

Desmoplastic small round cell tumor (DSRCT) is an aggressive, usually incurable, soft tissue sarcoma subtype that presents with diffuse abdominal sarcomatosis in adolescents and young adults. This article presents the largest retrospective analysis conducted to date of patients with DSRCT who were treated with an anti‐angiogenic therapy.
Keywords: Desmoplastic small round cell tumor, Pazopanib, EWS‐WT1, Translocation‐positive sarcoma
Abstract
Background.
Desmoplastic small round cell tumor (DSRCT) is an aggressive, often fatal soft tissue sarcoma that lacks an optimal salvage regimen. We retrospectively reviewed data from 29 pretreated DSRCT patients who received pazopanib at MD Anderson Cancer Center after failure of standard chemotherapies.
Subjects, Materials, and Methods.
Medical records of patients treated from January 2012 to December 2016 were reviewed and regression analyses were performed. Median progression‐free survival (PFS) and overall survival (OS) were estimated using the Kaplan‐Meier method and differences in survival were assessed by a log‐rank test. A landmark statistical analysis was used to assess OS at a predefined 12‐week time point following pazopanib initiation.
Results.
The mean age at pazopanib treatment was 27.5 years (range, 6.3–50.1 years). According to RECIST 1.1 criteria, 16 patients (55%) had stable disease, 1 patient (3%) had partial response, 1 patient (3%) had complete response, and 11 patients (38%) had progressive disease. Estimated median PFS was 5.63 months (95% confidence interval [CI]: 3.23–7.47). Median OS was 15.7 months (95% CI: 10.3–32.4). As of December 2016, 11 patients (38%) were still alive, with a median follow‐up time of 16.8 (range 3.8–30.1) months. Doses between 400 and 800 mg were included. Pazopanib was well tolerated and 23 (79%) of the patients continued it until progression or death, 4 discontinued because of side effects, and 2 were still on pazopanib at the time of data analysis.
Conclusion.
In the largest study conducted to date in DSRCT, pazopanib was well tolerated and clinically active in heavily pretreated patients who otherwise lack good treatment options.
Implications for Practice.
Desmoplastic small round cell tumor (DSRCT) is a rare, extremely aggressive soft tissue sarcoma subtype that most commonly occurs in adolescent and young adult males. No DSRCT‐specific therapies exist, and for lack of a better treatment approach, current therapies have relied upon U.S. Food and Drug Administration‐approved drugs like pazopanib that exhibit clinical activity in other sarcoma subtypes. This article describes the largest experience to date using pazopanib as salvage treatment in heavily pretreated DSRCT patients. Pazopanib was well tolerated and clinically active, surpassing predefined metrics proposed by the European Organization for Research and Treatment of Cancer indicative of "active" sarcoma drugs (5.63 months progression‐free survival [PSF], with 62% of the study population achieving progression‐free survival at 12 weeks).
Introduction
Desmoplastic small round cell tumor (DSRCT) is an aggressive, usually incurable soft tissue sarcoma subtype that generally presents with diffuse abdominal sarcomatosis in adolescents and young adults, with a mean age at diagnosis of 24.9 years. First described by Rosai et al. in 1989 after a pathognomonic EWSR1‐WT1 t(11;22)(p13:q12) translocation was discovered, this rare sarcoma subtype had previously been misclassified as an atypical germ cell tumor given DSRCT's poorly differentiated appearance, 9:1 male/female preponderance, and pattern of metastatic spread [1], [2], [3], [4], [5], [6].
The improved diagnostic accuracy enabled by detection of the characteristic translocation, and recognition that DSRCT molecularly resembles Ewing sarcoma (ES) and other EWSR1 translocation‐positive sarcomas, led to a paradigm shift over the last 2 decades in how these tumors are treated. In the few cancer centers that have expertise in caring for DSRCT patients, neoadjuvant and adjuvant chemotherapy using regimens typically reserved for ES treatment (e.g., VAI [vincristine, doxorubicin, ifosfamide], VDC/IE [vincristine, doxorubicin, cyclophosphamide/ifosfamide, etoposide], irinotecan/temozolomide) plays a central role. In select patients who have local‐regional chemosensitive disease confined to the abdomen or unifocal metastasis amenable to radiofrequency ablation or radiation, our institutional practice has also been to attempt complete cytoreductive surgery. Although still under clinical investigation, hyperthermic intraperitoneal chemotherapy (HIPEC) and adjuvant whole‐abdominal radiation (WART) are frequently offered to DSRCT patients who achieve complete macroscopic cytoreduction (CCR0 or CCR1; i.e., <2.5 mm of residual tumor) in an attempt to reduce the frequency of intra‐abdominal recurrence [7], [8], [9].
Although better chemotherapy options and refined surgical and radiation therapy techniques over the last decade have undoubtedly improved the 5‐year survival of DSRCT patients (approaching 25% at our center), the survival rate lags behind what can reasonably be expected for ES [8], [9] given DSRCT's diffuse intra‐abdominal presentation and high rate of treatment failure [4], [10]. Despite advances in the molecular profiling of DSRCT, these genomic and proteomic characterizations have not resulted in effective drug targets, and there remains no effective salvage therapy for this otherwise lethal disease [11], [12], [13], [14].
For lack of better chemotherapeutic options specifically catered to the unique genomic/proteomic aberrations present in DSRCT, the oncology community has continued to rely upon therapies generically used for other small round blue cell tumors (SRBCTs; e.g., ES, synovial sarcoma, rhabdomyosarcoma). In some sense, that strategy has worked, because DSCRT and those other SRBCT tumor types exhibit 70%–80% response rates to select cytotoxic chemotherapies. Yet, as biologically targeted therapies have increasingly entered the clinic, we are beginning to see divergent responses in activity between ES and DSRCT. As an example, striking tumor regression occurs in approximately 10%–14% of ES patients who receive IGF‐1R‐targeted therapies, whereas the response in DSRCT appears more subdued in the limited data that exist [15]. Conversely, although less than 5% of ES patients respond to pazopanib, anecdotal reports and a limited case series of nine patients have suggested that DSRCT is more sensitive [16], [17], [18], [19].
Pazopanib is a multitargeted receptor tyrosine kinase (RTK) inhibitor that parallels the antiangiogenic mechanisms of sunitinib in other metastatic tumors through inhibition of vascular endothelial growth factor receptors (VEGFR), platelet‐derived growth factor receptors (PDGFR), and c‐KIT, among others [20], [21]. It received U.S. Food and Drug Administration (FDA) approval in 2009 for renal cell carcinoma, and shortly thereafter in 2012 gained an indication for nonadipogenic soft tissue sarcoma (STS), primarily based on data from the European Organization for Research and Treatment of Cancer's (EORTC) phase III PALETTE study (NCT00753688), which was enriched for leiomyosarcoma, synovial sarcoma, and a basket cohort of “other” STSs. Together with an earlier phase II EORTC trial of pazopanib, two of six DSRCT patients were reported to respond [22], [23].
In an effort to explain this heightened sensitivity in DSRCT, and to more accurately characterize the magnitude of pazopanib's effect in a larger subset of patients, our current study presents the largest retrospective analysis conducted to date of DSRCT patients treated with an antiangiogenic therapy. Among our key findings, pazopanib showed encouraging clinical activity in heavily pretreated DSRCT patients, with 62% of our study population achieving prolonged progression‐free and overall survival without major induced toxicities.
Subjects, Materials, and Methods
Patients
Data from 38 patients with advanced DSRCT seen at MD Anderson Cancer Center from January 2012 to December 2016 treated with pazopanib were retrospectively reviewed from our electronic medical record (Fig. 1). Institutional Review Board approval was obtained, but no informed consent was required for this retrospective review, and all patient records were de‐identified prior to analysis. Nine of the thirty‐eight patients had received pazopanib with an additional chemotherapy and were, therefore, excluded from our analysis. Importantly, as clinical benefit (i.e., stable disease [SD], partial response [PR], and complete response [CR]) is assessed in the current study, all patients demonstrated tumor progression before initiating pazopanib. Archived tissue was available for all but one of the patients, and the initial diagnosis of DSRCT was confirmed by experienced sarcoma pathologists at MD Anderson using clinical information, immunohistochemistry, and cytogenetic analyses for the EWSR1‐WT1 fusion by fluorescence in situ hybridization or reverse transcription polymerase chain reaction.
Figure 1.

Consolidated Standards of Reporting Trials diagram of the retrospective study of DSRCT patients receiving pazopanib.
Abbreviations: DSRCT, desmoplastic small round cell tumor; HDAC, histone deacetylase; mTOR, mammalian target of rapamycin; RECIST, Response Evaluation Criteria In Solid Tumors.
Patient information was characterized by demographic factors (age at diagnosis, age at pazopanib treatment, sex, race) and clinical factors (translocation presence, metastatic disease at diagnosis, prior chemotherapy regimens, pazopanib dose, pazopanib combination with other drugs) and analyzed for clinical outcomes, including best response by Response Evaluation Criteria In Solid Tumors (RECIST) 1.1 criteria, progression‐free survival (PFS), and overall survival (OS) [24].
Statistical Analysis
Demographic and clinical characteristics were tabulated, and OS and PFS were calculated. OS was defined as the time from start of pazopanib treatment to the time of death or to the time of last contact for patients alive at the end of follow‐up. PFS was defined as the time from start of pazopanib treatment to the time of disease progression or death. Patients whose disease had not progressed at last follow‐up were censored for PFS at the time of last contact. Median OS and PFS were calculated using the Kaplan‐Meier method [25]. A log‐rank test was used to identify inequalities across strata based on patient characteristics [26]. Landmark analysis was performed based on clinical benefit status at 12 weeks as a prognostic factor for survival, with patients who died or were censored before the landmark timepoint excluded from analysis. Clinical benefit was defined as patients having stable disease, partial response (>30% reduction in tumor size), or complete response. No clinical benefit was defined as patients having progression (PD, >20% growth). Although causal inferences concerning treatment effectiveness based on the landmark analysis cannot be made, achievement of clinical benefit can be examined as a prognostic factor for survival. Regression analyses of survival data based on the Cox proportional hazards regression model were conducted for OS and PFS [26]. A two‐sided p value <.05 was considered statistically significant. SAS for Windows version 9.4 software (SAS Institute Inc., Cary, NC) was used for all analyses.
Results
Demographics and Baseline Characteristics
Demographics and baseline characteristics of 29 patients with advanced DSRCT are presented in Table 1. Consistent with prior reports, a majority of the patients were male (n = 23, 79%), and mean age at start of pazopanib treatment was 27.5 years (range, 6.3–50.1 years). Molecular confirmation of the diagnosis, using the EWSR‐WT1 translocation, was confirmed in 25 (86%) of the patients. The remaining patients (4 patients, 14%) failed to undergo translocation testing and were diagnosed using immunohistochemical techniques. Patients had tumors ranging in size from 0.9 to 20.6 cm. Fifteen (52%) patients had metastatic sites at diagnosis, with ten (67%) of these patients presenting with two or more distant metastatic sites and six (34%) of these patients presenting with extra‐abdominal disease.
Table 1. Demographic and clinical characteristics of desmoplastic small round cell tumor chemoresistant patients.
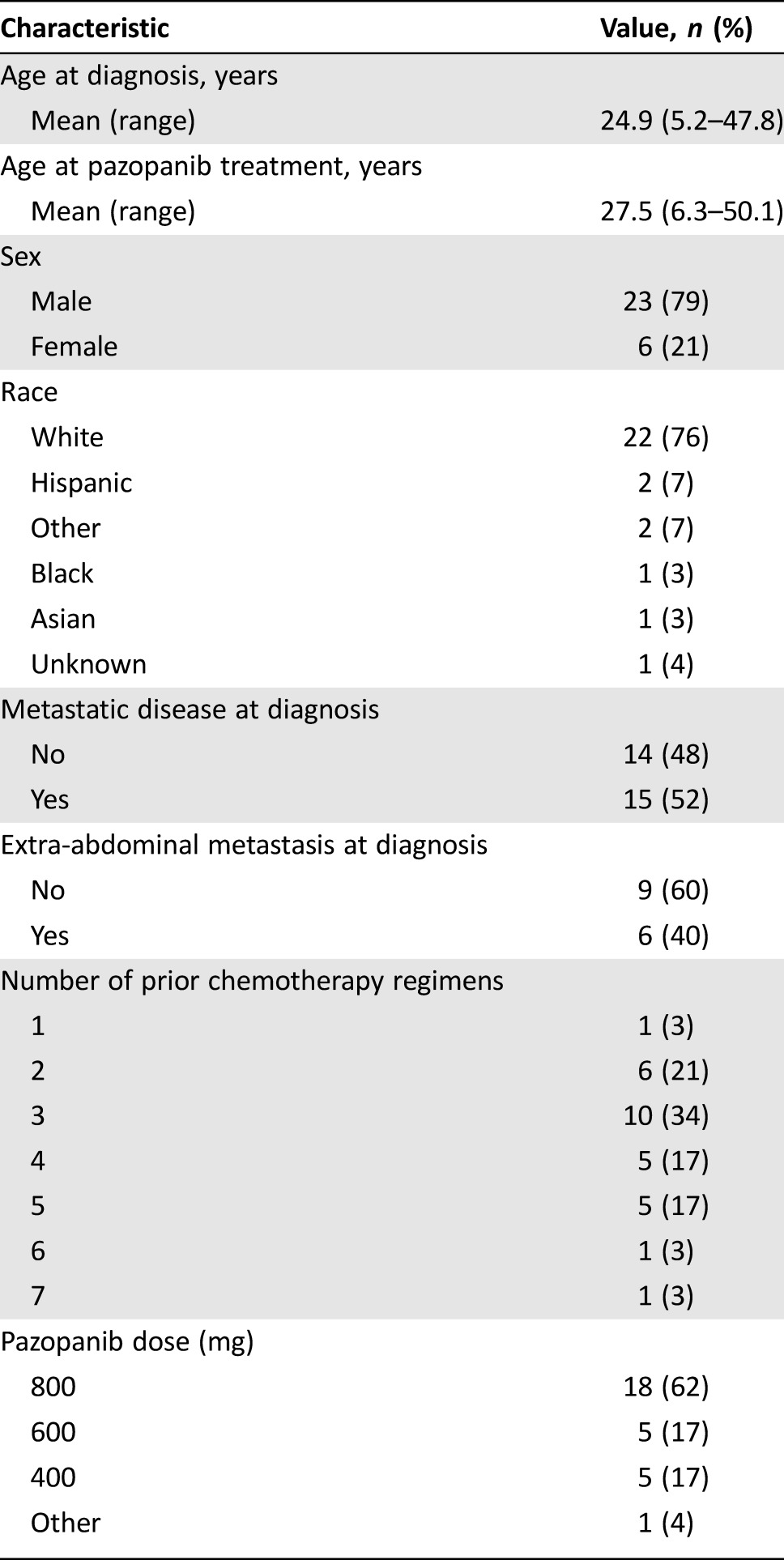
At the start of pazopanib treatment, all patients had been extensively pretreated, with 22 (76%) patients having had three or more prior chemotherapy regimens (range, 1–7; median, 3). Common regimens included Ewing's‐like alkylating therapy (i.e., P6, VDC/IE, VAI), irinotecan/temozolomide, high‐dose ifosfamide, cyclophosphamide/vinorelbine, and cyclophosphamide/topotecan.
Pazopanib Dosing and Side Effects
Although the FDA‐approved maximal dose of pazopanib for STS is 800 mg daily, not all patients tolerated that dose. As shown in Table 1, 18 patients (62%) received 800 mg pazopanib daily, whereas the remaining 11 patients received reduced dosages (range, 400–600 mg). Twenty‐one patients (72%) stopped pazopanib because of disease progression, two (7%) patients continued on pazopanib until their death, and four (14%) patients discontinued pazopanib due to well‐defined side effects; two exhibited hemorrhagic complications including hematuria, one developed hyperbilirubinemia, and one discontinued pazopanib to allow a fistula to heal.
Clinical Activity
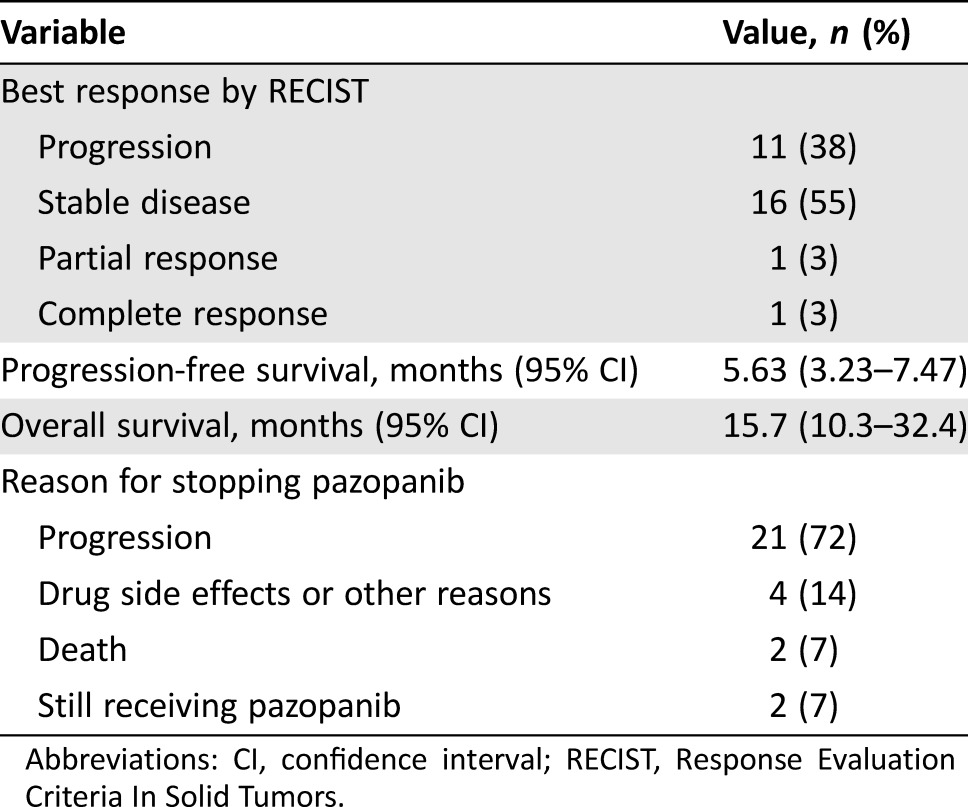
The clinical activity of pazopanib‐treated patients is summarized in Table 2. The average duration of pazopanib treatment was 3.4 months (range, 0.7–19.6 months). Using RECIST 1.1 criteria, more than half (n = 18; 62%) of the patients treated with pazopanib (irrespective of dose level) achieved a clinical benefit [24]. Consistent with the PALETTE data for STS [23] and the generally cytostatic nature of pazopanib, most of the clinical benefit could be ascribed to stable disease, as only one (3%) patient had a CR and one (3%) patient had a PR (Fig. 2). At the time of data analysis, two patients were still receiving pazopanib with SD and were censored on their dates of last follow‐up for PFS and OS analyses. Eighteen patients died during follow‐up, and the median follow‐up time for the surviving eleven patients was 16.8 months (range, 3.8–30.1 months).
Table 2. Clinical activity in pazopanib‐treated patients.
Abbreviations: CI, confidence interval; RECIST, Response Evaluation Criteria In Solid Tumors.
Figure 2.
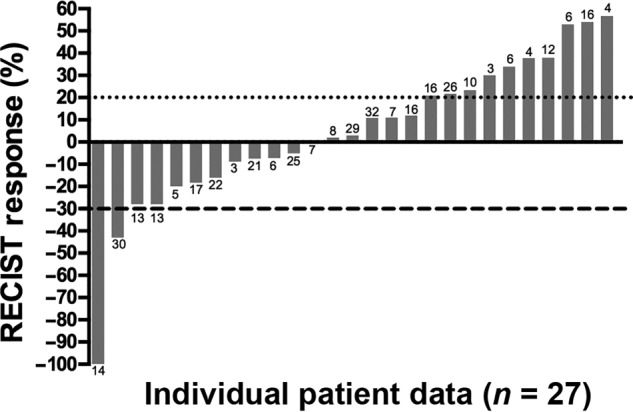
Antineoplastic effect of pazopanib in desmoplastic small round cell tumor per RECIST criteria. The percentage of reduction in tumor burden from baseline imaging in 26 patients to time of best response during the study period. Column labels indicate overall survival rounded to the nearest whole month for each patient from start of treatment. Cutoffs for partial response (>30% reduction in tumor volume) and progressive disease (>20% growth in tumor volume) as defined by RECIST 1.1 are noted with dotted (•) and dashed (‐) lines, respectively. Two patients with progressive disease are excluded from this graph: one patient who had immeasurable disease progression and one patient who died prior to follow‐up imaging.
Abbreviation: RECIST, Response Evaluation Criteria In Solid Tumors.
The estimated median PFS for patients receiving pazopanib was 5.63 months (95% confidence interval [CI]: 3.23–7.47), and median OS was 15.7 months (95% CI: 10.3–32.4).
Landmark analysis based on response status at 12 weeks after start of pazopanib treatment showed that median OS for patients achieving clinical benefit was 12.8 months (95% CI: 8.9–38.3) compared with 2.7 months (95% CI: 0.03–13.6) for patients with progressive disease (Fig. 3; log‐rank test, p = .004).
Figure 3.
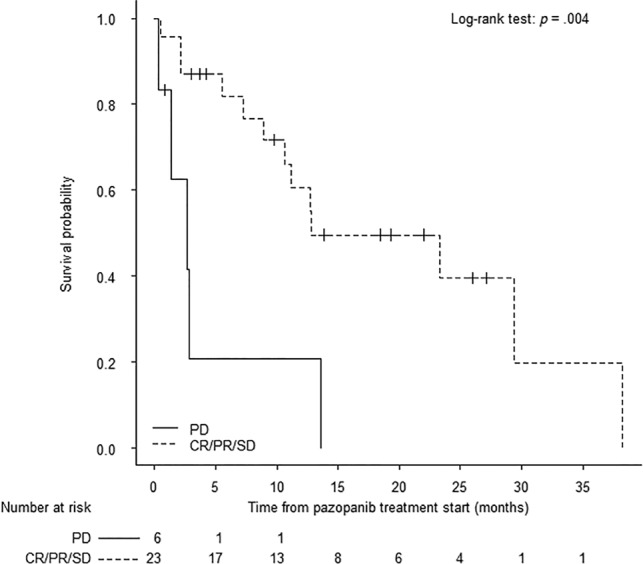
Overall survival (OS) at 12‐week landmark. The Kaplan‐Meier method was performed to evaluate OS based on response status at 12 weeks after the start of pazopanib treatment. Clinical benefit was defined as patients having SD, PR (>30% reduction in tumor size), or CR by Response Evaluation Criteria In Solid Tumors 1.1 criteria. No response was defined as patients having PD (>20% growth in tumor size).
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Discussion
In this article, we report the largest single‐institution review to date of patients with metastatic DSRCT treated with pazopanib. As a tertiary care and major referral center, our population included a heterogeneous subset of pediatric and adult patients with varying degrees of metastatic disease who underwent up to seven chemotherapy regimens prior to receiving the multitargeted tyrosine kinase inhibitor pazopanib. These prior regimens included distinctive combinations of multimodal therapy comprising standard‐of‐care Ewing's‐like alkylating therapies, high‐dose ifosfamide, and cyclophosphamide/vinorelbine and topoisomerase‐containing regimens, including irinotecan/temozolomide and cyclophosphamide/topotecan, that were utilized in combination with cytoreductive surgery, hyperthermic intraperitoneal chemotherapy, and whole‐abdominal radiation. Following pazopanib treatment, clinical benefit was achieved in 62% (n = 18) of the study population.
Consistent with prior studies, our patient population was predominately male (79%, n = 23). As in prior reports, the majority of our study population were adolescents and young adults, with 68% of all patients being younger than 30 years at treatment start. In general, patients with DSRCT typically present with advanced disease [3]. In our patient population, 52% had metastatic disease at diagnosis, with 34% of all patients presenting with two or more distant metastatic sites. Due to the typical presentation of DSRCT with diffuse abdominopelvic sarcomatosis, American Joint Committee on Cancer staging was not applied, as virtually the entire cohort would have been considered stage IV disease.
Patients treated with pazopanib as salvage therapy had an estimated median PFS of 5.63 months (95% CI: 3.23–7.47) and an estimated median OS of 15.7 months (95% CI: 10.3–30.24). These results compare favorably with the published results of the PALETTE study, which reported a PFS of 4.5 months and OS of 12.5 months in a group of STS patients that included just three DSRCT patients [23]. A smaller retrospective review of multi‐institutional experiences by Frezza et al. established comparable OS (15.4 months; 95% CI: 1.5–29.3) but a higher PFS of 9.2 months (95% CI: 0–23.2) in patients with advanced DSRCT treated with pazopanib [19]. Although it is difficult to directly compare results of these two studies, some of this variation may be due to differences in patient demographics, presenting clinical characteristics, and the heavily pretreated nature of our study population.
Unless drug‐related side effects warranted dose reductions, patients were treated using the FDA‐approved dose of pazopanib. A consensus has not been reached regarding the optimal pazopanib dose or schedule in DSRCT patients [27]; therefore, we strongly encourage readers to adhere to the pazopanib package insert. In the present series, a consistent 800‐mg daily dose of pazopanib was given to more than half of all patients (n = 18). Additionally, 17% of patients (n = 5) received 600 mg daily, and 17% of patients (n = 5) received 400 mg daily. Dosing information was not available for the remaining patient. Of the 10 patients (34%) receiving either 400 mg or 600 mg, 7 patients achieved clinical benefit on reduced dosages of pazopanib, including the patient who had a CR. Our analyses failed to demonstrate any difference in survival outcomes between dose levels (data not shown). The current study was underpowered to definitively answer whether a daily 800‐mg pazopanib dose is superior to lower doses in patients with DSRCT, and further studies, perhaps incorporating pharmacodynamic biomarkers linked to response, should seek to optimize the correct dose for patients to maximize clinical benefit while reducing side effects [27], [28].
Twenty‐three patients (79%) discontinued pazopanib treatment due to disease progression or death. An additional four patients (14%) discontinued treatment due to pazopanib‐associated side effects or other reasons. Of these, one patient who had previously been treated with HIPEC and WART discontinued pazopanib treatment in order to allow a fistula to heal at the site of a gastrostomy tube placement. Given DSRCT's typical diffuse intra‐abdominal presentation and widepsread use of WART and/or intraperitoneal chemotherapy, one might have anticipated a higher incidence of bowel perforation or fistulas than what was observed in the current study. Overall, the present series disclosed a low number of side effects compared with the prevalence found in prior studies (35%–72%) [19], [23], [27].
As a vascular tumor, DSRCT is characterized by an overexpression of vascular endothelial growth factor‐A (VEGFA) and its receptor, VEGF receptor‐2 (VEGFR‐2), which promote the angiogenic processes necessary for continued tumor growth and proliferation [29]. Pazopanib and sunitinib both inhibit angiogenesis by abrogating the VEGF‐induced phosphorylation of VEGF receptors as well as other RTKs including PDGFR, FGFR, and c‐KIT, affecting downstream activation of the PI3K/AKT, PKC, and other pathways that mediate cell proliferation, migration, and survival [30]. As monotherapies, both pazopanib and sunitinib have shown clinical benefit in treating renal cell carcinoma and STS with comparable outcomes, increasing PFS and OS on the order of weeks to months, although treatment with pazopanib typically resulted in fewer side effects [21].
Clinical benefit from pazopanib was observed in 62% of our patient population (CR in 1 patient, PR in 1 patient, SD in 16 patients). In general, this clinical benefit was transient, with 61% of these patients remaining progression‐free less than 6 months, an interval consistent with previous reports [31]. Although the majority of these responses were short‐lived, clinical benefit at 12 weeks was associated with increased OS as assessed using a landmark analysis. Although it cannot establish causality, the landmark approach used herein is one of several well‐validated statistical methods commonly used in observational studies to evaluate the relationship that clinical benefit has upon OS. Importantly, a 12‐week time‐to‐event landmark was selected a priori, because the progression‐free rate at 12 weeks has previously been shown to distinguish clinically active cytostatic drugs from inactive ones, particularly in STS [32]. Moreover, the landmark approach has been used by over 70 studies to evaluate therapeutic activity across a broad range of the more than 50 STS subtypes [22], [33], [34]. We note that the landmark statistical approach is dependent on selection of the landmark time and conclusions from the analysis can differ depending on which landmark is chosen, as patients who die before the landmark do not contribute to the analysis and patients who respond after the landmark are classified as nonresponders [35], [36]. Further, given the rarity of DSRCT and limited study size, attempts to stratify analysis to control for covariates such as tumor size or number of prior regimens were not feasible.
Although pazopanib is approved for treatment of nonadipogenic STS following the PALETTE study, currently little to no guidance or criteria exist to inform clinicians as to which STS subtypes would benefit the most from treatment, especially in relatively rare subtypes like DSRCT. To this end, as more than half the patients in our review achieved clinical benefit, further studies should seek to identify tissue‐based biomarkers that predict which patients are most likely to benefit. Comprehensive longitudinal profiling of the genomic and proteomic tumor landscape should be employed in future trials to help identify novel therapeutic targets and unearth potential mechanisms of drug resistance. These contributions, as part of a larger robust drug development pipeline, are critical in meaningfully improving the outcomes of patients with DSRCT.
Conclusion
As a rare orphan tumor with few proteomic or genetic targets, DSRCT has limited therapeutic options currently available beyond standard‐of‐care therapies employed for other SRBCTs [32]. In the salvage setting, our study demonstrated that pazopanib was well tolerated and has the potential to prolong OS, even in heavily pretreated patients. Although not curative, a 5.6‐month PFS is undoubtedly of value to the adolescent and young adult patients who struggle to combat this very aggressive malignancy. As such, pazopanib should be considered as an additional salvage regimen for patients with advanced‐stage DSRCT.
Acknowledgments
The University of Texas MD Anderson Cancer Center is supported by the National Institutes of Health through Cancer Center Support Grant CA016672 (used the Clinical Trials Support Resource and the Biostatistics Resource Group). J.A.L. is supported by R01‐CA180279‐01A1.
Contributed equally
Footnotes
For Further Reading: Silvia Stacchiotti, Olivier Mir, Axel Le Cesne et al. Activity of Pazopanib and Trabectedin in Advanced Alveolar Soft Part Sarcoma. The Oncologist 2018;23:62–70.
Implications for Practice: This retrospective study, conducted among the world reference centers for treatment of sarcoma, confirms the value of pazopanib in patients with advanced alveolar soft part sarcoma (ASPS), with dimensional and durable responses, whereas trabectedin shows a limited activity. Alveolar soft part sarcoma is resistant to conventional cytotoxic chemotherapy. Pazopanib and trabectedin are licensed for treatment of sarcoma from second line; in the lack of prospective clinical trials, these results are relevant to defining ASPS best management and strongly support initiatives aimed at obtaining the approval of pazopanib in the front line of the disease.
Author Contributions
Conception/design: Brian A. Menegaz, Branko Cuglievan, Jalen Benson, Joseph A. Ludwig
Provision of study material or patients: Branko Cuglievan, Pamela Camacho, Winston Huh, Vivek Subbiah, Robert S. Benjamin, Shreyaskumar Patel, Najat Daw, Andrea Hayes‐Jordan, Joseph A. Ludwig
Collection and/or assembly of data: Brian A. Menegaz, Branko Cuglievan, Jalen Benson, Pamela Camacho, Winston Huh, Vivek Subbiah, Robert S. Benjamin, Shreyaskumar Patel, Najat Daw, Andrea Hayes‐Jordan, Joseph A. Ludwig
Data analysis and interpretation: Brian A. Menegaz, Branko Cuglievan, Cheuk Hong Leung, Carla L. Warneke, Joseph A. Ludwig
Manuscript writing: Brian A. Menegaz, Branko Cuglievan, Jalen Benson, Pamela Camacho, Salah‐Eddine Lamhamedi‐Cherradi, Cheuk Hong Leung, Carla L. Warneke, Joseph A. Ludwig
Final approval of manuscript: Joseph A. Ludwig
Disclosures
Robert S. Benjamin: Novartis (C/A); Shreyaskumar Patel: Novartis, Janssen, Eisai, Bayer, Eli Lilly & Co., Epizyme, CytRx, EMD‐Serono (C/A). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Gerald WL, Haber DA. The EWS‐WT1 gene fusion in desmoplastic small round cell tumor. Semin Cancer Biol 2005;15:197–205. [DOI] [PubMed] [Google Scholar]
- 2. Jordan AH, Pappo A. Management of desmoplastic small round‐cell tumors in children and young adults. J Pediatr Hematol Oncol 2012;34(suppl 2):S73–S75. [DOI] [PubMed] [Google Scholar]
- 3. Hayes‐Jordan A, Anderson PM. The diagnosis and management of desmoplastic small round cell tumor: A review. Curr Opin Oncol 2011;23:385–389. [DOI] [PubMed] [Google Scholar]
- 4. Dufresne A, Cassier P, Couraud L et al. Desmoplastic small round cell tumor: Current management and recent findings. Sarcoma 2012;2012:714986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gerald WL, Rosai J. Case 2 Desmoplastic small cell tumor with divergent differentiation. Pediatr Pathol 1989;9:177–183. [DOI] [PubMed] [Google Scholar]
- 6. Gerald WL, Miller HK, Battifora H et al. Intra‐abdominal desmoplastic small round‐cell tumor. Report of 19 cases of a distinctive type of high‐grade polyphenotypic malignancy affecting young individuals. Am J Surg Pathol 1991;15:499–513. [PubMed] [Google Scholar]
- 7. Mora J, Modak S, Cheung NK et al. Desmoplastic small round cell tumor 20 years after its discovery. Future Oncol 2015;11:1071–1081. [DOI] [PubMed] [Google Scholar]
- 8. Hayes‐Jordan A, Green HL, Lin H et al. Complete cytoreduction and HIPEC improves survival in desmoplastic small round cell tumor. Ann Surg Oncol 2014;21:220–224. [DOI] [PubMed] [Google Scholar]
- 9. Hayes‐Jordan A, Green H, Fitzgerald N et al. Novel treatment for desmoplastic small round cell tumor: Hyperthermic intraperitoneal perfusion. J Pediatr Surg 2010;45:1000–1006. [DOI] [PubMed] [Google Scholar]
- 10. Lal DR, Su WT, Wolden SL et al. Results of multimodal treatment for desmoplastic small round cell tumors. J Pediatr Surg 2005;40:251–255. [DOI] [PubMed] [Google Scholar]
- 11. Ferrari S, del Prever AB, Palmerini E et al. Response to high‐dose ifosfamide in patients with advanced/recurrent Ewing sarcoma. Pediatr Blood Cancer 2009;52:581–584. [DOI] [PubMed] [Google Scholar]
- 12. Farhat F, Culine S, Lhomme C et al. Desmoplastic small round cell tumors: Results of a four‐drug chemotherapy regimen in five adult patients. Cancer 1996;77:1363–1366. [DOI] [PubMed] [Google Scholar]
- 13. Honoré C, Atallah V, Mir O et al. Abdominal desmoplastic small round cell tumor without extraperitoneal metastases: Is there a benefit for HIPEC after macroscopically complete cytoreductive surgery? PLoS One 2017;12:e0171639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ferrari A, Grosso F, Stacchiotti S et al. Response to vinorelbine and low‐dose cyclophosphamide chemotherapy in two patients with desmoplastic small round cell tumor. Pediatr Blood Cancer 2007;49:864–866. [DOI] [PubMed] [Google Scholar]
- 15. Naing A, LoRusso P, Fu S et al. Insulin growth factor‐receptor (IGF‐1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing's sarcoma family tumors. Clin Cancer Res 2012;18:2625–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamamoto Y, Nozawa M, Shimizu N et al. Pazopanib for recurrent extraosseous Ewing's sarcoma of the retroperitoneum. Int J Urol 2014;21:1183–1184. [DOI] [PubMed] [Google Scholar]
- 17. Attia S, Okuno SH, Robinson SI et al. Clinical activity of pazopanib in metastatic extraosseous Ewing sarcoma. Rare Tumors 2015;7:5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Alcindor T. Response of refractory Ewing sarcoma to pazopanib. Acta Oncol 2015;54:1063–1064. [DOI] [PubMed] [Google Scholar]
- 19. Frezza AM, Benson C, Judson IR et al. Pazopanib in advanced desmoplastic small round cell tumours: A multi‐institutional experience. Clin Sarcoma Res 2014;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoon SS, Segal NH, Olshen AB et al. Circulating angiogenic factor levels correlate with extent of disease and risk of recurrence in patients with soft tissue sarcoma. Ann Oncol 2004;15:1261–1266. [DOI] [PubMed] [Google Scholar]
- 21. Motzer RJ, Hutson TE, Cella D et al. Pazopanib versus sunitinib in metastatic renal‐cell carcinoma. N Engl J Med 2013;369:722–731. [DOI] [PubMed] [Google Scholar]
- 22. Sleijfer S, Ray‐Coquard I, Papai Z et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European organisation for research and treatment of cancer‐soft tissue and bone sarcoma group (EORTC study 62043). J Clin Oncol 2009;27:3126–3132. [DOI] [PubMed] [Google Scholar]
- 23. van der Graaf WT, Blay JY, Chawla SP et al. Pazopanib for metastatic soft‐tissue sarcoma (PALETTE): A randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2012;379:1879–1886. [DOI] [PubMed] [Google Scholar]
- 24. Eisenhauer EA, Therasse P, Bogaerts J et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247. [DOI] [PubMed] [Google Scholar]
- 25. Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc 1958;53:457–481. [Google Scholar]
- 26. Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966;50:163–170. [PubMed] [Google Scholar]
- 27. Abe K, Yamamoto N, Hayashi K et al. Balancing prolonged survival with qol using low‐dose pazopanib maintenance: A comparison with the PALETTE study. Anticancer Res 2016;36:2893–2897. [PubMed] [Google Scholar]
- 28. Miura H, Shirai H. Low‐dose administration of oral pazopanib for the treatment of recurrent angiosarcoma. Clin Exp Dermatol 2015;40:575–577. [DOI] [PubMed] [Google Scholar]
- 29. Magnan HD, Chou T, LaQuaglia MP et al. Elevated expression of VEGFR‐2 and VEGFA in desmoplastic small round cell tumor (DSRCT) and activity of bevacizumab and irinotecan in a xenograft model of DSRCT. J Clin Oncol (Meeting Abstracts) 2009;27:10016a. [Google Scholar]
- 30. Italiano A, Kind M, Cioffi A et al. Clinical activity of sunitinib in patients with advanced desmoplastic round cell tumor: A case series. Target Oncol 2013;8:211–213. [DOI] [PubMed] [Google Scholar]
- 31. Ikeue T, Ohi I, Noguchi S et al. Desmoplastic small round cell tumor of the pleura successfully treated with a lower dose of pazopanib. Intern Med 2016;55:2463–2467. [DOI] [PubMed] [Google Scholar]
- 32. Van Glabbeke M, Verweij J, Judson I et al. Progression‐free rate as the principal end‐point for phase II trials in soft‐tissue sarcomas. Eur J Cancer 2002;38:543–549. [DOI] [PubMed] [Google Scholar]
- 33. Benjamin RS. Observational studies: Goldmines of information on rare diseases. BMC Med 2017;15:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grunwald V, Litiere S, Young R et al. Absence of progression, not extent of tumour shrinkage, defines prognosis in soft‐tissue sarcoma ‐ An analysis of the EORTC 62012 study of the EORTC STBSG. Eur J Cancer 2016;64:44–51. [DOI] [PubMed] [Google Scholar]
- 35. Dafni U. Landmark analysis at the 25‐year landmark point. Circ Cardiovasc Qual Outcomes 2011;4:363–371. [DOI] [PubMed] [Google Scholar]
- 36. Anderson JR, Cain KC, Gelber RD. Analysis of survival by tumor response. J Clin Oncol 1983;1:710–719. [DOI] [PubMed] [Google Scholar]