and ErbB signaling pathways regulate diverse processes including proliferation, differentiation, apoptosis, glucose metabolism and OSR.
Correlation between Altered DNA Methylation and Altered Gene Expression
One of the important findings from this study is that there is very little correlation between differential methylation and gene expression. Similar observations have been made in other recent studies in different model systems [4, 5, 79–81] suggesting a complex relationship between DNA methylation and gene expression. Some of the potential reasons for the lack of high degree of correlation between DNA methylation and gene expression could be due to the experimental design and bisulfite sequencing approach used in this study. As DNA methylation changes precede gene expression changes, it is possible that one sampling time point may not be adequate to correlate the changes. In addition, primary and secondary gene expression changes in response to PCB exposure might be under different genetic and epigenetic regulatory mechanisms. Futhermore, the sample size is relatively small and biological variability might be confounding the results. Even though the eRRBS approach used in this study is powerful enough to capture the GC-rich genomic loci, there are several gene promoters that are GC-poor and are not captured using this approach. Future attempts to determine the correlation between DNA methylation and gene expression should consider sampling times carefully and use WGBS to capture majority of the GC loci in the genome.
Among the small number of genes that showed correlation include serotonin receptor (htr1aa), vitellogenin (vtg1), deiodinase 2(dio2), a monoxygenase (ywhag1) and a signaling adaptor molecule (baiap2a) in the liver. These genes play important roles not only in hepatic function but also regulate growth and reproduction. Similarly, the genes that showed correlation in the brain include a neurotransmitter receptor (grm8a), transcription factor (klf2b), chaperones (pfdn5, lrpap1), transmembrane glycoprotein (pmp22a) and a few uncharacterized genes. Some of these genes have been shown to be altered by PCBs, but there is no evidence suggesting their regulation by DNA methylation. Majority of these DMRs are located in the distal promoter regions, suggesting that DMRs are located in the regulatory elements such as enhancers and insulators, important players in epigenetic regulation. Even though distal regulatory elements can be located several kilobases from the core promoter, they can regulate gene expression via loops in the intervening DNA. Functional studies have demonstrated that transcription factor binding can cause demethylation of distal enhancers and activation of gene expression [82]. Similar studies need to be conducted to understand the functional relevance of toxicant-induced DMRs. Recently, thousands of DMRs were identified in the developing zebrafish genome and a significant portion were empirically shown to behave as developmental enhancers [27]. It is plausible that the DMRs identified in this study might also act as enhancers or insulators and regulate gene expression. Future studies should focus on the functional characterization of DMRs using CRISPR-Cas technology-based epigenome editing. The potential candidates for the functional studies would be those that alter gene expression patterns.
Conclusions
In this study, we demonstrated the effect of a potent AHR agonist (PCB126) on tissue-specific DNA methylation and concomitant gene expression changes in zebrafish. It is known that some toxicants can alter DNA methylation, but to our knowledge there have been no reports on toxicant effects on genome-wide DNA methylation and their relationship with gene expression. Our results suggest that the PCB effects on DNA methylation are distinct from changes in gene expression. Lack of correlation between PCB-induced changes in DNA methylation and gene expression suggests that the relationship between methylation and gene expression is dynamic and complex, involving multiple layers of regulation. As DNA methylation influences gene expression by altering the interaction between DNA, chromatin and transcription factors, understanding the chromatin state will provide insights into the mechanisms of action. Recent methods such as ATAC (Assay for Transposase-Accessible Chromatin using sequencing)-seq and NiCE (Nicking enzyme-assisted sequencing)-seq have enabled genome-wide chromatin profiling. Furthermore, time course studies should be conducted to determine the spatio-temporal dynamics of DNA methylation in response to toxicant exposure. Finally, as zebrafish is extensively used as a toxicological model, it is essential to develop database resources like ENCODE and an epigenomics portal to catalog genome-wide studies for future re-analysis and interpretation.
Supplementary Material
Acknowledgements
This work was supported by the National Institute of Health Outstanding New Environmental Scientist Award to NA (NIH R01ES024915) and Woods Hole Center for Oceans and Human Health [National Institutes of Health (NIH) grant P01ES021923 and National Science Foundation Grant OCE-1314642 to M. Hahn, J. Stegeman, NA and SK]. The authors would like to thank the toxicology lab group members at WHOI for their suggestions with data interpretation and Ms. Helena McMonagle for help with manuscript preparation.
Supplementary data
Supplementary data are available at EnvEpig online.
Conflict of interest statement. None declared.
References
- 1. Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R.. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005;33:5868–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ulahannan N, Greally JM.. Genome-wide assays that identify and quantify modified cytosines in human disease studies. Epigenetics Chromatin 2015;8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roforth MM, Farr JN, Fujita K, McCready LK, Atkinson EJ, Therneau TM, Cunningham JM, Drake MT, Monroe DG, Khosla S.. Global transcriptional profiling using RNA sequencing and DNA methylation patterns in highly enriched mesenchymal cells from young versus elderly women. Bone 2015;76:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fritz EL, Rosenberg BR, Lay K, Mihailovic A, Tuschl T, Papavasiliou FN.. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nat Immunol 2013;14:749–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Golzenleuchter M, Kanwar R, Zaibak M, Al Saiegh F, Hartung T, Klukas J, Smalley RL, Cunningham JM, Figueroa ME, Schroth GP.. et al. Plasticity of DNA methylation in a nerve injury model of pain. Epigenetics 2015;10:200–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nilsson E, Larsen Manikkam G, Guerrero-Bosagna MC, Savenkova MI, Skinner MK.. Environmentally induced epigenetic transgenerational inheritance of ovarian disease. PLoS One 2012;7:e36129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guerrero-Bosagna C, Savenkova M, Haque MM, Nilsson E, Skinner MK.. Environmentally induced epigenetic transgenerational inheritance of altered Sertoli cell transcriptome and epigenome: molecular etiology of male infertility. PLoS One 2013;8:e59922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Safe S. Molecular biology of the Ah receptor and its role in carcinogenesis. Toxicol Lett 2001;120:1–7 [DOI] [PubMed] [Google Scholar]
- 9. Mulero-Navarro S, Fernandez-Salguero PM.. New trends in aryl hydrocarbon receptor biology. Front Cell Dev Biol 2016;4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Consales C, Toft G, Leter G, Bonde JP, Uccelli R, Pacchierotti F, Eleuteri P, Jonsson BA, Giwercman A, Pedersen HS.. et al. Exposure to persistent organic pollutants and sperm DNA methylation changes in Arctic and European populations. Environ Mol Mutagen 2016;57:200–9 [DOI] [PubMed] [Google Scholar]
- 11. Itoh H, Iwasaki M, Kasuga Y, Yokoyama S, Onuma H, Nishimura H, Kusama R, Yoshida T, Yokoyama K, Tsugane S.. Association between serum organochlorines and global methylation level of leukocyte DNA among Japanese women: a cross-sectional study. Sci Total Environ 2014;490:603–9 [DOI] [PubMed] [Google Scholar]
- 12. Lee MH, Cho ER, Lim JE, Jee SH.. Association between serum persistent organic pollutants and DNA methylation in Korean adults. Environ Res 2017;158:333–41 [DOI] [PubMed] [Google Scholar]
- 13. Lind L, Penell J, Luttropp K, Nordfors L, Syvanen AC, Axelsson T, Salihovic S, van Bavel B, Fall T, Ingelsson E.. et al. Global DNA hypermethylation is associated with high serum levels of persistent organic pollutants in an elderly population. Environ Int 2013;59:456–61 [DOI] [PubMed] [Google Scholar]
- 14. Rusiecki JA, Baccarelli A, Bollati V, Tarantini L, Moore LE, Bonefeld-Jorgensen EC.. Global DNA hypomethylation is associated with high serum-persistent organic pollutants in Greenlandic Inuit. Environ Health Perspect 2008;116:1547–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bell MR, Hart BG, Gore AC.. Two-hit exposure to polychlorinated biphenyls at gestational and juvenile life stages: 2. Sex-specific neuromolecular effects in the brain. Mol Cell Endocrinol 2016;420:125–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Desaulniers D, Xiao GH, Cummings-Lorbetskie C.. Effects of lactational and/or in utero exposure to environmental contaminants on the glucocorticoid stress-response and DNA methylation of the glucocorticoid receptor promoter in male rats. Toxicology 2013;308:20–33 [DOI] [PubMed] [Google Scholar]
- 17. Desaulniers D, Xiao GH, Leingartner K, Chu I, Musicki B, Tsang BK.. Comparisons of brain, uterus, and liver mRNA expression for cytochrome p450s, DNA methyltransferase-1, and catechol-o-methyltransferase in prepubertal female Sprague-Dawley rats exposed to a mixture of aryl hydrocarbon receptor agonists. Toxicol Sci 2005;86:175–84 [DOI] [PubMed] [Google Scholar]
- 18. Desaulniers D, Xiao GH, Lian H, Feng YL, Zhu J, Nakai J, Bowers WJ.. Effects of mixtures of polychlorinated biphenyls, methylmercury, and organochlorine pesticides on hepatic DNA methylation in prepubertal female Sprague-Dawley rats. Int J Toxicol 2009;28:294–307 [DOI] [PubMed] [Google Scholar]
- 19. Kappil MA, Li Q, Li A, Dassanayake PS, Xia Y, Nanes JA, Landrigan PJ, Stodgell CJ, Aagaard KM, Schadt EE.. et al. In utero exposures to environmental organic pollutants disrupt epigenetic marks linked to fetoplacental development. Environ Epigenet 2016;2:dvv013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walker DM, Goetz BM, Gore AC.. Dynamic postnatal developmental and sex-specific neuroendocrine effects of prenatal polychlorinated biphenyls in rats. Mol Endocrinol 2014;28:99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK.. Dioxin (TCDD) induces epigenetic transgenerational inheritance of adult onset disease and sperm epimutations. PLoS One 2012;7:e46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jonsson ME, Orrego R, Woodin BR, Goldstone JV, Stegeman JJ.. Basal and 3, 3’, 4, 4’, 5-pentachlorobiphenyl-induced expression of cytochrome P450 1A, 1B and 1C genes in zebrafish. Toxicol Appl Pharmacol 2007;221:29–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glazer L, Hahn ME, Aluru N.. Delayed effects of developmental exposure to low levels of the aryl hydrocarbon receptor agonist 3, 3’, 4, 4’, 5-pentachlorobiphenyl (PCB126) on adult zebrafish behavior. Neurotoxicology 2016;52:134–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aluru N. Epigenetic effects of environmental chemicals: insights from zebrafish. Curr Opin Toxicol 2017;6:26–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang L, Zhang J, Wang JJ, Wang L, Zhang L, Li G, Yang X, Ma X, Sun X, Cai J.. et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell 2013;153:773–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Potok ME, Nix DA, Parnell TJ, Cairns BR.. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell 2013;153:759–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee HJ, Lowdon RF, Maricque B, Zhang B, Stevens M, Li D, Johnson SL, Wang T.. Developmental enhancers revealed by extensive DNA methylome maps of zebrafish early embryos. Nat Commun 2015;6:6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chatterjee A, Ozaki Y, Stockwell PA, Horsfield JA, Morison IM, Nakagawa S.. Mapping the zebrafish brain methylome using reduced representation bisulfite sequencing. Epigenetics 2013;8:979–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aluru N, Kuo E, Helfrich LW, Karchner SI, Linney EA, Pais JE, Franks DG.. Developmental exposure to 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin alters DNA methyltransferase (dnmt) expression in zebrafish (Danio rerio). Toxicol Appl Pharmacol 2015;284:142–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goldstone JV, Jonsson ME, Behrendt L, Woodin BR, Jenny MJ, Nelson DR, Stegeman JJ.. Cytochrome P450 1D1: a novel CYP1A-related gene that is not transcriptionally activated by PCB126 or TCDD. Arch Biochem Biophys 2009;482:7–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF.. Stages of embryonic development of the zebrafish. Dev Dyn 1995;203:253–310 [DOI] [PubMed] [Google Scholar]
- 32. Krueger F, Andrews SR.. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011;27:1571–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akalin A, Kormaksson M, Li S, Garrett-Bakelman FE, Figueroa ME, Melnick A, Mason CE.. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol 2012;13:R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang HQ, Tuominen LK, Tsai CJ.. SLIM: a sliding linear model for estimating the proportion of true null hypotheses in datasets with dependence structures. Bioinformatics 2011;27:225–31 [DOI] [PubMed] [Google Scholar]
- 35. Akalin A, Franke V, Vlahovi ek K, Mason CE, Schubeler D.. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics 2015;31:1127–9 [DOI] [PubMed] [Google Scholar]
- 36. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 2010;28:495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Andrews S. FastQC: a quality control tool for high throughput sequence data, 2010, http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 38. Aluru N, Karchner SI, Glazer L.. Early life exposure to low levels of AHR agonist PCB126 (3, 3’, 4, 4’, 5-pentachlorobiphenyl) reprograms gene expression in adult brain. Toxicol Sci 2017;160(2):386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L.. et al. Ensembl 2016. Nucleic Acids Res 2016;44:D710–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anders S, Pyl PT, Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson MD, McCarthy DJ, Smyth GK.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J.. g: profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Res 2016;44:W83–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Akalin A, Garrett-Bakelman FE, Kormaksson M, Busuttil J, Zhang L, Khrebtukova I, Milne TA, Huang Y, Biswas D, Hess JL.. et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet 2012;8:e1002781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chatterjee A, Stockwell PA, Horsfield JA, Morison IM, Nakagawa S.. Base-resolution DNA methylation landscape of zebrafish brain and liver. Genom Data 2014;2:342–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamstra JH, Sales LB, Alestrom P, Legler J.. Differential DNA methylation at conserved non-genic elements and evidence for transgenerational inheritance following developmental exposure to mono(2-ethylhexyl) phthalate and 5-azacytidine in zebrafish. Epigenetics Chromatin 2017;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang C, Hoshida Y, Sadler KC.. Comparative epigenomic profiling of the DNA methylome in mouse and zebrafish uncovers high interspecies divergence. Front Genet 2016;7:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yin C. Molecular mechanisms of Sox transcription factors during the development of liver, bile duct, and pancreas. Semin Cell Dev Biol 2017;63:68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi TY, Ninov N, Stainier DY, Shin D.. Extensive conversion of hepatic biliary epithelial cells to hepatocytes after near total loss of hepatocytes in zebrafish. Gastroenterology 2014;146:776–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Andreasen EA, Mathew LK, Tanguay RL.. Regenerative growth is impacted by TCDD: gene expression analysis reveals extracellular matrix modulation. Toxicol Sci 2006;92:254–69 [DOI] [PubMed] [Google Scholar]
- 50. Mathew Lk, Andreasen EA, Tanguay RL.. Aryl hydrocarbon receptor activation inhibits regenerative growth. Mol Pharmacol 2006;69:257–65 [DOI] [PubMed] [Google Scholar]
- 51. Hofsteen P, Plavicki J, Johnson SD, Peterson RE, Heideman W.. Sox9b is required for epicardium formation and plays a role in TCDD-induced heart malformation in zebrafish. Mol Pharmacol 2013;84:353–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xiong KM, Peterson RE, Heideman W.. Aryl hydrocarbon receptor-mediated down-regulation of sox9b causes jaw malformation in zebrafish embryos. Mol Pharmacol 2008;74:1544–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Garcia GR, Goodale BC, Wiley MW, La Du JK, Hendrix DA, Tanguay RL.. In vivo characterization of an AHR-dependent long noncoding RNA required for proper Sox9b expression. Mol Pharmacol 2017;91:609–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kaido T, Oe H, Yoshikawa A, Okajima A, Imamura M.. Expressions of molecules associated with hepatocyte growth factor activation after hepatectomy in liver cirrhosis. Hepatogastroenterology 2004;51:547–51 [PubMed] [Google Scholar]
- 55. Okajima A, Miyazawa K, Naitoh Y, Inoue K, Kitamura N.. Induction of hepatocyte growth factor activator messenger RNA in the liver following tissue injury and acute inflammation. Hepatology 1997;25:97–102 [DOI] [PubMed] [Google Scholar]
- 56. Itoh N, Nakayama Y, Konishi M.. Roles of FGFs as paracrine or endocrine signals in liver development, health, and disease. Front Cell Dev Biol 2016;4:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fernandez LP, Lopez-Marquez A, Santisteban P.. Thyroid transcription factors in development, differentiation and disease. Nat Rev Endocrinol 2015;11:29–42 [DOI] [PubMed] [Google Scholar]
- 58. Bano A, Chaker L, Plompen EP, Hofman A, Dehghan A, Franco OH, Janssen HL, Darwish Murad S, Peeters RP.. Thyroid function and the risk of nonalcoholic fatty liver disease: the Rotterdam study. J Clin Endocrinol Metab 2016;101:3204–11 [DOI] [PubMed] [Google Scholar]
- 59. Byrne JJ, Carbone JP, Hanson EA.. Hypothyroidism and abnormalities in the kinetics of thyroid hormone metabolism in rats treated chronically with polychlorinated biphenyl and polybrominated biphenyl. Endocrinology 1987;121:520–7 [DOI] [PubMed] [Google Scholar]
- 60. Goldey ES, Kehn LS, Lau C, Rehnberg GL, Crofton KM.. Developmental exposure to polychlorinated biphenyls (Aroclor 1254) reduces circulating thyroid hormone concentrations and causes hearing deficits in rats. Toxicol Appl Pharmacol 1995;135:77–88 [DOI] [PubMed] [Google Scholar]
- 61. Han DY, Kang SR, Park OS, Cho JH, Won CK, Park HS, Park KI, Kim EH, Kim GS.. Hypothyroidism induced by polychlorinated biphenyls and up-regulation of transthyretin. Bull Environ Contam Toxicol 2010;84:66–70 [DOI] [PubMed] [Google Scholar]
- 62. Pearce EN, Braverman LE.. Environmental pollutants and the thyroid. Best Pract Res Clin Endocrinol Metab 2009;23:801–13 [DOI] [PubMed] [Google Scholar]
- 63. Safe S. Hydroxylated polychlorinated biphenyls (PCBs) and organochlorine pesticides as potential endocrine disruptors In: Metzler M. (ed.), Endocrine Disruptors–Part I. Berlin, Heidelberg: Springer Berlin Heidelberg, 2001, 155–67 [Google Scholar]
- 64. Hisada A, Shimodaira K, Okai T, Watanabe K, Takemori H, Takasuga T, Koyama M, Watanabe N, Suzuki E, Shirakawa M.. et al. Associations between levels of hydroxylated PCBs and PCBs in serum of pregnant women and blood thyroid hormone levels and body size of neonates. Int J Hyg Environ Health 2014;217:546–53 [DOI] [PubMed] [Google Scholar]
- 65. Mani SR, Juliano CE.. Untangling the web: the diverse functions of the PIWI/piRNA pathway. Mol Reprod Dev 2013;80:632–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Czech B, Hannon GJ.. One loop to rule them all: the ping-pong cycle and piRNA-guided silencing. Trends Biochem Sci 2016;41:324–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stefani G, Slack FJ.. Small non-coding RNAs in animal development. Nat Rev Mol Cell Biol 2008;9:219–30 [DOI] [PubMed] [Google Scholar]
- 68. Roy J, Sarkar A, Parida S, Ghosh Z, Mallick B.. Small RNA sequencing revealed dysregulated piRNAs in Alzheimer’s disease and their probable role in pathogenesis. Mol Biosyst 2017;13:565–76 [DOI] [PubMed] [Google Scholar]
- 69. Chandran R, Mehta SL, Vemuganti R.. Non-coding RNAs and neuroprotection after acute CNS injuries. Neurochem Int 2017;111:12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Miousse IR, Chalbot MC, Lumen A, Ferguson A, Kavouras IG, Koturbash I.. Response of transposable elements to environmental stressors. Mutat Res Rev Mutat Res 2015;765:19–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goldstone HM, Tokunaga S, Schlezinger JJ, Goldstone JV, Stegeman JJ.. EZR1: a novel family of highly expressed retroelements induced by TCDD and regulated by a NF-kappaB-like factor in embryos of zebrafish (Danio rerio). Zebrafish 2012;9:15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liebner S, Czupalla CJ, Wolburg H.. Current concepts of blood-brain barrier development. Int J Dev Biol 2011;55:467–76 [DOI] [PubMed] [Google Scholar]
- 73. Reis M, Liebner S.. Wnt signaling in the vasculature. Exp Cell Res 2013;319:1317–23 [DOI] [PubMed] [Google Scholar]
- 74. Filbrandt CR, Wu Z, Zlokovic B, Opanashuk L, Gasiewicz TA.. Presence and functional activity of the aryl hydrocarbon receptor in isolated murine cerebral vascular endothelial cells and astrocytes. Neurotoxicology 2004;25:605–16 [DOI] [PubMed] [Google Scholar]
- 75. Reichard JF, Dalton TP, Shertzer HG, Puga A.. Induction of oxidative stress responses by dioxin and other ligands of the aryl hydrocarbon receptor. Dose Response 2005;3:306–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nault R, Fader KA, Lydic TA, Zacharewski TR.. Lipidomic evaluation of aryl hydrocarbon receptor-mediated hepatic steatosis in male and female mice elicited by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin. Chem Res Toxicol 2017;30:1060–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Aluru N, Karchner SI, Glazer L.. Early life exposure to low levels of AHR agonist PCB126 (3, 3’, 4, 4’, 5-Pentachlorobiphenyl) reprograms gene expression in adult brain. Toxicol Sci 2017;160:386–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhou X, Liao WJ, Liao JM, Liao P, Lu H.. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol 2015;7:92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lim YC, Li J, Ni Y, Liang Q, Zhang J, Yeo GSH, Lyu J, Jin S, Ding C.. A complex association between DNA methylation and gene expression in human placenta at first and third trimesters. PLoS One 2017;12:e0181155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schachtschneider KM, Liu Y, Rund LA, Madsen O, Johnson RW, Groenen MA, Schook LB.. Impact of neonatal iron deficiency on hippocampal DNA methylation and gene transcription in a porcine biomedical model of cognitive development. BMC Genomics 2016;17:856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Day SE, Coletta RL, Kim JY, Campbell LE, Benjamin TR, Roust LR, De Filippis EA, Dinu V, Shaibi GQ, Mandarino LJ.. et al. Next-generation sequencing methylation profiling of subjects with obesity identifies novel gene changes. Clin Epigenetics 2016;8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wiench M, John S, Baek S, Johnson TA, Sung MH, Escobar T, Simmons CA, Pearce KH, Biddie SC, Sabo PJ.. et al. DNA methylation status predicts cell type-specific enhancer activity. Embo J 2011;30:3028–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.