

SUMMARY
Chronic neuropathic pain is a major morbidity of neural injury, yet its mechanisms are incompletely understood. Hypersensitivity to previously non-noxious stimuli (allodynia) is a common symptom. Here, we demonstrate that the onset of cold hypersensitivity precedes tactile allodynia in a model of partial nerve injury, and this temporal divergence was associated with major differences in global gene expression in innervating dorsal root ganglia. Transcripts whose expression change correlates with the onset of cold allodynia were nociceptor related, whereas those correlating with tactile hypersensitivity were immune cell centric. Ablation of TrpV1 lineage nociceptors resulted in mice that did not acquire cold allodynia but developed normal tactile hypersensitivity, whereas depletion of macrophages or T cells reduced neuropathic tactile allodynia but not cold hypersensitivity. We conclude that neuropathic pain incorporates reactive processes of sensory neurons and immune cells, each leading to distinct forms of hypersensitivity, potentially allowing drug development targeted to each pain type.
In Brief
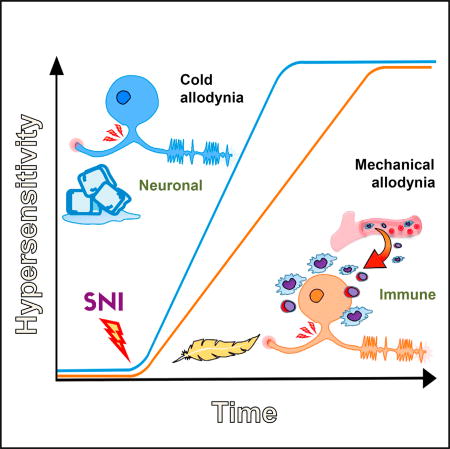
Cobos et al. correlated gene expression with behavior after nerve injury and found that two distinct processes contribute to neuropathic pain: one that occurs in neurons, leading to cold allodynia, and another that includes immune cells and neurons, leading to tactile allodynia.
INTRODUCTION
Peripheral neuropathic pain in animal models is associated with hypersensitivity to noxious and non-noxious stimuli in areas of tissue that neighbor those normally innervated by the damaged nerves. Changes both in the peripheral nervous system (PNS) and central nervous system (CNS) contribute to the development of this pain hypersensitivity (Costigan et al., 2009b). Global gene expression studies in the adult rodent dorsal root ganglia (DRG) in response to sciatic nerve injury have helped define the peripheral mechanisms likely to contribute to the changes in neuropathic pain-like hypersensitivity (Costigan et al., 2009b; LaCroix-Fralish et al., 2011), as well as novel targets for therapy (Dib-Hajj and Waxman, 2014; Tegeder et al., 2006).
Following sciatic nerve injury, the ipsilateral L3-5 lumbar DRGs contain the cell bodies of injured and non-injured primary sensory neurons, satellite cells, fibroblasts, and blood vessels, as well as resident immune cells and those recruited from the blood (Hu et al., 2007). Peripheral nerve injury induces transcriptional changes in each of these diverse cell types (Costigan et al., 2002, 2010; Watkins and Maier, 2002). Peripheral nerve injury induces pain-like hypersensitivity in rodents that develops over the first week or so following the axonal damage (Colleoni and Sacerdote, 2010; Jaggi et al., 2011). Here, we have determined the onset of two chronic pain-like sensory modalities (tactile and cold allodynia) in C57BL/6 mice at high temporal resolution (daily) over the first 10 days in the spared nerve injury (SNI) model (Decosterd and Woolf, 2000), and we found clear differences in their temporal evolution, with cold sensitivity developing quicker than tactile allodynia. The temporal separation of these two clinically important neuropathic pain modalities (Jensen and Finnerup, 2014) led us to design a global gene expression study in lumbar DRGs ipsilateral to the nerve injury to directly correlate the relative timing of transcript expression and sensory modality changes.
We demonstrate differences in the kinetics of early neuronal and late immune gene regulation events, changes which closely mirror the onset of cold and tactile allodynia respectively. These data indicate that different cellular and molecular mechanisms may be responsible for development of tactile and cold allodynia in the damaged PNS, which we confirmed by selectively targeting the immune and nervous systems. Understanding the differences in pain hypersensitivity features should allow us to develop new therapies tailored to their distinct underlying mechanisms.
RESULTS
Onset of Cold and Tactile Allodynia
Tactile and cold allodynia both develop subsequent to peripheral nerve injury and are major clinical concerns of neuropathic pain patients (Jensen and Finnerup, 2014). A high-density time-course analysis of neuropathic pain-related behavioral onset showed that cold and tactile hypersensitivity developed to maximal levels in the first week after nerve injury and persisted for at least 15 days. Cold allodynia developed relatively quickly, reaching a statistically significant increase at 3 days and achieving peak levels 4–5 days post-SNI (Figure 1A); however, tactile allodynia became established over a slower time frame, with a statistically significant decrease of the mechanical threshold at 5 days and reaching maximal levels 7–8 days post-SNI (Figures 1B and 1C). These data agree with previous reports in which cold allodynia develops faster than mechanical hypersensitivity (Decosterd and Woolf, 2000; Pertin et al., 2007; Wijnvoord et al., 2010). Sham-operated controls did not show alterations in either cold or tactile hypersensitivity (Figures 1A and 1B, respectively).
Figure 1. Differences in the Onset of Cold and Tactile Allodynia.

(A and B) Cold allodynia (A) develops relatively quickly post-injury, whereas tactile allodynia (B) develops at a slower pace.
(C) The onset time of cold hypersensitivity is illustrated by the blue line (cold) relative to the later onset of tactile hypersensitivity illustrated by the orange line (tactile).
Statistically significant differences between the values from mice after SNI and their basal measures in (A) and (B); *p < 0.01 (one-way repeated-measures ANOVA followed by Bonferroni post hoc test). There were no statistically significant differences between basal measures and values after sham surgery (gray lines, one-way repeated-measures ANOVA). Error bars indicate SEM (n = 13 or 14 per group; see Supplemental Experimental Procedures).
Transcripts Regulated in the DRG Post-SNI
The differences in the onset time of cold and tactile allodynia led us to consider if these pain modalities may have different mechanisms. To investigate this, we performed an expression array profiling experiment over a similar high-resolution time course to the behavioral studies. We evaluated global gene expression in the ipsilateral DRG daily for the first 10 days post-SNI and also at 8 and 16 hr post-SNI.
We initially assessed changes in gene expression in the DRG over 10 days post-SNI in all 1,704 probes differentially regulated over time between naive and injured conditions (moderated F-statistic, p < 0.01; Table S1) by a weighted gene co-expression network analysis (WGCNA) (Parikshak et al., 2015). By employing a module merger step that used Euclidean distance to cluster the average module regulation patterns across time (not shown), we were able to reveal eight distinct expression clusters (clusters I–VIII) comprising 1,699 of the regulated probes (Figure 2) and 5 additional probes that did not fit in any of the eight clusters (Table S1). Ingenuity Pathway Analysis (IPA) of content of each cluster annotated their general functional identity, which delineate the most overrepresented descriptors of gene function assigned to each transcript in each cluster (see “Function/Cell typ” and “Example Genes” in Figure 2).
Figure 2. WGCNA Analysis of All Regulated Transcripts in DRG.

Significantly regulated probes (moderated F-statistic, n = 1,704) were subject to WGCNA analysis to produce unbiased clusters of co-regulated transcripts representing the entire regulatory network of transcripts in the DRG following peripheral nerve injury (SNI) for 10 days sampled at least once a day over this period. The first column defines the clusters present, and the second shows heatmaps of each gene in each cluster. Blue represents low-level expression and red high-level expression. The third column gives a brief description of cluster function as defined by IPA software. Below this is the number of probes in each cluster and the p value IPA ascribed the function given. The final column gives example transcripts from each functional subdivision. See also Table S1.
A similarity plot of average gene expression across time for each cluster demonstrates that each has distinct expression patterns, although similarities are observed between clusters III and VI (two of the neuronal modules), and also between the clusters VII and VIII (mixed function and late immune) (Figure 3A). By plotting the relative expression of each gene cluster across time, we could identify four cluster groups with distinct kinetic patterns (Figures 3B–3E). Genes in cluster I (largely chemotaxis related) showed an immediate but short-lived expression change that peaked several hours post-SNI (Figure 3B). The next kinetic group contained the predominantly neuronal regulatory clusters II, III, and VI, which display a monophasic expression pattern (Figure 3C). Clusters II and III demonstrate a late decrease in relative expression, although average expression did not fall back to baseline by 10 days post-SNI. In contrast, the expression level of cluster VI remained at or around maximal levels until 10 days after SNI. Next, genes in clusters IV and V initially showed a down-regulation in reaction to the injury and then displayed a relatively strong increase that peaked at ~3 days after nerve injury before settling to approximately naive levels at later time points (Figure 3D). The final kinetically distinct group contained two clusters that demonstrated sustained increases of expression, reaching maximal levels over 10 days, with the rise of cluster VII (mixed neuronal/support cell and immune) preceding that of cluster VIII (late immune) (Figure 3E). Each cluster contains not only genes regulated in the direction depicted in Figures 3B–3E but also genes regulated in a pattern mirroring that shown (reciprocal regulation events). Figure 2 shows all gene regulation patterns both positive and negative.
Figure 3. Transcript Regulation in the DRG after SNI Follows Distinct Patterns.

(A) Similarity plot of the module eigengenes of each cluster showing very little overlap in pattern regulation.
(B–E) Representations of the regulation of each cluster given as singles line plots with intensity of regulation on the y axis and time on the x axis. Cluster I (B), clusters II, III, and VI (C), clusters IV and V (D), and clusters VII and VIII (E). Each cluster contains not only genes regulated in the fashion drawn but also reciprocal regulation events.
See also Figures S1, S2, and S3 and Tables S1, S2, and S3.
We performed a transcription factor binding site (TFBS) enrichment analysis for each of the eight clusters to uncover the potential regulatory network contributing to the observed gene regulation patterns after nerve injury. To avoid confounders and identify only the most statistically robust sites, we used 3 different control datasets as background (1,000 bp sequences upstream of all mouse genes, mouse CpG islands, and the mouse chromosome 19 sequence). We identified 210 TFs (Figure S1A) whose DNA binding motifs were over represented in the promoters of each gene cluster set (Table S2). Interestingly, hierarchical clustering of the TFBS enrichment score for each cluster revealed a separation of neuronal and immune-associated gene modules, suggesting distinct regulatory control by transcription of these gene sets after nerve injury (Figure S1A).
To identify potential protein signaling pathways operational after nerve injury, we determined the protein-protein interaction (PPI) network represented by all the differentially regulated genes (see Experimental Procedures). We screened for experimentally validated PPI among all possible combinations of gene pairs present in the DRG regulated gene set, obtaining a PPI network consisting of 310 nodes and 442 edges. This revealed certain key signaling molecules such as ATF3, JUN, BDNF, mitogen-activated protein kinase 1/3 (MAPK1/3), transforming growth factor β1 (TGF-β1), STAT3, TCF3, CCR5, and interleukin-1β (IL-1β) (Figure S2) and certain transcription factors (TFs) as major hubs, potentially regulating many of the genes present in the global injury response; including SP1, ESR1, SMAD3, TP53, and STAT5A (Figure S2). Next, by screening for signaling pathways in the PPI network, we observed enrichment of several important signaling pathways that may contribute to nerve injury response, including the neurotrophin, MAPK, TGF-β, chemokine, and ErbB signaling pathways (Table S3). To identify signaling cascades activated over time after nerve injury, we also examined for the presence of signaling pathway genes in each cluster. These analyses show that there is a group of strongly represented neuronal signaling pathways, including NGF, EGFR, p38 MAPK, and TGF-β in clusters III, IV, and VII (Figure S3), suggesting co-activation of multiple signaling pathways in response to nerve injury (Abe and Cavalli, 2008).
Analysis of the full gene expression dataset suggested a generalized kinetic separation of groups of neuronal and immune-rich transcripts across time, such that overall neuronal gene changes preceded alterations in immune transcript expression in the DRG over the first 10 days after nerve damage (Figures 3C and 3E, respectively). This led us to consider whether such expression differences could reveal information on the functional origins of the diverse behavioral manifestations of neuropathic pain.
Genes Correlated with Cold and Tactile Allodynia in the DRG
To define those transcripts with expression changes most correlated with cold and tactile allodynia, we chose transcripts with a Pearson correlation coefficient greater than 0.85 (directly correlated) or less than −0.85 (inversely correlated) with each behavioral hypersensitivity onset curve (Figures 4A and 4B). The cold allodynia time course closely correlated with the temporal pattern of 145 probes, corresponding to 137 distinct transcripts, of which 107 (78%) were upregulated and 30 (22%) were downregulated (Figure 4A). The list of the transcripts correlated with cold allodynia and their Pearson correlation values can be found in Table S4. For tactile allodynia, we identified 40 probes, corresponding to 36 distinct transcripts whose expression closely correlated to the onset of mechanical sensitivity. Of these, 33 transcripts (92%) were upregulated and 3 (8%) were downregulated (Figure 4B). The list of the transcripts correlated with tactile allodynia and their Pearson correlation values can be found in Table S5.
Figure 4. Transcript Expression in the DRG Correlating with the Temporal Profile of Cold Allodynia Was Nociceptor Related, whereas That with Tactile Hypersensitivity Was Immune Cell Centric.

Gene expression was correlated with the time courses of cold and tactile allodynia development using the Pearson coefficient of similarity.
(A) Heatmap of the relative expression of the probes that most tightly correlate with cold allodynia onset.
(B) Relative expression of the probes that most tightly correlate with tactile allodynia.
(C and D) Representative functional characteristics using IPA of these cold- (C) and tactile-related (D) transcripts.
(E and F) Strongest GO terms for transcripts correlated with cold (E) and tactile (F) allodynia.
(G) Cross-comparison of transcripts present in each cluster with transcript lists derived from isolated DRG nociceptors and isolated macrophages/T cells. Orange represents genes contained only in the immune gene list, blue represents genes contained only in the nociceptor list, gray represents genes present in both lists, and white represents genes not contained in either list.
See also Tables S4, S5, S6, S7, S8, and S9 and Figure S4.
We performed a biological validation of the gene lists that made up the cold- and tactile- correlated groups by comparing these lists with results from DRG transcriptome profiling obtained using RNA sequencing (RNA-seq) (naive and 1, 3, and 7 days after SNI) from biologically distinct tissue to that used in the array studies. We found 116 common genes in the acetone (neuronal) list and 36 common genes in the von Frey (immune) list in both platforms. The heatmaps of common genes from the arrays and RNA-seq for each sensory modality are shown in Figures S2A–S2D, and these data demonstrate that the overall expression patterns of the constituent genes were virtually identical regardless of the platform used to define gene regulation. This RNA-seq dataset also allows validation of the other regulated transcripts described in Figure 2.
When the group of genes correlated with cold allodynia was processed by IPA, “Neurological disease” was the top biological function defined for 33 genes (24%). “Seizures,” “Migraines,” and “Neuropathic pain” were also among functional subgroups identified as present (Figure 4C). (See Table S6 for the content of genes in each annotated function.) In contrast, for the group of genes significantly correlated with tactile allodynia, with IPA, “Immune disease” was identified as the top biological function, with 19 transcripts (53%). “Systemic autoimmune syndrome,” “Antigen presentation activation,” “Insulin-dependent diabetes mellitus,” and “Rheumatoid arthritis” were other functional subgroups identified as present (Figure 4D). (See Table S7 for the content of genes in each annotated function.)
To further assay genes associated with the development of cold and tactile allodynia, we investigated a more inclusive group of transcripts including those genes directly and inversely correlated with a Pearson coefficient of correlation >0.75 (see Experimental Procedures). These gene sets were then subjected to Gene Ontology (GO) analysis to find the most related functional terms (Figures 4E and 4F). Transcripts in this set whose expression over time correlated with the onset of cold allodynia (375 probes) were almost entirely related to neuronal function (Figure 4E). (See Table S8 for the content of genes in each annotated function.) The genes most related to the development of tactile allodynia in the GO analysis were almost entirely related to immune function (130 probes). (See Table S9 for the content of genes in each annotated function.) Both GO analyses accord with the IPA-based data (shown in Figures 4E and 4F).
An enrichment of a particular IPA functional annotation or GO category signifies that multiple genes participating in the same process correlate with the phenotype in question. Using two thresholds of correlation allowed us to span the expression data more exhaustively than using one. The fact that each of these lists result in very complementary functional descriptions using two different pathway search tools with different correlated transcript inclusion criteria represents an internal control for the validity of the associations.
We also compared the cold- and tactile-allodynia-correlated transcripts to previously published analyses of genes specifically expressed either in nociceptor DRG neurons (Chiu et al., 2014) or in activated macrophages and T cells (Brown et al., 2015; Rosas et al., 2014) (see Experimental Procedures). This analysis revealed that 37% of the genes identified as correlated with cold allodynia are expressed specifically in nociceptors, with 16% specific to leukocytes. Using the same approach to assay genes whose expression correlates with tactile allodynia, 51% are specifically expressed in leukocytes, with only 6% specific to nociceptors. Therefore, transcripts correlated with the onset of tactile allodynia have a stronger immune component than neuronal, with the reverse holding true for cold allodynia (Figure 4G).
TrpV1 Lineage Neurons and Neuropathic Allodynia
To determine if changes in a specific set of DRG sensory neurons are responsible for development of cold allodynia post-SNI, we examined TrpV1CreDTAflox-stop mice that lack TrpV1-lineage nociceptors but retain mechanoreceptors (Mishra et al., 2011). We first assessed cold sensitivity in naive (uninjured) TrpV1CreDTAflox-stop mice and their wild-type litter-mates in a dynamic thermal place cold aversion challenge. While the littermate control mice rapidly manifested a preference for moderate (11°C–25°C) rather than noxious cold (4°C–11°C) temperatures, TrpV1CreDTAflox-stop mice displayed no aversion to noxious cold. These data indicate that TrpV1 lineage nociceptors are essential for cold detection under baseline conditions (Figure 5A).
Figure 5. Trpv1 Lineage Neuronal Deletion Mice Develop Tactile, but Not Cold, Allodynia after SNI Injury.

(A) When naive mice are given the choice between two opposing temperatures, wild-type control littermate (LM) mice move toward the more ambient temperature, whereas Trpv1 DTA mice do not. Each point on the graph measures the amount of time spent on plate A in a 30-s window (y axis) when the plate was set to the temperature given on the lower x axis. The top x axis gives the temperature of the alternate plate for that time bin.
(B) TrpV1 lineage DTA mice and their LM control counterparts develop tactile allodynia post-SNI. Statistically significant differences between the values from mice after SNI and their basal measures are shown (**p < 0.01). However, there was no significant difference between the two curves (two-way repeated-measures ANOVA).
(C) TrpV1 lineage DTA mice develop very weak levels of cold allodynia relative to their LM controls. Statistically significant differences between the values from mice after SNI and their basal measures (**p < 0.01) and between wild-type LM controls and TrpV1 DTA mice in cold sensitivity (##p < 0.01) are shown (two-way repeated-measures ANOVA, Bonferroni post hoc test).
For (A), n = 9 (TrpV1-DTA), n = 10 (LM controls); for (B) and (C), n = 8 (both groups). Error bars indicate SEM. See also Figure S5.
Next, we determined the temporal development of tactile and cold allodynia after SNI in the TrpV1 neuron ablated mice relative to their littermate controls. TrpV1 DTA mice showed a total loss of TrpV1 expression in the DRG (Figure S5A), indicating the efficiency of the ablation. The tactile behavioral hypersensitivity was fully apparent in these mice at 7 days post-SNI injury and continued at similar levels until at least 21 days post-SNI (Figure 5B). In contrast, TrpV1 DTA mice failed to develop cold allodynia 7 or 14 days after nerve injury (Figure 5C). The lack of normal cold- or injury-induced cold sensitivity in these animals may reflect the absence of TrpM8 expression in the DRG relative to wild-type controls (Figure S5B). At later time points (21 days), injured TrpV1 DTA mice developed a slight level of cold allodynia relative to naive TrpV1 DTA mice (Figure 5C). The mechanism for this late and muted response is unclear, although it likely represents compensatory changes in gene expression/function in the remaining (non TRPV1 lineage) intact sensory neurons that enable them to develop sufficient cold thermoception to drive the central circuits that produce cold allodynia. Some residual TrpA1 (but not TrpM8) expression is present in the DRGs of TrpV1 DTA mice (~20% of wild-type levels) (Figure S5C), and TrpA1 is implicated in neuropathic cold allodynia (del Camino et al., 2010), suggesting this channel maybe the source of this late-onset low-level activity. The early loss in cold allodynia in TrpV1 DTA mice is consistent with the primarily neuronal gene expression changes in the DRG that correlate with its onset. Based on these findings, we conclude that an alteration in TrpV1 lineage sensory neurons is required for early development of this phenotype.
Peripheral Macrophages Are Crucial to the Development of Tactile, but Not Cold, Allodynia
Genes whose expression pattern matched the time course of tactile allodynia onset are primarily expressed in immune cells. Specifically, they appear to be enriched in macrophages and T cells (Figure 4G), two leukocyte populations present in DRGs after nerve injury (Hu and McLachlan, 2002; Moalem and Tracey, 2006) (see also Figures 7A and 7B). To assess whether circulating macrophages have a role in mediating allodynia, we transiently depleted them in C57BL/6 mice using liposomal clodronate. Macrophages naturally phagocytose these liposomes within the blood; this releases clodronate intracellularly and results in their death. Following a single liposomal clodronate dose, animals regenerate a full macrophage blood count from bone marrow precursors over the next 2 weeks (Camilleri et al., 1995). Animals administered with liposomal clodronate 1 day before nerve injury (SNI) developed minimal tactile allodynia at 7 and 10 days post-SNI relative to control mice treated with empty liposomes (Figure 6A). However, clodronate-treated and control animals developed cold allodynia to a similar extent post-SNI (Figure 6B). By assaying the blood at 7 days post-SNI, we found markedly reduced myeloid cells in clodronate-treated mice relative to liposome controls (Figure 6C). These changes also translated to the DRG tissue (Figures 6D and 6E), which may explain the large differences in mechanical threshold in the same mice (Figure 6F). We confirmed the decrease in macrophages/monocytes by immunohistochemistry; liposome control mice showed a marked IBA1 immunoreactivity in the ipsilateral L3-5 DRGs 7 days post-SNI, and this was markedly decreased in clodronate-treated mice (Figures 6G and 6H, respectively). We further quantified the relative expression within the injured DRG by qPCR of the macrophage/monocyte markers CD68 and CD11b, which both showed significant decreases relative to liposome control mice (Figures 6I and 6J, respectively). In addition, we quantified the transcript of CD163 (a marker of tissue resident macrophages), which was strongly decreased following clodronate treatment (Figure 6K).
Figure 7. T and B Cell-Deficient Rag1−/− Mice Develop Normal Neuropathic Cold Allodynia, but Not Complete Tactile Allodynia, following SNI Injury.

T cell reintroduction into Rag1−/− mice abolishes tactile sensitivity differences present between Rag1−/− mice and wild-type control littermates (LM) but leaves cold allodynia unaltered.
(A and B) Immunohistochemistry for the monocyte/macrophage marker IBA1 (red) in noninjured (A) and 7-day SNI-injured DRG in Lck Cre-zsGreen mice, which express labeled T cells (green) (B). Scale Bar: 50 µm.
(C) Representative FACS plots of CD4 versus CD8 cell counts from splenic preparation showing cells positive for both markers in the wild-type and in Rag1−/− and T mice, but not Rag1−/− mice (top). Representative FACS plots of CD4 versus B220 counts showing the presence of B cells in wild-type LMs, but not Rag1−/− or reconstituted Rag1−/− mice.
(D) Rag1−/− mice develop less tactile allodynia post-SNI than their LM controls (Rag versus WT, p = 0.003; Rag versus Rag and T, p = 0.002). Reconstituted Rag1−/− mice (Rag1−/− and T) showed full levels of tactile sensitivity (WT versus Rag and T, not significantly different). Significant differences between the values after SNI and their basal measures are shown (**p < 0.01, Rag1−/−mice versus WT littermates [blue #]; Rag1−/− mice versus Rag and T [red #]; #p < 0.05, ##p < 0.01, two-way repeated-measures ANOVA, Bonferroni post hoc test).
(E) Wild-type LMs, Rag1−/−, and T cell-reconstituted mice develop similar neuropathic cold allodynia (no significant differences among the three curves). For (D) and (E), error bars indicate SEM (wild-type LM, n = 15; Rag1−/−, n = 10; Rag1−/− and T, n = 15).
Figure 6. Mice Depleted of Peripheral Macrophages/Monocytes Using Clodronate Develop Delayed Tactile Allodynia but Normal Neuropathic Cold Allodynia.

(A) Clodronate-treated mice develop markedly less tactile allodynia post-peripheral nerve injury than empty-liposome-treated controls.
(B) Clodronate-treated mice develop significant levels of cold allodynia post-SNI. Statistically significant differences between the values from mice after SNI and their basal measure (**p < 0.01) and between mice treated with clodronate or vehicle in tactile allodynia (##p < 0.01) are shown (two-way repeated-measures ANOVA, Bonferroni post hoc test). Clodronate-treated C57BL/6 mice develop significant levels of cold allodynia post-SNI (*p< 0.05, **p<0.01), but there were no significant differences in cold allodynia between clodronate- and vehicle-treated mice (p = 0.323; two-way repeated-measures ANOVA).
For (A) and (B), n = 10 clodronate, n = 13 vehicle.
(C–E) Levels of myelocytes (CD45+CD11b+CD11c−SiglecF−CD3−) measured by FACS in blood (C) and DRG (D and E) 7 days after SNI in mice treated with clodronate or vehicle.
(F) Mechanical threshold in these mice.
For (C)–(F), p values are given (unpaired Student’s t test).
(G and H) Iba1 immunoreactivity in the DRG from SNI mice treated with vehicle liposomes (G) or clodronate liposomes (H). Scale Bar: 100 µm.
(I–K) Real-time qPCR of the macrophage/monocyte markers CD68 (I), CD11b (J), and CD163 (K) in the DRG of SNI mice treated with liposomes or clodronate. p values are given (n = 5 per group; unpaired Student’s t test).
Error bars indicate SEM.
Consequence of Absence of T and B Cells on Nerve-Injury-Induced Tactile and Cold Hypersensitivity
To demonstrate whether in addition to macrophages, activated T cells were also present in the ipsilateral DRG 7 days post-SNI injury, we utilized a combination of a Lck-zsGreen mouse line, which expresses the fluorescent marker zsGreen in activated T cells (Zhang et al., 2005), and immunostaining for IBA1. In the uninjured Lck-zsGreen transgenic DRG, very few labeled T cells were present and there was little IBA1 staining (Figure 7A). In ipsilateral DRGs 7 days post-SNI, there were in contrast many labeled T cells and a strong IBA1 signal (red, Figure 7B). To test whether the absence of T cells had any effect on cold and tactile allodynia, we exploited Rag1−/− mice that lack T and B cells (Figure 7C, left and middle). In agreement with our previous observations (Costigan et al., 2009a), we found a marked reduction in tactile hypersensitivity in Rag1−/− animals relative to their littermate controls (Figure 7D). We extended this by showing that introducing CD4+/CD8+ T cells into Rag1−/−mice (Figure 7C) could rescue the tactile allodynia phenotype for at least 4 weeks post-SNI (Figure 7D). The full rescue of tactile allodynia by T cells in reconstituted Rag1−/− mice, which still lack B cells, argues against a role for this latter immune cell type in this phenotype (Figures 7C and 7D). In contrast, we witnessed little difference in the extent of cold allodynia in Rag1−/−animals (Figure 7E), suggesting that both T and B cells are dispensable for that response.
Taken together, these data strongly support a role for of each of these two leukocyte cell types (macrophages and T cells) in the establishment of tactile allodynia after peripheral nerve damage but demonstrate that they are dispensable for the development of cold allodynia.
DISCUSSION
By assaying the development of two common clinical manifestations of stimulus-evoked neuropathic pain (cold and tactile allodynia) (Jensen and Finnerup, 2014) at high temporal resolution, we demonstrate that they develop over different time courses in the first week post-surgery in the SNI model. This difference led us to hypothesize that there may be mechanistic differences underlying these two pain modalities and that these could perhaps be revealed by correlating the changes in global transcript expression in the PNS in response to nerve injury with the temporal evolution of the behavior. To investigate this, we generated DRG expression array profiles beginning immediately after the nerve injury and at a higher temporal definition than in previous investigations (Costigan et al., 2010; Li et al., 2015).
To assay global transcript expression changes in the DRG following SNI, we separated all of the potentially regulated genes into co-regulated clusters using WGCNA, since groups of genes with similar expression patterns across large datasets are often functionally connected as part of the same tissue, cell type, or biological pathway (Parikshak et al., 2015). There were relatively few clustered groups of co-regulated transcripts in the injured DRG over the 10 days following the injury (eight in total), with a temporal analysis suggesting an early pattern of neuronal gene regulation followed later by regulation of immune transcripts. Correlation of the distinct trajectories of cold and tactile hyper-sensitivity onset respectively with gene expression further suggested a link between the two. The patterns suggest predominant involvement of sensory neurons in the onset of cold allodynia and a contribution of activated peripheral immune cells in the generation of tactile allodynia. Next, we used a combination of genetically targeted and cell depletion protocols to test these predictions.
TrpV1-mediated Cre expression in the embryo occurs in all thermo-sensing progenitor neurons, which are consequently deleted by DTA expression. This removes all TrpM8 expression and, therefore, all normo-cold sensation in the adult mouse, as shown here and in previous studies (McKemy, 2011; Mishra et al., 2011). We demonstrate that ablating TrpV1 lineage nociceptors results in a lack of cold allodynia after nerve injury, consistent with this process occurring primarily through neuronal mechanisms. In addition, recent independent data are entirely consistent with cold allodynia occurring through neuronal signaling pathways (Lippoldt et al., 2016).
Cold allodynia, like tactile allodynia following SNI, must be the consequence of input to the CNS transmitted by uninjured sural nerve axons. This raises the question of whether something changes in these “uninjured” peripheral neurons due to the SNI procedure, such that previously innocuous stimuli can now activate a set of nociceptors or if there is an abnormal reaction in the CNS, such that the ‘normal input’ generated by innocuous temperatures in spared fibers now elicits pain-like behavior? For the former, one might speculate that injured sensory neurons may produce paracrine signals in the DRG that somehow alter the sensitivity of neighboring non-injured thermo-nociceptors to cold. For the latter, as injured C-nociceptors become ectopically active early following peripheral nerve injury (4–13 hr) (Kirillova et al., 2011), this abnormal afferent drive could maintain central sensitization altering the central processing of low-threshold thermoceptor inputs in the dorsal horn, leading to them being perceived as painful (Latremoliere and Woolf, 2009).
In spite of the marked reduction in cold allodynia seen in the TrpV1 DTA line, we found relatively normal levels of tactile allodynia in these mice, which rules out the specific need for this nociceptor lineage in the production of this form of stimulus-evoked pain hypersensitivity after nerve injury. These findings are in agreement with previous reports showing that ablation of either TrpV1 or Nav1.8 lineage nociceptor neurons does not alter neuropathic tactile allodynia (Abrahamsen et al., 2008; Lager-ström et al., 2011; Mishra et al., 2011). Tactile allodynia must require other sensory neurons for its manifestation, and indeed many studies have indicated that it is carried by low-threshold mechanoreceptors (Campbell and Meyer, 2006; Xu et al., 2015).
The correlation of immune related transcripts with the development of tactile allodynia suggested that the immune system may play a role in the development of this hypersensitivity, but that these cells are largely dispensable for cold allodynia. To test this prediction, we targeted two leukocyte populations, macrophages/monocytes and T cells, as these cells represent a large portion of the immune reaction within the DRG following nerve injury (Hu and McLachlan, 2002; Moalem and Tracey, 2006), and found that both peripheral macrophages and T cells contribute to the development of neuropathic tactile sensitivity but minimally to cold allodynia.
T cells and macrophages play a major role in orchestrating the actions of one another during the early and late phases of the immune response (Biswas et al., 2012; Roberts et al., 2015). The complex interplay between innate and adaptive components of the immune system is a major component of the reaction of the PNS to damage, with functional roles in clearance of debris and promotion of regeneration (DeFrancesco-Lisowitz et al., 2015). However, recruitment of these leukocytes following nerve injury can lead to effects on sensory neurons that activate or sensitize them, leading to neuropathic pain (Costigan et al., 2009b).
Following nerve injury, macrophage-derived signaling molecules such as IL-1β, TNF-α, and CCL2 likely contribute to pain-like hypersensitivity (Andrade et al., 2014; Schuh et al., 2014; Zhu et al., 2014) as well as the initiation of axonal regrowth (Dubový et al., 2013). In turn, Th1 T cell-derived interferon-γ (IFN-γ) can recruit macrophages to sites of inflammation and cause pain (Liou et al., 2011), whereas type 2 inflammatory cytokines, such as IL-4, IL-10, and TGF-β, can ameliorate neuropathic pain-like behavior (Chen et al., 2015; Dengler et al., 2014; Kiguchi et al., 2015). The balance between these pro- and anti-inflammatory subsets in different experimental settings may explain the differences seen between studies focused on the role of T cells on neuropathic pain (Austin et al., 2012; Kiguchi et al., 2015); some studies did not find any impact on the pain phenotype of T cell actions (Sorge et al., 2015), whereas we and several others have (Cao and DeLeo, 2008; Costigan et al., 2009a; Kobayashi et al., 2015; Leger et al., 2011; Zhang et al., 2014).
While the mechanisms by which macrophages and T cells interact to co-produce a convergent set of changes in the DRG that lead to tactile allodynia now need to be explored, our data indicate that they must act on sensory fibers other than TrpV1 lineage nociceptors. Activated macrophages are seen preferentially around injured large-diameter A-fiber sensory neuron cell bodies in the DRG after sciatic nerve injury (Vega-Avelaira et al., 2009), but the functional consequence of this on these sensory neurons is still unknown. It is possible that activated immune cells in the DRG may stimulate injured A-fibers into initiating ectopic activity, which contributes to the maintenance of central sensitization in the dorsal horn, such that central pain neurons begin to be activated by low-threshold mechanoreceptors. In the naive state, only C-fibers can initiate central sensitization, but after nerve injury, A-fibers develop this capacity by a phenotypic switch (Decosterd et al., 2002), and this shift may be triggered by immune cell activation in the DRG. Consistent with this, ectopic firing of injured A-fiber neurons manifests later after nerve injury than in C-fibers (ectopic A-fiber activity is seen at 4–7 days) (Kirillova et al., 2011). Alternatively, spared high-threshold A-fiber nociceptors may be sensitized by immune cell action following peripheral nerve damage. Either of these mechanisms, or possibly a combination of both, may lead to mechanical allodynia. What we now show conclusively, however, is that the mechanism responsible is dependent on peripheral immune cells.
Distinct mechanistic etiologies for these two neuropathic pain symptoms may in consequence require different treatment strategies, if our findings translate to humans, one targeted at a particular set of TRPV1 lineage nociceptors for patients whose primary symptoms are cold allodynia and another for patients with predominant tactile allodynia, which could incorporate either targeting the actions of the immune component of nerve injury (macrophages and/or T cells) or non-TRPV1-expressing afferents (mechanoreceptors). Identifying the key elements of immune-neural signaling that underlie the development of allodynia after nerve damage using cell-specific profiling technologies will be important for developing targeted therapies for peripheral neuropathic pain.
EXPERIMENTAL PROCEDURES
Experiments were performed in adult (9–10 weeks old) male C57BL/6J mice (Jackson Laboratory [Jax], ME). Heterozygous TrpV1-Cre (strain 017769) and heterozygous DTA stop animals (strain 010527) were bred together to produce TrpV1 DTA animals. Rag1 null (strain 2216) mice were also used. To reveal T cell infiltration in the injured DRG, we bred Lck-Cre (Jackson Laboratory [Jax] 3802) mice with zsGreen reporter mice (Rosa-CAG-LSL-ZsGreen1-WPRE) (Jax 7906). All studies performed in USA were reviewed and approved by the IACUC at Boston Children’s Hospital under animal protocols 15-04-2928R and 16-01-3080R. All experimental procedures performed in Spain were conducted in strict accordance to European standards (European Communities Council Directive 2010/63) and after approval of the animal protocols by regional (Junta de Andalucía) and institutional (Research Ethics Committee of the University of Granada, Spain) authorities.
Detailed description of the animal strains used and the experimental procedures for surgeries (SNI), behavioral tests (von Frey test, acetone test and dynamic thermal place aversion test), immune cell depletion or reconstitution, gene expression analysis by microarray, RNA-seq and real-time qPCR, bioinformatics (weighted gene co-expression network analysis, correlation between expression changes and the pain phenotype, functional enrichment analysis, transcription-factor-binding site enrichment, and PPI network analyses); fluorescence-activated cell sorting to determine myeloid cells or T and B cells, and immunohistochemistry are described in Supplemental Experimental Procedures.
Statistical Significance
Gene regulation in the expression microarrays was determined by a moderated F-statistics using Bioconductor packages. In the rest of the experiments, multiple comparisons were analyzed using repeated-measures ANOVA with Bonferroni post-test, and single comparisons were analyzed using an unpaired Student’s t test. Statistical analyses were performed with SigmaPlot 12.0 software (Systat Software, CA), with significance defined as p < 0.05.
Supplementary Material
Highlights.
Peripheral processes leading to neuropathic cold and tactile allodynia differ
TrpV1-lineage neurons participate in cold, but not tactile, allodynia
Immune system activation contributes to tactile allodynia but minimally to cold allodynia
Acknowledgments
This study was supported by NIH grants R01NS074430 (M.C.), R01NS58870, and R37NS039518 (C.J.W.); the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (C.J.W., G.C., and D.G.); NINDS Informatics Center for Neurogenetics and Neurogenomics (P30 NS062691) (F.G. and G.C.); Neuro-developmental Behavior Core, grant CHB IDDRC, 1U54HD090255 (N.A.A.); and grants SAF2013-47481P and SAF2016-80540-R from the Spanish Ministry of Economy and Competitiveness (MINECO) and the European Regional Development Fund (FEDER) (E.J. Cobos). E.J. Cobos was supported by the Research Program of the University of Granada. I.B.-C. was supported by an FPU grant from MINECO. R.G.C. was supported by the Alfonso Martin Es-cudero fellowship. F.R.N. was supported by a Juan de la Cierva postdoctoral grant from MINECO. M.R. was supported by a Junior-1 salary support award from the Fonds de recherche du Québec - Santé (FRQS).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for all microarray and RNA-seq datasets reported in this paper is GEO: GSE102937.
Supplemental Information includes Supplemental Experimental Procedures, five figures, and nine tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.006.
AUTHOR CONTRIBUTIONS
Conceptualization, M.C. and C.J.W.; Methodology, M.C., C.J.W., E.J. Cobos, and E.J. Chesler; Formal Analysis, E.J. Cobos, C.A.N., F.G., V.C., M.W.P., D.H.G., G.C., M.R., C.J.W., and M.C.; Investigation, E.J. Cobos, C.A.N., I.B.-C., R.G.-C., N.A.A., A.L., C.R.S., G.P., F.R.N., N.J., M.W.P., P.R., M.R., and M.C.; Resources, G.C. and D.H.G.; Data Curation, E.J. Cobos, G.C., F.G., D.H.G., and M.C.; Writing – Original Draft, M.C., E.J. Cobos, and C.J.W.; Writing – Review & Editing, E.J. Cobos, C.A.N., F.G., V.C., I.B.-C., R.G.-C., P.R., N.A.A., A.L., C.R.S., G.P., F.R.N., N.J., M.W.P., C.H.E.M., T.O., E.J. Chesler, D.H.G., G.C., M.R., C.J.W., and M.C.; Visualization, E.J. Cobos, I.B.-C., A.L., V.C., and M.C., Supervision, M.C., G.C., and C.J.W.; Project Administration, E.J. Cobos, G.C., M.C., and C.J.W.; Funding Acquisition, E.J. Cobos, M.C., and C.J.W.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Abe N, Cavalli V. Nerve injury signaling. Curr. Opin. Neurobiol. 2008;18:276–283. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Andrade P, Hoogland G, Del Rosario JS, Steinbusch HW, Visser-Vandewalle V, Daemen MA. Tumor necrosis factor-α inhibitors alleviation of experimentally induced neuropathic pain is associated with modulation of TNF receptor expression. J. Neurosci. Res. 2014;92:1490–1498. doi: 10.1002/jnr.23432. [DOI] [PubMed] [Google Scholar]
- Austin PJ, Kim CF, Perera CJ, Moalem-Taylor G. Regulatory T cells attenuate neuropathic pain following peripheral nerve injury and experimental autoimmune neuritis. Pain. 2012;153:1916–1931. doi: 10.1016/j.pain.2012.06.005. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012;53:11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma-Garcia I, Al-Bader R, Ortiz C, Elgueta R, Arno M, et al. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity. 2015;42:499–511. doi: 10.1016/j.immuni.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camilleri JP, Williams AS, Amos N, Douglas-Jones AG, Love WG, Williams BD. The effect of free and liposome-encapsulated clodro-nate on the hepatic mononuclear phagocyte system in the rat. Clin. Exp. Immunol. 1995;99:269–275. doi: 10.1111/j.1365-2249.1995.tb05544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, DeLeo JA. CNS-infiltrating CD4+ T lymphocytes contribute to murine spinal nerve transection-induced neuropathic pain. Eur. J. Immunol. 2008;38:448–458. doi: 10.1002/eji.200737485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Park CK, Xie RG, Ji RR. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-β secretion. J. Clin. Invest. 2015;125:3226–3240. doi: 10.1172/JCI80883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Barrett LB, Williams EK, Strochlic DE, Lee S, Weyer AD, Lou S, Bryman GS, Roberson DP, Ghasemlou N, et al. Transcriptional profiling at whole population and single cell levels reveals somatosensory neuron molecular diversity. eLife. 2014;3:e04660. doi: 10.7554/eLife.04660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleoni M, Sacerdote P. Murine models of human neuropathic pain. Biochim. Biophys. Acta. 2010;1802:924–933. doi: 10.1016/j.bbadis.2009.10.012. [DOI] [PubMed] [Google Scholar]
- Costigan M, Befort K, Karchewski L, Griffin RS, D’Urso D, Allchorne A, Sitarski J, Mannion JW, Pratt RE, Woolf CJ. Replicate high-density rat genome oligonucleotide microarrays reveal hundreds of regulated genes in the dorsal root ganglion after peripheral nerve injury. BMC Neurosci. 2002;3:16. doi: 10.1186/1471-2202-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Moss A, Latremoliere A, Johnston C, Verma-Gandhu M, Herbert TA, Barrett L, Brenner GJ, Vardeh D, Woolf CJ, Fitzgerald M. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J. Neurosci. 2009a;29:14415–14422. doi: 10.1523/JNEUROSCI.4569-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009b;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costigan M, Belfer I, Griffin RS, Dai F, Barrett LB, Coppola G, Wu T, Kiselycznyk C, Poddar M, Lu Y, et al. Multiple chronic pain states are associated with a common amino acid-changing allele in KCNS1. Brain. 2010;133:2519–2527. doi: 10.1093/brain/awq195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Allchorne A, Woolf CJ. Progressive tactile hypersensitivity after a peripheral nerve crush: non-noxious mechanical stimulus-induced neuropathic pain. Pain. 2002;100:155–162. doi: 10.1016/s0304-3959(02)00275-0. [DOI] [PubMed] [Google Scholar]
- DeFrancesco-Lisowitz A, Lindborg JA, Niemi JP, Zigmond RE. The neuroimmunology of degeneration and regeneration in the peripheral nervous system. Neuroscience. 2015;302:174–203. doi: 10.1016/j.neuroscience.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino D, Murphy S, Heiry M, Barrett LB, Earley TJ, Cook CA, Petrus MJ, Zhao M, D’Amours M, Deering N, et al. TRPA1 contributes to cold hypersensitivity. J. Neurosci. 2010;30:15165–15174. doi: 10.1523/JNEUROSCI.2580-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengler EC, Alberti LA, Bowman BN, Kerwin AA, Wilkerson JL, Moezzi DR, Limanovich E, Wallace JA, Milligan ED. Improvement of spinal non-viral IL-10 gene delivery by D-mannose as a transgene adjuvant to control chronic neuropathic pain. J. Neuroinflammation. 2014;11:92. doi: 10.1186/1742-2094-11-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Waxman SG. Translational pain research: Lessons from genetics and genomics. Sci. Transl. Med. 2014;6:249sr4. doi: 10.1126/scitranslmed.3007017. [DOI] [PubMed] [Google Scholar]
- Dubový P, Jančálek R, Kubek T. Role of inflammation and cytokines in peripheral nerve regeneration. Int. Rev. Neurobiol. 2013;108:173–206. doi: 10.1016/B978-0-12-410499-0.00007-1. [DOI] [PubMed] [Google Scholar]
- Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112:23–38. doi: 10.1016/s0306-4522(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Hu P, Bembrick AL, Keay KA, McLachlan EM. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav. Immun. 2007;21:599–616. doi: 10.1016/j.bbi.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Jaggi AS, Jain V, Singh N. Animal models of neuropathic pain. Fundam. Clin. Pharmacol. 2011;25:1–28. doi: 10.1111/j.1472-8206.2009.00801.x. [DOI] [PubMed] [Google Scholar]
- Jensen TS, Finnerup NB. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. Lancet Neurol. 2014;13:924–935. doi: 10.1016/S1474-4422(14)70102-4. [DOI] [PubMed] [Google Scholar]
- Kiguchi N, Kobayashi Y, Saika F, Sakaguchi H, Maeda T, Kishioka S. Peripheral interleukin-4 ameliorates inflammatory macrophage-dependent neuropathic pain. Pain. 2015;156:684–693. doi: 10.1097/j.pain.0000000000000097. [DOI] [PubMed] [Google Scholar]
- Kirillova I, Rausch VH, Tode J, Baron R, Jänig W. Mechano-and thermosensitivity of injured muscle afferents. J. Neurophysiol. 2011;105:2058–2073. doi: 10.1152/jn.00938.2010. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kiguchi N, Fukazawa Y, Saika F, Maeda T, Kishioka S. Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J. Biol. Chem. 2015;290:12603–12613. doi: 10.1074/jbc.M115.636506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCroix-Fralish ML, Austin JS, Zheng FY, Levitin DJ, Mogil JS. Patterns of pain: meta-analysis of microarray studies of pain. Pain. 2011;152:1888–1898. doi: 10.1016/j.pain.2011.04.014. [DOI] [PubMed] [Google Scholar]
- Lagerström MC, Rogoz K, Abrahamsen B, Lind AL, Olund C, Smith C, Mendez JA, Wallén-Mackenzie Å, Wood JN, Kullander K. A sensory subpopulation depends on vesicular glutamate transporter 2 for mechanical pain, and together with substance P, inflammatory pain. Proc. Natl. Acad. Sci. USA. 2011;108:5789–5794. doi: 10.1073/pnas.1013602108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J. Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger T, Grist J, D’Acquisto F, Clark AK, Malcangio M. Glatiramer acetate attenuates neuropathic allodynia through modulation of adaptive immune cells. J. Neuroimmunol. 2011;234:19–26. doi: 10.1016/j.jneuroim.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Li S, Xue C, Yuan Y, Zhang R, Wang Y, Wang Y, Yu B, Liu J, Ding F, Yang Y, Gu X. The transcriptional landscape of dorsal root ganglia after sciatic nerve transection. Sci. Rep. 2015;5:16888. doi: 10.1038/srep16888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JT, Liu FC, Mao CC, Lai YS, Day YJ. Inflammation confers dual effects on nociceptive processing in chronic neuropathic pain model. Anesthesiology. 2011;114:660–672. doi: 10.1097/ALN.0b013e31820b8b1e. [DOI] [PubMed] [Google Scholar]
- Lippoldt EK, Ongun S, Kusaka GK, McKemy DD. Inflammatory and neuropathic cold allodynia are selectively mediated by the neurotrophic factor receptor GFRa3. Proc. Natl. Acad. Sci. USA . 2016;113:4506–4511. doi: 10.1073/pnas.1603294113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKemy DD. A spicy family tree: TRPV1 and its thermoceptive and nociceptive lineage. EMBO J. 2011;30:453–455. doi: 10.1038/emboj.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra SK, Tisel SM, Orestes P, Bhangoo SK, Hoon MA. TRPV1-lineage neurons are required for thermal sensation. EMBO J. 2011;30:582–593. doi: 10.1038/emboj.2010.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res. Brain Res. Rev. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Parikshak NN, Gandal MJ, Geschwind DH. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015;16:441–458. doi: 10.1038/nrg3934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertin M, Allchorne AJ, Beggah AT, Woolf CJ, Decosterd I. Delayed sympathetic dependence in the spared nerve injury (SNI) model of neuropathic pain. Mol. Pain. 2007;3:21. doi: 10.1186/1744-8069-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CA, Dickinson AK, Taams LS. The interplay between monocytes/macrophages and CD4(+) T cell subsets in rheumatoid arthritis. Front. Immunol. 2015;6:571. doi: 10.3389/fimmu.2015.00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas M, Davies LC, Giles PJ, Liao CT, Kharfan B, Stone TC, O’Donnell VB, Fraser DJ, Jones SA, Taylor PR. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344:645–648. doi: 10.1126/science.1251414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuh CD, Pierre S, Weigert A, Weichand B, Altenrath K, Schreiber Y, Ferreiros N, Zhang DD, Suo J, Treutlein EM, et al. Prostacyclin mediates neuropathic pain through interleukin 1β-expressing resident macro-phages. Pain. 2014;155:545–555. doi: 10.1016/j.pain.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci. 2015;18:1081–1083. doi: 10.1038/nn.4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I, Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, et al. GTP cyclohydrolase and tetrahydrobiopterin regulate pain sensitivity and persistence. Nat. Med. 2006;12:1269–1277. doi: 10.1038/nm1490. [DOI] [PubMed] [Google Scholar]
- Vega-Avelaira D, Géranton SM, Fitzgerald M. Differential regulation of immune responses and macrophage/neuron interactions in the dorsal root ganglion in young and adult rats following nerve injury. Mol. Pain. 2009;5:70. doi: 10.1186/1744-8069-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol. Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Wijnvoord N, Albuquerque B, Häussler A, Myrczek T, Popp L, Tegeder I. Inter-strain differences of serotonergic inhibitory pain control in inbred mice. Mol. Pain. 2010;6:70. doi: 10.1186/1744-8069-6-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZZ, Kim YH, Bang S, Zhang Y, Berta T, Wang F, Oh SB, Ji RR. Inhibition of mechanical allodynia in neuropathic pain by TLR5-mediated A-fiber blockade. Nat. Med. 2015;21:1326–1331. doi: 10.1038/nm.3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, Killeen N. Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J. Immunol. 2005;174:6725–6731. doi: 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- Zhang X, Wu Z, Hayashi Y, Okada R, Nakanishi H. Peripheral role of cathepsin S in Th1 cell-dependent transition of nerve injury-induced acute pain to a chronic pain state. J. Neurosci. 2014;34:3013–3022. doi: 10.1523/JNEUROSCI.3681-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Cao S, Zhu MD, Liu JQ, Chen JJ, Gao YJ. Contribution of chemokine CCL2/CCR2 signaling in the dorsal root ganglion and spinal cord to the maintenance of neuropathic pain in a rat model of lumbar disc herniation. J. Pain. 2014;15:516–526. doi: 10.1016/j.jpain.2014.01.492. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.