

Abstract
Background
Mosquitoes are the potential vectors for a variety of viruses that can cause diseases in the human and animal populations. Viruses in the order Picornavirales infect a broad range of hosts, including mosquitoes. In this study, we aimed to characterize a novel picorna-like virus from the Culex spp. of mosquitoes from the Zambezi Valley of Mozambique.
Methods
The extracted RNA from mosquito pools was pre-amplified with the sequence independent single primer amplification (SISPA) method and subjected to high-throughput sequencing using the Ion Torrent platform. Reads that are classified as Iflaviridae, Picornaviridae and Dicistroviridae were assembled by CodonCode Aligner and SPAdes. Gaps between the viral contigs were sequenced by PCR. The genomic ends were analyzed by 5′ and 3′ RACE PCRs. The ORF was predicted with the NCBI ORF finder. The conserved domains were identified with ClustalW multiple sequence alignment, and a phylogenetic tree was built with MEGA. The presence of the virus in individual mosquito pools was detected by RT-PCR assay.
Results
A near full-length viral genome (9740 nt) was obtained in Culex mosquitoes that encoded a complete ORF (3112 aa), named Culex picorna-like virus (CuPV-1). The predicted ORF had 38% similarity to the Hubei picorna-like virus 35. The sequence of the conserved domains, Helicase-Protease-RNA-dependent RNA polymerase, were identified by multiple sequence alignment and found to be at the 3′ end, similar to iflaviruses. Phylogenetic analysis of the putative RdRP amino acid sequences indicated that the virus clustered with members of the Iflaviridae family. CuPV-1 was detected in both Culex and Mansonia individual pools with low infection rates.
Conclusions
The study reported a highly divergent, near full-length picorna-like virus genome from Culex spp. mosquitoes from Mozambique. The discovery and characterization of novel viruses in mosquitoes is an initial step, which will provide insights into mosquito-virus interaction mechanisms, genetic diversity and evolution.
Electronic supplementary material
The online version of this article (10.1186/s12985-018-0981-z) contains supplementary material, which is available to authorized users.
Keywords: Culex, Mosquitoes, Picorna-like virus, Iflavirus, RNA virus
Background
Viruses in the order Picornavirales infect a variety of plants, animal hosts and insects. The order consists of five viral families, Dicistroviridae, Iflaviridae, Marnaviridae, Picornaviridae, Secoviridae, and an unassigned group [1]. Viruses in this order have a single-stranded, positive-sense RNA genome (+ssRNA) of approximately 9 KB in length and are non-enveloped. Most of the genomes in this order have a single open reading frame (ORF) that is flanked with a genome-linked, virus-encoded protein (VPg) at the 5′ end and a poly (A) tail at the 3′ end. The ORF encodes both structural and non-structural proteins having a conserved organization of the helicase-protease-RNA-dependent RNA polymerase (RdRp) domains [2]. The translation process is controlled by an internal ribosomal entry site (IRES) and the translated proteins are proteolytically cleaved into mature viral proteins. Dicistroviridae and Iflaviridae mainly consist of insect picorna-like viruses and are rapidly expanding families in the order Picornavirales. Some of these viruses are pathogenic and cause severe mortality to the host, such as deformed wing virus [3] and slow bee paralysis virus in honeybees [4].
Culex mosquitoes are known as potential vectors for several pathogenic viruses, such as Japanese encephalitis virus, West Nile virus and Zika virus [5–7] and are often associated with human infections and can cause mortality worldwide. In contrast, several mosquito-only viruses identified in Culex spp., have only been isolated from mosquitoes or mosquito cell lines [8, 9] and have no known association with vertebrates.
High-throughput sequencing (HTS) technologies have enabled the identification and genetic characterization of many novel viruses, including picorna-like viruses, from various insect hosts, such as plant hoppers [10], bean bugs [11], cotton bollworms [12], spiders [13, 14], as well as Armigeres and Anopheles mosquitoes [15, 16]. However, the picorna-like viruses in mosquitoes are poorly characterized. Previously, we have through viral metagenomics on Culex mosquitoes from Mozambique discovered a large number of sequences related to the Iflaviridae viral family [17]. In this study, we used these sequences to assemble and characterize a full-length picorna-like viral genome from the Culex mosquito pool using PCR and Sanger sequencing.
Methods
Mosquito collection, viral metagenomics and bioinformatics analysis
The mosquito collection and viral metagenomic analysis were performed in our previous study on Culex mosquitoes [17]. Briefly, mosquitoes were collected from Cuacua village in the Zambezi Valley of Mozambique from October–November 2014 using CDC light traps, and the genus was determined morphologically. The mosquito pools (up to 20 mosquitoes per pool) were homogenized mechanically. The total RNA was extracted with TRIzol LS reagent (Invitrogen, Life Technologies, USA) according to the manufacturer’s instructions. The samples were pre-amplified by Sequence Independent Single Primer Amplification (SISPA) and submitted to the SciLifeLab for library preparation and sequencing. The sample was sequenced on an Ion Torrent PGM sequencer with an Ion 318™ chip v2 and a max read length of 400 bp. The high-quality reads were mapped to the host genomes (Anopheles, Aedes and Culex genomes), and unmapped reads were then classified using BLASTn and BLASTx with an E-value cutoff of 1e-03. The virus-related sequences were extracted and assembled with the de novo assembler SPAdes [18].
Sequencing of the viral genome and sequence analysis
Extracted RNA was used to synthesize cDNA using Superscript III (Invitrogen) as recommended by the supplier. First-strand synthesis was initiated with random primers. To sequence the genome gaps between the HTS assembled contigs, primers were designed, and the PCR reactions were carried out in a 25-μl reaction using the thermal profile as follows: 95 °C for 10 min; followed by 35 cycles of 95 °C for 30 s, 60–62 °C for 30 s and 72 °C for 1–1.5 min; 72 °C for 7 min. The list of the primers used in this study is enclosed as a Additional file 1. The positive PCR products were sequenced at Macrogen Europe (Macrogen Inc.) using Sanger sequencing. After the amplification and genetic characterization, Bowtie2 [19] was used to assess the genome coverage in the original HTS dataset by mapping the viral metagenomic reads back to the assembled, near full-length viral genome.
RACE analysis of 5′ and 3′ UTRs
The extreme genomic 5′ and 3′ ends were analyzed using rapid amplification of the cDNA ends (RACE). To determine the 3′ end of the viral genome, a total of 1.2 μg of total RNA were annealed with a 0.5 μM of poly (A) specific primer (AP) in a 20 μl volume by heating at 65 °C for 5 min and chilling on ice for 2 min. First-strand cDNA was synthesized with Superscript III at 42 °C for 50 min followed by an enzyme inactivation at 70 °C for 15 min. A gene-specific forward primer (GSP-F) and universal amplification primer (UAP) were used to amplify the viral 3′ end sequences using AmpliTaq Gold DNA polymerase. For the 5′ genomic ends, 1.6 μg of total RNA and 0.5 μM of GSP-RT primer were mixed in a total volume of 20 μl, incubated at 65 °C for 5 min and chilled on ice for 2 min. First-strand synthesis was carried out by Superscript III as described previously. The RNA templates from the cDNA:RNA hybrids were degraded with 1.5 U of RNase H at 37 °C for 20 min. The excess primers from the cDNA reaction were removed using the GeneJet PCR purification kit (Thermo Fisher Scientific). The cDNA was recovered in 15 μl of elution buffer and mixed with 5 μl of 5× tailing reaction buffer, 2.5 μl of 2.5 mM dCTP and 2 μl of terminal deoxynucleotidyl transferase (Tdt) (Thermo Fisher Scientific) for 5′ end tailing at 37 °C for 15 min and then 10 min at 65 °C to inactivate the reaction. For the first round of amplification, Platinum SuperFi DNA Taq polymerase (Thermo Fisher Scientific) was used. In short, 2 μl of cDNA was used in a 25-μl reaction with 10 pmols of each GSP-reverse primer and an AUAP forward primer. The following thermo profile was used to carry out the amplification reactions: 95 °C for 2 min; 35 cycles of 95 °C for 10 s, 60–62 °C for 10 s and 68 °C for 2 min; 68 °C for 5 min and then cooling to 4 °C. The amplification products from the first round were diluted 20-fold in 1 mM EDTA. For the second round of amplification, 2 μl of diluted product and 10 pmols of GSP2- reverse primer and AUAP forward primer were used in a final volume of 25 μl, and the reaction was carried out as before. The amplified PCR products were visualized, and the bands were purified using GeneJet Gel Extraction Kit (Thermo Fisher Scientific). The purified PCR products were cloned into the pJET1.2 vector using the CloneJET PCR cloning kit (Thermo Fisher Scientific) and sequenced at Macrogen Europe.
Analysis of the nucleotide sequence and evolutionary relationships
The open reading frame (ORF) of the viral genome was predicted using ORF finder at NCBI (https://www.ncbi.nlm.nih.gov/orffinder). The conserved domains (helicase, protease and RNA-dependent RNA polymerase (RdRP)) in the predicted ORF amino acid sequence were analyzed by multiple sequence alignment with other members of the order Picornavirales using ClustalW. The pairwise identity percentage matrix was generated with MegaAlign 9.0.4 (DNASTAR). To determine the phylogenetic relationships, the predicted RdRP region of different viruses belonging to the order Picornavirales were obtained from GenBank. ClustalW alignment of 421 amino acids (aa) corresponding to 2650–3071 aa positions were used and the phylogeny was generated using the Maximum Likelihood method based on the JTT matrix-based model with MEGA 7.0.26 software [20]. All positions containing gaps and missing data were eliminated, resulting a total of 256 aa positions of RdRP region in the final dataset. The statistical significance of the tree topologies was evaluated by 500 bootstraps. Viral sequences used in the multiple sequence alignment and phylogeny are summarized in Table 1.
Table 1.
Summary of viruses used in the multiple sequence alignment and phylogentic analysis
| Host | Virus name | Abbreviation | Genbank accession no. | Reference |
|---|---|---|---|---|
| Plant | Cowpea mosaic virus | CPMV | CAA25029.1 | [33] |
| Parsnip yellow fleck virus | PYFV | BAA03151.1 | [34] | |
| Algae | Heterosigma akashiwo RNA virus | HaRNAV | AAP97137.1 | [35] |
| Mammals | Human poliovirus | PV | NP_041277.1 | [36] |
| Foot-and-mouth disease virus | FMDV | CAA25416.1 | [37] | |
| Encephalomyocaditis virus | EMCV | NP_056777.1 | [38] | |
| Human rhinovirus 1B | HRV | BAA00168.1 | [39] | |
| Insect | Slow bee paralysis virus | SBPV | ABS84820.1 | [40] |
| Brevicoryne brassicae virus | BrBV | YP_001285409.1 | [41] | |
| Deformed wing virus | DWV | NP_853560.2 | [23] | |
| Varroa destructor virus | VDV-1 | YP_145791.1 | [42] | |
| Ectropus obliqua picorna-like virus | EoV | AAQ64627.1 | [43] | |
| Kakugo virus | KV | BAD06930.1 | [44] | |
| Aphid lethal paralysis virus | ALPV | NP_733845.1 | [45] | |
| Perina nuda virus | PnV | NP_277061.1 | [22] | |
| Black queen cell virus | BQCV | NP_620564.1 | [24] | |
| Cricket paralysis virus | CrPV | AAF80998.1 | [46] | |
| Acute bee paralysis virus | ABPV | AAN63804.2 | [47] | |
| Triatoma virus | TrV | AAF00472.1 | [48] | |
| Hematobi P virus | HiPV | BAA32553.1 | [49] | |
| Sacbrood virus | SBV | NP_049374.1 | [50] | |
| Plaitia stali intestine virus | PSIV | BAA21898 | [51] | |
| Drosophila C virus | DCV | AAC58807.1 | [25] | |
| Rhapalosiphum padi virus | RhPV | NP_046155.1 | [52] | |
| Infectious flacherie virus | IFV | ADP24157.1 | [53] | |
| Hubei picorna-like virus 35 | HplV-35 | YP_009337666.1 | [28] | |
| Hubei picorna-like virus 34 | HplV-34 | YP_009337152.1 | [28] | |
| Hubei arhropod virus 1 | HuAV 1 | YP_009336629.1 | [28] | |
| Lasiun niger virus 1 | LniV-1 | ASK12212.1 | [54] | |
| Shuangao insect virus 8 | ShiV-8 | APG77945.1 | [28] | |
| Solenopsis invicta virus 4 | SINV-4 | ASK12194.1 | [54] | |
| Solenopsis invicta virus 2 | SINV-2 | ASK12217.1 | [54] | |
| Hubei picorna-like virus 81 | HplV-81 | APG77984.1 | [28] | |
| Lasius neglectus virus 1 | LneV-1 | ASK12212.1 | [54] | |
| Armigeres iflavirus | AaIFV | LC310707 | [15] | |
| Moku virus | MV | KU645789 | [55] | |
| Shrimp | Taura syndrome virus | TSV | ABB17263.2 | [56] |
| Crab | Mud crab dicistrovirus | MCDV | YP_004063985.1 | [57] |
Detection of the virus by RT-PCR assays
Extraction of the total RNA from Culex and Mansonia mosquito pools was performed as described above. Up to one microgram of total RNA was used for first-strand cDNA synthesis with Superscript III and random hexamers, and RT-PCR was performed with AmpliTaq Gold DNA polymerase. The PCR primers used in the assay were 8F (5’-CGACCTAGGACTTATCCAGC-3′) and 7R (5’-ACAATCTAGTGCCTCCTTCTG-3′), with an expected amplicon size of 577 bp. The PCR program was used as follows: 95 °C for 10 min, 35 cycles at 95 °C for 30 s, 60 °C for 30 s and 72 °C for 1 min, and a final extension at 72 °C for 5 min.
Results
The viral metagenomic analysis on Culex spp. mosquitoes showed that the majority of the viral sequences (94.6%) were classified as the Iflaviridae, Dicistroviridae and Picornaviridae viral families [17]. The sequences were assembled into four longer contigs ranging from 578 to 2240 nt, and these contigs showed the closest similarity to Hubei picorna-like virus 35 with an amino acid identity of 37–49% (Table 2). Together, they covered approximately 60% of the Hubei picorna-like virus 35 genome (YP009337666.1). By filling the gaps between the contigs and through the RACE analysis, a nearly full-length viral genome was obtained. Unfortunately, the 3′ RACE of the viral genome was unsuccessful, while the partial 5′ UTR including complete coding sequence was obtained through a series of three successive 5′ RACE reactions. In summary, the near-full length genome, containing the entire coding region, was obtained. This sequence is 9740 nt in length and was tentatively named Culex picorna-like virus 1 (CuPV-1). The genome sequence was submitted to GenBank under the accession number MG833031.
Table 2.
Best BLAST hits for the contigs assembled from Iflaviridae, Dicistroviridae and Picornaviridae viral reads from Culex spp.
| Contig name | Contig length in nt |
Closest relative | % Identity | Accession number |
|---|---|---|---|---|
| contig1 | 2240 | Hubie picrona-like virus 35 | 41 | YP009337666.1 |
| contig2 | 1456 | Hubie picrona-like virus 35 | 37 | YP009337666.1 |
| contig3 | 869 | Hubie picrona-like virus 35 | 41 | YP009337666.1 |
| contig4 | 578 | Hubie picrona-like virus 35 | 49 | YP009337666.1 |
Open reading frame analysis and genome organization
The viral genomic sequence was found to be A/U rich (A- 32.23%, U- 31.02%, G- 21.79% and C- 14.94%). An in silico analysis of the identified nucleotide sequence of CuPV-1 showed that the genomic RNA contains a single large open reading frame (ORF) oriented from the 5′ to 3′ end. This large ORF consists of 9339 nt, encoding a 3112-amino acid protein and accounting for 95.58% of the CuPV-1 genome (Fig. 1). It has a predicted molecular mass of 352.21 kDa and theoretical isoelectric point (pI) of 5.78. The ORF was appended by a partial 5′ and 3′ UTRs which are 361 and 41 nt respectively. No large ORFs were found in the inverse orientation of the CuPV-1 genome, suggesting that the CuPV-1 genome is a positive-strand RNA virus.
Fig. 1.
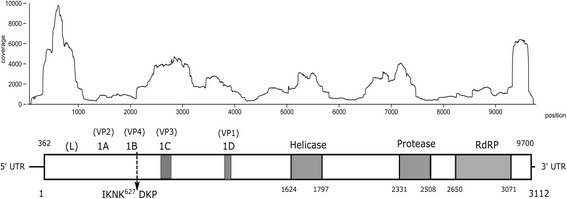
Schematic presentation of the genome of CuPV-1. The HTS reads were mapped back to the sequenced genome using Bowtie2 to display the coverage and sequence depth (upper panel). The ORF corresponds to the entire open box (lower panel). The numbers above the ORF indicate the nucleotide positions and below are the amino acid positions. In the box, the position of the putative structural proteins (1A to D) and the non-structural proteins (Leader peptide, L; Helicase; Protease; RNA-dependent RNA polymerase, RdRP) are shown. The dark areas in the ORF represent regions containing conserved sequences in the viral structural or the non-structural proteins. The dotted-arrow represents the identified cleavage site. The approximate positions of the structural and non-structural proteins were identified by the sequence similarity of other picorna-like viruses
Structural proteins
Conserved domains of structural proteins were found on 5′ end of the CuPV-1 ORF. An rhv_like domain (Picornavirus capsid protein domain_like, cd00205) was identified by NCBI BLAST conserved domain suite with an E-value of 3.73e-19, at amino acids 682–876. The deduced amino acid multiple sequence analysis of the insect picorna-like viruses, including CuPV-1, revealed that CuPV-1 contains key motifs that are known to be present in the capsid proteins of picornaviruses. The conserved motifs identified on the amino acid sequences were: YXGX8VX4HX9F for 1C (VP3), FXRG and DDFX7GXP for 1D (VP1) (Additional file 2, A and B). The cleavage site for 1B/1C (NX/DXP) was also detected in CuPV-1 (Fig. 1). No conserved motifs for 1A (VP2) (NXNXFQXG) and the leader protein, the most variable region of the insect picorna-like virus genomes, were identified.
Non-structural proteins
The comparison of the CuPV-1 non-structural proteins with other picorna-like viral proteins was performed to identify the similarities and conserved regions of the putative helicase, protease and RdRP (Fig. 2).
Fig. 2.

Comparison of the deduced amino acid sequences of the non-structural proteins of CuPV-1 and 11 other picorna-like viruses. a Alignment of the conserved regions of the putative RNA helicase region. (Full names and references of these viruses are shown in Table 1). The motifs identified by Koonin and Dolja (1993) are labeled A, B, and C. b Alignment of the putative protease domain of CuPV-1 with those of other viruses. c Alignment of the putative RNA-dependent RNA polymerase domain of CuPV-1 with those of other viruses, which are labeled I-VIII. The conserved residues are marked with asterisks (*) and residues that are identical in more than 60% of the sequences are shown in dark shades
Helicase: Three conserved helicase regions were recognized in the deduced amino acid sequences of the predicted CuPV-1 ORF ranging from 1624 to 1797. A conserved RNA-helicase domain (pfam00910, E-value 2.83E-11) was identified in this region. The highly conserved consensus sequence within the first domain, GXXGXGKS, was found between amino acid positions 1650–1657, although ‘K’ was substituted for ‘G’ at the 1653 amino acid position. The last two conserved domains deviated somewhat from the consensus. The highly conserved amino acids were QX5DD and KGX4SX5STN, while the equivalents in CuPV-1 were HX5DD and KDX4PX5TSN, respectively (Fig. 2a).
Protease: The deduced amino acid sequence of the CuPV-1’s ORF from 2331 to 2508 is similar to the protease sequence of the other picorna-like viruses [10, 21]. The conserved motif, GXCG, was found at 2469–2472, and the equivalent motif of GXHXXG, SXHXXG was found at 2485–2490. The amino acids are thought to form a catalytic triad of the protease, with the presence of H2349, D2388 and C2471 in this region (Fig. 2b).
RNA-dependent RNA polymerase (RdRP): Eight conserved domains, found between 2650 and 3071 in the deduced amino acid sequence of CuPV-1, correspond to those recognized previously, and an RNA_dep_RNAP domain (cd01699; E-value 2.52e-44) was also found in this region (Fig. 2c). This showed that the CuPV-1 RdRP belongs to a superfamily of positive-strand RNA eukaryotic viruses. The conserved or equivalent domains I-VIII in the RdRP of CuPV-1 are located in amino acids between 2743 and 3041 (Table 3). The RdRP amino acid sequence identity between CuPV-1 and closely related virus (HplV-35) was found to be 57.2%. The amino acid sequence identity matrix with RdRP regions and complete polyproteins was described in the Additional file 3.
Table 3.
Conserved domains of RdRP amino acid sequences identified in CuPV-1
| RNA-dependent RNA polymerase domains | Amino acid positions | Equivalent conserved domains in CuPV-1 |
|---|---|---|
| I | 2743–2748 | DCLKDE |
| II | 2764–2790 | FSMSNIEGSIALRRFTLSLTNHIKNNR |
| III | 2795–2807 | VCVGINPESPEWT |
| IV | 2823–2833 | DFTNFGAGLNY |
| V | 2886–2905 | GSPSGACITVEINSFVHLMY |
| VI | 2955–2965 | TACVYGDDGIF |
| VII | 3007–3019 | EECTFLKRRFVTH |
| VIII | 3027–3041 | AAEMEWLSIEECVKW |
Phylogenetic relationship of CuPV-1 to other viruses in Picornavirales order
To determine the phylogenetic relationship of CuPV-1 in other members of the Picornavirales order, phylogenetic analysis was performed using the highly conserved RdRP region including I-VIII domains. The virus formed a clade with known members of iflaviruses, such as Hubei picorna-like virus 35 and Hubei picorna-like virus 34 (unclassified picorna-like viruses), Sacbrood virus (iflavirus) and Hubei arthropod virus 1 (HuAV-1) (Fig. 3). Other clades consisted of viruses that belonged to the families, such as Dicistroviridae, Picornaviridae, Secoviridae, Marnaviridae and newly proposed Polycipiviridae. This suggests that CuPV-1 belongs to the Iflaviridae viral family.
Fig. 3.
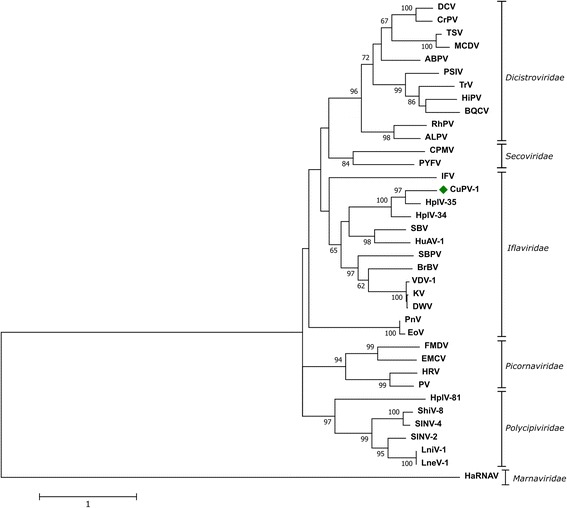
Phylogenetic analysis of the putative RdRP region. The Maximum Likelihood tree was produced, and bootstrapped at 500 replicates using MEGA7 software and involved 37 amino acid sequences, including CuPV-1. Viruses and references are listed in Table 1
Detection of CuPV-1 in Culex and Mansonia mosquito pools
A total of 340 mosquitoes were collected in the Zambezi Valley, a central region of Mozambique. The specimens included mosquitoes of two species, Culex (159) and Mansonia (181), in 23 pools (13 Culex and 10 Mansonia). These mosquito pools were screened with RT-PCR, using primers designed for the selected CuPV-1 RdRP region. Seven pools were positive for CuPV-1, 5 from Culex and 2 from Mansonia. Each pool included 1–20 individuals and included both male and female mosquitoes (Table 4). The overall minimal infection rate (MIR), which was expressed as the number of positive pools per 1000 mosquitoes, was 0.20 (7/340), while the specific MIR for Culex was 0.31 (5/159) and for Mansonia was 0.11 (2/181). The Sanger sequencing of the positive PCR products showed that pools 9, 12, and 13 were identical to CuPV-1, however, Culex pools 3 and 5 showed minor sequence variation compared to both CuPV-1 (97% identity) and each other (97% identity).
Table 4.
Mosquito pools belong to Culex and Mansonia spp. screened for CuPV-1 by RT-PCR with primers specific to RdRP region
| Pool no. | Mosquito spp. | No. of mosquitoes | Positive/Negative |
|---|---|---|---|
| 1 | Culex | 1 | Negative |
| 2 | Culex | 9 | Negative |
| 3 | Culex | 18 | Positive |
| 4 | Culex | 9 | Negative |
| 5 | Culex | 20 | Positive |
| 6 | Culex | 20 | Negative |
| 7 | Culex | 20 | Negative |
| 8 | Culex | 4 | Negative |
| 9 | Culex | 1 | Positive |
| 10 | Culex | 13 | Negative |
| 11 | Culex | 20 | Negative |
| 12 | Culex | 20 | Positive |
| 13 | Culex | 4 | Positive |
| 14 | Mansonia | 20 | Negative |
| 15 | Mansonia | 20 | Negative |
| 16 | Mansonia | 20 | Negative |
| 17 | Mansonia | 12 | Positive |
| 18 | Mansonia | 20 | Negative |
| 19 | Mansonia | 20 | Negative |
| 20 | Mansonia | 20 | Negative |
| 21 | Mansonia | 20 | Negative |
| 22 | Mansonia | 16 | Negative |
| 23 | Mansonia | 13 | Positive |
Mapping of Culex metagenomic reads to CuPV-1 genome
The Culex metagenomic dataset, after the quality check, was mapped to the CuPV-1 genome using Bowtie2 to estimate the read coverage in the original data set. Among the 1,684,319 reads in the dataset, 80,636 reads (4.83% of total reads) were aligned throughout the genome with a coverage range of 9–9784 (Fig. 1), with a lower coverage towards the ends of the genome.
Discussion
With the advances in high-throughput sequencing technologies, the number of novel viruses detected and genetically characterized has rapidly increased. Here, we report a novel and highly divergent virus sequence named Culex picorna-like virus 1 that was characterized from the Culex spp. mosquitoes by viral metagenomics and Sanger sequencing. The monopartite, monocistronic, near full-length single-stranded RNA genome (9.7 kb) was obtained. The genome encodes as single ORF coding for a 3112-amino acid polyprotein. RACE analysis was used; however, a partial 5′ UTR was obtained, which may be due to the presence of complex secondary structures of RNA, the IRES sequence and genome-linked viral proteins that interfere with cloning and sequencing [10, 22].
Multiple sequence alignments of the CuPV-1 ORF with other Picornavirales ORFs revealed that CuPV-1 possesses three functional motifs of helicase, protease and RNA-dependent RNA polymerases that are conserved in all members of the order Picornavirales. These motifs were located at the 5′ end, as observed in other picorna-like viruses [23, 24]. The non-structural protein sequences located at the 3′ end have also been found in other picorna-like viruses [24, 25]. These are involved in different functions, such as the unwinding of nucleic acids, polyprotein processing and the replication of viral genome [26, 27]. The equivalent conserved motifs for 1C and 1D were observed at the 5′ end of the ORF and showed similarities with iflavirus genome organization, suggesting that the CuPV-1 may belong to the Iflaviridae viral family. However, the cleavage sites of the structural and non-structural proteins of CuPV-1 need to be confirmed.
Phylogenetic analysis showed that the CuPV-1 clustered with the members of iflaviruses, further suggesting that CuPV-1 is a novel member of the Iflaviridae family. CuPV-1 showed closest evolutionary relationship to the unclassified picorna-like viruses Hubei picorna-like virus 35 and 34 as well as to Sacbrood virus (SBV) and HuAV-1. These viruses have all been identified in different species, Odonata, Coleoptera, honey bee and from an arthropod mix respectively, suggesting that the evolutionary relationship of different iflaviruses is not always connected to the host. This have also been seen for other iflaviruses. Novel iflaviruses from a wide range of hosts may be required to understand the evolutionary relationships of the family Iflaviridae. Mosquito pools that were positive for CuPV-1 in the current study suggested that CuPV-1 can infect both Culex and Mansonia mosquito species with varying infection rates. The sequence variation between the mosquito pools also indicate the presence of different CuPV-1 variants in nature. Widespread screening of mosquito species in different areas may reveal the prevalence and host range of CuPV-1.
Picorna-like viruses have been identified in a broad range of insects from the class Insecta. These insect viruses were classified by their genome organization, and most of them are assigned to Iflaviridae and Dicistroviridae in the order Picornavirales. By the recent metagenomic analyses, several viruses related to this order were also identified in the class Mammalia [28–30]. Insect picorna-like viruses are maintained in the nature by horizontal or vertical transmissions. For example, DWV can transmit from the queen honey bee to the offspring by vertical transmission and from Varroa mites to bees by horizontal transmission [3, 31]. Both, transovarial and horizontal transmissions have also been observed in SBV and Helicoverpa armigera iflavirus [12, 32]. A few of these viruses are pathogenic to the insect host and are also economically important, such as the infectious flacherivirus of silkworm, acute bee paralysis virus and the SBV of honeybees. However, the host range and pathogenicity of CuPV-1 needs to be further investigated. Previously, picorna-like virus (Armigeres iflavirus) has been isolated from asymptomatic Armigeres spp. mosquitoes [15], and, in a different study, dicistroviruses were found and believed to be members of the natural virome of Anopheles spp. mosquitoes [16].
Conclusions
In the current study, the near full-length genome of a novel picorna-like virus, CuPV-1, was characterized from Culex spp. mosquitoes from Mozambique. The genome organization and phylogenetic analysis indicated that CuPV-1 is a novel member in the order Picornavirales, most likely belonging to the Iflaviridae family, and exhibit great divergence from currently known genera. The discovery and characterization of novel viruses in mosquitoes is an initial step that will facilitate studies on mosquito-virus interactions and pathogenesis.
Additional files
Primers used in this study. (PDF 12 kb)
Conserved domains corresponding to 1C(VP3) and 1D(VP1) in CuPV-1. (PDF 489 kb)
The pairwise amino acid identity matrix of CuPV-1 with other iflaviruses. (PDF 60 kb)
Acknowledgements
The authors would like to acknowledge the support of the National Genomics Infrastructure (NGI) / Uppsala Genome Center and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure. Work performed at the NGI / Uppsala Genome Center has been funded by RFI / VR and Science for Life Laboratory in Sweden. The authors also thank SLU Global Bioinformatics Centre at the Swedish University of Agricultural Sciences in Uppsala for providing the computational time for data analysis. The authors would also like to thank the Swedish Research council VR (grant number: SWE-2012-138 and SWE-2016-01251) and FORMAS (grant number: 2012-586) for the financial support to conduct this study. The funder had no role in study design, data collection and analysis, or the decision to publish or reprint the manuscript.
Funding
The study was funded by the Swedish Research council VR (grant number: SWE-2012-138 and SWE-2016-01251) and FORMAS (grant number: 2012–586). The funder had no role in study design, data collection and analysis, or the decision to publish or reprint of the manuscript.
Availability of data and materials
The metagenomic dataset used in the analysis during the current study is available in the NCBI Sequence read archive (SRA)(http://www.ncbi.nlm.nih.gov/sra) under the accession number PRJNA325205. The CuPV-1 nucleotide sequence was deposited in GenBank under the accession number: MG833031.
Abbreviations
- BLAST
Basic local alignment search tool
- CuPV-1
Culex picorna-like virus 1
- HTS
High-throughput sequencing
- MIR
Minimal infection rate
- ORF
Open reading frame
- RACE
Rapid amplification of cDNA ends
- RdRP
RNA-dependent RNA polymerase
- SISPA
Sequence independent single primer amplification
Authors’ contributions
ALB, MB and JMF participated in the study conception, design and supervision. HC and JMF conducted the sampling of the mosquitoes. HC, JH, ALB, and MB analyzed the sequencing data. HC conducted the full genome sequencing and analysis and drafted manuscript. ALB, MB, JH and JMF reviewed and edited the manuscript. ALB, MB, JH and JMF provided resources for the project. All the authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have declared no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12985-018-0981-z) contains supplementary material, which is available to authorized users.
Contributor Information
Harindranath Cholleti, Email: harindranath.cholleti@slu.se.
Juliette Hayer, Email: juliette.hayer@slu.se.
Jose Fafetine, Email: jfafetine@yahoo.com.
Mikael Berg, Email: mikael.berg@slu.se.
Anne-Lie Blomström, Email: anne-lie.blomstrom@slu.se.
References
- 1.Valles SM, Chen Y, Firth AE, Guerin DMA, Hashimoto Y, Herrero S, et al. ICTV virus taxonomy profile: Iflaviridae. The Journal of general virology. 2017;98:527–528. doi: 10.1099/jgv.0.000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koonin EV, Dolja VV. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit Rev Biochem Mol Biol. 1993;28:375–430. doi: 10.3109/10409239309078440. [DOI] [PubMed] [Google Scholar]
- 3.Wilfert L, Long G, Leggett HC, Schmid-Hempel P, Butlin R, Martin SJ, et al. Deformed wing virus is a recent global epidemic in honeybees driven by Varroa mites. Science. 2016;351:594–597. doi: 10.1126/science.aac9976. [DOI] [PubMed] [Google Scholar]
- 4.Bonning BC, Miller WA. Dicistroviruses. Annu Rev Entomol. 2010;55:129–150. doi: 10.1146/annurev-ento-112408-085457. [DOI] [PubMed] [Google Scholar]
- 5.de Wispelaere M, Despres P, Choumet V. European Aedes albopictus and Culex pipiens are competent vectors for Japanese encephalitis virus. PLoS Negl Trop Dis. 2017;11:e0005294. doi: 10.1371/journal.pntd.0005294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colpitts TM, Conway MJ, Montgomery RR, Fikrig E. West Nile virus: biology, transmission, and human infection. Clin Microbiol Rev. 2012;25:635–648. doi: 10.1128/CMR.00045-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guedes DR, Paiva MH, Donato MM, Barbosa PP, Krokovsky L, Rocha S, et al. Zika virus replication in the mosquito Culex quinquefasciatus in Brazil. Emerging microbes & infections. 2017;6:e69. doi: 10.1038/emi.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolling BG, Weaver SC, Tesh RB, Vasilakis N. Insect-specific virus discovery: significance for the arbovirus community. Viruses. 2015;7:4911–4928. doi: 10.3390/v7092851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolling BG, Eisen L, Moore CG, Blair CD. Insect-specific flaviviruses from Culex mosquitoes in Colorado, with evidence of vertical transmission. The American journal of tropical medicine and hygiene. 2011;85:169–177. doi: 10.4269/ajtmh.2011.10-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murakami R, Suetsugu Y, Kobayashi T, Nakashima N. The genome sequence and transmission of an iflavirus from the brown planthopper, Nilaparvata lugens. Virus research. 2013;176:179–187. doi: 10.1016/j.virusres.2013.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Yang YT, Nai YS, Lee SJ, Lee MR, Kim S, Kim JS. A novel picorna-like virus, Riptortus pedestris virus-1 (RiPV-1), found in the bean bug, R. Pedestris, after fungal infection. J Invertebr Pathol. 2016;141:57–65. doi: 10.1016/j.jip.2016.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Yuan H, Xu P, Yang X, Graham RI, Wilson K, Wu K. Characterization of a novel member of genus Iflavirus in Helicoverpa armigera. J Invertebr Pathol. 2017;144:65–73. doi: 10.1016/j.jip.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 13.Shean RC, Makhsous N, Crawford RL, Jerome KR, Greninger AL. Draft genome sequences of six novel Picorna-like viruses from Washington state spiders. Genome announcements. 2017;5 10.1128/genomeA.01705-16. [DOI] [PMC free article] [PubMed]
- 14.Li CX, Shi M, Tian JH, Lin XD, Kang YJ, Chen LJ, et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. elife. 2015;4 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed]
- 15.Kobayashi D, Isawa H, Fujita R, Murota K, Itokawa K, Higa Y, et al. Isolation and characterization of a new iflavirus from Armigeres spp. mosquitoes in the Philippines. The Journal of general virology. 2017;98:2876–2881. doi: 10.1099/jgv.0.000929. [DOI] [PubMed] [Google Scholar]
- 16.Carissimo G, Eiglmeier K, Reveillaud J, Holm I, Diallo M, Diallo D, et al. Identification and characterization of two novel RNA viruses from Anopheles gambiae species complex mosquitoes. PLoS One. 2016;11:e0153881. doi: 10.1371/journal.pone.0153881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cholleti H, Hayer J, Abilio AP, Mulandane FC, Verner-Carlsson J, Falk KI, et al. Discovery of novel viruses in mosquitoes from the Zambezi Valley of Mozambique. PLoS One. 2016;11:e0162751. doi: 10.1371/journal.pone.0162751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of computational biology : a journal of computational molecular cell biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liljas L, Tate J, Lin T, Christian P, Johanson JE. Evolutionary and taxonomic implications of conserved structural motifs between picornaviruses and insect picorna-like viruses. Arch Virol. 2002;147:59–84. doi: 10.1007/s705-002-8303-1. [DOI] [PubMed] [Google Scholar]
- 22.Wu CY, Lo CF, Huang CJ, Yu HT, Wang CH. The complete genome sequence of Perina nuda picorna-like virus, an insect-infecting RNA virus with a genome organization similar to that of the mammalian picornaviruses. Virology. 2002;294:312–323. doi: 10.1006/viro.2001.1344. [DOI] [PubMed] [Google Scholar]
- 23.Lanzi G, de Miranda JR, Boniotti MB, Cameron CE, Lavazza A, Capucci L, et al. Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L.) J Virol. 2006;80:4998–5009. doi: 10.1128/JVI.80.10.4998-5009.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leat N, Ball B, Govan V, Davison S. Analysis of the complete genome sequence of black queen-cell virus, a picorna-like virus of honey bees. The Journal of general virology. 2000;81:2111–2119. doi: 10.1099/0022-1317-81-8-2111. [DOI] [PubMed] [Google Scholar]
- 25.Johnson KN, Christian PD. The novel genome organization of the insect picorna-like virus Drosophila C virus suggests this virus belongs to a previously undescribed virus family. The Journal of general virology. 1998;79(Pt 1):191–203. doi: 10.1099/0022-1317-79-1-191. [DOI] [PubMed] [Google Scholar]
- 26.Cheng Z, Yang J, Xia H, Qiu Y, Wang Z, Han Y, et al. The nonstructural protein 2C of a Picorna-like virus displays nucleic acid helix destabilizing activity that can be functionally separated from its ATPase activity. J Virol. 2013;87:5205–5218. doi: 10.1128/JVI.00245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Porter AG. Picornavirus nonstructural proteins: emerging roles in virus replication and inhibition of host cell functions. J Virol. 1993;67:6917–6921. doi: 10.1128/jvi.67.12.6917-6921.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature. 2016; 10.1038/nature20167. [DOI] [PubMed]
- 29.Sakuna K, Elliman J, Owens L. Discovery of a novel Picornavirales, Chequa iflavirus, from stressed redclaw crayfish (Cherax quadricarinatus) from farms in northern Queensland. Australia Virus research. 2017;238:148–155. doi: 10.1016/j.virusres.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Yinda CK, Zell R, Deboutte W, Zeller M, Conceicao-Neto N, Heylen E, et al. Highly diverse population of Picornaviridae and other members of the Picornavirales, in Cameroonian fruit bats. BMC Genomics. 2017;18:249. doi: 10.1186/s12864-017-3632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yue C, Genersch E. RT-PCR analysis of deformed wing virus in honeybees (Apis mellifera) and mites (Varroa destructor) The Journal of general virology. 2005;86:3419–3424. doi: 10.1099/vir.0.81401-0. [DOI] [PubMed] [Google Scholar]
- 32.Shen M, Cui L, Ostiguy N, Cox-Foster D. Intricate transmission routes and interactions between picorna-like viruses (Kashmir bee virus and sacbrood virus) with the honeybee host and the parasitic varroa mite. The Journal of general virology. 2005;86:2281–2289. doi: 10.1099/vir.0.80824-0. [DOI] [PubMed] [Google Scholar]
- 33.Lomonossoff GP, Shanks M. The nucleotide sequence of cowpea mosaic virus B RNA. EMBO J. 1983;2:2253–2258. doi: 10.1002/j.1460-2075.1983.tb01731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turnbull-Ross AD, Reavy B, Mayo MA, Murant AF. The nucleotide sequence of parsnip yellow fleck virus: a plant picorna-like virus. The Journal of general virology. 1992;73(Pt 12):3203–3211. doi: 10.1099/0022-1317-73-12-3203. [DOI] [PubMed] [Google Scholar]
- 35.Lang AS, Culley AI, Suttle CA. Genome sequence and characterization of a virus (HaRNAV) related to picorna-like viruses that infects the marine toxic bloom-forming alga Heterosigma akashiwo. Virology. 2004;320:206–217. doi: 10.1016/j.virol.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Racaniello VR, Baltimore D. Molecular cloning of poliovirus cDNA and determination of the complete nucleotide sequence of the viral genome. Proc Natl Acad Sci U S A. 1981;78:4887–4891. doi: 10.1073/pnas.78.8.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forss S, Strebel K, Beck E, Schaller H. Nucleotide sequence and genome organization of foot-and-mouth disease virus. Nucleic Acids Res. 1984;12:6587–6601. doi: 10.1093/nar/12.16.6587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelsen-Salz B, Zimmermann A, Wickert S, Arnold G, Botta A, Eggers HJ, et al. Analysis of sequence and pathogenic properties of two variants of encephalomyocarditis virus differing in a single amino acid in VP1. Virus Res. 1996;41:109–122. doi: 10.1016/0168-1702(95)01257-5. [DOI] [PubMed] [Google Scholar]
- 39.Hughes PJ, North C, Jellis CH, Minor PD, Stanway G. The nucleotide sequence of human rhinovirus 1B: molecular relationships within the rhinovirus genus. The Journal of general virology. 1988;69(Pt 1):49–58. doi: 10.1099/0022-1317-69-1-49. [DOI] [PubMed] [Google Scholar]
- 40.de Miranda JR, Dainat B, Locke B, Cordoni G, Berthoud H, Gauthier L, et al. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.) The Journal of general virology. 2010;91:2524–2530. doi: 10.1099/vir.0.022434-0. [DOI] [PubMed] [Google Scholar]
- 41.Ryabov EV. A novel virus isolated from the aphid Brevicoryne brassicae with similarity to Hymenoptera picorna-like viruses. The Journal of general virology. 2007;88:2590–2595. doi: 10.1099/vir.0.83050-0. [DOI] [PubMed] [Google Scholar]
- 42.Ongus JR, Peters D, Bonmatin JM, Bengsch E, Vlak JM, van Oers MM. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. The Journal of general virology. 2004;85:3747–3755. doi: 10.1099/vir.0.80470-0. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Zhang J, Lu J, Yi F, Liu C, Hu Y. Sequence analysis and genomic organization of a new insect picorna-like virus, Ectropis obliqua picorna-like virus, isolated from Ectropis obliqua. The Journal of general virology. 2004;85:1145–1151. doi: 10.1099/vir.0.19638-0. [DOI] [PubMed] [Google Scholar]
- 44.Fujiyuki T, Takeuchi H, Ono M, Ohka S, Sasaki T, Nomoto A, et al. Novel insect picorna-like virus identified in the brains of aggressive worker honeybees. J Virol. 2004;78:1093–1100. doi: 10.1128/JVI.78.3.1093-1100.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Munster M, Dullemans AM, Verbeek M, Van Den Heuvel JF, Clerivet A, Van Der Wilk F. Sequence analysis and genomic organization of aphid lethal paralysis virus: a new member of the family Dicistroviridae. The Journal of general virology. 2002;83:3131–3138. doi: 10.1099/0022-1317-83-12-3131. [DOI] [PubMed] [Google Scholar]
- 46.Wilson JE, Powell MJ, Hoover SE, Sarnow P. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol Cell Biol. 2000;20:4990–4999. doi: 10.1128/MCB.20.14.4990-4999.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Govan VA, Leat N, Allsopp M, Davison S. Analysis of the complete genome sequence of acute bee paralysis virus shows that it belongs to the novel group of insect-infecting RNA viruses. Virology. 2000;277:457–463. doi: 10.1006/viro.2000.0616. [DOI] [PubMed] [Google Scholar]
- 48.Czibener C, La Torre JL, Muscio OA, Ugalde RA, Scodeller EA. Nucleotide sequence analysis of Triatoma virus shows that it is a member of a novel group of insect RNA viruses. The Journal of general virology. 2000;81:1149–1154. doi: 10.1099/0022-1317-81-4-1149. [DOI] [PubMed] [Google Scholar]
- 49.Nakashima N, Sasaki J, Toriyama S. Determining the nucleotide sequence and capsid-coding region of himetobi P virus: a member of a novel group of RNA viruses that infect insects. Arch Virol. 1999;144:2051–2058. doi: 10.1007/s007050050726. [DOI] [PubMed] [Google Scholar]
- 50.Ghosh RC, Ball BV, Willcocks MM, Carter MJ. The nucleotide sequence of sacbrood virus of the honey bee: an insect picorna-like virus. The Journal of general virology. 1999;80(Pt 6):1541–1549. doi: 10.1099/0022-1317-80-6-1541. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki J, Nakashima N, Saito H, Noda H. An insect picorna-like virus, Plautia stali intestine virus, has genes of capsid proteins in the 3′ part of the genome. Virology. 1998;244:50–58. doi: 10.1006/viro.1998.9094. [DOI] [PubMed] [Google Scholar]
- 52.Moon JS, Domier LL, McCoppin NK, D'Arcy CJ, Jin H. Nucleotide sequence analysis shows that Rhopalosiphum padi virus is a member of a novel group of insect-infecting RNA viruses. Virology. 1998;243:54–65. doi: 10.1006/viro.1998.9043. [DOI] [PubMed] [Google Scholar]
- 53.Isawa H, Asano S, Sahara K, Iizuka T, Bando H. Analysis of genetic information of an insect picorna-like virus, infectious flacherie virus of silkworm: evidence for evolutionary relationships among insect, mammalian and plant picorna(−like) viruses. Arch Virol. 1998;143:127–143. doi: 10.1007/s007050050273. [DOI] [PubMed] [Google Scholar]
- 54.Olendraite I, Lukhovitskaya NI, Porter SD, Valles SM, Firth AE. Polycipiviridae: a proposed new family of polycistronic picorna-like RNA viruses. The Journal of general virology. 2017;98:2368–2378. doi: 10.1099/jgv.0.000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mordecai GJ, Brettell LE, Pachori P, Villalobos EM, Martin SJ, Jones IM, et al. Moku virus; a new Iflavirus found in wasps, honey bees and Varroa. Sci Rep. 2016;6:34983. doi: 10.1038/srep34983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Côté I, Navarro S, Tang KFJ, Noble B, Lightner DV. Taura syndrome virus from Venezuela is a new genetic variant. Aquaculture. 2008;284:62–67. doi: 10.1016/j.aquaculture.2008.07.059. [DOI] [Google Scholar]
- 57.Guo ZX, He JG, Xu HD, Weng SP. Pathogenicity and complete genome sequence analysis of the mud crab dicistrovirus-1. Virus Res. 2013;171:8–14. doi: 10.1016/j.virusres.2012.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Primers used in this study. (PDF 12 kb)
Conserved domains corresponding to 1C(VP3) and 1D(VP1) in CuPV-1. (PDF 489 kb)
The pairwise amino acid identity matrix of CuPV-1 with other iflaviruses. (PDF 60 kb)
Data Availability Statement
The metagenomic dataset used in the analysis during the current study is available in the NCBI Sequence read archive (SRA)(http://www.ncbi.nlm.nih.gov/sra) under the accession number PRJNA325205. The CuPV-1 nucleotide sequence was deposited in GenBank under the accession number: MG833031.