

Summary
Solvent molecules interact intimately with proteins and can profoundly regulate their structure and function. However, accurately and efficiently modeling protein solvation effects at the molecular level has been challenging. Here, we present a method that improves the atomic-level modeling of soluble and membrane protein structures and binding by efficiently predicting de novo protein-solvent molecule interactions. The method predicted with unprecedented accuracy buried water molecule positions, solvated protein conformations, and challenging mutational effects on protein binding. When applied to homology modeling, solvent-bound membrane protein structures, pockets, and cavities were recapitulated with near-atomic precision even from distant homologs. Blindly refined atomic-level structures of evolutionary distant G protein-coupled receptors imply strikingly different functional roles of buried solvent between receptor classes. The method should prove useful for refining low-resolution protein structures, accurately modeling drug binding sites in structurally-uncharacterized receptors, and designing solvent-mediated protein catalysis, recognition, ligand binding, and membrane protein signaling.
Keywords: protein modeling, explicit solvation model, protein-solvent molecule interactions, protein binding energy prediction, protein homology modeling, ligand binding cavities, protein-protein complexes, transmembrane receptors, G protein-coupled receptors
eTOC Blurb
Lai et al present a method to improve atomic-level modeling of protein structures and binding by efficiently predicting protein-solvent molecule interactions. The approach recapitulated mutational effects on protein binding, modeled solvated cavities from distant homologs, and buried solvent networks in membrane proteins with unprecedented accuracy.
Introduction
Solvent water and ion molecules constitute the intimate environment of proteins and nucleic acids and critically regulate their structure and function. For example, solvent-protein interactions contribute to protein fold stability, participate to enzyme catalysis, signal transduction, and mediate protein-ligand, protein-DNA, and protein-protein recognition (Ahmad et al., 2011; Angel et al., 2009; Breiten et al., 2013; Levy and Onuchic, 2006; Nygaard et al., 2010; Papoian et al., 2004).
Solvent molecules can in principle be detected in proteins using a wide range of experimental techniques (Garczarek and Gerwert, 2006; Rath et al., 1998), (Gupta et al., 2012; Orban et al., 2010), (Amann-Winkel et al., 2016), (Carugo, 2016). However, because solvent molecules move with a broad range of dynamics, their observation can be challenging especially for weakly bound molecules which dissociate rapidly from the protein (Persson and Halle, 2008; Persson and Halle, 2013). The ability to detect solvent molecules in protein X-ray structures also depends on the resolution and the interpretation of X-ray crystallographic diffraction patterns (Richardson et al., 2013; Wlodawer et al., 2008).
Most membrane protein structures are solved at low-resolution, which prevents the reliable observation of solvent molecules. In the few available high-resolution structures, solvent is found in the transmembrane (TM) core region interacting with bound ligands and buried polar residues, frequently performing critical functions (Angel et al., 2009; Liu et al., 2012). Not surprisingly, perturbing buried solvent molecules often profoundly affected protein stability and signaling activity (Gutiérrez-de-Terán et al., 2013; Valentin-Hansen et al., 2015). Buried solvent weakly interacting with protein cavities and pockets can also reveal the presence of putative drug binding sites (Hollenstein et al., 2014; Mason et al., 2012). Therefore, predicting protein structures at atomic level with interacting solvent molecules starting from either low-resolution or homolog TM protein structures would considerably leverage drug and protein design approaches, and structure/function studies. However, while protein structure prediction and design have made great strides in recent years (Boyken et al., 2016; Chen et al., 2014a; Fleishman et al., 2011), high-resolution protein modeling with explicit solvent still represents a great notable challenge.
Modeling protein structure and solvent environment at atomic resolution represents one key application of molecular dynamic simulations techniques (Karplus and McCammon, 2002). Starting usually from static protein X-ray structure snapshots, these approaches can reveal invaluable relationships between protein conformational dynamics, solvent, and ligand molecule interactions, and protein function(Miao and McCammon, 2016). However, the large number of protein-interacting solvent molecules and associated movements considerably increase computational modeling time and complexity. Consequently, modeling both protein and solvent with molecular detail has remained intractable in most protein design and structure prediction applications which involve the extensive rebuilding of protein structures through the exploration of immense protein conformational spaces(Das and Baker, 2008). Alternatively, solvent molecules can be represented implicitly as a continuum liquid greatly improving computational efficiency (Kleinjung and Fraternali, 2014). However, implicit solvent approaches often lack accuracy in predicting and designing protein-protein, protein-ligand binding and protein catalysis which involve specific protein-solvent molecule interactions (Dragan et al., 2016; Lensink et al., 2014; Mobley and Dill, 2009; Sousa et al., 2006). A compromise is to isolate and model only specific regions with explicit solvent molecules, such as the ligand-binding site (Lemmon and Meiler, 2013; Li and Bradley, 2013) but current implementations present several limitations (Jiang et al., 2005; Lensink et al., 2014; Li and Bradley, 2013). First, limited improvement in protein and DNA structure modeling was observed over implicit solvent approaches (Jiang et al., 2005; Li and Bradley, 2013). Second, solvent molecules were modeled from the knowledge of their consensus location in similar structures. Such knowledge-based approaches are not suitable when homolog structures are not available or only solved at low-resolution such as for membrane proteins. In these systems, the number, position and interactions of solvent molecules need to be predicted de novo, which remains a considerable challenge. Because the optimal positions of solvent molecules are unknown, de novo modeling approaches are not only computationally more expensive but also necessitate a physical model calculating accurately the free energy of moving solvent molecules between protein-bound and bulk solvent positions.
To address these limitations, we developed SPaDES, a Structure Prediction and Design with Explicit Solvent approach, for modeling and designing soluble and membrane protein structures at atomic resolution with explicit solvent protein interactions. The method implemented in the software Rosetta is based on a hybrid implicit-explicit solvation model which efficiently predicts de novo the geometry, positions, and energetics of protein interacting solvent molecules. We validated SPaDES in challenging protein structure and protein-protein binding predictions. We then applied our approach in homology modeling applications and showed for the first time that membrane protein structures, solvated drug binding cavities and pockets can be accurately modeled even from distant protein homologs. In all cases, SPaDES outperformed alternative methods. Our approach should prove particularly useful for modeling complex protein structures at atomic resolution and designing challenging solvent-mediated protein functions.
Results
Approach
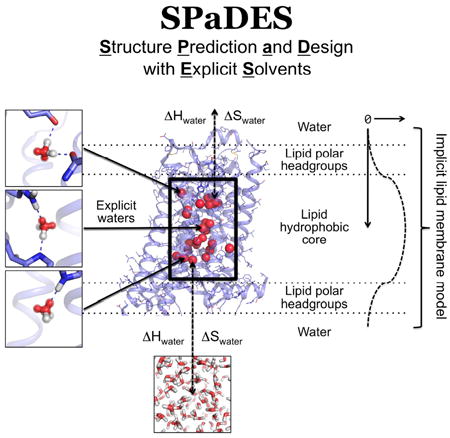
To enable protein structure prediction and design with explicit solvent, we developed a physical model that represents an effective compromise between modeling accuracy and calculation efficiency. For that purpose, we focused on functionally and structurally important protein-solvent interactions and developed a hybrid implicit-explicit solvation model. In this method, membrane protein interiors or protein binding interfaces are modeled at atomic resolution with explicit solvent molecules while protein interactions with the lipid or bulk solvent environment are treated implicitly using Rosetta’s energy functions for membrane and soluble proteins (Barth et al., 2009; Leaver-Fay et al., 2011) (Figure 1).
Figure 1. SPaDES framework for high-resolution membrane protein modeling.
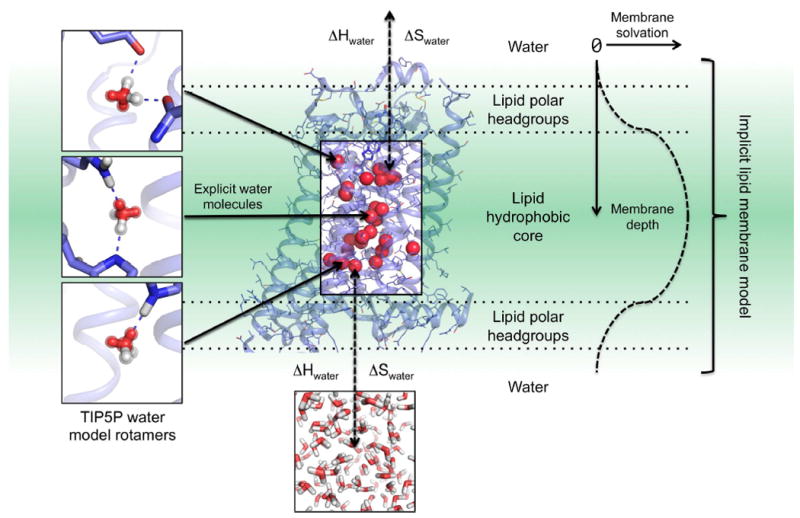
Transmembrane protein structures are modeled with a hybrid solvent representation where buried water molecules are modeled explicitly in the protein core (middle box) and the protein environment (lipid, water, and ion molecules) is modeled implicitly (green background). Explicit buried solvent molecules are modeled with Tip5p water rotamers anchored by hydrogen bonds to unsatisfied protein polar atoms in either a bridging configuration (top and middle left panels) or a single bond (bottom left panel). Water molecules are predicted to stay within the interior of the protein when the enthalpic (ΔH) and entropic (ΔS) cost of removing them from the solvent (ΔH and ΔS) is lower than the binding energy with the protein (dotted arrows). Otherwise, water molecules move back to the bulk solvent (bottom panel). Protein interactions with the implicit lipid membrane are described by the RosettaMembrane energy function, which accounts for the membrane depth and solvation (right diagrams and labels).
To ensure that the method can be applied to even proteins without available structural homologs or knowledge of solvent positions, we modeled protein-solvent interactions de novo. The method calculates the optimal location of water molecules based only on the physical interactions that these molecules can form with protein atoms. The structure modeling starts by placing water molecules in the protein to create optimal hydrogen bonds with a pair of polar atoms not yet fully coordinated to polar atoms from the protein or other water molecules. This step is performed for all possible discrete conformations of the protein amino-acid side-chains (i.e. protein rotamers) and generates millions of possible discrete water molecule placements and orientations around potential water sites (i.e. water rotamers). To efficiently produce accurate configurations of water and contacting residue rotamers, water molecules are modeled with electron clouds (i.e. Tip5p water models (Mahoney and Jorgensen, 2000)). This enables the rapid construction of energetically optimal water mediated interactions using geometric criteria only (Figure 1, S1, methods). Because protein residue rotamers overlap in space, this approach generates water rotamers with highly redundant locations (Figure S1A). Additionally, the water rotamer positions are frequently incompatible with neighboring protein atoms not involved in the polar contacts with the water molecule. To address these shortcomings, water rotamers are rapidly clustered and energetically filtered, which considerably reduce the rotamer space to be searched and minimizes the runtime increase despite the additional model complexity (Figure S1B, methods).
Starting from an ensemble of the above-mentioned constructed protein and water rotamers, the energetically optimal solvated protein conformation is identified using a stochastic Monte Carlo simulated annealing protein structure sampling protocol followed by a gradient-based energy minimization of the system over all conformational degrees of freedom. Solvated protein structure energies are calculated using the knowledge- and physically-based energy function of the Rosetta software (Das and Baker, 2008) supplemented with specific protein-solvent molecule interaction energy terms (methods). Since the optimal number of water molecules interacting with the protein is unknown, each water molecule is given the choice to reside in the protein core or to move back to the solvent during conformational sampling (Figure 1). The water molecule will remain bound to the protein if the protein-water interactions overcome the loss in water-water interactions (ΔHwater) and entropy (ΔSwater) upon burial in the protein core (Figure 1, methods). To identify the energetically optimal conformation of the hydrated protein from the astronomical number of starting rotamer combinations, around a hundred independent simulations are performed. This ensures that at least 10% of the simulations converge to within 1 Rosetta energy unit (REU) standard error of mean (SEM) for the lowest energy models. 1 REU equates to approximately 0.3 kcal/mol and 0.4 kcal/mol in SPaDES and Rosetta, respectively (Table S3). As in blind prediction contests, the representative models analyzed and reported in the study were selected from the lowest energy structures (methods). We estimate that our method, SPaDES, calculates solvated protein structures with 5-orders of magnitude increased efficiency compared to standard all-atom molecular dynamics simulations while only decreasing efficiency by 1-order of magnitude over the traditional Rosetta scoring function despite the explicit modeling of solvent molecules (methods).
Energy function training
To develop an accurate hybrid implicit-explicit solvent energy function, we needed to ensure that the interactions involving explicit solvent molecules were compatible with the terms describing intraprotein interactions and implicit solvation from the Rosetta energy functions. We trained the contributions (i.e. weights) of the explicit solvent (e.g. solvent-protein, solvent-solvent interaction energy terms, methods) in the hybrid energy function to optimize its accuracy in protein structure and protein interaction energy prediction tests. Because high-resolution structure and energetic information on membrane proteins remain scarce, we trained the explicit solvation terms on a dataset of soluble protein-protein bound complexes that bear a large number of buried protein residues, cavities, and water molecules at the binding interface. We reasoned that short-range interactions between protein atoms and buried water molecules at protein-protein binding interfaces or in membrane proteins should be similar and, to a first approximation, not dependent on the exact physical properties of the environment around the protein. Also, most water molecules in membrane proteins (e.g. G protein-coupled receptors (GPCRs)) are believed to exchange directly with the polar phases of the protein environment (i.e. lipid headgroup and water phase) similarly to soluble proteins (Figure 1) (Yuan et al., 2014; Yuan et al., 2015). Therefore, the aqueous phase is an appropriate reference state for the solvent molecules in membrane proteins.
Because our goal was to develop an energy function that could be used in both structure prediction and design approaches, we combined both structure and energy prediction tests in our training. The structure prediction tests consisted of recovering native side-chain conformations and native water molecule positions from a large dataset of non-redundant protein structures. The energy prediction tests consisted of recapitulating a large dataset of challenging mutational effects on the binding energies of protein complexes. A subset of the data (referred below as “high-resolution hydrated set”), for which high-resolution wild-type protein-protein complex structures with experimentally detected water molecules at the binding interface were available, was used to train the energy function. The resulting energy function was then validated on the complete dataset (referred below as “low- and high-resolution set”) (methods). To assess the accuracy of our method, we compared its performance to that of leading protein modeling and design techniques using knowledge-based or physically-based energy functions and implicit or explicit solvation models. Specifically, these techniques are: Rosetta (protein modeling and design software with implicit solvation (Das and Baker, 2008)), Fold-X (protein modeling and design software with knowledge-based explicit solvent model (Schymkowitz et al., 2005)), HADDOCK (protein modeling refinement step using physically-based Molecular Dynamics simulations (MD) with explicit solvent (de Vries et al., 2007; Dominguez et al., 2003)) and Modeller (homology modeling technique using knowledge-based implicit solvation (Eswar et al., 2008)).
Native water position recovery
Reliable detection of protein-bound solvent molecules is limited to the small fraction of very high-resolution (i.e. < 2.0 Å) protein X-ray structures. Therefore, most protein structures would benefit from an atomic-level refinement accurately predicting the conformations and interactions of protein-bound solvent molecules, which has remained challenging. We stringently tested SPaDES’s ability to predict the position and number of native protein-bound water molecules observed in a high-resolution protein structure set. Starting from the experimentally determined protein backbone structures, we predicted de novo the conformations of amino-acid side-chains and protein-bound water molecules. As shown in Figure 2 and Table S1, 77% of native water molecules observed in protein binding interfaces and membrane protein structures were recovered within 2 Å with de novo modeled water molecules (Figure 2A). Likewise, 75% of the predicted waters overlapped within 2 Å with the position of native water molecules (i.e. “coverage”). Similar accuracy was observed even with a very stringent distance threshold criteria (1.0 Å) (Figure S2). By contrast, both Fold-X and HADDOCK MD-based refinement displayed substantially lower accuracy predictions with only 44%, 57% true positive and only 65%, 50% coverage. The combined high native water recovery and true positive rates suggest that, unlike Fold-X and HADDOCK, SPaDES correctly captures both the water-protein interactions and the energetic balance describing the propensity of a water molecule to preferentially reside in the protein instead of the bulk solvent. Lastly, since the test set includes diverse protein structures (protein binding interfaces, ligand-bound transmembrane receptors) and environment (water or lipid), our results support the universality of our hybrid energy function (Figure 2B-E).
Figure 2. Accurate de novo prediction of protein-bound water molecule positions.
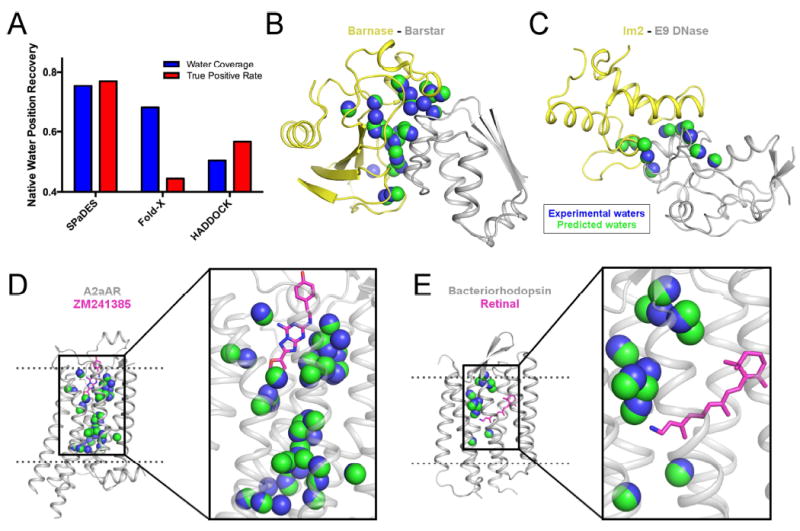
(A) Recovery of experimentally-observed water molecules (blue) and true positive rates of predicted water molecules (red) are shown with SPaDES, Fold-X with explicit water molecule modeling, and HADDOCK refinement with a 2.0 Å distance cutoff critera between predicted and experimentally-resolved waters. Higher fractional rates, ranging from zero to one, indicate increased accuracy of the predictions. (B-E) Examples of predicted water positions are shown for two protein-protein complexes and two integral ligand-bound membrane proteins: (B) barnase in complex with barstar, (C) colicin E2 immunity protein in complex with colicin E9 DNase, (D) A2a adenosine receptor bound to the ZM241385 ligand, and (E) bacteriorhodopsin bound to the retinal ligand. Blue spheres: water molecules observed in protein X-ray structures (“experimental waters”). Green spheres: de novo predicted water molecules (“predicted waters”). Ligands are shown in magenta sticks. See also Figure S2, Table S1.
Native side-chain conformation recovery
Protein side-chain conformations at protein surfaces and binding interfaces constitute important protein interaction sites and drug binding pockets but remain difficult to model with high-accuracy because interacting solvent molecules often dictate their conformation. We assessed whether SPaDES improved the prediction of more than 400 amino-acid side-chain conformations at diverse protein-protein binding interfaces (Figure 3A,B) when compared to the standard Rosetta software using implicit solvation. We found that a large fraction (i.e. 35.4%) of the native side-chains modeled incorrectly by Rosetta was recovered when including explicitly modeled water molecules with SPaDES (Table S2), while a considerably smaller fraction (i.e. 9.5%) of side-chains incorrectly modeled by SPaDES was recovered by Rosetta. The largest improvements were observed for polar amino-acids and protein-protein complexes with highly hydrated binding interfaces (Figure 3). Many native polar side-chain conformations stabilized by hydrogen bonding to specific water molecules and correctly predicted using SPaDES could not be recapitulated using Rosetta (Figure 3C-F). These examples highlight the critical role played by water-mediated interactions in shaping the structure of protein surfaces and binding interfaces. Consistent with the lower accuracy prediction of water molecule positions, Fold-X was also substantially outperformed by SPaDES in de novo modeling solvent-interacting side-chain conformations (Figure 3A,B). The HADDOCK refinement step does not build de novo protein side-chain conformations and therefore could not be used in this test.
Figure 3. Improved de novo prediction of side-chain conformations with explicit solvent.
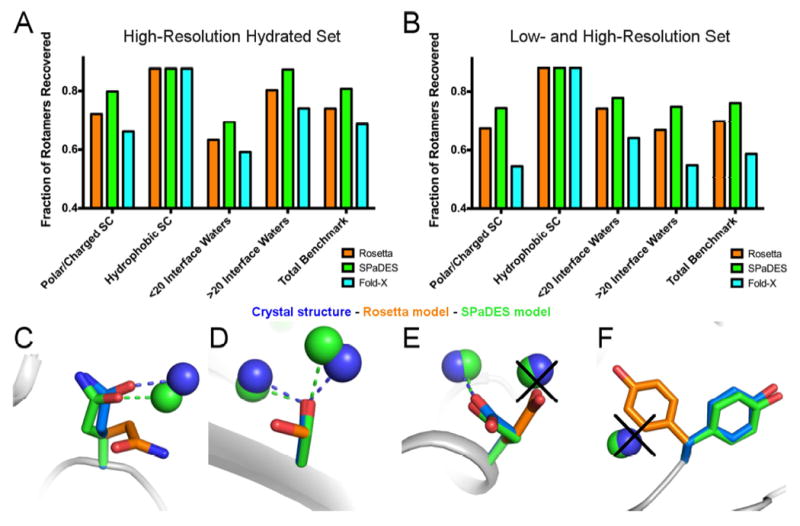
(A, B) Fraction of experimentally-observed side-chain conformations recovered using either Rosetta (orange), SPaDES (green), Fold-X with explicit water molecule modeling (cyan) for the high-resolution hydrated (A) and low- and high- resolution (B) set of protein-protein complex structures. Side-chain rotamer recovery is reported for specific classes of residues: polar and charged side-chains, hydrophobic side-chains, side-chains at protein-protein binding interfaces with less than 20 waters, and side-chains at protein-protein binding interfaces with more than 20 interface waters. (C-F) Improved side-chain conformation predictions using SPaDES are shown for (C) a glutamine in UCH-L3, (D) a serine in SHV-1 beta-lactamase, (E) a glutamic acid in TEM-1 beta-lactamase, and (F) a tyrosine in BLIP. Dotted lines indicate hydrogen bonds for the corresponding colored structures and black crosses indicate clashes between the Rosetta prediction and the experimentally-resolved waters. See also Table S2.
Prediction of mutational effects on protein-protein binding energies
Solvent molecules often mediate critical protein-protein interactions governing binding affinity and specificity. Modeling protein binding without accounting for solvent molecules can lead to poor prediction of the effects of amino-acid substitutions and failure in designing novel protein binding complexes. We assessed whether SPaDES improves the prediction of mutational effects on protein-protein binding when compared to Rosetta and Fold-X. We selected a large dataset of soluble protein-protein binding interfaces with more than 500 experimentally measured binding affinity changes upon amino-acid substitution from the curated SKEMPI database (Moal and Fernandez-Recio, 2012). Part of the data was gathered into the “hydrated high-resolution” set while the complete dataset is referred below as the “low- and high-resolution” set (methods). We modeled the effects of point mutations on the binding energy of the protein complexes by calculating the energy difference between the bound complex and the unbound proteins for the wildtype and mutant proteins. The conformations of the protein side-chains and water molecules at the binding interface were predicted de novo starting from the structure of the bound wildtype complex (methods).
We observed a substantially increased prediction accuracy of SPaDES versus Rosetta (Figure 4A,B). Predicted energies displayed much stronger correlations with the experimental binding energies (pearson correlation coefficient R, 0.72 versus 0.56 and 0.60 versus 0.33 in the high-resolution hydrated and low- and high-resolution sets, respectively) and lower standard errors. The qualitative classification of mutational effects on the protein binding interface into stabilizing, destabilizing, and neutral classes was also significantly improved (methods, Figure 4A,B, Table S3). Considering that the majority of the protein-protein complexes in the low- and high-resolution dataset were not used during the training of the hybrid energy function, the level of improvement observed in this test set is remarkable. Our results strongly suggest that the hybrid energy function in SPaDES captures important universal properties of solvent-mediated protein interactions. By contrast, Fold-X predicted mutational effects with significantly lower accuracy (pearson correlation coefficient R=0.47 and R=0.39 in the high-resolution hydrated and low- and high-resolution sets, respectively, Figure 4C,D). Interestingly, we observed no significant improvement upon explicitly modeling water molecules in Fold-X, when compared to Fold-X implemented with implicit solvation. These results further stress the challenge of accurately modeling solvent molecule effects on protein structures and energies.
Figure 4. Improved prediction of protein-protein binding energies with explicit solvation.
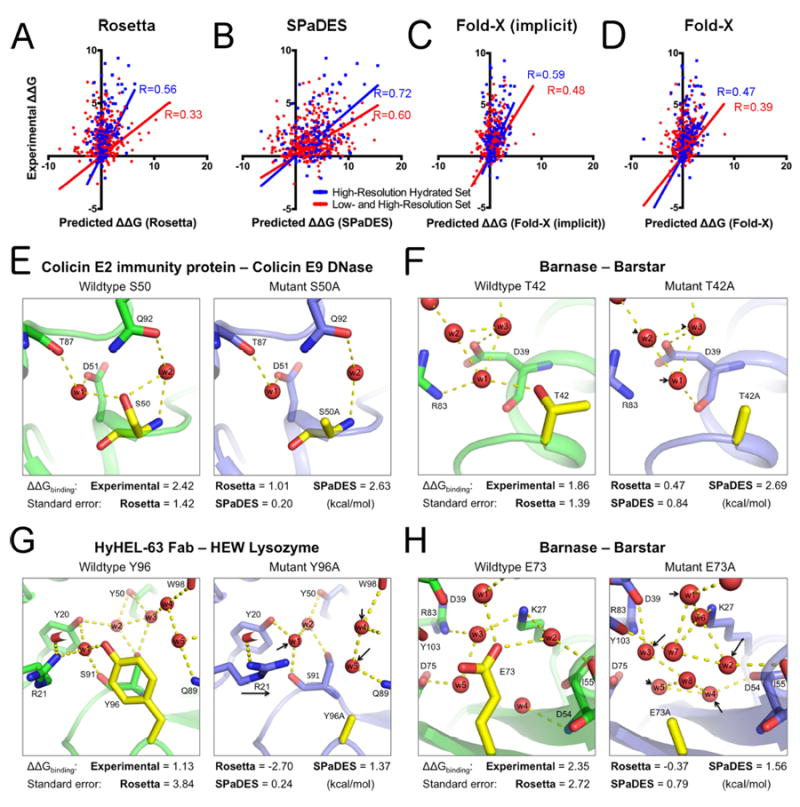
(A-D) Correlations between experimentally determined mutation-induced free energy changes of protein binding ΔΔGbinding and predicted values using Rosetta (A), SPaDES (B), Fold-X with implicit solvation (Fold-X implicit) (C), and Fold-X with explicit water molecule modeling (Fold-X) (D). Correlations are calculated for the high-resolution hydrated set of mutations selected near an experimentally determined water molecule (blue) and the high- and low-resolution set (red) of protein-protein complexes with the best linear correlation fit constrained to pass through the origin as a reference state (methods). Predicted ΔΔGbinding on the x-axis are in Rosetta Energy Units for Rosetta and SPaDES and kcal.mol-1 for the Fold-X results while the experimental ΔΔGbinding on the y-axis are all in kcal.mol-1. Pearson correlation coefficients R are color-coded and reported on each respective panel. (E-H) Examples of improved predictions of mutational effects on protein binding. Green: wild-type protein predicted structure; Blue: mutant protein predicted structure; red sphere: predicted water molecule; yellow: mutated amino-acid; dotted line: strong hydrogen bond (<-0.5 REU). Black arrow: mutation-induced shift in the position of a water molecule or side-chain conformation. (E-H) Experimentally determined and predicted ΔΔGbinding and standard errors of the predictions. (E) S50A mutant of the colicin E2 immunity protein complexed with the colicin E9 DNase. (F) T42A mutation for barnase bound to barstar. (G) Y96A mutation for HyHEL-63 Fab bound to HEW lysozyme. (H) E73A mutation for barnase bound to barstar. Overall and hydrogen bond energetics are described in Table S4. ΔΔGbinding are reported in kcal.mol-1 for both predicted and experimental values. Predicted values in Rosetta Energy Units were translated into kcal.mol-1 using the slope of the best linear correlation fit between predicted and experimental values (Table S3, S5). See also Table S3-S5.
Explicitly modeled water molecules improved mutational binding energy predictions in various ways (Figure 4E-H). For example, our model predicted that Ser50Ala mutation at the colicin E2 immunity protein-colicin E9 DNase interface led to the loss of water-mediated hydrogen bonds without perturbing the position of the water molecules due to strong remaining hydrogen bonds with other protein side-chains (Figure 4E, Table S4). On the other hand, Thr42Ala mutation at the barnase-barstar interface led to a loss of a water-mediated hydrogen bond and slight movement of neighboring waters to fill in the new space, recoordinating the water’s hydrogen bond to the protein backbone (Figure 4F). More drastic water-mediated interaction network changes were observed for Tyr96Ala at the HyHel-63 Fab-HEW lysozyme interface and Glu73Ala at the barnase-barstar interface. For these mutants, both the change in hydration sites and the size difference between wildtype and mutant amino-acid side-chains led to large reorganization of the overall water networks and neighboring amino-acids (Figure 4G,H). In all four scenarios, unlike Rosetta, SPaDES correctly predicted the energetic effect of the mutation (Figure 4, Table S4).
The high quality prediction of mutational effects suggests that SPaDES can select aminoacid substitutions that alter protein binding and stability and therefore can be used in de novo protein design applications involving solvent-mediated protein interactions.
High-resolution membrane protein homology modeling
Structure prediction represents an important complementary approach to the difficult experimental membrane protein structure determination. However, accurately predicting membrane protein structures at atomic resolution remains a challenge in part because water, ion, and lipid molecules stabilize protein conformations by forming strong and specific interactions that cannot be recapitulated using implicit solvent models. Additionally, solvated protein cavities and pockets revealing the presence of putative binding sites for drug ligands and allosteric regulators (Hollenstein et al., 2014; Mason et al., 2012) are often poorly recapitulated when structures are predicted in absence of explicitly modeled solvent (Chen et al., 2014a). To address this limitation, we implemented the hybrid energy function of SPaDES in RosettaMembrane’s homology modeling, a top performing structure prediction approach (Figure 1, (Chen et al., 2014a)). In this application, a target protein is modeled starting from a homolog protein structure that serves as a template. The two protein sequences are aligned, the target sequence is threaded onto the homolog template structure, and the poorly aligned loop regions are rebuilt de novo using coarse-grained peptide fragment insertion. To fully accommodate the target sequence, loop rebuilt template structures then undergo extensive all-atom backbone and side-chain relaxation with concurrent de novo explicit solvent modeling in the entire transmembrane region (methods). Incorporating the explicit modeling of water molecules in homology modeling is challenging, because the protocol uses both coarse-grained and all-atom representations of the protein and cycles between different smoothened versions of the all-atom energy function to avoid the structure to expand during relaxation (methods).
We tested our homology modeling protocol with SPaDES on several target/template G-protein coupled receptor pairs. Twenty relatively distant homolog pairs were selected with sequence identities ranging between 20% and 40% to stringently test the effects of modeling explicit solvent molecules (Table S5). Due to low sequence homology, the structures of the solvated regions in the homolog template and target were often significantly different, which justified the full structure relaxation protocol (Table S5).
We first characterized the accuracy of our models using a fine-grained metric calculating the geometry of the solvated cavities within the receptor TM region (Figure 5A). We compared the accuracy of models generated with explicit solvent using SPaDES or with implicit solvation using the methods RosettaMembrane (Chen et al., 2014a) and Modeller (Webb and Sali, 2014). We observed that, for most targets, the geometry and volume of the cavities modeled using water molecules were substantially closer to those of the native structures (i.e. native cavity point recovery increased over 80% in the best case, Figure 5B, Table S5). The improved modeling of protein cavities was also usually accompanied by higher accuracy of the protein structure in the vicinity of the cavities (Figure S3A, Table S5).
Figure 5. High-resolution prediction of solvated transmembrane regions in G protein-coupled receptor homology models.
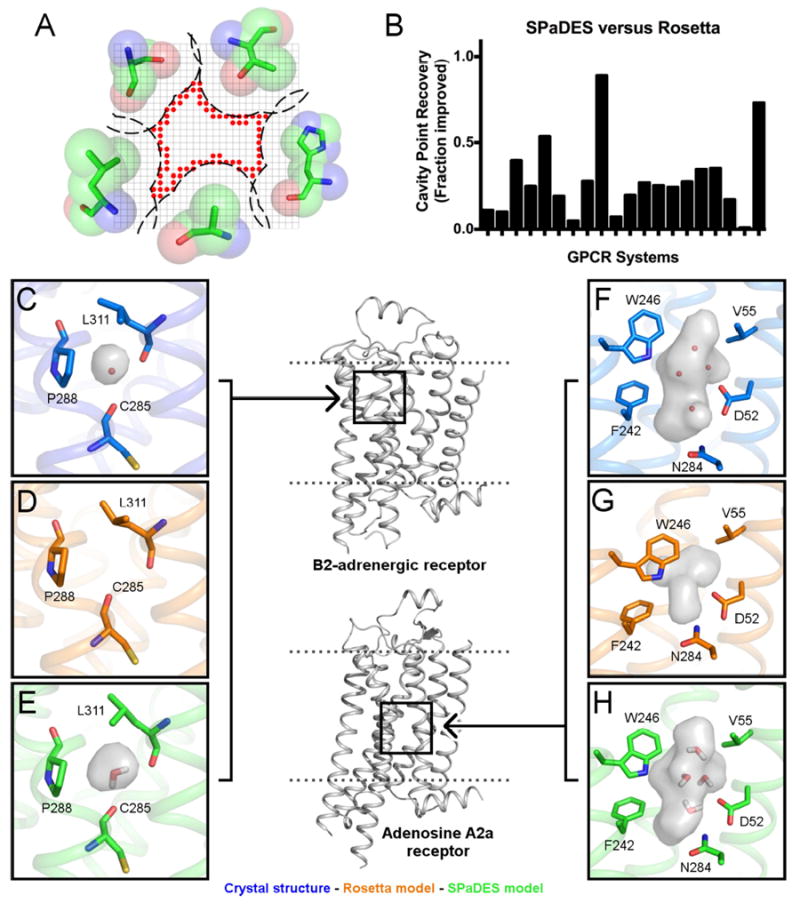
The structures of twenty GPCRs were modeled starting from distant homologs (sequence identity between 20 and 40%) using RosettaMembrane implemented with either the implicit membrane solvation model or the hybrid solvation model of SPaDES. (A) Schematic representation of the grid-based metric calculating the geometry of the hydrated cavities. Red sphere: cavity lattice point. (B) Recovery of cavity lattice points was used to determine the fraction improvement of SPaDES predictions over Rosetta for each system (p<0.001, Student’s t-test). (C-H) Accurate prediction of hydrated cavities and buried solvent molecules in the TM core of the beta 2 adrenergic receptor modeled from the adenosine A2A receptor (E) and the adenosine A2A receptor modeled from the beta 1 adrenergic receptor (H). These cavities are mostly lost when modeled using Rosetta (D, G). The positions of the water molecules observed in the X-ray structures (C, F) are predicted accurately (E, F). The cavities are shown as gray surfaces, neighboring side-chains that affect the cavity shape are shown in sticks with text labels, and water molecules predicted by SPaDES are shown as red sticks in (E). Panels have been rotated to show all marked components. See also Figure S3-S4, Table S5.
Remarkably, SPaDES recovered several cavities that almost completely collapsed during relaxation with RosettaMembrane (Figure 5D,G, S4) or that were not accurately predicted by Modeller (Webb and Sali, 2014) (Figure S3C). Additionally, most experimentally-observed water molecules in the target GPCRs were accurately predicted de novo even without the knowledge of their positions in homolog structures (Figure 5E,H). The recovered cavities comprise solvated pockets close to the extracellular ligand or intracellular effector binding sites and the conserved solvated channel undergoing large conformational changes during the activation of GPCRs (Angel et al., 2009) (Figure 5C,E, S4). Solvent molecules buried in the latter cavity have been shown to regulate allosterically the signaling responses to ligand agonist binding (Gutiérrez-de-Terán et al., 2013; Liu et al., 2012; Nygaard et al., 2010; Valentin-Hansen et al., 2015). Therefore, predicting their locations and interactions with the protein is critical to rationally engineer the function of receptors even those without solved experimental structures.
Blind prediction of solvent-mediated interactions in GPCRs
High-resolution structural information provides critical mechanistic insights into protein structure-function relationships. Low-resolution membrane protein structures can therefore benefit from the high-resolution structural refinement with explicit solvent provided by SPaDES.
To better understand the relationships between structure, solvation, and function in the large GPCR family, we targeted receptor classes B, C, and F, which, unlike class A Rhodopsin-like GPCRs, have not yet been structurally characterized at high-resolution (i.e. <2.0 Å). We selected representative ligand-bound receptor structures for each class (i.e. 1 class A, 2 class B, 2 class C, and 1 class F) and, starting from the backbone structures, predicted de novo the residue side-chain and buried solvent molecule conformations. Previous analysis of high-resolution class A GPCR structures highlighted the high level of hydration and structural conservation of buried solvent molecules in the receptor inactive state (Angel et al., 2009) (Figure 6A). Water molecules connect the bound extracellular ligand to allosteric residues (e.g. Trp 6.48, toggle switch), which interact with a large solvated cavity in the TMH core lined by conserved polar residues from TMHs 1, 2, 6, and 7. This cavity is connected to the intracellular side by a gate (i.e. Tyr 7.53) from the conserved NPxxY motif. Receptor activation upon ligand agonist and G protein binding involves a major rearrangement of solvent-mediated interactions in the receptor TMH region, remodeling the solvated cavities and solvent penetration in the receptor (Yuan et al., 2014; Yuan et al., 2015). These observations strongly suggest that solvent molecules participate to the allosteric pathways transmitting structural and dynamic changes from the extracellular to the intracellular sides of the receptor structure.
Figure 6. Blind prediction of buried solvent networks in evolutionary distant G protein-coupled receptors from classes A, B, C, and F.
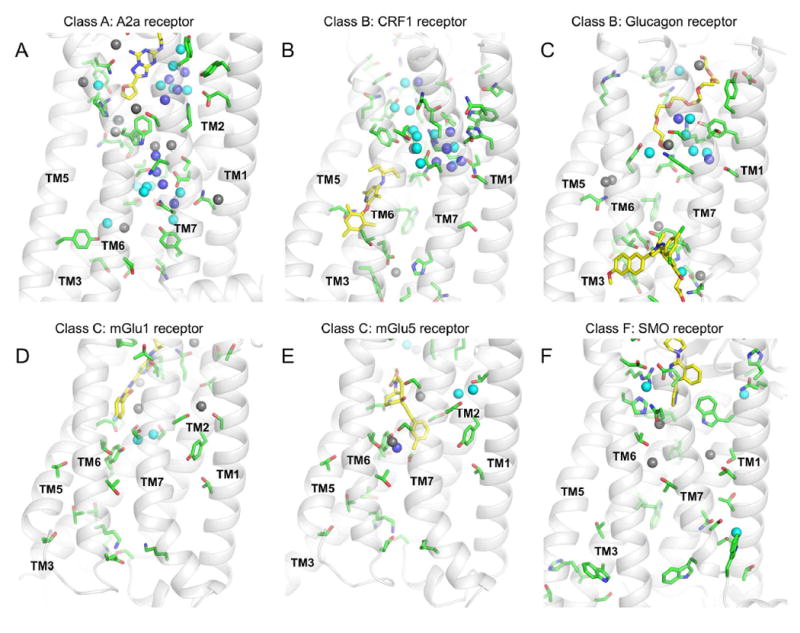
The conformations of amino-acid side-chains and buried solvent molecules were predicted de novo in the inactive state ligand-bound structures of evolutionary distant GPCRs using SPaDES. (A-F) Six representative GPCRs for four distinct classes were considered: (A) adenosine A2a receptor (PDBid:4EIY) from class A, (B) corticotropin-releasing factor receptor type 1 (PDBid:4K5Y) from class B, (C) glucagon receptor (PDBid:5EE7) from class B, (D) metabotropic glutamate receptor 1 (PDBid:4OR2) from class C, (E) metabotropic glutamate receptor 5 (PDBid:4OO9) from class C, and (F) smoothened receptor (PDBid:4JKV) from class F. Green sticks indicate buried polar amino-acids (some interacting directly with waters), yellow sticks indicate bound ligands, and spheres indicate de novo predicted water molecules. Water molecules are color-coded according to their relative calculated energy: dark blue indicates lower energy (i.e. more stable), cyan indicates average energy, and grey indicates higher energy (i.e. less stable). See also Table S6.
High-resolution refinements of GPCR structures from evolutionary distant classes using SPaDES now provide unique insights into the role of solvent-mediated interactions across GPCR families. As shown in Table S6 and Figure 6, class A GPCRs primarily differ from the other classes by the high level of solvation of the TMH domain, which buries up to 5-fold more predicted water molecules than for the other classes. Interestingly, all receptor classes bury similar number of polar residues, implying that the conformation of the TMHs and cavities, and the specific position of polar residues primarily determine the level of TMH solvation (Table S6, Figure 6). In class B, most predicted water molecules were found on the large extracellular hormone binding site bound to residues critical for peptide binding and receptor activation (Figure 6B,C). A few structural water molecules were predicted on the intracellular side mediating interactions with conserved polar residues (i.e. His2.50, Glu3.50, Tyr7.57 mimicking the class A ionic lock) and with the cytosolic allosteric negative modulator stabilizing the receptor inactive state. The only waters predicted to be deeply buried in the TMH core were found at the binding site for the antagonist bridging TMHs 3, 5, and 6 in the corticotropin-releasing factor receptor type 1.
Interestingly, these molecules had higher calculated energy (i.e. not fully engaged in stabilizing interactions) which are typical signatures of drug binding sites (Hollenstein et al., 2014; Mason et al., 2012). The orthosteric ligand binding site for class C GPCRs lies in the extracellular domain separated from the TMH region, which only binds allosteric modulators. Consequently, class C TMH domains are more compact than in other classes and display 4-fold lower solvation than class A (Figure 6D,E). As for class B, mostly high energy water molecules were predicted to be buried at the allosteric modulator binding site. Our predictions suggest also the presence of an additional putative ligand binding site bridging the extracellular side of TMHs 1, 2, and 7, which bind a few relatively high energy water molecules. Class F TMH is the most hydrophobic among all classes. It buries only a few relatively high-energy isolated water molecules in addition to more stable water molecules mediating ligand receptor interactions on the extracellular side (Figure 6F). Overall, our predictions suggest that buried solvent molecules share a common role in all receptor classes by mediating ligand-receptor interactions or providing putative drug ligand binding sites in the TM region. The especially high solvation in class A GPCRs enables a solvent mediated interaction network to connect the entire receptor structure across the membrane, suggesting a unique additional allosteric role for the buried solvent in this receptor family. Interestingly, the allosteric ligand binding sites in class A are substantially less buried in the TMH core and farther from the intracellular G protein binding site than in class B and C GPCRs (Jazayeri et al., 2016). Therefore, it is tempting to speculate that the corresponding allosteric pathways in class A connecting allosteric ligands to the intracellular side may be longer-distance, requiring more complex networks of interactions with solvent molecules providing key dynamic interactions to facilitate the propagation of structural changes.
Limitations of the approach
Despite numerous improvements over alternative techniques, SPaDES presents a few limitations. Unlike molecular dynamics simulations, our Monte Carlo simulations do not directly provide dynamic information on the solvent molecules. Because SPaDES is designed to recapitulate highly coordinated solvent molecules, our model may not be well suited for protein surfaces where low occupancy waters only transiently interact. Also, because it relies on polar interactions with the protein, the technique is not implemented to model pure hydrophobic (i.e. entropic) effects and solvent filled hydrophobic cavities. We provide in Figure S5A a typical example of this limitation. In the SHV-1 beta-lactamase-BLIP complex X-ray structure, a water molecule is observed in a small cavity located more than 5Å away from any polar side-chain atom. It interacts with the protein through Van der Waals (VDW) contacts and only one weak hydrogen bond with a nearby protein backbone carbonyl oxygen. During water rotamer construction, SPaDES filters out water rotamers, which do not form at least two significant hydrogen bond interactions (i.e. of energy of at least -0.5 Rosetta Energy Unit. Optimal hydrogen bond energy is -1.5 Rosetta Energy Unit; see Methods) with its direct environment. Therefore, SPaDES did not select any water rotamers to repack at that location. This limitation likely applies to highly transient (low occupancy) water molecules, which only form relatively weak hydrogen bonds with the protein. Expanding SPaDES to model solvent molecules transiently interacting with protein surfaces would require modification of the approach and taking into account multiple types of weak interactions during solvent rotamer construction.
SPaDES builds complex networks of buried water molecules in an iterative manner (i.e. one hydration shell at a time starting from solvent molecules directly interacting with the protein) to avoid a combinatorial explosion of water rotamers. However, using this procedure, slight inaccuracies in the first shell of hydration can propagate to the next shell preventing the necessary space for placing and stabilizing additional solvent molecules. Consequently, our approach sometimes fails to recapitulate water molecules that are stabilized through solvent-solvent interactions only. We provide an example of this limitation in Figure S5B. In the Subtilisin Calsberg-Eglin C complex X-ray structure, an experimentally-resolved water molecule is observed at the center of a cavity mostly coordinated by surrounding water molecules. While the water molecules making polar interactions with the protein were correctly predicted by SPaDES, the experimentally-resolved molecule at the center of the cavity was not recapitulated.
While SPaDES does not yet model ion-protein interactions, it should be straightforward to implement these interactions. Ions perturb bulk water properties through strong charge-dipole interactions (Chen et al., 2016). Similarly, protein-ion interactions (Rembert et al., 2012) are expected to influence the configurations and dynamics of protein-bound water molecules networks. Finally, while water-mediated ligand-protein interactions are accurately recapitulated, performing ligand and protein docking with explicit solvation on the fly remains to be implemented in our method.
Discussion
Solvent molecules profoundly affect protein conformation, stability, catalysis, signaling, and ligand binding. However, these molecules are small and bind in large numbers to proteins with a broad range of dynamics. Therefore, their effects on proteins remain very challenging to model accurately and efficiently at atomic resolution. To address this problem, we developed and applied SPaDES, a novel method based on a hybrid implicit-explicit solvation model (Figure 1), which efficiently predicts de novo protein-interacting solvent molecules and improves the atomic-level structure modeling and design of soluble and membrane proteins (Figures 2-6). Overall, because of its accuracy, efficiency, and generality, our method should prove useful in a large range of protein modeling, design, and drug screening applications.
Unlike many alternative explicit solvation approaches, our de novo solvation model is very general and does not rely on any prior knowledge of solvent position. Therefore, it can be applied to any protein with buried cavities or binding sites (e.g. soluble protein-protein complex and membrane proteins) as long as protein polar sites are not fully satisfied and available for interacting with solvent molecules. The balance between solvent-solvent, solvent-protein, and protein-protein interaction energy terms was optimized during the training of our energy function, enabling solvent free energies to be correctly estimated. Consequently, the number and position of protein-bound solvent molecules, as well as mutational effects on protein binding energies involving alterations of solvent network mediating important protein interactions could be recapitulated. By contrast, alternative techniques tested in our study displayed substantially higher false positive rate in solvent molecule placement. Including explicit water molecule modeling in these alternative techniques also did not increase the accuracy of mutational effects predictions. These observations suggest that solvent free energies may not be correctly estimated and highlight the challenge of developing efficient yet accurate explicit solvation models for protein structure prediction and design applications.
Many examples where our model considerably outperformed implicit solvation involved perturbation of highly coordinated water molecules strongly interacting with the protein, which can not be approximated by bulk solvent properties assuming uniformly high solvent molecule dynamics (Figure 4, Table S4). As another demonstration of the generality of our model, accurate predictions were obtained for membrane proteins and solvent mediated ligand-receptor interactions. Considering our strong performance in predicting mutational effects on hydrated protein-protein binding interfaces, our efficient atom-based de novo predictions of solvent mediated protein interactions should prove particularly useful for the rational de novo design of solvated protein functional and binding sites in enzymes, protein-protein, protein-ligand complexes, and membrane proteins.
The high-resolution structure prediction of membrane proteins has been challenging because highly dynamic solvent and lipid molecules influence protein conformations by interacting at many protein sites but are rarely observed in experimental membrane protein structures. Traditional protein structure prediction methods modeling protein atoms only, such as Modeller and RosettaMembrane, often result in a critical loss of structural information near key protein functional sites (Figure 5). By contrast, our hybrid solvent homology modeling technique recapitulated native protein local conformations and interacting water networks critically involved in ligand binding or membrane receptor signaling (Gutiérrez-de-Terán et al., 2013; Liu et al., 2012; Nygaard et al., 2010; Valentin-Hansen et al., 2015). The high-resolution prediction of many key functional solvated sites in transmembrane proteins even starting from distant homologs suggest that our homology models should provide reliable starting templates for rationally designing novel functions in many structurally-uncharacterized proteins. Of particular interest are the numerous solvated cavities, which were recapitulated using our approach but lost when using alternative techniques. Since these cavities or pockets often represent key protein regulatory target sites for drug molecules (Gutiérrez-de-Terán et al., 2013; Liu et al., 2012; Ngo et al., 2017; Zheng et al., 2016), recapitulating their geometry should considerably leverage in silico drug screening and design approaches. Lastly, as we describe for the large family of GPCRs (Figure 6), SPaDES should prove particularly useful for refining low-resolution membrane protein structures and provide key atomic-level insights into the role of solvent mediated interactions in ligand binding, allosteric pathways, and structure-function relationships in general.
As demonstrated in this study, we have developed and applied SPaDES, a very general high-resolution protein structure modeling and design approach which predicts de novo protein-solvent molecule interactions. SPaDES should prove particularly useful in the refinement of low-resolution protein structures, prediction of missense mutational effects on protein stability and binding, prediction and design of protein-protein complexes, and of membrane protein structures and interaction with ligands, which remain difficult to characterize at high-resolution.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for datasets can be directed to the corresponding and lead author: Patrick Barth, patrickb@bcm.edu
METHOD DETAILS
Energy function
We developed our method within the Rosetta modeling suite (Leaver-Fay et al., 2011), and including the explicit modeling of water molecules required a number of modifications that are described in the following sections.
Buried explicit water terms
The Rosetta energy function, by default, treats solvent water and ion molecules implicitly, and calculates solvation energies using the Lazaridis-Karplus EEF1 solvation model (Lazaridis and Karplus, 1999). Similarly, the RosettaMembrane energy function (Barth et al., 2007) models the lipid membrane implicitly using a hydrophobic phase (for the lipid acyl chain region), a polar phase (for the lipid headgroup / water region), and a transition phase in between. In both cases, solvation energies of protein atoms are calculated as a desolvation cost from a fully solvated reference state where single isolated amino-acids are only interacting with the solvent (i.e. water or organic solvent mimicking lipids). The desolvation energies are calculated using experimentally determined free energy of transfer of amino-acid analogs from water or hydrophobic organic solvent (i.e. fully solvated state) to vacuum (i.e. fully desolvated state).
In our approach, “buried” water molecules bound to the protein are modeled explicitly as any protein residue. The interactions of a “buried” water molecule within the protein are approximated by its Van der Waals (VDW) and polar contacts with protein atoms and other buried water molecules. Such interactions are calculated using the Lennard-Jones (LJ) and hydrogen bond potentials in Rosetta. However, because water molecules can constantly exchange with the bulk solvent, their propensity to reside in the protein depends on the free energy difference between their interactions with the protein and their interactions with other solvent molecules in the bulk. Therefore, to calculate the free energy of a water molecule, its interactions in the protein are supplemented by a water desolvation and water entropy loss term, which calculate the cost of removing the molecule from the bulk solvent. The free energy of the water molecule is used to determine during the Monte Carlo simulation its propensity to reside in the protein or to remain in the bulk.
To estimate the water desolvation cost, we ran a Monte Carlo simulation of bulk water with standard pressure and temperature using 512 water molecules under infinite boundary conditions. After the system energy converged, we calculated the energy of removing a water molecule and used the resulting value of 4.8 Rosetta Energy Unit (R.E.U) as the water desolvation energy (ΔHwater in Figure 1). To calculate the entropic cost of bringing a water molecule from bulk to the protein environment (ΔSwater in Figure 1), we considered the study by Dunitz (Dunitz, 1994) reporting a maximal entropy difference between water in liquid versus water in ice or in a hydrated salt of 2 kcal mol-1 K-1. This quantity defined the maximal entropic cost of moving a water molecule from the bulk to a fully coordinated (i.e. engaged in four hydrogen bonds) and stable position in the protein. To estimate the entropy cost for non-fully coordinated buried water molecules, we considered the study by Yu and Rick (Yu and Rick, 2010) who calculated the entropy of a buried water molecule in various protein environments and found a linear correlation with the number of hydrogen bonds. Given the aforementioned information, we assigned an entropic cost of 0.5 kcal mol-1 K-1 for each energetically favorable protein-water hydrogen bond. In our model, the entropic cost of burying a water molecule can be as low as 0.5, which is consistent with the findings reported independently by Huggins (Huggins, 2015). The combination of terms that have different intrinsic units to the same energy function is accomplished by weighing and optimizing each individual term with a scaling factor, as described with more details in later method sections.
To enable pre-calculation of protein energies for all possible discrete protein conformations sampled by Rosetta in structure prediction or design simulations, protein interactions are approximated by one body (interaction of an amino-acid with itself) and two body (interaction between two amino-acids) interaction energies. The total interaction energy of an amino-acid with its environment is then equal to the sum of two-body interactions over all pairs of interacting residues. However, water molecules can donate or accept only a limited number of hydrogen bonds. Since buried water molecules are often found within hydrogen bonding distance of several polar protein atoms, summing over all pairs of polar interactions can overestimate the number of physically possible water-mediated polar interactions. We corrected this multi-body overcoordination effect by decreasing the energy of water bifurcated hydrogen bonds (i.e. when the same polar atom is engaged in more than one hydrogen bond) (Rozas et al., 1998). If the acceptor atom is an oxygen atom and engaged in more than two hydrogen bonds, the two strongest interaction energies are decreased by 20% (as described in Rozas (Rozas et al., 1998)) while the reminder are ignored. When the energy of the strongest hydrogen bond is five times lower than that of the second strongest, the strongest bond is kept without decreasing its strength. If the acceptor is a nitrogen atom, only the strongest bond is considered.
Hybrid implicit-explicit score function
Our energy function was developed to model entire protein structures including regions exposed to the external bulk solvent or tightly packed, which do not form strong and stable interactions with buried solvent molecules. The computationally expensive explicit treatment of solvent molecules is unnecessary for these regions, which are best modeled using implicit solvent models. Therefore, we developed a hybrid energy function that can model concurrently distinct protein regions using implicit and explicit solvent representations depending on their structural properties and location.
In our hybrid model, we defined two canonical regions: 1. “Hydrated” regions comprising buried water molecules and protein residues exposed to the space occupied by solvent molecules, which are either predefined or within a specific distance from water molecules, modeled using the explicit solvent part of the hybrid energy function, 2. “Implicit” regions comprising all protein residues not directly in contact with solvent molecules and modeled using the standard implicit part of the energy function (Barth et al., 2007; Barth et al., 2009; Leaver-Fay et al., 2011).
Since only short-range interactions are modeled in our hybrid energy function, the boundary conditions describing the transition between explicit and implicit functions are simple and defined as follows. The one-body (i.e. self-interaction) energy term is calculated for each protein residue and defined based on the assignment of the protein residue to the “hydrated” or “implicit” regions. For the two body interactions, hydrogen bond energies between protein and buried water molecules were calculated using the explicit energy function weights and were corrected for over-coordinated hydrogen bonds as described above. Additionally, the environment dependency of the hydrogen bond weight describing implicitly the competition between protein-protein and protein-water hydrogen bonds at the protein surface in the implicit energy function was turned off for any hydrogen bond in the vicinity (i.e. within 4 Å) of an explicit water molecule. This modification was necessary because the competition between protein-protein and protein-water interactions is modeled explicitly in the “hydrated region”. Lastly, the VDW and polar interactions between buried solvent molecules and protein atoms calculated using the Lennard-Jones (LJ) and hydrogen bond potentials directly and explicitly accounted for the protein solvation in the “hydrated” region. Therefore, the weight for the implicit solvation Lazaridis-Karplus term was set to zero in that region of the protein to avoid double counting protein solvation effects.
Hybrid implicit-explicit score function adapted for membrane proteins
The hybrid score function adapted for membrane proteins followed that described above except the implicit solvent model which is taken from RosettaMembrane. Unlike for water-soluble proteins, the solvation of a residue in a membrane protein involves two components.
The first component measures the desolvation cost of transferring the individual residue (stripped of the rest of the protein) from the water phase to its specific depth in the membrane bilayer. This term is calculated from experimentally measured transfer free energies of amino acid analogs from water to hydrophobic organic solvents and approximates the desolvation cost of transferring an individual amino-acid from a high dielectric to a low dielectric environment that would be calculated using Poisson Boltzmann based approaches. As described in Barth et al., 2007, an interpolation function models the transition between the high dielectric lipid headgroup and low dielectric hydrophobic core regions of the membrane for estimating the desolvation costs at different depths in the membrane.
The second component measures the desolvation cost of transferring the individual residue from a lipid-exposed position to its final position in the protein; i.e. the solvation effect of the membrane protein environment. This solvation component depends on the position of the residue in the protein and the environment of the residue in the protein that define the dielectric properties around that residue.
In the following, we consider the hypothetical case of two glutamate residues both at a depth corresponding to the center of the membrane. The first one is exposed to the hydrophobic core of the lipid membrane and the second one is buried in the protein but exposed to a solvated cavity.
For the first glutamate residue, the desolvation cost will be dominated by the first solvation component described above, which for a glutamate corresponds to a large energetic desolvation penalty (even with the glutamate in the neutral protonated state).
For the second glutamate residue, the second solvation component will depend on its interactions with explicitly modeled solvent molecules in the protein cavity. If the glutamate residue can establish several strong VDW and polar interactions with buried solvent molecules, the solvation effects of transferring the residue from a lipid exposed to a protein buried position will be favorable and may compensate the desolvation cost of moving the residue in the lipid membrane (first solvation term).
In conclusion, when modeled using SPaDES, the glutamate facing the lipid molecules will be characterized by a large total desolvation penalty if it is located in the hydrophobic core of the lipid bilayer. On the other hand, the glutamate located at the same membrane depth but exposed to a hydrated cavity in the protein core will undergo a considerably reduced desolvation cost. Therefore, SPaDES can capture distinct protein location specific desolvation penalties.
Sampling explicit water molecule conformations
To identify low energy hydrated protein structures, we explored the protein conformational space using a Monte Carlo simulated annealing protocol followed by a Quasi-Newton (“dfpmin”) energy minimization. In the Rosetta modeling suite (Leaver-Fay et al., 2011), the simulated annealing is referred to as packing: during this step, the backbone of the protein is kept fixed, while the side-chains explore multiple discrete pre-calculated conformations called rotamers. Rotamers are built according to a library (developed by Dunbrack et al. (Shapovalov and Dunbrack, 2011)) of consensus amino-acid conformations recurrently observed in high-resolution experimental protein structures. To accurately calculate protein-water interactions during protein packing, water molecules were modeled as any protein residues using discrete rotamer conformations.
De novo water rotamer modeling
Since high-resolution experimental structures capable of providing reliable positional information about the water molecules are not readily available for membrane proteins, de novo water molecules were built without prior knowledge of their specific locations. We developed our technique to model and design buried solvent molecules interacting strongly with the protein. Therefore, we built and selected water rotamers that form optimal hydrogen bond and VDW contacts with pairs of or single “unsatisfied” polar protein atoms (i.e. not yet engaged in polar contacts). Such protein polar atoms are defined as anchor sites and are used to construct de novo water rotamers with energetically optimal conformation. Since water rotamers can in principle be anchored to any (or pair) of polar residue rotamer, several millions of possible water conformations need to be built (see below). To efficiently construct these rotamers without having to calculate hydrogen bond energies, we used a TIP5P water model (Mahoney and Jorgensen, 2000) (i.e. characterized by explicitly represented lone electron pairs on the oxygen atom with a tetrahedral-like geometry) enabling the identification of energetically optimal protein-water interactions from geometric criteria only. The idealized dimensions for the TIP5P water geometry are defined as follows: a H-O-H angle of 104.5°, O-H distance of 0.9572 Å, lone electron pairs to oxygen angle of 109.5°, and lone electron pair to oxygen distance of 0.7 Å. To construct water rotamers anchored to protein rotamers, an idealized donor-acceptor distance of 2.75 Å was considered. The optimal location of placing a water oxygen with respect to a donor protein heavyatom is directly 2.75 Å from the donor protein heavyatom, with the angle direction determined by the donor hydrogen. After placing the water oxygen, water rotamers can be generated by reorienting the molecule at equal angular steps (5 steps of size 72 degrees) about the oxygen while maintaining the idealized TIP5P geometry with a lone electron pair pointing to the protein donor. The placement of a water oxygen with respect to an acceptor protein heavyatom, on the other hand, is determined by placing the oxygen at different locations around the acceptor depending on the orbital hybridization type (i.e. sp2 or sp3) and the idealized donor-acceptor distance. Multiple water rotamers can again be generated by reorienting the molecule about the oxygen with respect to a water hydrogen pointing to the acceptor with idealized TIP5P geometry.
To preferentially select water molecules forming strong polar contacts with the protein, the de novo water rotamers previously described were constructed in two steps. In step 1, only water rotamers were built that form two hydrogen bonds with protein anchors (“bridging” water). Then, protein side-chains and “bridging” water were repacked together to identify their energetically optimal conformations. In step 2, additional protein-bound water molecules were built forming either only one hydrogen bond with protein anchors or contacting both a protein anchor and a “bridging” water selected in step 1. These additional water molecules were then repacked concurrently with protein and bridging water rotamers followed by a minimization of all the aforementioned molecules. This approach can lead to a substantial redundancy in the position of water rotamers. For example, consider a scenario where water a, associated to polar atom A, uses neighboring polar atom B to optimize its second hydrogen bond. Similarly, water b, associated to atom B, will use atom A to optimize its second hydrogen bond. This will, in turn, generate a redundant set of rotamers characterizing the conformations of the exact same water molecule. Additionally, several rotamers of water molecule a (e.g. interacting with a different polar atom C) will likely overlap with rotamers of water molecule b, potentially biasing the phase space search of the entire system. To address these problems, we did not repack all the de novo water molecules during the first step, but instead only attempted 75% of the potential hydratable sites, which were randomly selected for each independent Monte Carlo simulation.
Size of the water rotamers set
A large fraction of polar atoms used as anchors to build water rotamers are side-chain atoms considered flexible during the simulation, which multiplies considerably the number of positions for building water rotamers in the protein and can lead to an explosion in the number of water rotamers. Considering that a polar amino-acid can have up to ~100 rotamers and a standard of nine optimal hydrogen bond orientations to locate the water molecule, there would be close to a million (9×100×9×100) possible combinations for a “bridging” water molecule linking two flexible protein side-chain atoms. Moreover, for each combination of anchor sites, we typically construct three water rotamers, two with the oxygen located at each optimal position with respect to each of the two protein polar atoms and a third in the middle.
Besides the intrinsic computational problems (i.e. time and memory constraints) associated with a very large number of water rotamers, using it concurrently with the amino-acid rotamer sets, four orders of magnitude smaller, would make convergence of almost any Monte Carlo procedure practically unattainable. Therefore, the sizes of the water molecule rotamers sets were reduced, which we accomplished through a series of filters. The first filter discarded any water rotamers that do not have at least two hydrogen bonds with energy lower than -0.5 REU. The second filter removed water rotamers sterically clashing with fixed parts of the system (i.e. backbone or non-flexible side-chains). In many cases, these two energy-based filters were enough to reduce the rotamer set to a few hundreds. However, several water molecules were still characterized by thousands of rotamer conformations. To further reduce the rotamer set size, a maximal number of 500 rotamers were randomly selected for a given water molecule. To test the validity of this filter, a uniformly distributed sample of 500 water rotamers was extracted from the already culled set (using the energetic filters). In a drastic example, even when we performed a 30-fold size reduction (i.e. from 15,000 rotamers to 500), space coverage was not significantly reduced as we still had rotamers in most of the original space (Figure S1); instead, we find that mainly redundant rotamers were removed using this additional filter.
Analyzing the hybrid solvation model
Experimental benchmarks of water-soluble proteins and transmembrane proteins were selected to assess the performance of SPaDES in energy prediction of protein-protein interactions, prediction of native water molecule positions, and prediction of protein conformations. In addition, the accuracies of these predictions from SPaDES were compared to alternative approaches: Fold-X(Schymkowitz et al., 2005) HADDOCK(de Vries et al., 2007; Dominguez et al., 2003) and Modeller (Eswar et al., 2008).
Water-soluble protein benchmark construction
The benchmark was initially built from the SKEMPI database (Moal and Fernandez-Recio, 2012), which is a curated dataset containing free energy binding changes upon mutation extracted from previously published scientific literature for protein-protein complexes. A number of filters based on information provided by the SKEMPI database were applied to select a dataset appropriate for our study. First, complexes with more than one mutated position were removed from the dataset and the measured binding energies for duplicate entries from multiple sources were averaged. Second, we only considered entries published in a peer-reviewed journal and excluded those generated using “unorthodox” experimental techniques. Third, mutations at the edges of the binding interface were discarded. Specifically, only “core” and “support” positions according to a criteria defined by Levy (Levy, 2010) were considered; these are positions that have relative accessible solvent accessibility (rASA) of less than 25% and exhibited a change in rASA at that particular position when comparing the unbound monomers with the bound complex. Lastly, a small fraction of the mutations displaying predicted destabilization considerably larger than the experimentally measured ΔΔG values (i.e. ΔΔE > 5 kcal/mol from the experimental values) using both the implicit and the hybrid solvation were also removed. These mutations likely induced large structural changes at the protein-protein binding interfaces that could not be predicted using our fixed backbone protocol. The remaining entries constituted a large benchmark of 532 mutations on 39 protein-protein complexes (referred to “low- and high-resolution set” in the main text) (Table S7). To train the hybrid energy function, a subset of this benchmark was selected consisting of high-resolution protein structures (2.0 Å or lower) where water molecules were experimentally detected at the binding interface near each mutational position. This smaller subset (referred to “high-resolution hydrated set” in the main text) contains 120 mutations from 12 protein-protein complexes.
Prediction of mutational effects on protein binding energies
We assessed whether the hybrid solvation model could recapitulate mutational effects on protein binding energies (ΔΔG binding) in both the large mutagenesis benchmark of 532 mutations and the high-resolution hydrated benchmark of 120 mutations. For this analysis, both the wildtype and the mutant structures of each entry were predicted using only the experimental structure of the wildtype complex as starting information. Following the procedures previously described, structures were generated by Monte Carlo repacking and minimization for each protein sequence (i.e. wildtype and mutant) in the bound and unbound states. Multiple structures (several hundreds) were generated to ensure that the simulation converged, as defined by the lowest 10% energy models reaching a SEM of less than 1 REU. Since explicit solvent molecules were only modeled at buried positions of the protein-protein binding interface, monomers were modeled with the default implicit solvation score function of Rosetta since all positions of the binding interface were solvent exposed in the unbound state. Wildtype and mutant structures of the bound complexes were modeled using the hybrid energy function. In addition, all predictions were performed under the assumption that the mutations would only perturb their local environment. Therefore, only side-chain rotamers from amino-acids with at least one heavy atom within a threshold distance of 8 Å of a heavy atom in the amino-acid to be mutated, were repacked. The remaining protein backbone and side-chain conformations were kept fixed to their crystallographic coordinates. To construct de novo water rotamers, the potential anchor sites at the binding interfaces were limited to the polar atoms of amino-acids with a heavy atom within 6 Å of the mutated residue. This procedure limited the number of de novo waters to be built without reducing significantly the accuracy of our predictions.
Predicted binding energies were calculated from a representative repacked structure for each state selected from the lowest energy conformations generated by multiple independent Monte Carlo simulations. Specifically, for each state, the 10% lowest energy structures were selected and clustered; then, the center member of the lowest energy cluster populated with a size of at least a tenth of all the clustered structures was selected as the representative structure. From the representative structures for each state, the binding ΔΔG was calculated as follows:
where AB is the bound state containing monomers A and B, and ΔΔG describe the change in ΔG between the mutant (mut) and the wildtype (wt) states. The resulting binding ΔΔG was therefore the difference between the change in ΔG for the bound complex and the summed change in ΔG for the unbound monomers. All structure predictions of protein bound complexes were also performed using the Rosetta’s default implicit solvent energy function (Lazaridis and Karplus, 1999; Leaver-Fay et al., 2011) as well as Fold-X with and without explicit water molecule modeling (described in the Fold-X predictions section) to assess the difference in accuracy between SPaDES and alternative techniques based on implicit or explicit solvation. Pearson’s correlation coefficients were calculated with the best-fit linear regression constrained to pass through the origin reference state (ΔΔG=0) to ensure that the values capture the correct qualitative relationships between predicted and experimental data.
Side-chain rotamer and water recovery
To assess the accuracy of the hybrid energy function in protein structure predictions, recovery rates of diverse structural features of high-resolution protein X-ray structures were considered. For the protein amino- acids, native side-chain rotamer recovery was used to judge the quality of the prediction. The recovery rate of side-chain rotamers was defined as the fraction of residues with side-chain dihedral angles (i.e. X1 to X5) within 30° from that of the X-ray structure. Since the rotamer does not change for residue conformations that are fixed, only repacked side-chain residues were considered in this calculation. In addition, to capture the effects of the inclusion of buried water molecules, the benchmark side-chain rotamers were limited to those in direct vicinity to a predicted water molecule (i.e. with at least one heavy atom within 3 Å of a water oxygen). Only buried interface amino- acids, as defined by Rosetta’s InterfaceAnalyzer (Leaver-Fay et al., 2011) and the relative solvent-accessible calculations from DSSP (Joosten et al., 2011), were allowed to be repacked and hydrated as an anchor site. Multiple independent simulations were performed for each protein-protein complex to ensure convergence. As in blind structure prediction contests, the native side-chain rotamer recovery was calculated from the most accurate structure among the five lowest energy generated model. As a benchmark against other computational approaches, Fold-X was considered for comparison (described in the Fold-X predictions section).
To assess the prediction accuracy of the position of native water molecules in protein structures, we considered two metrics: native water coverage and true positive rates. The water coverage rate described the fraction of native water positions that were correctly recapitulated by a de novo water molecule. Specifically, a native water position was defined recovered if a predicted water molecule was within a distance cutoff of 1.0 to 2.5 Å of the native water position in the X-ray structure. To assess the rate of false positive predictions, we also considered a true positive water recovery rate, which was defined as the fraction of predicted de novo water molecules that were within a distance threshold of 1.0 to 2.5 Å of a water position in the X-ray structure. All buried interface amino-acids (as previously described) were allowed to be repacked and hydrated. Multiple independent simulations were performed using either the implicit or the hybrid implicit-explicit solvation models to ensure convergence. As in blind structure prediction contests, the water recovery was calculated from the most accurate structure (i.e. with the highest water recovery) among the five lowest energy generated model. Similar predictions were made with Fold-X and HADDOCK for comparison (described in the Fold-X predictions and HADDOCK predictions sections).
Homology modeling of GPCRs
The hybrid implicit-explicit solvation scoring function was implemented into the homology modeling protocol of RosettaMembrane (Chen et al., 2014b) and tested on challenging transmembrane protein targets. A benchmark of twenty relatively distant homolog G protein coupled receptor pairs was identified using HHPred (Soding et al., 2005) with sequence identities between 20% and 40%, and no gaps in the aligned transmembrane helical regions (Table S6). In each pair, one protein was designated as the target to model, while the other was used as a starting template for the homology model prediction.
Based on the aligned amino-acid sequences, the target sequence was threaded onto the homolog template structure. The poorly aligned loop regions, and missing densities resulting from this threading step were rebuilt de novo by coarse-grained peptide fragment insertion of the loop modeling protocol (Wang et al., 2007) and scored using the coarse-grained energy function of RosettaMembrane (Barth et al., 2009; Yarov-Yarovoy et al., 2006). The reminder of the template structure (including the transmembrane (TM) region) remained fixed during this step and no attempt of side-chain and water rotamer modeling was performed. The rebuilt loop conformations were clustered and the cluster centers were ranked based on loop energies scored by the default RosettaMembrane scoring function using implicit solvation (Chen et al., 2014b).
Third, the resulting loop rebuilt coarse-grained models were extensively relaxed at all-atom with de novo hydration of the TM structure. The protocol performed several cycles of relaxation with distinct smoothed versions of the all-atom scoring function to prevent coarse-grained structures to lose compactness upon accommodation of all-atom contacts. Specifically, the repulsive term of the Lennard-Jones potential was scaled down during three of four cycles of relaxation using the following factor: 0.02, 0.25, 0.55, and 1.00 (in the final cycle, the potential was fully considered). Each relaxation cycle involved protein side-chain and de novo constructed water rotamer repacking followed by energy minimization over all protein and water conformational degrees of freedom. Water molecules were allowed to move to and equilibrate with the bulk solvent in each step of the relaxation. Multiple (at least 100) independent all-atom relaxation simulations were performed for each GPCR target to guarantee convergence using RosettaMembrane implemented with implicit or hybrid solvation. As in blind structure prediction contests, the most accurate among the top 5 lowest energy models was selected as a representative model for structural analysis.
To analyze the cavity geometry recovery, we identified cavities by using a 1.2 Å probe over a cubic lattice (0.25 Å vertex). In this test, only heavy atoms were assumed to occupy volume as determined by their VDW radius. In addition, only buried lattice points were considered, which was determined by dividing the solid angle around each lattice point in 98 uniform sections. If more than 70% of the sections have a heavy atom within 10 Å, the point is considered buried and thus kept. Using these filtered lattice cavity points in both the native and modeled structures, we assessed the cavity point coverage and cavity point true positive rates. The coverage was defined as the rate of native cavity points that have at least one modeled cavity point within a distance threshold of 0.8 Å. The true positive rate was defined as the rate of modeled cavity points with at least one native cavity point within the same distance threshold. The cavity point recovery score is the product of the cavity point coverage and true positive rate, and is subsequently used to calculate the fraction improved between different solvation models when compared to the native cavities of the target structure. When comparing RosettaMembrane with implicit or hybrid solvation, the reported fraction improved is determined from subtracting the cavity recovery score of the implicit solvation prediction from the hybrid solvation prediction, and then normalizing the difference by the cavity recovery of the implicit. The resulting fractional value describes the extent of which hybrid solvation improved over implicit solvation in recapitulating the experimental cavity geometry. The same comparison was performed to assess the improved structural accuracy of RosettaMembrane with hybrid solvation compared to the starting template structure or the structures generated by the standard default mode of the homology modeling software Modeller (Eswar et al., 2008).
In addition to the cavity point recovery analysis, the structural prediction accuracy of the amino-acids in the vicinity of the above-described cavities were assessed using the global distance calculation for all atoms (GDC-all) metric calculated with the LGA package (Zemla, 2003). For the GDC-all calculation, we choose four 0.5 Å bins (i.e. four bins from 0.5 Å to 2 Å) to measure the similarity between the cavity amino-acids for the structural predictions against the X-ray structure of the target. Fractional improvements between the GDC-all calculations were reported for the hybrid and implicit solvation scoring functions as previously described.
Fold-X predictions
To compare the performance of SPaDES in the water recovery, sidechain recovery, and mutagenesis ΔΔGbinding benchmarks, Fold-X 4, a protein modeling and design software with knowledge-based explicit solvent model, was considered. The BuildModel protocol of Fold-X reconstructs amino-acid sidechains according to a probability-based rotamer library and an empirical force field, which is comparable to repacking in Rosetta. As a comparison with SPaDES on the mutagenesis benchmark, the BuildModel protocol of Fold-X which enables in silico mutagenesis was run with or without the water prediction options flags (–water –PREDICT –pdbWaters true) on the mutagenesis benchmark on the bound protein-protein complex and the unbound protein monomers. Only the mutant position and amino-acid were specified as input to Fold-X in the “individual_list.txt” mutant-file since the protocol, as a builtin feature, automatically moves neighboring sidechains around the mutant positions. The default Fold-X settings were used for all other options. Following the same analysis as previously described, the binding ΔΔG was calculated from the difference between the change in the Fold-X total energy for the bound complex and the summed change in Fold-X total energy for the unbound monomers.
Likewise, the BuildModel protocol of Fold-X was also used for comparison against the SPaDES water recovery and sidechain recovery benchmarks. The protocol was run with and without water prediction option flags where the amino-acids of the hydrated interface region, as previously described, were specified with their wildtype amino-acids in the “individual_list.txt” mutant-file of Fold-X so that only those sidechain configurations were de novo rebuilt while neighboring sidechains are automatically allowed to move, which is the built-in behavior of Fold-X. All other Fold-X options were kept at their respective defaults. The resulting output generated structures and corresponding water molecule positions from Fold-X were compared against experimental structures and water positions with the same procedures as previously described.
HADDOCK predictions
HADDOCK 2.2 is an ab initio protein docking and refinement software suite performing an all-atom protein structure refinement with explicit solvent modeling which consist of a series of Molecular Dynamics Simulations trajectories in a TIP3P water box. This refinement step provides an additional structure relaxation technique with explicit solvent for comparison with SPaDES. Following the recommended default settings, HADDOCK was used to simulate 200 separate trajectories with MD refinement starting from the X-ray structure. The first two docking stages of HADDOCK, rigid body energy minimization and semi-flexible simulated annealing, were turned off through their respective options while all other options were kept as default for the final flexible explicit solvent refinement stage. Similar to SPaDES, the results for the recovery tests were calculated from the most accurate model among the five lowest scoring HADDOCK refined structures.
Modeller predictions
Modeller 9.17 is a premier protein modeling software for performing homology or comparative modeling to produce atomic-resolution model of target proteins from their amino acid sequences and template protein structures. As a comparison to SPaDES implemented into the homology modeling protocol of RosettaMembrane, the sequence alignments and template structures from the benchmark described in the Homology modeling of GPCRs section were input into Modeller while all other input options were kept as the defaults set in the MPI bioinformatics Toolkit (Alva et al., 2016). The same steps outlined in the Homology modeling of GPCRs section were used to analyze the cavity geometry recovery and to compare against the performance of SPaDES.
Weight optimization
To ensure that the new energy terms describing water-mediated interactions were compatible with the other terms describing protein energies, we optimized the weights describing their contribution to the hybrid scoring function. Since the hybrid energy function was designed to model proteins using both implicit and explicit solvent models, the weights describing all terms from the previously optimized implicit energy function RosettaMembrane were kept unchanged. The weights for the following five terms were optimized: hydrogen bond between protein backbone and side-chain atoms, hydrogen bond between protein side-chain atoms, hydrogen bond between water and protein or water atoms, entropy loss of water upon interaction with protein atoms, desolvation cost of water upon interaction with protein atoms. The weight for the hydrogen bond between backbone atoms was kept fixed to that of the regular Rosetta scoring function because it is mostly designed to guarantee the formation and stability of protein secondary structures. These weights were optimized against the specific high-resolution training dataset described in detail in the Water-soluble protein benchmark construction section, which consisted of experimentally measured mutational effects on protein-protein binding energies (ΔΔGbinding) and water positions in multiple high-resolution protein structures.
Objective function definition
To optimize the weights, we considered an objective function that quantitatively describes the performance of the energy function through various component tests. The objective function used to indicate performance of the weights is a combination of different components describing the performances on the binding energy dataset and the native water recovery. The resulting function takes the form,
where the component weighing factor w scales the component C for the binding energy correlation coefficient R (ΔΔG), the native water coverage rate (cov), and the native water true positive rate (TP). The native water coverage rate is defined as the fraction of native water molecules defined by their X-ray structure that were accounted for by a predicted de novo water molecule within a distance of 1.0 Å; the native water true positive rate is defined as the fraction of predicted de novo water molecules that were within a distance threshold of 1.0 Å of a native water position. The weighing factor for the binding energy component was set to 0.5, while the weighing factor for the native water coverage and native water true positive components were both set to 0.25. The specific components were evaluated as described in previous sections.
Optimization routines
We implemented two optimization routines to find the set of weights that maximizes the defined objective function. Five adjustable weight parameters were considered while all other terms were fixed to their default weight values from the default Rosetta scoring function. The first optimization routine considered was the OptE protocol (Leaver-Fay et al., 2013), which is based on a particle-swarm optimization (PSO) method. Briefly, the protocol considers two steps in optimizing the weights. First, preliminary optima are rapidly determined through the use of the PSO method on fixed input structures. In our case, these structures consisted of our protein-protein complex benchmark whose structures were generated using weights from previous optimization cycles. In this step, the weight parameterization was based on the mutagenesis binding ΔΔG correlation using 1000 swarm particles and 100 swarm cycles. Next, the results from the PSO step are mixed at varying levels with the starting weights of the cycle, and used to repack and minimize the benchmark input structures, which are subsequently used to further optimize the weights based on various metrics; in our case, these criteria consisted of the native water coverage and native water true positive rates previously described. The resulting weights are collected and used in the following cycles (i.e. as starting weights for the next PSO run) where the entire routine repeats until convergence is observed. Specific details on this protocol has been published elsewhere (Leaver-Fay et al., 2013).
As a cross-validation of the OptE protocol, we also performed the weight fitting using a different optimization algorithm. For this alternative approach, we considered the simplex (a.k.a. Nelder-Mead or convex) method (Nelder and Mead, 1965), which is a numerical heuristic search algorithm used to find the maximum of an objective function in multi-parameter space. Due to the computational cost of predicting hydrated structures at every evaluation of the objective function, we decided to repack the protein structures at only every six cycles of weight optimizations. This ensured that the optimization process was more efficient while guaranteeing optimal convergence of the weights. To further increase the efficiency of the protocol, we also reduced the number of parallel repacking simulations for each structure to 10. This number was sufficient to gain a stable correlation coefficient (i.e. standard deviation of approximately 0.01 for the R coefficients when running multiple independent repacking simulations) in the binding energy predictions. The lowest energy conformations were then saved and used for six cycles of weight optimization where these structures were just rescored with the new weights without repacking or rehydrating. After six iterations of weight optimization, the weights that gave the best evaluation of the objective function were used to predict the next set of hydrated protein structures.
Due to the stochastic nature of the components used in the objective function of these optimization routines, multiple optimization runs from different initial starting points had to be repeated to confirm the convergence and robustness of the solution. In addition, the weight optimization was performed on the smaller subset benchmark containing high-resolution experimentally-determined water positions, and the resulting weights were validated with the larger complete benchmark, all of which has been described in previous sections.
Optimized scoring function weights
The final optimized scoring function weights for the main energy terms affected by the explicit modeling of water molecules in SPaDES are defined as follows. hbond_bb_sc: hydrogen bonding interaction between backbone and side-chain residue atoms (weight = 2.427). hbond_sc: hydrogen bonding interaction between residue side-chain atoms (weight = 1.251). hbond_wat*: hydrogen bonding interaction involving at least a water atom (i.e. water-residue or water-water) (weight = 1.027). wat_entropy*: entropy cost of moving a water molecule away from the bulk solvent (weight = 0.529). wat_desolv*: enthalpic desolvation cost of moving a water molecule away from the bulk solvent (weight = 0.436). Astericks (*) correspond to water-specific terms added to the regular Rosetta energy function.
SPaDES efficiency
The efficiency of SPaDES was compared against standard all-atom molecular dynamics simulations in explicit lipid membrane on a typical GPCR structure relaxation benchmark. David Shaw and colleagues showed that molecular dynamics simulation trajectories of at least one-microsecond (Dror et al., PNAS 2011) are necessary to reach roughly similar level of structural relaxation and conformational sampling than that achieved by SPaDES (i.e. relaxing an active like GPCR conformation to an inactive like conformation). A typical GPCR structure relaxation trajectory using SPaDES in hybrid implicit lipid membrane with explicit buried solvent molecules requires around 4 CPU hours. This runtime is approximately three orders of magnitude more efficient than a one-microsecond classical molecular dynamics simulation with explicit water and lipid molecules on specialized hardware from the D.E. Shaw Research group (e.g. CPU hours reported by D.E. Shaw for the hardware Anton 2). After accounting for the speed improvements of the specialized optimized hardware, similar simulations on general-purpose hardware are estimated to require 2×105 CPU hours, which is almost 5-orders of magnitude less efficient than the SPaDES approach.
Due to the increased sampling complexity associated with the explicit modeling of water molecules as compared to the default implicit solvent model of the Rosetta software, SPaDES does exhibit longer runtimes. In typical packing simulations performed for predicting the effects of mutations at protein binding interfaces (6 Å sphere of hydration and an 8 Å sphere of side-chain repacking), SPaDES requires only 1-order of magnitude more CPU hours than Rosetta (using the same 8 Å sphere of side-chain repacking). Similar difference in efficiency is observed for the membrane protein structure relaxation application of SPaDES.
DATA AND SOFTWARE AVAILABILITY
The SPaDES method and source code described in this work will be made available free to academic users as part of the Rosetta Software Suite (https://www.rosettacommons.org/).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and Algorithms | ||
| Rosetta | Das and Baker, 2008 | http://rosettacommons.org/software |
| Fold-X | Schymkowitz et al., 2005 | http://foldxsuite.crg.eu/ |
| HADDOCK | de Vries et al., 2007; Dominguez et al., 2003 | http://www.bonvinlab.org/software/haddock2.2/ |
| Modeller | Eswar et al., 2008 | https://salilab.org/modeller/ |
Highlights.
Improved atomic-level protein structure modeling with solvent-protein interactions
Quantitatively predicted challenging mutational effects on protein-protein binding
Predicted solvated cavity structures of membrane receptors from distant homologs
Blindly predicted buried solvent networks in GPCR classes A, B, C, and F
Acknowledgments
We thank the members of the Barth lab for insightful discussions during this study and critical comments on the manuscript. This work was supported by a grant from the National Institute of Health (1R01GM097207) and by a supercomputer allocation from XSEDE (MCB120101) to P.B.
Footnotes
Author contributions:
P.B. designed the study; J.L., J.A., Y.W. and P.B. developed the method; J.L., J.A. optimized the parameters and performed the blind and benchmark structure predictions; J.L., J.A. and P.B. analyzed and discussed the results; J.L., P.B. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahmad M, Gu W, Geyer T, Helms V. Adhesive water networks facilitate binding of protein interfaces. Nature Communications. 2011;2:261. doi: 10.1038/ncomms1258. [DOI] [PubMed] [Google Scholar]
- Alva V, Nam SZ, Soding J, Lupas AN. The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic acids research. 2016;44:W410–415. doi: 10.1093/nar/gkw348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amann-Winkel K, Bellissent-Funel MC, Bove LE, Loerting T, Nilsson A, Paciaroni A, Schlesinger D, Skinner L. X-ray and Neutron Scattering of Water. Chemical reviews. 2016;116:7570–7589. doi: 10.1021/acs.chemrev.5b00663. [DOI] [PubMed] [Google Scholar]
- Angel TE, Chance MR, Palczewski K. Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc Natl Acad Sci USA. 2009;106:8555–8560. doi: 10.1073/pnas.0903545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Schonbrun J, Baker D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104:15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Wallner B, Baker D. Prediction of membrane protein structures with complex topologies using limited constraints. Proceedings of the National Academy of Sciences. 2009;106:1409–1414. doi: 10.1073/pnas.0808323106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyken SE, Chen Z, Groves B, Langan RA, Oberdorfer G, Ford A, Gilmore JM, Xu C, DiMaio F, Pereira JH, et al. De novo design of protein homo-oligomers with modular hydrogen-bond network-mediated specificity. Science. 2016;352:680–687. doi: 10.1126/science.aad8865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiten B, Lockett MR, Sherman W, Fujita S, Al-Sayah M, Lange H, Bowers CM, Heroux A, Krilov G, Whitesides GM. Water Networks Contribute to Enthalpy/Entropy Compensation in Protein–Ligand Binding. Journal of the American Chemical Society. 2013;135:15579–15584. doi: 10.1021/ja4075776. [DOI] [PubMed] [Google Scholar]
- Carugo O. Statistical survey of the buried waters in the Protein Data Bank. Amino Acids. 2016;48:193–202. doi: 10.1007/s00726-015-2064-4. [DOI] [PubMed] [Google Scholar]
- Chen KYM, Sun J, Salvo JS, Baker D, Barth P. High-Resolution Modeling of Transmembrane Helical Protein Structures from Distant Homologues. PLoS Computational Biology. 2014a;10:e1003636. doi: 10.1371/journal.pcbi.1003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen KY, Sun J, Salvo JS, Baker D, Barth P. High-resolution modeling of transmembrane helical protein structures from distant homologues. PLoS Comput Biol. 2014b;10:e1003636. doi: 10.1371/journal.pcbi.1003636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Okur HI, Gomopoulos N, Macias-Romero C, Cremer PS, Petersen PB, Tocci G, Wilkins DM, Liang C, Ceriotti M, et al. Electrolytes induce long-range orientational order and free energy changes in the H-bond network of bulk water. Science advances. 2016;2:e1501891. doi: 10.1126/sciadv.1501891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Baker D. Macromolecular modeling with rosetta. Annual review of biochemistry. 2008;77:363–382. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- Dragan AI, Read CM, Crane-Robinson C. Enthalpy–entropy compensation: the role of solvation. European Biophysics Journal. 2016 doi: 10.1007/s00249-016-1182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Arlow DH, Maragakis P, Mildorf TJ, Pan AC, Xu H, Borhani DW, Shaw DE. Proceedings of the National Academy of Sciences. 2011 Nov 15;108(46):18684–9. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunitz JD. The Entropic Cost of Bound Water in Crystals and Biomolecules. Science. 1994;264:670–670. doi: 10.1126/science.264.5159.670. [DOI] [PubMed] [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garczarek F, Gerwert K. Functional waters in intraprotein proton transfer monitored by FTIR difference spectroscopy. Nature. 2006;439:109–112. doi: 10.1038/nature04231. [DOI] [PubMed] [Google Scholar]
- Gupta S, D’Mello R, Chance MR. Structure and dynamics of protein waters revealed by radiolysis and mass spectrometry. Proceedings of the National Academy of Sciences. 2012;109:14882–14887. doi: 10.1073/pnas.1209060109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez-de-Terán H, Massink A, Rodríguez D, Liu W, Han Gye W, Joseph Jeremiah S, Katritch I, Heitman Laura H, Xia L, Ijzerman Adriaan P, et al. The Role of a Sodium Ion Binding Site in the Allosteric Modulation of the A2A Adenosine G Protein-Coupled Receptor. Structure. 2013;21:2175–2185. doi: 10.1016/j.str.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, de Graaf C, Bortolato A, Wang MW, Marshall FH, Stevens RC. Insights into the structure of class B GPCRs. Trends in pharmacological sciences. 2014;35:12–22. doi: 10.1016/j.tips.2013.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huggins David J. Quantifying the Entropy of Binding for Water Molecules in Protein Cavities by Computing Correlations. Biophysical Journal. 2015;108:928–936. doi: 10.1016/j.bpj.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri A, Dore AS, Lamb D, Krishnamurthy H, Southall SM, Baig AH, Bortolato A, Koglin M, Robertson NJ, Errey JC, et al. Extra-helical binding site of a glucagon receptor antagonist. Nature. 2016;533:274–277. doi: 10.1038/nature17414. [DOI] [PubMed] [Google Scholar]
- Jiang L, Kuhlman B, Kortemme T, Baker D. A “solvated rotamer” approach to modeling water-mediated hydrogen bonds at protein-protein interfaces. Proteins. 2005;58:893–904. doi: 10.1002/prot.20347. [DOI] [PubMed] [Google Scholar]
- Joosten RP, te Beek TAH, Krieger E, Hekkelman ML, Hooft RWW, Schneider R, Sander C, Vriend G. A series of PDB related databases for everyday needs. Nucleic Acids Research. 2011;39:D411–D419. doi: 10.1093/nar/gkq1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nature structural biology. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Kleinjung J, Fraternali F. Design and application of implicit solvent models in biomolecular simulations. Current Opinion in Structural Biology. 2014;25:126–134. doi: 10.1016/j.sbi.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T, Karplus M. Effective energy function for proteins in solution. Proteins. 1999;35:133–152. doi: 10.1002/(sici)1097-0134(19990501)35:2<133::aid-prot1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Leaver-Fay A, O’Meara MJ, Tyka M, Jacak R, Song Y, Kellogg EH, Thompson J, Davis IW, Pache RA, Lyskov S, et al. Scientific Benchmarks for Guiding Macromolecular Energy Function Improvement. Methods in enzymology (Elsevier) 2013:109–143. doi: 10.1016/B978-0-12-394292-0.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman KW, Renfrew PD, Smith CA, Sheffler W, et al. Rosetta3. Methods in enzymology (Elsevier) 2011:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon G, Meiler J. Towards Ligand Docking Including Explicit Interface Water Molecules. PLoS ONE. 2013;8:e67536. doi: 10.1371/journal.pone.0067536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Moal IH, Bates PA, Kastritis PL, Melquiond ASJ, Karaca E, Schmitz C, van Dijk M, Bonvin AMJJ, Eisenstein M, et al. Blind prediction of interfacial water positions in CAPRI: Blind Prediction of Interfacial Water Positions. Proteins: Structure, Function, and Bioinformatics. 2014;82:620–632. doi: 10.1002/prot.24439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy ED. A Simple Definition of Structural Regions in Proteins and Its Use in Analyzing Interface Evolution. Journal of Molecular Biology. 2010;403:660–670. doi: 10.1016/j.jmb.2010.09.028. [DOI] [PubMed] [Google Scholar]
- Levy Y, Onuchic JN. Water mediation in protein folding and molecular recognition. Annual Review of Biophysics and Biomolecular Structure. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- Li S, Bradley P. Probing the role of interfacial waters in protein-DNA recognition using a hybrid implicit/explicit solvation model: Interfacial Waters in Protein-DNA Recognition. Proteins: Structure, Function, and Bioinformatics. 2013;81:1318–1329. doi: 10.1002/prot.24272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, Han GW, Roth CB, Heitman LH, Ijzerman AP, et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney MW, Jorgensen WL. A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. The Journal of Chemical Physics. 2000;112:8910. [Google Scholar]
- Mason JS, Bortolato A, Congreve M, Marshall FH. New insights from structural biology into the druggability of G protein-coupled receptors. Trends in pharmacological sciences. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- Miao Y, McCammon JA. G-protein coupled receptors: advances in simulation and drug discovery. Curr Opin Struct Biol. 2016;41:83–89. doi: 10.1016/j.sbi.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moal IH, Fernandez-Recio J. SKEMPI: a Structural Kinetic and Energetic database of Mutant Protein Interactions and its use in empirical models. Bioinformatics. 2012;28:2600–2607. doi: 10.1093/bioinformatics/bts489. [DOI] [PubMed] [Google Scholar]
- Mobley DL, Dill KA. Binding of small-molecule ligands to proteins: “what you see” is not always “what you get”. Structure. 2009;17:489–498. doi: 10.1016/j.str.2009.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelder JA, Mead R. A Simplex Method for Function Minimization. The Computer Journal. 1965;7:308–313. [Google Scholar]
- Ngo T, Ilatovskiy AV, Stewart AG, Coleman JL, McRobb FM, Riek RP, Graham RM, Abagyan R, Kufareva I, Smith NJ. Orphan receptor ligand discovery by pickpocketing pharmacological neighbors. Nature chemical biology. 2017;13:235–242. doi: 10.1038/nchembio.2266. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nygaard R, Valentin-Hansen L, Mokrosinski J, Frimurer TM, Schwartz TW. Conserved Water-mediated Hydrogen Bond Network between TM-I, -II, -VI, and -VII in 7TM Receptor Activation. Journal of Biological Chemistry. 2010;285:19625–19636. doi: 10.1074/jbc.M110.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orban T, Gupta S, Palczewski K, Chance MR. Visualizing Water Molecules in Transmembrane Proteins Using Radiolytic Labeling Methods. Biochemistry. 2010;49:827–834. doi: 10.1021/bi901889t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papoian GA, Ulander J, Eastwood MP, Luthey-Schulten Z, Wolynes PG. From The Cover: Water in protein structure prediction. Proceedings of the National Academy of Sciences. 2004;101:3352–3357. doi: 10.1073/pnas.0307851100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson E, Halle B. Nanosecond to Microsecond Protein Dynamics Probed by Magnetic Relaxation Dispersion of Buried Water Molecules. Journal of the American Chemical Society. 2008;130:1774–1787. doi: 10.1021/ja0775873. [DOI] [PubMed] [Google Scholar]
- Persson F, Halle B. Transient Access to the Protein Interior: Simulation versus NMR. Journal of the American Chemical Society. 2013;135:8735–8748. doi: 10.1021/ja403405d. [DOI] [PubMed] [Google Scholar]
- Rath P, Delange F, Degrip WJ, Rothschild KJ. Hydrogen bonding changes of internal water molecules in rhodopsin during metarhodopsin I and metarhodopsin II formation. Biochem J. 1998;329(Pt 3):713–717. doi: 10.1042/bj3290713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembert KB, Paterova J, Heyda J, Hilty C, Jungwirth P, Cremer PS. Molecular mechanisms of ion-specific effects on proteins. J Am Chem Soc. 2012;134:10039–10046. doi: 10.1021/ja301297g. [DOI] [PubMed] [Google Scholar]
- Richardson JS, Prisant MG, Richardson DC. Crystallographic model validation: from diagnosis to healing. Current Opinion in Structural Biology. 2013;23:707–714. doi: 10.1016/j.sbi.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas I, Alkorta I, Elguero J. Bifurcated Hydrogen Bonds: Three-Centered Interactions. The Journal of Physical Chemistry A. 1998;102:9925–9932. [Google Scholar]
- Schymkowitz JWH, Rousseau F, Martins IC, Ferkinghoff-Borg J, Stricher F, Serrano L. Prediction of water and metal binding sites and their affinities by using the Fold-X force field. Proceedings of the National Academy of Sciences. 2005;102:10147–10152. doi: 10.1073/pnas.0501980102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapovalov Maxim V, Dunbrack Roland L. A Smoothed Backbone-Dependent Rotamer Library for Proteins Derived from Adaptive Kernel Density Estimates and Regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research. 2005;33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- Valentin-Hansen L, Frimurer TM, Mokrosinski J, Holliday ND, Schwartz TW. Biased Gs Versus Gq Proteins and β-Arrestin Signaling in the NK1 Receptor Determined by Interactions in the Water Hydrogen Bond Network. Journal of Biological Chemistry. 2015;290:24495–24508. doi: 10.1074/jbc.M115.641944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Bradley P, Baker D. Protein–Protein Docking with Backbone Flexibility. Journal of Molecular Biology. 2007;373:503–519. doi: 10.1016/j.jmb.2007.07.050. [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15. doi: 10.1007/978-1-4939-0366-5_1. [DOI] [PubMed] [Google Scholar]
- Wlodawer A, Minor W, Dauter Z, Jaskolski M. Protein crystallography for non-crystallographers, or how to get the best (but not more) from published macromolecular structures: Protein crystallography for non-crystallographers. FEBS Journal. 2008;275:1–21. doi: 10.1111/j.1742-4658.2007.06178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy V, Schonbrun J, Baker D. Multipass membrane protein structure prediction using Rosetta. Proteins. 2006;62:1010–1025. doi: 10.1002/prot.20817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Rick SW. Free Energy, Entropy, and Enthalpy of a Water Molecule in Various Protein Environments. The Journal of Physical Chemistry B. 2010;114:11552–11560. doi: 10.1021/jp104209w. [DOI] [PubMed] [Google Scholar]
- Yuan S, Filipek S, Palczewski K, Vogel H. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat Commun. 2014;5:4733. doi: 10.1038/ncomms5733. [DOI] [PubMed] [Google Scholar]
- Yuan S, Hu Z, Filipek S, Vogel H. W246(6.48) opens a gate for a continuous intrinsic water pathway during activation of the adenosine A2A receptor. Angew Chem Int Ed Engl. 2015;54:556–559. doi: 10.1002/anie.201409679. [DOI] [PubMed] [Google Scholar]
- Zemla A. LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Research. 2003;31:3370–3374. doi: 10.1093/nar/gkg571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Qin L, Zacarias NV, de Vries H, Han GW, Gustavsson M, Dabros M, Zhao C, Cherney RJ, Carter P, et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature. 2016;540:458–461. doi: 10.1038/nature20605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.