

Abstract
Motivation
Proteomics profiling is increasingly being used for molecular stratification of cancer patients and cell-line panels. However, systematic assessment of the predictive power of large-scale proteomic technologies across various drug classes and cancer types is currently lacking. To that end, we carried out the first pan-cancer, multi-omics comparative analysis of the relative performance of two proteomic technologies, targeted reverse phase protein array (RPPA) and global mass spectrometry (MS), in terms of their accuracy for predicting the sensitivity of cancer cells to both cytotoxic chemotherapeutics and molecularly targeted anticancer compounds.
Results
Our results in two cell-line panels demonstrate how MS profiling improves drug response predictions beyond that of the RPPA or the other omics profiles when used alone. However, frequent missing MS data values complicate its use in predictive modeling and required additional filtering, such as focusing on completely measured or known oncoproteins, to obtain maximal predictive performance. Rather strikingly, the two proteomics profiles provided complementary predictive signal both for the cytotoxic and targeted compounds. Further, information about the cellular-abundance of primary target proteins was found critical for predicting the response of targeted compounds, although the non-target features also contributed significantly to the predictive power. The clinical relevance of the selected protein markers was confirmed in cancer patient data. These results provide novel insights into the relative performance and optimal use of the widely applied proteomic technologies, MS and RPPA, which should prove useful in translational applications, such as defining the best combination of omics technologies and marker panels for understanding and predicting drug sensitivities in cancer patients.
Availability and implementation
Processed datasets, R as well as Matlab implementations of the methods are available at https://github.com/mehr-een/bemkl-rbps.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Large-scale profiling studies using multiple omics technologies are providing increasingly accurate views of the molecular and genomic landscapes of many cancer subtypes, with the eventual aim to improve selection of treatment strategies for the distinct cancer subtypes (so-called stratified medicine or precision oncology). However, treatment response-predictive biomarkers are currently available only for a few FDA-approved therapies (Meric-Bernstam et al., 2015). In particular, despite of many large-scale cancer-sequencing efforts, only a few genomically informed personalized cancer therapies have made it to the clinical practice for specific cancer classes. Some of the best-known examples of clinically actionable genomic alterations include HER2 amplification in breast and gastric cancers, EGFR mutations and ALK fusions in non-small cell lung cancer (NSCLC), and BRAF V600 mutations in melanoma (Druker et al., 2006; Flaherty et al., 2012; Maemondo et al., 2010; Shaw et al., 2013). For most cancer types or genomic alterations, however, evidence is either absent or insufficient to support clinical implementation of biomarker-based therapies.
Despite their critical role in the pathophysiology of many cancers, genomic alterations (point mutations or copy number variation, CNV) provide only one layer of biological information, and it still remains unclear how much the other layers of molecular information could contribute to drug response predictions. To this end, NCI/DREAM7 challenge carried out an extensive comparison of the predictive power of currently available genomic, molecular and epigenetic profiles in the task of predicting the sensitivity of 28 drugs across 53 breast cancer cell lines (Costello et al., 2014). The omics technologies included genome-wide CNV, exome/RNA-seq, DNA methylation and microarray gene expression arrays, as well as reverse phase protein arrays (RPPA). Even though the NCI/DREAM7 RPPA dataset covered only 66 proteins (Costello et al., 2014), it was shown to provide second-largest contribution to predictive power, after the genome-wide transcriptomics profiles, suggesting that proteomic profiling is important for drug sensitivity prediction, at least in the studied subtypes of breast cancer cell lines.
Recently, mass spectrometry (MS)-based proteomic profiling is increasingly being carried out in multiple human tissues and cell types (Gholami et al., 2013; Kim et al., 2014; Lawrence et al., 2015; Wilhelm et al., 2014). Compared to RPPA technology, which allows quantitative measurement of protein abundance in a large number of biological samples when high-quality antibodies are available (Gautam et al., 2016; Li et al., 2013), MS-based proteomics provide opportunity for more global, quantitative profiling of post-translational modifications, in terms of yielding proteome-wide information about cancer cell signaling activity that is not accessible by genomics or transcriptomics alone. Accordingly, it has been shown that MS-based proteomic and phosphoproteomics profiles enable identification of functional differences between cancer subtypes (Casado et al., 2013; Lawrence et al., 2015; Tyanova et al., 2016), as well as protein markers and pathway activities associated with drug sensitivity and mechanisms of drug resistance (Gholami et al., 2013; Lawrence et al., 2015; Wilhelm et al., 2014). However, how to best use proteomics data in predictive modeling remains currently unknown.
To systematically investigate the predictive power gained from large-scale proteomics, we carried out, to our knowledge, the first pan-cancer, multi-omics comparative investigation of the relative contribution and optimal use of RPPA and MS-based proteomics profiles to predicting the sensitivity of both FDA-approved chemotherapeutics and molecularly targeted compounds in 58 cell lines spanning over nine cancer types. To assess the predictive power of the omics datasets, either separately or in combinations, we used the predictive model that was found to perform best in the NCI/DREAM7 challenge, namely the Bayesian Efficient Multiple Kernel Learning (BEMKL) (Gönen, 2012). In this study, we focus on predictive modeling of individual and combinations of omics profiles, rather than comparing the prediction performance of various machine learning models. We demonstrate that the global MS-based proteomic profiling (8113 proteins) provides improved predictive power, but only when treated optimally and combined with the other omics datasets (gene and miRNA expression, point mutations and CNV). However, the maximal predictive power was obtained when also the RPPA dataset (162 proteins) was combined into the BEMKL model, suggesting a complementary signal from these two proteomic technologies for drug sensitivity prediction.
2 Materials and methods
2.1 Predictive modeling
2.1.1 BEMKL
We used the state-of-the-art BEMKL model (Gönen, 2012) for drug response prediction from multi-omics datasets, integrated in a biologically meaningful way. BEMKL was the top-performing model in the NCI/DREAM7 drug sensitivity prediction challenge (Costello et al., 2014), among various classes of machine learning models. BEMKL (Fig. 1, grey area) belongs to a class of nonlinear regression models, which employs kernelized regression, multi-view and multi-task learning and Bayesian inference to solve the drug responses prediction problem.
Fig. 1.

Data modeling approach, employing BEMKL method, applied on NCI60 genomics (point mutations and CNV), molecular (gene and miRNA expression) and proteomics (MS and RPPA) profiles across 58 pan-cancer cell lines to predict drug response of selected drugs. The BEMKL method learns the multi-view kernel weights to form a joint kernelized representation of the data which is used with the multi-task drug weights to model the response profile across the cell lines to each individual drug (the outcome matrix at the right)
To predict the drug response of an unseen cell line , BEMKL models the datasets using a kernel-based decision function:
| (1) |
where kernel captures pair-wise similarities between samples (here, cell lines) in the omics profiles, while a and b represent the unknown weight vector for samples and the error term, respectively. The kernel function is used to calculate pair-wise similarities, such that , given independent identically distributed training data , where N = number of cell lines and D = number of features. The kernelized regression formulation of Equation (1) models nonlinear relationships between cell-lines by capturing similarities from omics profiles (called views) to predict drug responses.
BEMKL employs multiple kernel learning (MKL) (Gönen and Alpaydin, 2011) to simultaneously integrate feature information coming from multiple views as kernels and effectively yields an increased signal-to-noise ratio and predictive accuracy. The combined kernel is calculated as a weighted sum of M input kernels as . Thus, kernel in Equation (1) can be replaced with a combined kernel using MKL algorithm:
| (2) |
where e represents the vector of kernel weights and represents the kernels for each view. The view specific kernel weights are learned based on the view’s relevance for the response predictions, making it possible for the model to efficiently integrate multiple heterogeneous views by learning their joint weighted representation.
Additionally, BEMKL leverages upon multi-task learning (MTL) to simultaneously model drug response predictions across multiples drugs (also referred to as tasks, where drug response prediction of an individual drug alone is a single task t). Specifically, the model assumes that the kernel weights em are shared across all the tasks. The distributional assumptions of the model are defined and explained below as:
Here, denotes normal distribution with mean μ and covariance Σ, while, is gamma distribution with shape parameter α and the scale parameter β. For N training samples and M input kernels, Km represents the N × N kernel matrices for , while G represents the M × N matrix of intermediate outputs. Parameters a, b denote the weight vectors, whereas e and w are vectors of kernel weights and their priors and y is a vector of outputs. and ε represent the precision parameters for intermediate and target outputs.
To summarize, BEMKL can be seen as a two-step procedure. In the first step, intermediate variables for each task are estimated from view-specific kernels, using weight vector for samples (here, cell lines). In the second step, intermediate variables are combined to estimate the output (drug-response) matrix, using the vector of shared kernel weights across selected set of drugs.
BEMKL is implemented in a Bayesian formulation to overcome sample specific uncertainty in learning the model parameters, attributing each parameter to a specific probability distribution. Since, the exact inference is intractable and Gibbs sampling requires rather large computational resources, the model has been formulated using deterministic variational Bayesian (VB) approximation for efficient inference of the model parameters resulting into point estimates for the posterior mean and covariance of the model parameters. Details of constraints applied on MT-MKL algorithm used in BEMKL and inference of approximate posterior distributions for BEMKL can be found in the original paper (Costello et al., 2014; Gönen, 2012).
2.1.2 Rule based protein selection
In order to identify combinations of protein abundances that best explain the drug sensitivity profiles (and which eventually could be used as predictive biomarkers for clinical translation), we carried out a two-step procedure. First the normalized abundance of D proteins from the MS and RPPA datasets were binarized to represent up and down regulated protein activity for , where Pj is a vector over the samples (here, cell lines). In the absence of a ground truth, we used scores above mean as up-regulated and below mean as down-regulated. Pairwise interactions between the binarized protein abundances were then computed as
| (3) |
where is the interaction between proteins j and l.
In the second step, an exhaustive search was carried out among all the pairwise interactions between the proteins () for identifying such marker combinations whose expression explains drug sensitivity profile. Specifically, each pairwise interaction was scored as a fraction of normalized mean of (where GI50 refers to the concentration required to inhibit 50% of maximal cell growth) values for drug t over N cell lines:
| (4) |
where is average for cell lines , and is average for cell lines for the given drug t. The scaling factor, , emphasizes the interactions with high abundances across multiple cell lines. As a result, interactions with rule based protein selection (RBPS) scores close to 0 indicate that the corresponding protein abundances do not explain the drug sensitivity profile, while those close to 1 identify protein activity patterns that perfectly explain the drug sensitivity outcome.
2.2 Publically available datasets
The primary dataset comprised of genomic, molecular and proteomics profiles of 59 human pan-cancer cell lines from the National Cancer Institute (NCI), also referred to as NCI-60 cell lines panel (Shoemaker, 2006), along with their drug responses reported as () values (Supplementary Fig. S1). The other dataset comprised of 53 breast cancer cell lines, extracted from NCI/DREAM7 project (Costello et al., 2014), where along with the omic profiles, pathway and other prior biological knowledge was also exploited to predict drug responses. Clinical data from The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov/docs/publications/tcga/, http://www.cbioportal.org) patients was used to investigate the clinical relevance of the RBPS-selected proteins.
2.3 Experimental setup
In NCI-60 cell lines, six omics views were integrated (Supplementary Table S1), for 58 cell lines with omics profiles available. Frequent feature-wise data missingness was observed in MS-based proteomics dataset (on average 55% partially measured proteins across NCI-60 cell-lines panel). To avoid data-sparsity-induced noise issues, only the completely available MS-features (505 proteins) were considered. We used scores as drug sensitivity responses, averaged over multiple five dose-assays, tested over different concentration ranges, for selected sets of 47 FDA-approved cytotoxic drugs and 24 targeted agents with known targeted mechanism of action (MoA, Supplementary Tables S2 and S3). Owing to the multi-view and multi-task nature of BEMKL, the outcome of the model is a matrix of scores with cell lines as rows and selected set of drugs as columns. Although we cannot assume that an in vitro estimate is predictive of an in vivo response to the same drug, we chose to use in the current work as it was also used in the original NCI/DREAM7 challenge, so that we can compare our results also against the original NCI/DREAM7 results. In NCI/DREAM7 cell lines, all 22 views were integrated into the BEMKL model to predict drug sensitivities for 28 drugs across 53 breast cancer cell lines. The sample size in DREAM7 data was further reduced to 30 cell lines considering the availability of detailed RPPA-based proteomics data from The Cancer Protein Atlas (TCPA, Li et al., 2013, http://tcpaportal.org/tcpa/index.html).
In the predictive modeling, drug responses were mean-normalized, whereas all the other omic views were z-transformed. In BEMKL model, Gaussian kernels were used for real-valued views and Jaccard similarity coefficients for binary-valued views. The prior hyperparameter values were set analogous to those in the NCI/DREAM7 study (Costello et al., 2014).
To evaluate the predictive accuracy, we performed leave-one-out cross validation (LOO-CV) with both of the drug sets, repeated three times and computed average prediction accuracies. We used several metrics to compare the model performance, including drug-wise Spearman’s correlation, Root Mean Square Error (RMSE), concordance index and area under curve (AUC, full results are provided in Supplementary Tables S4 and S5).
The purpose of this work was to compare the predictive performance of the two proteomic platforms, MS and RPPA, using the state-of-the-art BEMKL method, rather than comparison of several machine learning methods. However, we additionally performed a comparison, using standard, single-task, kernel-based SVM regression method, since SVM-based kernel methods performed next to the winning BEMKL method in the original NCI/DREAM7 Drug Sensitivity Prediction Challenge (Costello et al., 2014). The outcome, of SVM, was drug-specific scores as vector , N = number of cell lines. To mimic the multi-view nature of BEMKL, omics profiles were aggregated feature-wise in SVM. Further information on the datasets and experimental setup is available in Supplementary Material.
3 Results
We first evaluated the relative performance of the various omics profiles in the NCI-60 datasets, both individually and in combinations, for the drug sensitivity predictions. In these comparisons, we primarily investigated the added value of the proteomics profiles, as compared to the other omics datasets (gene and miRNA expressions, point mutations and CNV, collectively referred below to as GM4 views, whereas adding proteomics profiles to the GM4 views are referred to as GMP6 views).
3.1 Cytotoxic drugs
In the drug set of 47 FDA-approved, cytotoxic agents, we observed that the completely measured MS-based proteomics dataset (505 proteins) significantly improved the response prediction beyond the GM4 datasets (P < 0.01, one-sided, paired t-test; Fig. 2A). The original MS data (8113 proteins), with 55% missing value rate, did not lead to increased prediction power, having predictive accuracy similar to the limited RPPA data (162 proteins) (Fig. 2A). The missing value distribution of MS data is highly non-uniform (Supplementary Fig. S2), since missing values depend on the MS levels themselves. Even though the kernel-based models can deal with noise to some degree, the current MS data has so many missing values that the quite extreme removal of all proteins with missing values led to significant improvement in the results (Fig. 2). However, the maximal predictive power for the cytotoxic drugs was obtained after adding both the completely measured MS and the RPPA dataset, combined with further focusing on the protein abundance of the 42 overlapping COSMIC cancer census genes from completely measured MS data (P < 0.01, one-sided, paired t-test; Fig. 2A). These results not only demonstrated the importance of the proteomic data for the prediction accuracy, but also the critical role of missing MS values and their treatment in predictive modeling to minimize the effect of data-sparsity-induced noise, as well as the complementary nature of the MS and RPPA profiles.
Fig. 2.

Average Spearman correlation, with standard error of the mean, between experimental and predicted drug sensitivity levels over 58 pan-cancer NCI-60 cell lines, using different omics data combinations for selected set of (A) 47 cytotoxic and (B) 24 targeted compounds. The red horizontal line indicates the baseline GM4 prediction accuracy. Statistical significance of the difference against the GM4 predictions was assessed with one-sided, paired t-test for the cytotoxic drugs and Wilcoxon signed-rank test for the targeted compounds. Statistical testing method was chosen based on the normality of the drug response distribution with Chi-square test
Individually, the RPPA and MS datasets alone resulted in similar predictive accuracies, in between of those from genomics profiles (point mutations and CNV) and molecular profiles (gene and miRNA expression). We note that even if the point mutations alone led to negative individual accuracy (Supplementary Fig. S3A), combining the exome-seq-based mutations with the other omics profiles, using the multi-view approach, makes it possible to extract the shared signal that contributes positively to the combined accuracy (Supplementary Tables S4 and S5). A similar trend was observed among the targeted compounds, for which the use of completely measured MS profiles was even more important (Supplementary Fig. S3B); however, even though the overall improvement of adding the proteomics profiles was significant (P < 0.03, Wilcoxon signed-rank test; Fig. 2B), there appeared much more variability in the prediction accuracies among the 24 targeted compounds (described in the next section).
We next explored among the individual cytotoxic drugs the effects of adding proteomics profiles on their response prediction (Fig. 3). Although, on average, the prediction performance increased 75% across the whole set of 47 cytotoxic drugs, we observed marked inter-drug differences; drugs with most and least improvement in their predictive accuracy are listed boldfaced in Supplementary Table S2 (marked in bold in Fig. 3). We note that mode of action (MoA) information of the cytotoxic drugs was not a determinant of their predictability, with or without proteomics data (Supplementary Fig. S6B).
Fig. 3.

Drug-specific comparison between the baseline GM4 and best GMP6 predictions, based on average Spearman correlation, for 47 cytotoxic drugs. The dotted vertical line distinguishes drugs with improved prediction using proteomics data (left-hand side). The dotted horizontal line indicates well-predicted drugs (correlation 0.5). Boldfacing marks the example drugs selected for further study (Supplementary Table S2). MoA details are shown in Supplementary Figure S6B
Doxorubicin is a clinically used cytotoxic drug therapy for multiple cancers, however, our analyses showed its best efficacy in leukemia, melanoma, breast and lung cancer NCI-60 cell lines (Fig. 4A). The ROC-AUC calculation also supported the added value of proteomics profiles along with other genomic and molecular views for doxorubicin response prediction (AUC increased from 0.58 to 0.61). Asparaginase is another positive example, with increased sensitivity especially in leukemia, renal and prostate cancers cells (Supplementary Fig. S7B). Procarbazine is a negative example, in which proteomics profiles did not improve the sensitivity predictions (Supplementary Fig. S8A). However, ranking of the cell lines for procarbazine sensitivity shows that the drug response is dominated by the cancer type, rather than by their molecular or genomic features. This explains why the pan-cancer model cannot predict well the cell-specific procarbazine response patterns.
Fig. 4.

Ranked cell-specific prediction of (A) doxorubicin and (B) midostaurin response using the baseline GM4 and best GMP6 model. Value x = 0 is set to equal rank for both the measured and predicted drug responses. Asterisk indicates the case closer to the measured rank for a particular cell line. Color bar quantifies the measured cell line drug sensitivity responses (GI50). ROC-AUC calculation was based on quantile value of 0.55
3.2 Targeted compounds
Compared to the cytotoxic drugs, the set of 24 molecularly targeted compounds showed higher variability in their predictive accuracy with and without proteomics (Fig. 5). The maximal predictive power was obtained after focusing only on 55 overlapping COSMIC cancer census genes from both the completely measured MS (42 overlapping genes) and the RPPA (13 overlapping genes) datasets. In particular, HSP90 inhibitors, antifolates (folic acid antagonists, Df), MEK and mTOR inhibitors showed improved performance with the optimal treatment of the proteomics data (left-hand side compounds in Fig. 5). However, similar to the cytotoxic drugs, the predictability of the drug responses was seriously limited by the high proportion of missing MS and RPPA data for many of the primary targets (grey squares in Supplementary Fig. S9B). Midostaurin, selumetinib and staurosporine were selected as positive examples, which showed improved compounds response predictions with proteomics data, whereas alvocidib and lapatinib are negative examples, i.e. compounds that have better prediction accuracy when modeled without the proteomics datasets (Supplementary Table S3, boldfaced).
Fig. 5.
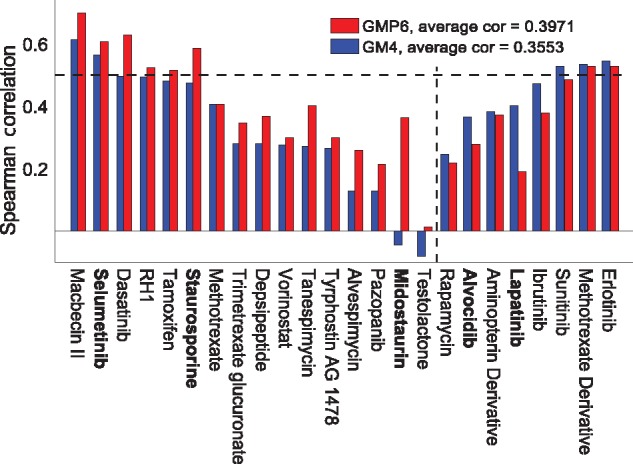
Drug-specific comparison between baseline GM4 and best GMP6 predictions, based on average Spearman correlation, for 24 targeted compounds. The dotted vertical line distinguishes compounds with improved prediction using proteomics data (left-hand side). The dotted horizontal line indicates well-predicted drugs (correlation 0.5). Boldfacing marks the example compounds selected for further study (Supplementary Table S3). Primary drug target details are available in Supplementary Figure S9B
Midostaurin, a multi-targeted kinase inhibitor, showed especially high relative improvement after adding the proteomics profiles (Fig. 4B). Multiple cancer types among the NCI-60 cell line panel, including TNBC, leukemia, CNS, melanoma, prostate and ovarian cancer, showed high sensitivity to midostaurin. It has been suggested that midostaurin suppresses the proliferation of TNBC cells through inhibition of the Aurora kinase family, especially AURKA (Kawai et al., 2015), which is breast tumor-amplified kinase involved in the phosphorylation of BRCA1 in breast cancer cells (Ouchi et al., 2004). Further, midostaurin received recently a Breakthrough Therapy designation from the FDA for newly diagnosed FLT3-mutated acute myeloid leukemia (AML). In leukemic cells, midostaurin inhibits c-KIT and FLT3 kinases (Gallogly and Lazarus, 2016; Stein and Tallman, 2016).
To further study the role of the primary targets in sensitivity predictions, we selected seven targeted compounds (macbecin II, selumetinib, tamoxifen, tanespimycin, alvespimycin alvocidib and lapatinib), with completely measured primary target proteins in the RPPA and/or MS datasets. Removing their primary target proteins from the proteomics datasets resulted in systematic decrease in the accuracy of the response predictions for each of these compounds (P < 0.01, one-sided, paired t-test; Supplementary Fig. S10). This result further supports the importance of having detailed proteomics data for all the primary targets for understanding of their MoA, and for improved response prediction of targeted compounds in cancer cells.
As a specific example, we focused on lapatinib, a clinically approved dual EGFR-ERBB2 tyrosine kinase inhibitor for breast cancers, especially for women with HER2+ breast cancer. Since we took here the pan-cancer modeling approach, the overall association between the lapatinib response and proteomics profile takes precedence over the selective lapatinib sensitivity in a specific cancer subtypes. This led to a poor overall correlation between ERBB2 and lapatinib response across all the 58 cell lines considered in the model (Fig. 6, solid black line). However, when focusing on the breast and ovarian cancer cell lines only, we observed a significant positive correlation (Fig. 6, dotted line), supporting the role of ERBB2 as a predictive biomarker for lapatinib sensitivity. The absence of any HER2+ breast cancer cell lines in the NCI-60 cell line panel also partly explains the poor prediction accuracy for lapatinib.
Fig. 6.
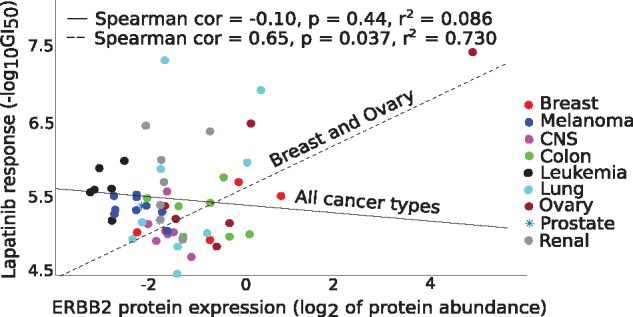
Correlation between measured lapatinib response and ERBB2 protein abundance (available in RPPA only) across all 58 cell lines (solid black line), and in breast and ovarian cancer cells (dotted line). ERBB2 is a known molecular predictor of lapatinib response in breast cancer patients. Statistical significance was assessed with two-sided t-test
3.3 Predictive biomarkers
Finally, we applied the RBPS algorithm to identify the optimal combination of biomarkers explaining response of the seven targeted compounds (Table 1). Pair-wise interactions were computed between all the 55 proteins (42 proteins from the completely measured MS and 13 proteins from RPPA dataset). For four of the compounds (macbecin II, alvespimycin, lapatinib and tanespimycin), the primary targets were identified amongst the top 10 rules explaining their sensitivity, again showing the importance of primary target abundance for response prediction. For those compounds whose primary targets did not appear to have a relationship with their response, when studied through RBPS, the top first rule with most abundantly expressed protein was listed in Table 1. For instance, NPM1 was identified as the most informative protein for selumetinib sensitivity, even though it is not its primary target. NPM1 has been associated with initiation of AML (Noren et al., 2016), and the cell line response results also identified HL60 leukemic cell line as the most sensitive cell line (Supplementary Fig. S11A). Below, we investigate the clinical relevance of both the target and non-target markers for selected compounds.
Table 1.
Optimal protein combinations identified using the RBPS algorithm for explaining the response of the selected compounds
| Drug name | Protein selection | RBPS score |
|---|---|---|
| Alvespimycin | HSP90AB1UP OR HSP90AA1DOWN | 0.74 |
| Macbecin II | HSP90AB1UP OR HSP90AA1DOWN | 0.90 |
| HSP90AB1 UP OR RHOADOWN | 0.86 | |
| Selumetinib | NPM1 UP OR U2AF1DOWN | 0.93 |
| Tamoxifen | SF3B1UP OR DDX5DOWN | 0.89 |
| Tanespimycin | HSP90AB1UP OR HSP90AA1DOWN | 0.81 |
| Lapatinib | ERBB2UP | 0.96 |
| Alvocidib | NPM1UP OR FHDOWN | 0.88 |
Note: For instance, alvespimycin response can be explained accurately when HSP90AB1 is up-regulated or HSP90AA1 is down-regulated. For macbecin II, alvespimycin and tanespimycin, the primary targets (bold) were identified amongst the top 10 rules. For lapatinib, the primary target was identified only when modeled for breast and ovarian cancers (see Fig. 6).
Tamoxifen is a commonly used hormonal therapy for breast cancers. Based on the TCGA patient data, the tamoxifen-predictive RBPS rule (SF3B1UP OR DDX5DOWN) was associated with poor survival in breast cancer patients (P = 0.0067, log-rank test; Supplementary Fig. S13A), even though neither of these non-target proteins showed significant effect on survival when tested individually. Further support for these RBPS-selected biomarker proteins comes from a recent study that showed high levels of SF3B1 in tamoxifen-resistant ER+ breast cancer cells (Gökmen-Polar et al., 2015), suggesting MoA of tamoxifen through splicing factor 3b subunits. Further, DDX5, which is required for DNA regulation and cell proliferation, has been found overexpressed in breast cancer cells, and suggested as a novel therapeutic target for breast cancer treatment (Mazurek et al., 2012)
Similarly, the RBPS rule (HSP90AB1UP OR HSP90AA1DOWN) for the three HSP90 inhibitors, alvespimycin, tanespimycin and macbecin II, was associated in TCGA data with poor survival in papillary renal-cell carcinoma cancer (PRCC) patients (P = 0.023, log-rank test; Supplementary Fig. S13B). A recent study linked high expression of HSP90 in clear-cell renal cell carcinoma (Massari et al., 2014). This suggests the prognostic role of HSP90 in renal cancer, although deeper understanding is still lacking for specific renal cancer subtypes. Interestingly, RBPS also selected both target and non-target protein markers for macbecin II (HSP90 inhibitor) and alvocidib (multi-serine threonine cyclin-dependent kinase inhibitor), associated with poor survival in PRCC patients: HSP90AB1UP (P = 0.0014, log-rank test), RHOADOWN (P = 0.0064, log-rank test), NPM1UP (P = 0.036, log-rank test) and FHDOWN (P = 0.044, log-rank test; Supplementary Fig. S13C–F). Previous studies have also implicated these proteins or related pathways in renal cancer (Cheng et al., 2016; Llamas-Velasco et al., 2016). Our cell line response predictions for alvocidib also identified renal cancer cell lines as most sensitive and well predicted with proteomics (Supplementary Fig. S12A).
4 Discussion
Global proteomic profiling is increasingly being carried out both in cancer cell line panels as well as in patient-derived samples to provide insights into cancer type classification and potential treatment options. However, systematic assessment of the predictive power of proteomics for a wide spectrum of drug compounds and cancer types has been lacking. To that end, we carried out the first pan-cancer, multi-omics comparative analysis of the relative performance of two proteomic technologies, RPPA and MS, in terms of their accuracy for predicting the sensitivity of both FDA-approved chemotherapeutics and molecularly targeted anticancer compounds in the NCI-60 cell line panel. The key findings were: (i) global MS data are more informative for drug response prediction as compared to targeted RPPA technology, (ii) missing values (i.e. partially measured proteins) in MS data complicate its use in predictive modeling and require non-standard treatment of the MS profiles, (iii) rather surprisingly, reducing the number of proteins from 8113 (original MS data) to 42 (COSMIC cancer census genes with no missing values) resulted in maximal predictive accuracy for both cytotoxic and targeted compounds, (iv) MS and RPPA profiles provide complementary signal for the response prediction and (v) primary target information is critical when predicting targeted compounds. These results provide important insights into the relative performance of the two widely applied proteomic technologies, which should prove useful in many practical applications, such as for choosing the best combination of omics technologies for understanding and predicting drug sensitivities in cancer cells.
In comparison with kernel-based SVM regression model, we observed that the BEMKL method was efficient in capturing the available predictive signal from the proteomics profiles, as there were no overall performance improvements by SVM. The prediction performance of kernel-based SVM regression method supports the overall results obtained with BEMKL (Supplementary Figs S4 and S5), in terms of the importance of the different omics datasets for drug response prediction, for both cytotoxic and targeted compounds. However, there were a few notable differences due to inherent differences between BEMKL and SVM models. SVM showed improved predictions using mutation profile alone for the targeted compounds (Supplementary Fig. S4B). However, as predictive accuracy of mutation data alone was very low, compared to the other views, and since no similar result was seen for the cytotoxic drugs (Supplementary Fig. S4A), we believe this is more like a technical artefact. Moreover, the prediction performance with the combination views, and after adding complete MS or both of proteomics data was significantly improved with BEMKL (Supplementary Fig. S5), whereas SVM shows only a marginal improvement. This difference can be attributed to the multi-view and multi-task nature of BEMKL, which is absent in the standard SVM implementation and thus affected the SVM performance. Although both BEMKL and SVM are kernel-based regression methods, BEMKL has more principled ways of dealing with noise (Gönen, 2012) through hyper-parameters and error term (also known as bias). We believe that this difference in noise treatment resulted in markedly poorer performance of SVM for the GM4 + MS view combination, for both cytotoxic and targeted compounds (Supplementary Fig. S5).
In addition to the NCI-60 cell line panel, we further confirmed the importance of comprehensive proteomic coverage in 30 additional NCI/DREAM7 breast cancer cell lines using the same BEMKL model. We observed that a more detailed RPPA data (102 proteins) led to an improved drug response prediction as compared to the original NCI/DREAM7 challenge RPPA data (66 proteins) (P < 0.05, one-sided, paired t-test; Supplementary Fig. S14). Although our results support the conclusions from the NCI/DREAM7 challenge (Costello et al., 2014), these results extend to pan-cancer setting and also demonstrate the increased performance gained from the global MS profiles, when treated adequately in the BEMKL model. We note that there were no comprehensive MS data available for most of the NCI/DREAM breast cancer cell lines. Previous studies on drug response prediction have often focused only on gene expression and genomics profiles, and the main focus has often been on comparative assessment of improvements, as compared to existing predictive models (Azuaje, 2016). A notable exception is the recent work (Cortés-Ciriano et al., 2016), where the authors used all different profiles available for the NCI-60 cell line panel. Our results are in agreement with their results, showing that protein abundance together with gene and miRNA expression provides the highest predictive signal. However, instead of using the limited RPPA dataset (89 proteins), we demonstrated here the added value of the more detailed RPPA and global MS proteomic profiling.
There are only a few comparative analyses between RPPA and MS measurements in overlapping samples. One study noticed that RPPA misses part of the MS-detected phosphorylation events (Zhang et al., 2016), which can partly explain the increased predictive signal from the global MS data. Our results show relatively low-correlated pattern between the MS and RPPA profiles observed across the NCI-60 cell lines and overlapping proteins (cor = 0.053, P = 0.025, two-sided t-test; Supplementary Fig. S15). It is possible that combining RPPA data can compensate to some degree the missing MS data for key predictive proteins, for instance, through correlation links between the protein abundance levels of related proteins in target pathways, consequently leading to improved joint predictive power. Perhaps not surprisingly, the availability of proteomic data for primary targets was proven important for the response of targeted compounds; however, also the non-target features were required for accurate predictions. Off-target and other indirect mechanisms also play an important role in determining cell line response to a particular drug treatment.
The dual EGFR-ERBB2 inhibitor lapatinib serves as a good example to illustrate the various lessons learned from this study. First, only one of its primary targets (HER2/ERBB2) was completely measured in the RPPA, whereas the other (EGFR/ERBB1) was absent in the RPPA and partially measured in MS, which likely explains why the present proteomics datasets were unable to predict its response accurately. More comprehensive mapping of the target protein abundance, together with other target mechanisms, should vastly improve the future drug response predictions. In contrast to the pan-cancer approach taken here, however, it may also be beneficial to stratify the prediction models according to different cancer types, as was illustrated in the lapatinib example (Table 1 and Fig. 6). Lapatinib is an effective inhibitor of HER2+ cells, which unfortunately are not included in NCI-60 cell lines panel. Similarly, ibrutinib and sunitinib responding-cell lines are also not covered by the NCI-60 panel, partly explaining why the protein information does not improve their sensitivity prediction (Fig. 5). The cancer-type-specific models would naturally require much larger set of panels from each cancer type (Azuaje, 2016), something that may not be feasible especially in clinical applications.
As a potential extension of the current work, other means to deal with the missing MS data could lead to even better prediction accuracies. The missing value pattern in the MS data across the NCI60 cell lines is MNAR (missing not at random), often due to low abundance peptides in a sample (below the limit of detection of the MS instrument), resulting in highly non-uniform missing value distribution. Improved MS protocols for lower-abundance protein activity measurement as well as missing data imputation methods, potentially already at the peptide level before data normalization, should be implemented to avoid so large percentage of missing data values in the future studies, and to make the best use of all the MS data points for the drug response prediction. There are many approaches to imputing missing proteomic values before the actual data modeling (Webb-Robertson et al., 2015); however, ideally, the predictive model itself should be able to treat the missing data values as part of the modeling process (Aittokallio, 2010). In order to make maximal use of the MS information, the BEMKL model could be coupled with kernel completion approaches (Bhadra et al., 2017).
Further possible approach would be using component-wise kernel method (Ammad-Ud-Din et al., 2016), along with information on chemical properties of drug compounds. Such extension might potentially also enhance our knowledge of drug MoA, and predictive biomarkers, via pathway-response associations. Since the BEMKL method integrates the datasets in the kernel space instead of directly operating on the feature space, one future modeling approach could be to formulate an extension of BEMKL method that learns the kernels from the feature space while integrating out the missing values in the features; however, developing such a model is a non-trivial machine learning task and beyond the scope of current study. Further, the point mutation data should ideally be modeled using other than the standard binary coding, which could also lead to its enhanced contribution to the prediction power. Improved feature selection methods might also lead to identification of protein marker combinations, resulting in novel associations between cancer types and drugs. Experimental validation is, however, of core importance for pre-clinical support of the marker panels.
An important future translational step will be the transition from in vitro cell-line panels to patient-derived ex-vivo or clinical in-vivo response prediction, enabling the identification of novel proteomics markers for choosing individualized therapies, in line with the current clinical evidence; for instance, erlotinib has a FDA-label for EGFR (ERBB1) protein expression in NSCLC and pancreatic cancer (Dienstmann et al., 2015). Clinical response prediction studies have also observed that mRNA and miRNA expression show better predictive accuracy than CNV or DNA methylation (Ding, 2016), in line with the cell line results, and also the importance of RPPA proteomic data to predict standard chemotherapy responses in AML patient (Noren et al., 2016). Moreover, the goal of the present study was to establish biomarker profiles that are predictive of in vitro responses, which makes it possible to start exploring in-vivo drug responses, since without the linked predictive biomarkers, one cannot even explore whether the in vitro drug response patterns translate to the clinical setting in most cancer types. The lessons learned form this work could therefore become useful also for future clinical proteomic-based precision oncology strategies.
Supplementary Material
Acknowledgements
We thank Matti Kankainen for the help with selection of appropriate data types, Zia Ur Rehman and Balaguru Ravikumar for drug primary and off-target information, and Muhammad Ammad-ud-Din for his help in setting up the model for analysis.
Funding
This work was supported by the Academy of Finland (grants 269862, 272437, 279163, 292611, 295504 and 310507 to TA, grants 272577 and 277293 to K.W. and 296516 to S.K.), the Sigrid Jusélius Foundation (K.W.) and the Cancer Society of Finland (T.A. and K.W.).
Conflict of Interest: none declared.
References
- Aittokallio T. (2010) Dealing with missing values in large-scale studies: microarray data imputation and beyond. Brief. Bioinformatics, 11, 253–264. [DOI] [PubMed] [Google Scholar]
- Ammad-Ud-Din M. et al. (2016) Drug response prediction by inferring pathway-response associations with kernelized Bayesian matrix factorization. Bioinformatics, 32, i455–i463. [DOI] [PubMed] [Google Scholar]
- Azuaje F. (2016) Computational models for predicting drug responses in cancer research. Brief. Bioinform., 18, 820–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra S. et al. (2017) Multi-view kernel completion. Mach. Learn., 106, 713–739. [Google Scholar]
- Casado P. et al. (2013) Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol., 14, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. et al. (2016) Honokiol inhibits migration of renal cell carcinoma through activation of RhoA/ROCK/MLC signaling pathway. Int. J. Oncol., 49, 1525–1530. [DOI] [PubMed] [Google Scholar]
- Cortés-Ciriano I. et al. (2016) Improved large-scale prediction of growth inhibition patterns using the NCI60 cancer cell line panel. Bioinformatics, 32, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello J.C. et al. (2014) A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol., 32, 1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienstmann R. et al. (2015) Database of genomic biomarkers for cancer drugs and clinical targetability in solid tumors. Cancer Discov., 5, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z. et al. (2016) Evaluating the molecule-based prediction of clinical drug responses in cancer. Bioinformatics, 32, 2891–2895. [DOI] [PubMed] [Google Scholar]
- Druker B. et al. (2006) Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N. Engl. J. Med., 355, 2408–2417. [DOI] [PubMed] [Google Scholar]
- Flaherty K.T. et al. (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med., 367, 1694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gökmen-Polar Y. et al. (2015) Expression levels of SF3B3 correlate with prognosis and endocrine resistance in estrogen receptor-positive breast cancer. Mod. Pathol., 28, 677–685. [DOI] [PubMed] [Google Scholar]
- Gönen M., Alpaydin E. (2011) Multiple kernel learning algorithms. J. Mach. Learn. Res., 12, 2211–2268. [Google Scholar]
- Gönen M. (2012). Bayesian efficient multiple kernel learning. In: Langford, J. and Pineau J. (eds.) 29th International Conference on Machine Learning (ICML-12). ACM, New York, USA, pp. 1–8.
- Gallogly M.M., Lazarus H.M. (2016) Midostaurin: an emerging treatment for acute myeloid leukemia patients. J. Blood Med., 7, 73.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam P. et al. (2016) Identification of selective cytotoxic and synthetic lethal drug responses in triple negative breast cancer cells. Mol. Cancer, 15, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholami A.M. et al. (2013) Global proteome analysis of the NCI-60 cell line panel. Cell Rep., 4, 609–620. [DOI] [PubMed] [Google Scholar]
- Kawai M. et al. (2015) Midostaurin preferentially attenuates proliferation of triple-negative breast cancer cell lines through inhibition of Aurora kinase family. J. Biomed. Sci., 22, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S. et al. (2014) A draft map of the human proteome. Nature, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence R.T. et al. (2015) The proteomic landscape of triple-negative breast cancer. Cell Rep., 11, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. et al. (2013) TCPA: a resource for cancer functional proteomics data. Nat. Methods, 10, 1046–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llamas-Velasco M. et al. (2016) Loss of fumarate hydratase and aberrant protein succination detected with S-(2-Succino)-Cysteine staining to identify patients with multiple cutaneous and uterine leiomyomatosis and hereditary leiomyomatosis and renal cell cancer syndrome. Am J. Dermatopathol., 38, 887–891. [DOI] [PubMed] [Google Scholar]
- Maemondo M. et al. (2010) Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med., 362, 2380–2388. [DOI] [PubMed] [Google Scholar]
- Massari F. et al. (2014) Quantitative score modulation of HSP90 and HSP27 in clear cell renal cell carcinoma. Pathology, 46, 523–526. [DOI] [PubMed] [Google Scholar]
- Mazurek A. et al. (2012) DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov., 2, 812–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meric-Bernstam F. et al. (2015) A decision support framework for genomically informed investigational cancer therapy. J. Natl. Cancer Inst., 107, pii: djv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noren D.P. et al. (2016) A crowdsourcing approach to developing and assessing prediction algorithms for AML prognosis. PLoS Comput. Biol., 12, e1004890.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi M. et al. (2004) BRCA1 phosphorylation by Aurora-A in the regulation of G2 to M transition. J. Biol. Chem., 279, 19643–19648. [DOI] [PubMed] [Google Scholar]
- Shaw A. et al. (2013) Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med., 368, 2385–2394. [DOI] [PubMed] [Google Scholar]
- Shoemaker R.H. (2006) The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer, 6, 813–823. [DOI] [PubMed] [Google Scholar]
- Stein E.M., Tallman M.S. (2016) Emerging therapeutic drugs for AML. Blood, 127, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S. et al. (2016) Proteomic maps of breast cancer subtypes. Nat. Commun., 7, 10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb-Robertson B.J. et al. (2015) Review, evaluation, and discussion of the challenges of missing value imputation for mass spectrometry-based label-free global proteomics. J. Prot. Res., 14, 1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M. et al. (2014) Mass-spectrometry-based draft of the human proteome. Nature, 509, 582–587. [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. (2016) Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell, 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.