

Abstract
Laminin polymerization is a key step of basement membrane self-assembly that depends on the binding of the three different N-terminal globular LN domains. Several mutations in the LN domains cause LAMA2-deficient muscular dystrophy and LAMB2-deficient Pierson syndrome. These mutations may affect polymerization. A novel approach to identify the amino acid residues required for polymerization has been applied to an analysis of these and other laminin LN mutations. The approach utilizes laminin-nidogen chimeric fusion proteins that bind to recombinant non-polymerizing laminins to provide a missing functional LN domain. Single amino acid substitutions introduced into these chimeras were tested to determine if polymerization activity and the ability to assemble on cell surfaces were lost. Several laminin-deficient muscular dystrophy mutations, renal Pierson syndrome mutations, and Drosophila mutations causing defects of heart development were identified as ones causing loss of laminin polymerization. In addition, two novel residues required for polymerization were identified in the laminin γ1 LN domain.
Keywords: LN domains, Chimeric proteins, Basement membrane, LAMA2-MD, Pierson syndrome, Drosophila
Introduction
Members of the laminin family [1] are essential for basement membrane (BM) assembly [2–4], a likely consequence of the unique ability of laminins to bind to cells, to self, and to other BM components. A general model of BM assembly is as follows: Laminins adhere to cell surfaces and become anchored to the cytoskeleton through cognate integrin and dystroglycan transmembrane receptors [5–12]. Laminin attachment to a cell, by locally increasing laminin above its critical concentration of polymerization, facilitates self-polymerization to create a solid-phase polymer [13]. The laminin matrix binds to nidogens and the heparan sulfate proteoglycan (HSPG) agrin, while nidogens bind to collagen-IV [14,15] and the HSPG perlecan [16,17]. Coincidently, collagen-IV polymerizes to form a second network to stabilize the nascent laminin-based BM [7,18–23]. Non-neural agrin forms collateral linkages with α-dystroglycan (αDG) and integrins, acting to increase cytoskeletal anchorage and along with perlecan present heparan sulfates for growth factor and morphogen binding/activation [16,17,24–30].
It is thought that laminin polymerization is important not only because of its role in BM assembly, but because mutations in the LN polymerization domain of several laminins are causative of muscle, nerve, and kidney diseases [13]. Earlier studies revealed that that only laminins with three short arms self-polymerize [31], that assembly of the ternary complex of α, β, and γ N-terminal LN domains creates the polymer node [32–34], and that calcium bound to the γ1LN domain is required for the trimerization step of laminin self-assembly [32,35]. Assembly of the α-β-γ-LN node was later confirmed by Hohenester [33,34,36] in study employing N-terminal fragments that consisted of the LN and adjacent LEa domains. Recently, Hohenester crystallized and determined the structures of the Lmα5 (α1 homologue), Lmβ1 and Lmγ1 LN and adjacent LEa domains. He also identified a putative α5LN polymerization patch in the region of the sequence PLENGE located on the conserved β6-β3-β8 LN face that we will refer to as the “front” face [35,36]. The “rear” face, in contrast, is less conserved and contains N-linked carbohydrate. Mutation of the PLENGE glutamates to lysines (E231K, E234K) in Lmα5LN fragments was found to inhibit laminin polymerization [35,36].
LN domain mutations have been found to be causative of laminin α2-deficient muscular dystrophy (LAMA2-MD), β2 subunit-dependent renal/ocular Pierson syndrome, and β1 subunit-dependent fly heart-associated defects [37–42]. LAMA2-MD mutations in the α2 LN domain include Q167P, Y138H, G284R on the α2 LN surface and C86Y, W152G, S157F, S277L, S204F, L243P in the LN interior [43]. Pierson syndrome mutations in the β2 LN domain include S80R, L139P, H157R, D167Y, S179F and R246 Q/W [41,44]. Drosophila cardiac alary muscle attachment defects result from β1 E215K, V226E, and G286R, all mapping to the front face of the LN domain [40]. We asked, “Do these mutations cause a failure of polymerization?” To begin to approach this question, and a more general goal of determining the residues that mediate polymerization, we analyzed laminin complexes that contained disease-associated and other mutations to determine if polymerization was impeded by in vitro means. While this goal could have been accomplished by introducing the mutations into full laminin heterotrimers, the approach was deemed cumbersome because of the time and cost needed to modify the large corresponding cDNAs. Instead, we choose an alternative means by binding a synthetic short arm containing an LN mutation of interest to a non-polymerizing laminins lacking the LN domain. The added arm had the potential to enable polymerization if it contained the active residues. The analysis was two-fold: a direct polymerization determination accompanied by an evaluation of the laminin complex to form an extracellular matrix on cell surfaces.
The short arm proteins consist of a laminin α1, β1 and γ1 LN-LEa domain segment fused to the nidogen-1C-terminal G2-G3 moiety (Fig. 1A). Collectively we refer to these proteins as “xLNNds” where x refers to the α1, β1 or γ1subunit origin of the LN-LEa domains. The shared G3 domain allows the xLNNd to bind with high affinity (~1 nM Kd) to the laminin γ1 short arm at domain α1LEb3, near the intersection of the laminin cross [45,46]. The G2 domain allows collagen-IV and perlecan to bind to the laminin complex as normally occurs with nidogen [34,47,48]. The LN domain is required for laminin polymerization with the adjacent LE domains needed for LN folding. The LE and EGF-like rod-forming domains act to separate laminins from each other within the polymer and to separate laminin-binding from collagen- and perlecan-binding. While relevant for complete BM assembly, the G2 domain is not needed for assessment of the ability of an xLNNd to restore polymerization activity or assemble a laminin-based ECM on a cell surface.
Fig. 1.
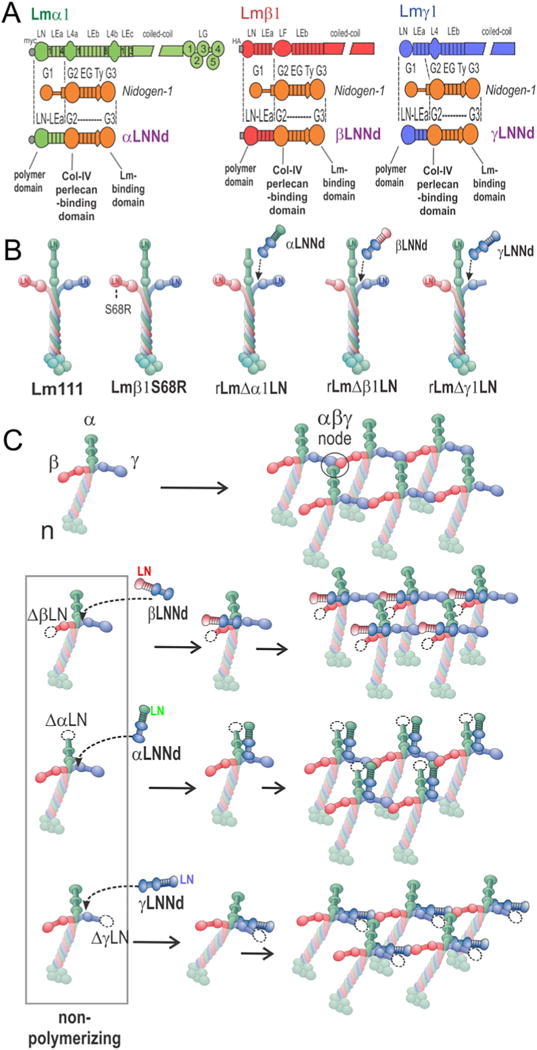
xLNNd and laminin proteins. A. Construct design for chimeric hybrids consisting of β1, α1 and γ1 laminin LN-LEa domains fused to the nidogen-1 G2 through G3 domains. B. Recombinant α1β1γ1 laminins, wild-type (WT) and bearing an S68R point mutation or with α, β or γ1 mutations used in the study. C. Laminin polymerizes through the binding of α, β and γ LN domains, forming a ternary node complex. Laminins lacking any of the three LN domains are unable to polymerize. Polymerization can be restored to these proteins by binding to corresponding α, β, and γLNNd proteins that provide the missing domain in an artificial short arm.
The xLNNd proteins provide a test cassette for functional restoration. The linker proteins are bound to a recombinant laminin-111 (Lm111) that lacks the corresponding subunit class LN domain (e.g. laminin lacking an α-LN for coupling to αLNNd). WT xLNNd was expected to restore polymerization to the appropriate laminin in contrast to inactivating mutations in xLNNd. These differences should be manifest in polymerization and cell surface assembly assays. The cells increase the effective surface laminin concentration and hence assembly, allowing one to study BM assembly at concentrations not achievable in solution and functionally closer to that occurring in vivo [33]. Rat high-passage Schwann cells (SCs) were employed to evaluate cell surface assembly because (a) they present large exposed surfaces for laminin accumulation and (b) they are very sensitive to laminin polymerization [33,34]. The assembly of Lm111 β1S68R and Lm121-β2S83R was initially tested to confirm loss of polymerization shown earlier with laminin fragments [49] and to compare and validate the approach of using mutated chimeric linker proteins. Given the highly conserved LN sequences among subunits, it was found that mutations discovered in a subunit isoform can be introduced into the LN domains of Lmα1, β1 and γ1 LN domains [36]. Thus β2 mutations can be introduced into β1, and α2 and α5 mutations can be placed in α1 for analysis.
We found that the xLNNd polymerization analysis, combined with the available crystal structures for the LN-LEa domains, can also be used to identify novel residues that participate in the formation of the ternary node. Mapping of surface-exposed residues for this purpose is a logical extension of the evaluation of LN disease mutations for the long-term goal of solving the polymer node molecular organization and interactions. An initial evaluation of disease-causing, developmentally important, and novel LN residues is reported in this study.
Results
Laminin-nidogen chimeric proteins and non-polymerizing laminins
The αLNNd (α1), βLNNd (β1) and γLNNd (γ1) proteins, without and with single amino acid LN substitutions, were prepared as described in the methods section (Fig. 1A). Five laminin heterotrimers were used in the study (Fig. 1B). These were wild-type (WT) Lm111, Lm111 bearing an S68R substitution in the β1 LN domain, and non-polymerizing laminins with a deletion of either an α1LN, β1LN or γ1LN domain [33,49]. The expectation was that the xLNNd proteins, if they contained functionally active LN domains and bound to their corresponding LN-deleted laminins, would polymerize and assemble matrices on cell surfaces (Fig. 1C).
β2S80R LN Pierson syndrome mutation causes loss of polymerization and basement membrane assembly: assessment with intact Lm111
The human mutation of Lmβ2 S80R causes a variant form of Pierson syndrome characterized by childhood-onset severe myopia, proteinuria, and renal pathology showing mild diffuse mesangial sclerosis [50]. Previous analysis of a laminin fragment containing the homologous S68R mutation in Lmβ1 prevented protein heterotrimerization [51] and laminin containing the mutation adversely affected cell surface assembly [49]. To formally determine if this mutation prevents polymerization we evaluated assembly with a standard polymer sedimentation assay [31,33] and compared it to that of cell assembly (Fig. 2). Control Lm111 polymerized in the expected fashion (Fig. 2A, C) with increasing polymer detected at increasing laminin concentrations. In contrast, Lm111 bearing the β1S68R mutation (Lm111β1S68R) did not polymerize (Fig. 2B, C).
Fig. 2.
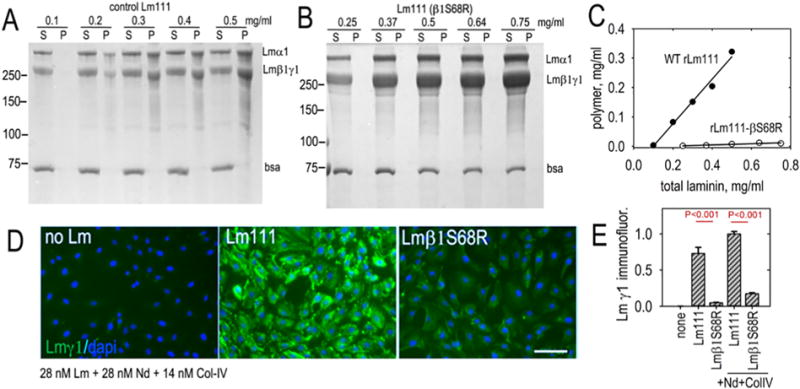
Laminin containing an S68R amino acid substitution in the β-LN domain does not polymerize and does not assemble a basement membrane. A-C. β2-S80 is a conserved residue and patients with a laminin β2-S80R mutation develop Pierson syndrome. The ability of laminin-111 bearing the corresponding mutation in β1LN to polymerize was evaluated in a polymerization sedimentation assay. Effectively, no polymer was detected in the indicated concentration range. D–E. Laminin assembly was evaluated on Schwann cell surfaces at 28 nM laminin alone or in the presence of 28 nM nidogen-1 and 14 nM collagen. WT laminin accumulated substantially while Lmβ1 S68R did not.
These same proteins were compared for their ability to accumulate on cultured SC surfaces both without and with nidogen-1 and collagen-IV (Fig. 2D, E). Control Lm111 exhibited substantial accumulation while the laminin with S68R did not, even in the presence of nidogen-1 and collagen-IV. Subsequent assays were restricted to assembly with laminin and laminin complexes alone.
Analysis of laminin polymerization and cell surface assembly with βLNNd proteins
If laminin-nidogen chimeric proteins can be used to determine which mutations adversely affected polymerization, then wild-type βLNNd bound to its corresponding non-polymerizing laminin should polymerize while βLNNdS68R bound to the same non-polymerizing laminin should not. Similarly, cultured SCs used to model laminin assembly on cell surfaces should reflect the alternations seen in polymerization assays [33,34].
S68R and other βLN mutations were evaluated in βLNNd proteins by the polymerization assay that used to evaluate WT Lm111 and Lm111β1S68R (Fig. 3A and Supplemental Figs. 1–3). WT βLNNd coupled to Lmβ1ΔLN polymerized substantially, albeit with a lower slope than WT Lm111, while Lmβ1ΔLN alone did not polymerize. βLNNd containing the S68R mutation and coupled to Lmβ1ΔLN, similar to Lmβ1ΔLN alone, did not polymerize. We also evaluated β1S68A to assess whether the gained basic charge in S68R prevented polymerization, or whether loss of the serine was more likely responsible for the change in polymerization activity. S68A produced a loss of polymerization that, however, was not as great as that observed with S68R. The difference suggests that gain of a charge increases disruption beyond that due to loss of the serine residue. Two other causes of Pierson syndrome are the mutations β2H147R (corresponds to β1H135R) and β2R246Q (corresponds to β1 R234Q). These were also evaluated in the βLNNd chimeric protein coupled to non-polymerizing Lmβ1ΔLN. These complexes polymerized substantially below the WT βLNNd but still retained some activity as seen by the low concentration-dependent slope. A Drosophila βLN mutation, E215K (E198K in mouse Lmβ1), found to cause a heart developmental defect [40], introduced into βLNNd, also greatly reduced the polymerization slope. On the other hand, a less-conserved mutation located on the side of the LN domain (K184E) polymerized to a similar degree as WT βLNNd when coupled to Lmβ1ΔLN.
Fig. 3.
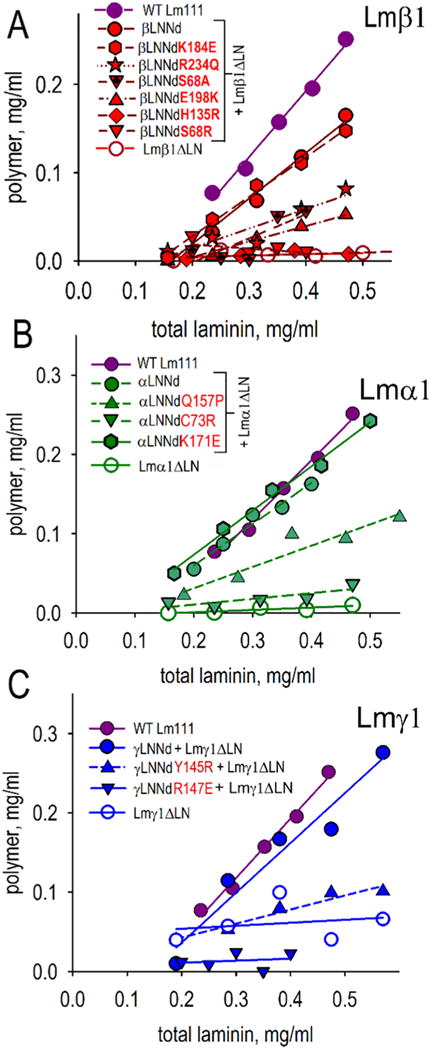
Ability of xLNNd linker proteins to enable laminin polymerization. The indicated β (A), α (B) and γ LNNd (C) proteins were bound to corresponding immobilized non-polymerizing laminins (Lmβ1ΔLN, Lmα1ΔLN, Lmγ1ΔLN) and eluted as an equimolar complex. Aliquots (50 μl) were incubated at 37 °C for 3 h, centrifuged to separate supernatant (S) from polymer (P), and analyzed by SDS-PAGE. Scans of the Coomassie blue stained gels were quantitated by densitometry and plotted as total concentration against polymer concentration. Non-polymerizing laminins coupled to the corresponding WT xLNNd proteins polymerized. LN mutations reduced or fully prevented polymerization.
The next experimental phase for xLNNd-laminin complexes focused on cell surface assembly. Given evidence that laminin polymerization requires three different LN domains, we first sought to assess the subunit LN specificity of xLNNd rescue of cell surface laminin assembly using laminins with α1, β1 and γ1 LN deletions (Fig. 4A–C). It was found that this prediction held for xLNNd-laminin complexes in that βLNNd only enabled Lmβ1ΔLN assembly, αLNNd only enabled Lmα1ΔLn assembly, and γLNNd only enabled Lmγ1ΔLN assembly.
Fig. 4.
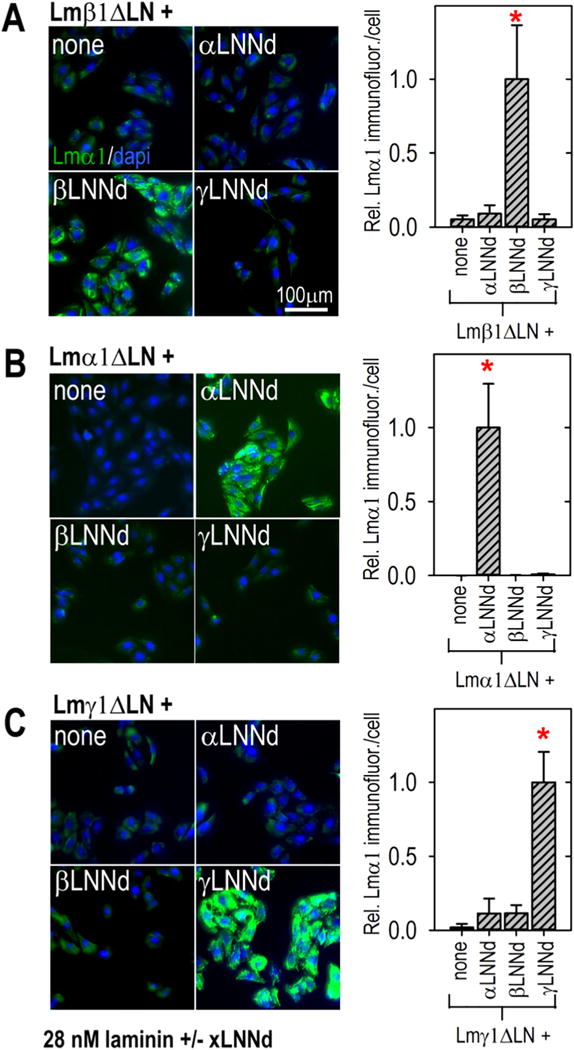
LN domain subunit specificity of assembly on cells. SCs were incubated with 28 nM Lmβ1ΔLN (A), Lmα1ΔLN (B) or Lmγ1ΔLN (C) alone or coupled to βLNNd, αLNNd or γLNNd. After 1 h the cells were washed, fixed and immunostained with Lmα1-specific antibody with a dapi counterstain. Immunofluorescence intensities/cell were determined and are shown in adjacent plots (average ± SD, n = 7–11 fields/condition). Bars marked with asterisks within each graph significantly differ (P < 0.0001) from unmarked bars. Laminin accumulation was dependent on pairing the βLNNd with Lm β1ΔLN, αLNNd with Lmα1ΔLN, and γLNNd with Lmγ1ΔLN, i.e. assembly of complexes required a full complement of α, β and γ LN domains.
The β-subunit mutations examined in polymerization assays were then evaluated for cell surface assembly in an equimolar βLNNd–Lmβ1ΔLN concentration series with cultured SCs (Fig. 5A,B). The highest level of laminin accumulation was seen with βLNNd-Lmβ1ΔLN followed closely by βLNNdK184E-Lmβ1ΔLN. The Pierson mutations β2S80R/β1S68R, β2H147R/β1H135R, and β2R246Q/β1R234Q in the protein complexes accumulated poorly over the same concentration range. Similarly the Drosophila mutation βE215K/β1E198K coupled with non-polymerizing laminin accumulated poorly. The results qualitatively reflected the polymerization loss and provided evidence that the disease and developmental phenotypes resulted from a failure of laminin polymerization. The βLNNd-Lmβ1ΔLN complex was also compared to WT Lm111 (Fig. 5C,D). While laminin accumulation was well above that of Lmβ1ΔLN alone, it was not as great as that seen with WT laminin.
Fig. 5.
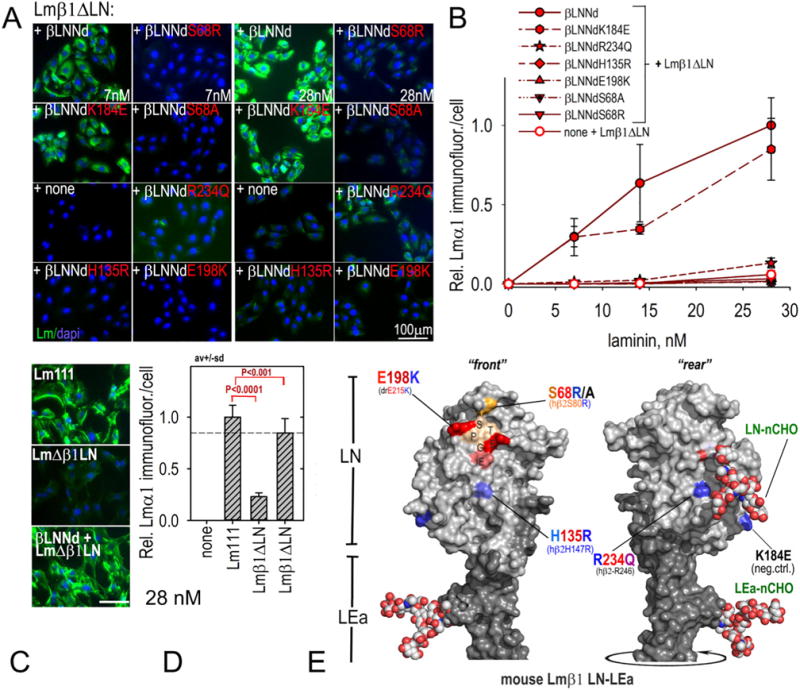
Assembly of βLNNd–Lmβ1ΔLN complexes on cell surfaces. Panels A–D. βLNNd proteins, WT and bearing LN point mutations and coupled to Lmβ1ΔLN were added to the medium of SCs for 1 h, washed, fixed and immunostained to detect cell-adherent laminin through detection of laminin α1. Representative images (panel A) and plots of average (±S.D., n = 6–7 fields/condition) intensity/cell as a function of laminin/βLNNd concentration (panel B) are shown. WT Lm111 accumulation was compared to that of the βLNNd-Lmβ1ΔLN complex at 28 nM laminin + βLNNd in panels C and D. Panel E. Surface contour of mouse laminin β1LN and adjacent LEa domain segment with attached N-linked carbohydrates (N-CHO) showing location of the mutations examined in βLNNd proteins (Pymol rendition of Protein Data Bank (PDB) 4AQS shown [35]). Polymerization-deficient mutations S68R/A, H135R, and E198K map to the LN tip and front face while R234Q maps to the LN rear face.
The human β2R246Q mutation, like that of human β2S80R, has been examined in mouse models of renal disease [49,52,53]. The residue is located on the rear side of the LN domain, unlike other residues involved in assembly (Fig. 5E). Secretion of R246Q, unlike S80R, was found to be reduced, suggesting a subunit folding problem. This was not observed in the linker protein βLNNdR234Q, although of note we were unable to isolate clones with high fully intact Lm111R234Q expression (data not shown). Collectively the evidence suggests that human β2R246Q suffers from both a secretion and polymerization deficiency. While it is conceivable that R234 directly participates in polymerization, the location of the residue on the rear surface near a glycan whose mass would likely sterically hinder access, and the reduction of secretion observed in vivo, collectively argue that R246Q prevents proper folding of the domain.
Analysis of laminin polymerization and cell surface assembly with αLNNd proteins
As reported previously [34], αLNNd selectively restores polymerization to laminins lacking the αLN domain. Three mutations were evaluated. One of these, α2Q167P/α1Q157P, is a cause of LAMA2-deficient ambulatory muscular dystrophy [38]. Another, α2C79R/α1C73R is a cause of a mouse LAMA2-deficient dystrophy that serves as a model for ambulatory dystrophy [39]. A third (α1K171E) was chosen as a residue unlikely to affect polymerization because of its lack of strict conservation and location on the side of the LN domain. The polymerization assays (Fig. 3B and Supplemental Figs. 2, 3) revealed that Q157P caused a ~60% drop in polymerization while C73R caused a more substantial loss of polymerization. Evaluation of cell surface assembly (Fig. 6A,B) revealed that Q157P and C73R caused a substantial but incomplete reduction in laminin accumulation while K171E caused no reduction. The αLNNd-Lmα1ΔLN complex was compared to WT Lm111 (Fig. 6C,D). While laminin accumulation was well above that of Lmα1ΔLN alone, it was not as great as seen with wild-type laminin.
Fig. 6.
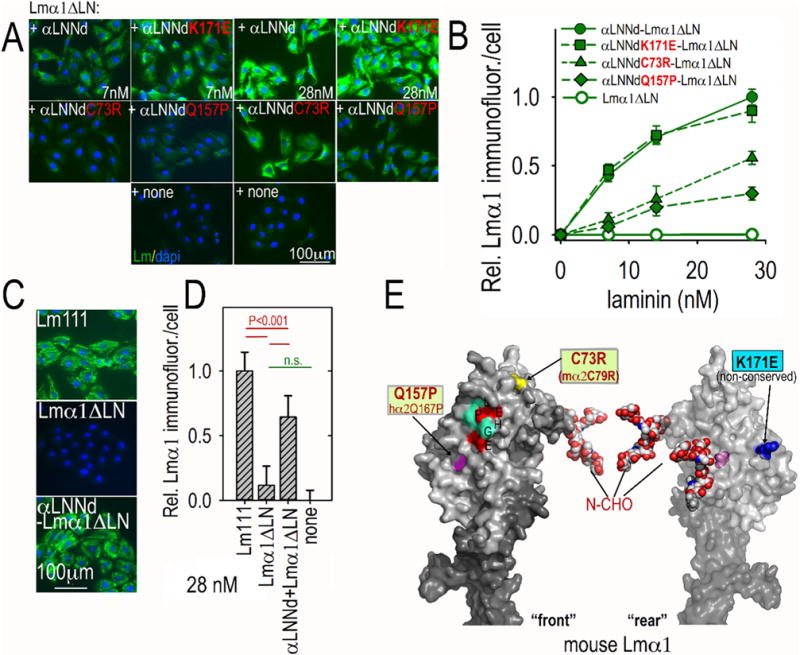
Assembly of αLNNd–Lmα1ΔLN complexes on cell surfaces. Panels A–D. αLNNd proteins, WT and bearing LN point mutations and coupled to Lmα1ΔLN, were added to the medium of SCs for 1 h, washed, fixed and immunostained to detect cell-adherent laminin through detection of α1 LG domains. For the concentration series, representative images (A, B) and plots of average (±S.D., n = 6–7 fields/condition) intensity/cell as a function of laminin/βLNNd concentration are shown. C73R and Q157P substantially but incompletely suppressed laminin assembly on SCs. Co-purified equimolar αLNNd-Lmα1ΔLN accumulated to ~65% of WT Lm111 at 28 nM (C,D). Panel E. Surface contour of mouse laminin α1LN and adjacent LEa domain segment with N-linked carbohydrates (N-CHO) attached to α1 LN domain. The C73 location may not be precise because of disorder in that region of the Lmα5 structure. Locations of the mutations examined are shown in a Pymol rendition of homology-fitted mouse Lmα1.
Q157 maps to the ‘front’ face of the LN domain near residues that correspond to the putative polymerization patch found in Lmα5 (Fig. 6E). The C73 residue is seen from the front view near the tip. As the C73R mutation causes a loss of one member of a cysteine pair, the effect may be to destabilize the domain fold rather than represent a direct loss of a node-binding residue.
Analysis of laminin polymerization and cell surface assembly with γLNNd proteins
Absence of the Lmγ1 is incompatible with mouse survival beyond the peri-implantation period and diseases resulting from mutations within the subunit may not exist [54]. To explore LN residues that might be required for polymerization, two conserved residues located in the central portion of the ‘front’ face were chosen for examination based on central front face location and conservation. One is γ1Y145R and the other is γ1R147E. Both mutations caused a substantial loss of polymerization (Fig. 3C and Supplemental Fig. 2). When examined in the SC assay, these same proteins prevented cell surface assembly (Fig. 7A,B). The γLNNd-Lmγ1ΔLN complex was compared to WT Lm111 (Fig. 7C,D). Unlike the other linker-laminin combinations, cell surface assembly of the γLNNd-Lmγ1ΔLN complex was higher than that observed with wild-type laminin. It is thought that this may be a consequence of the γ-linker protein extending out from the γ-short arm, rendering it more accessible to the other LN domains when on a cell surface.
Fig. 7.
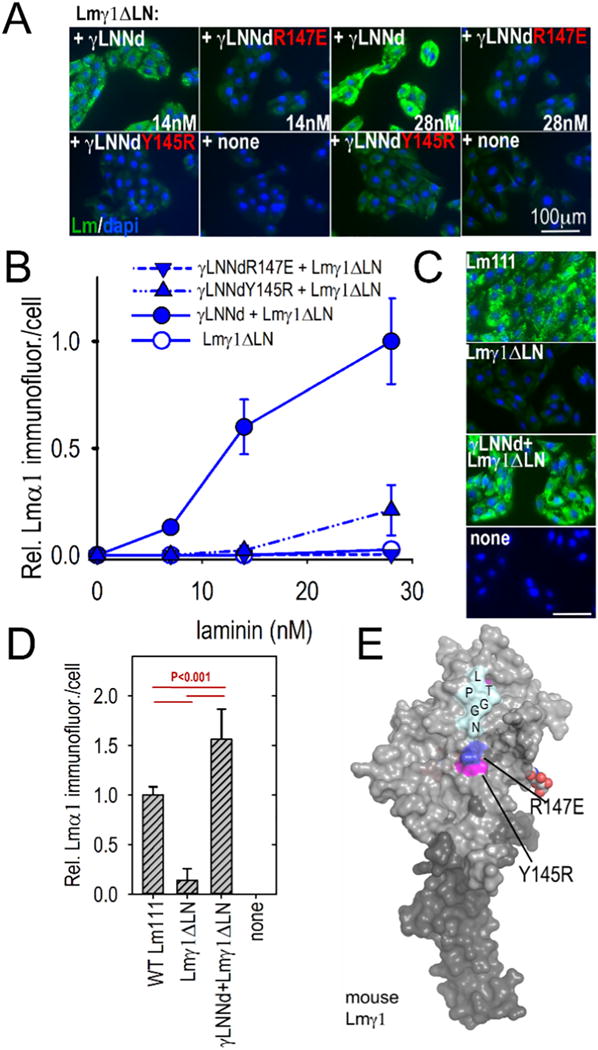
Assembly of γLNNd–Lmγ1ΔLN complexes on cell surfaces. Panels A–B. γLNNd proteins, WT and bearing LN point mutations and coupled to Lmγ1ΔLN, were added to the medium of SCs for 1 h, washed, fixed and immunostained to detect cell-adherent laminin through detection of α1 LG domains. For the concentration series, representative images and plots of average (±S.D., n = 6–7 fields) intensity/cell as a function of laminin/γLNNd concentration are shown. Laminin assembly on SCs was abolished by R147E and almost completely abolished by Y145R. Panels C–D. WT Lm111 accumulation was compared to that of the γLNNd-Lmγ1ΔLN complex at 28 nM laminin. Panel E. Surface contour of mouse laminin γ1LN and adjacent LEa domain segment with attached N-linked carbohydrates showing location of the mutations examined in γLNNd proteins (Pymol surface contour rendition of mouse Lmγ1, PDB # 4AQT, [36]).
The two mutations map to the front surface of the LN domain (Fig. 7E) and represent, as best as we can determine, the first mutations that substantially reduce polymerization in the Lmγ1 LN domain. Given the near-universality of the laminin γ1 subunit in tissues and its essential role in development, we suspect that these mutations would be lethal in vivo.
Discussion
Laminin polymerization was first reported in 1985 [55] while evidence to support assembly through ternary binding of three different LN domain was described several years later [32]. The first findings to suggest a biological role for polymerization came from an analysis of laminin extracted from the dy2J dystrophic mouse [42]. However, a generalized understanding of the functions of polymerization has been elusive. The more recent identification of LN domain mutations as a cause of human diseases affecting kidney and eye (Pierson syndrome) and muscle, peripheral nerve and brain (LAMA2-MD) implicated a widespread importance of laminin polymerization. That an LN mutation is also a cause of invertebrate developmental defects extends this even further. However, unresolved was whether these mutations actually affected polymerization and whether they were likely to diminish BM assembly.
The possibility of using laminin-binding proteins to provide an alternative arm for polymerization started with αLNNd. This protein could enable polymerization in a laminin that lacked an αLN domain, but not one that lacked a βLN or γLN domain. The question arose whether βLNNd and γLNNd proteins could enable polymerization in laminins that lacked the corresponding βLN and γLN domains. We report here that βLNNd and γLNNd have these properties. This is actually a remarkable finding. Assembly of the modified laminin likely depends upon the flexibility of the arms of laminin molecules [56] as it is difficult to envision such assembly being permissible with a rigid short arm structure given the shorter arm lengths and orientations of xLNNd proteins all extending from a single locus on the γ1LEb3 domain. Indeed, the finding that γLNNd bound to the nidogen binding locus in the γ1-short arm is more effective for assembly rescue compared to βLNNd reveals that that the nidogen-locus is not equally beneficial bound to different xLNNd proteins. This may reflect some degree of strain in the allowed spatial degrees of freedom of the different xLNNd synthetic arms due to interference arising from the adjacent native arms.
Having discovered that the three xLNNd proteins can enable polymerization of non-polymerizing laminins, the next question was whether they could be used as cassettes to introduce mutations for analysis by biochemical and cell culture means. This was also found to be the case. While the extent of polymerization did not quantitatively predict the degree of assembly on cell surfaces (possibly due to small changes in LN adhesion resulting from the mutations), a loss of polymerization correlated with a loss of cell surface assembly.
Thus the chimeric fusion analysis of laminin self-assembly provided an opportunity to determine if a given LN mutation results in a partial or complete loss of polymerization, affecting BM assembly. Using this approach, we were able to confirm that several Pierson and LAMA2 deficiency mutations reduced or ablated laminin polymerization. This in turn supported the conclusions that such mutations are likely to be causative of the diseases. Other LN mutations may similarly prove to be causative of diseases as well. Recently we reported that transgenic expression of αLNNd ameliorated the dy2J muscular dystrophy and that, in combination with a protein that enhanced receptor binding, ameliorated the more severe dyW dystrophy [57,58]. It may be possible to treat LAMA2 dystrophic patients with somatic gene delivery of αLNNd [43]. Such an approach would likely demand a prior determination of the likely efficacy of αLNNd treatment, i.e. whether the mutation would respond to the chimeric protein. This could be accomplished by evaluating the mutation in αLNNd as described herein. The same may prove to hold for Pierson syndrome in which βLNNd proteins could be used to restore polymerization to glomerular Lm521 bearing β2LN mutations.
Overall, conserved residues in the LN front face and tip appear to be involved in polymerization (Figs. 3,5,6,7 and Supplemental Figs. 1–5). Applied more broadly to the LN surfaces, modification of xLNNd provides an opportunity to map the residues required for formation of the ternary polymer node, to determine whether charge changes caused loss of function, and whether loss of non-charged residues were involved. The finding that S68R and S68A cause a loss of activity argues that the gain of a basic charge contributed, but was not the sole cause of the polymerization defect. Such expanded mapping would likely focus on conserved surface residues rather than those causing cysteine mismatches or interior residues required for proper folding of the domain. Mapping could greatly aid the elucidation of the ternary node structure.
Experimental procedures
DNA constructs
Expression vectors for the mouse αLNNd, mouse α1, human β1, and human γ1-subunits as well as deletions of the α1LN, β1LN, γ1LN and the S68R point mutation in β1 laminin were previously described [33,49]. To generate point mutations in the αLNNd protein, overlapping PCR (Jumpstart Taq, Sigma; see Supplemental table 1) followed by either HindIII-XagI or HindIII-Eco91I digests were performed and inserted into the αLNND pcDNA3.1 Zeo construct. βLNNd was generated by overlapping PCR using human β1 laminin and the αLNNd templates (Supplemental table 1) and inserting a HindIII-BstEII product into the αLNND pcDNA3.1 Zeo plasmid. Likewise, γLNNd was made using overlapping PCR with the human γ laminin and αLNNd templates, followed by HindIII-BsrG1 digest and insertion into the αLNNd construct. All βLNNd point mutations except R246Q (Supplemental table 1) were generated by overlapping PCR and insertion into the HindIII-EcoN1 site of the βLNNd plasmid. Similarly, overlapping PCR generated γLNNd mutations which were inserted into the HindIII-BsrG1 site of γLNNd plasmid. The R246Q mutation in βLNNd was first generated in the full length human β1 laminin chain, hβ1pcDNA3.1 Zeo, using overlapping PCR and HindIII-ClaI insertion of the product. The R246Q and S68R point mutations were moved into the βLNNd construct with a HindIII-EcoN1 digest of the β1 laminin pcDNA3.1 Zeo plasmids.
Basement membrane proteins
HEK293 cells stably expressing WT and LN deletions of laminin were Flag-affinity purified as previously described [33]. xLNNd co-purified laminins were first co-incubated at 4 °C overnight in a molar ratio of ~6 to 1 (xLNNd:laminin) followed by Flag-affinity purification. xLNNd proteins were produced in stably expressing HEK293 cells grown under low serum conditions (1%) after reaching confluency and purified on Colbalt Hispur agarose (Thermo Fisher). Nidogen-1 and collagen-IV were purified as described [33]. All proteins were dialyzed at 4 °C overnight in TBS50 (Tris-HCL pH 7.4, 90 mM NaCl, 0.125 mM EDTA).
Protein determinations
Molar laminin concentrations were determined by densitometry of Coomassie blue–stained acrylamide gels compared with an EHS-laminin (710-kDa protein mass) standard, corrected for changes in the calculated mass (LmΔxLN, ~683 kDa) as previously described [33]. Absorbance at 280 nm was used to measure the concentration of xLNNd (~157 kDa).
Culturing, immunostaining, and analysis of BM assembly in Schwann cells
Schwann cells isolated from sciatic nerves from newborn Sprague-Dawley rats were the kind gift of Dr. James Salzer (New York University). These cells were maintained in Dulbecco’s modified Eagle’s medium, 10% fetal calf serum (Gibco, Thermo Fisher Scientific), neuregulin (0.5 μg/ml, Sigma), forskalin (0.2 μg/ml, Sigma), and 1% penicillin/streptomycin. Cells at passages 26–39 were plated onto 24-well dishes 50,000 cells/well; (Denville) and treated with the indicated proteins for 1 h at 37 °C. Adherent SC cultures were washed 3 times with PBS, followed by fixation in 3.5% paraformaldehyde in PBS for 20 min at room temperature (RT). Cultures were blocked overnight at 4 °C with 5% goat serum and 0.5% BSA in PBS. The cells were stained with primary polyclonal antibody specific for laminin G4–5 subunits (anti-E3) as described and characterized [57]. Detection of bound primary antibody was accomplished with Alexa Fluor 647 (far red) goat anti-rabbit IgG secondary antibody (Molecular Probes) and nuclear counterstaining with 4′,6-diamidino-2-phenylindole (dapi). SC cultures were viewed by indirect immunofluorescence using an inverted microscope (IX70; Olympus) fitted with an IXFLA fluorescence attachment and a CoolSnap digital camera (Roper Scientific) controlled by IP Lab 4.08 (Scanalytics). Images were recorded (5–12 fields, each 1300 × 1030 pixels) using a ×10 objective, with the same exposure time within an experimental set. Note that laminin immunofluorescence images have been displayed in faux green rather than red in the figures. Fluorescence intensity levels were estimated from the digital images with ImageJ software (NIH), with calculations performed in Microsoft Excel. Laminin staining intensities were calculated based on a protocol described for the measurement of laminin on Schwann cells [34]. Briefly, a single segmentation cut-off value was set to exclude non-cellular regions for all images being compared. The summed pixel intensities overlying the treated cells in each field were then divided by the cell number as determined from dapi-stained nuclei counts. Values were expressed as the mean ± SD of summed intensities from different fields normalized to control, with plotting in SigmaPlot 12.5 (Systat Software).
Laminin polymerization assay
Aliquots (50 μl) of laminin without or with xLNNd in polymerization buffer were incubated at 37 °C in a series of concentrations. Small aliquots of albumin (BSA) were included to control for non-specific protein adhesion to the tubes. Eppendorf tubes containing the aliquots were then centrifuged to separate polymerized protein. Supernatants (S) and pellets (P) were analyzed by SDS-PAGE with Coomassie blue staining, scanned with GE Image Scanner III, and quantitated as described [33,34]. Background-subtracted summed pixel values fitted by linear regression were plotted in SigmaPlot 12.5.
Homology fitting and protein structure imaging
(a) The mouse laminin α1 LN-LEa FASTA sequence (residues 1–481) was modeled by homology fitting online in the program HHpred (Max-Planck-Gesellschaft, Tübingen, Germany; https://toolkit.tuebingen.mpg.de) [59]. Following multiple sequence alignment, the domains were modeled against mouse Lmβ1 (4AQS), mouse netrin-1 (4OVE), mouse Lmα5 (2Y38), mouse netrin-4 (4WNX) and mouse Lmγ1 (4AQT) [35,36,60,61]. (b) Protein Data Bank (PDB) files for the LN to LEa domains of mouse Lmβ1 (4AQS), mouse Lmγ1 (4AQT), and mouse Lmα1 (from homology fit) were rendered as images showing the protein surfaces in the program Pymol (version 2.0, Schrödinger, LLC). Glycans were added to the structures at the asn-x-ser/thr residues considered as N-glycosylation sites in order to enable the viewer to appreciate likely regions of glycan-dependent steric hindrance. Png image files of suitable views of the surface structures were saved and labeled in Adobe Photoshop version CS6.
Statistics
The average and standard deviation (SD) of immunofluorescence images and analysis by one way ANOVA were calculated in SigmaPlot 12.5. Probability (P) values were considered significant if <0.05.
Supplementary Material
Acknowledgments
This study was supported by a grant (R01-DK36425) to P.D.Y. from the National Institutes of Health. We thank Samantha Hobbs for technical assistance. We also thank Dr. Erhard Hohenester (Imperial College, London) for his recommendations of γLN candidate mutations.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.matbio.2018.01.012.
Footnotes
Disclosure
The authors have no conflicting financial interests.
References
- 1.Macdonald PR, Lustig A, Steinmetz MO, Kammerer RA. Laminin chain assembly is regulated by specific coiled-coil interactions. J Struct Biol. 2010;170(2):398–405. doi: 10.1016/j.jsb.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li S, Harrison D, Carbonetto S, Fassler R, Smyth N, Edgar D, Yurchenco PD. Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J Cell Biol. 2002;157(7):1279–1290. doi: 10.1083/jcb.200203073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray P, Edgar D. Regulation of programmed cell death by basement membranes in embryonic development. J Cell Biol. 2000;150(5):1215–1221. doi: 10.1083/jcb.150.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pozzi A, Yurchenco PD, Iozzo RV. The nature and biology of basement membranes. Matrix Biol. 2017;57–58:1–11. doi: 10.1016/j.matbio.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol. 2005;169(1):179–189. doi: 10.1083/jcb.200501098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bose K, Nischt R, Page A, Bader BL, Paulsson M, Smyth N. Loss of nidogen-1 and -2 results in syndactyly and changes in limb development. J Biol Chem. 2006;281:11573–11581. doi: 10.1074/jbc.M607886200. [DOI] [PubMed] [Google Scholar]
- 7.Poschl E, Schlotzer-Schrehardt U, Brachvogel B, Saito K, Ninomiya Y, Mayer U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development. 2004;131(7):1619–1628. doi: 10.1242/dev.01037. [DOI] [PubMed] [Google Scholar]
- 8.Costell M, Gustafsson E, Aszodi A, Morgelin M, Bloch W, Hunziker E, Addicks K, Timpl R, F R. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. 1999;147(5):1109–1122. doi: 10.1083/jcb.147.5.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85(4):525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 10.Leiton CV, Aranmolate A, Eyermann C, Menezes MJ, Escobar-Hoyos LF, Husain S, Winder SJ, Colognato H. Laminin promotes metalloproteinase-mediated dystroglycan processing to regulate oligodendrocyte progenitor cell proliferation. J Neurochem. 2015;135(3):522–538. doi: 10.1111/jnc.13241. [DOI] [PubMed] [Google Scholar]
- 11.Rooney JE, Gurpur PB, Yablonka-Reuveni Z, Burkin DJ. Laminin-111 restores regenerative capacity in a mouse model for alpha7 integrin congenital myopathy. Am J Pathol. 2009;174(1):256–264. doi: 10.2353/ajpath.2009.080522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McKee KK, Yang DH, Patel R, Chen ZL, Strickland S, Takagi J, Sekiguchi K, Yurchenco PD. Schwann cell myelination requires integration of laminin activities. J Cell Sci. 2012;125(19):4609–4619. doi: 10.1242/jcs.107995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yurchenco PD. Integrating activities of laminins that drive basement membrane assembly and function. Curr Top Membr. 2015;76:1–30. doi: 10.1016/bs.ctm.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Fox JW, Mayer U, Nischt R, Aumailley M, Reinhardt D, Wiedemann H, Mann K, Timpl R, Krieg T, Engel J, Chu ML. Recombinant nidogen consists of three globular domains and mediates binding of laminin to collagen type IV. EMBO J. 1991;10:3137–3146. doi: 10.1002/j.1460-2075.1991.tb04875.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohfeldt E, Sasaki T, Gohring W, Timpl R. Nidogen-2: a new basement membrane protein with diverse binding properties. J Mol Biol. 1998;282(1):99–109. doi: 10.1006/jmbi.1998.2004. [DOI] [PubMed] [Google Scholar]
- 16.Hopf M, Gohring W, Kohfeldt E, Yamada Y, Timpl R. Recombinant domain IV of perlecan binds to nidogens, laminin-nidogen complex, fibronectin, fibulin-2 and heparin. Eur J Biochem. 1999;259(3):917–925. doi: 10.1046/j.1432-1327.1999.00127.x. [DOI] [PubMed] [Google Scholar]
- 17.Hopf M, Gohring W, Mann K, Timpl R. Mapping of binding sites for nidogens, fibulin-2, fibronectin and heparin to different IG modules of perlecan. J Mol Biol. 2001;311(3):529–541. doi: 10.1006/jmbi.2001.4878. [DOI] [PubMed] [Google Scholar]
- 18.Timpl R, Wiedemann H, van Delden V, Furthmayr H, Kuhn K. A network model for the organization of type IV collagen molecules in basement membranes. Eur J Biochem. 1981;120(2):203–211. doi: 10.1111/j.1432-1033.1981.tb05690.x. [DOI] [PubMed] [Google Scholar]
- 19.Yurchenco PD, Furthmayr H. Self-assembly of basement membrane collagen. Biochemistry. 1984;23(8):1839–1850. doi: 10.1021/bi00303a040. [DOI] [PubMed] [Google Scholar]
- 20.Yurchenco PD, Ruben GC. Basement membrane structure in situ: evidence for lateral associations in the type IV collagen network. J Cell Biol. 1987;105(6 Pt 1):2559–2568. doi: 10.1083/jcb.105.6.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Glanville RW, Qian RQ, Siebold B, Risteli J, Kuhn K. Amino acid sequence of the N-terminal aggregation and cross-linking region (7S domain) of the alpha 1 (IV) chain of human basement membrane collagen. Eur J Biochem. 1985;152(1):213–219. doi: 10.1111/j.1432-1033.1985.tb09186.x. [DOI] [PubMed] [Google Scholar]
- 22.Siebold B, Qian RA, Glanville RW, Hofmann H, Deutzmann R, Kuhn K. Construction of a model for the aggregation and cross-linking region (7S domain) of type IV collagen based upon an evaluation of the primary structure of the alpha 1 and alpha 2 chains in this region. Eur J Biochem. 1987;168(3):569–575. doi: 10.1111/j.1432-1033.1987.tb13455.x. [DOI] [PubMed] [Google Scholar]
- 23.Vanacore R, Ham AJ, Voehler M, Sanders CR, Conrads TP, Veenstra TD, Sharpless KB, Dawson PE, Hudson BG. A sulfilimine bond identified in collagen IV. Science. 2009;325(5945):1230–1234. doi: 10.1126/science.1176811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gesemann M, Cavalli V, Denzer AJ, Brancaccio A, Schumacher B, Ruegg MA. Alternative splicing of agrin alters its binding to heparin, dystroglycan, and the putative agrin receptor. Neuron. 1996;16(4):755–767. doi: 10.1016/s0896-6273(00)80096-3. [DOI] [PubMed] [Google Scholar]
- 25.Yamada H, Denzer AJ, Hori H, Tanaka T, Anderson LV, Fujita S, Fukuta-Ohi H, Shimizu T, Ruegg MA, Matsumura K. Dystroglycan is a dual receptor for agrin and laminin-2 in Schwann cell membrane. J Biol Chem. 1996;271(38):23418–23423. doi: 10.1074/jbc.271.38.23418. [DOI] [PubMed] [Google Scholar]
- 26.Denzer AJ, Brandenberger R, Gesemann M, Chiquet M, Ruegg MA. Agrin binds to the nerve-muscle basal lamina via laminin. J Cell Biol. 1997;137(3):671–683. doi: 10.1083/jcb.137.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denzer AJ, Schulthess T, Fauser C, Schumacher B, Kammerer RA, Engel J, Ruegg MA. Electron microscopic structure of agrin and mapping of its binding site in laminin-1. EMBO J. 1998;17(2):335–343. doi: 10.1093/emboj/17.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gesemann M, Brancaccio A, Schumacher B, Ruegg MA. Agrin is a high-affinity binding protein of dystroglycan in non-muscle tissue. J Biol Chem. 1998;273(1):600–605. doi: 10.1074/jbc.273.1.600. [DOI] [PubMed] [Google Scholar]
- 29.Smirnov SP, Barzaghi P, McKee KK, Ruegg MA, Yurchenco PD. Conjugation of LG domains of agrins and perlecan to polymerizing laminin-2 promotes acetylcholine receptor clustering. J Biol Chem. 2005;280(50):41449–41457. doi: 10.1074/jbc.M508939200. [DOI] [PubMed] [Google Scholar]
- 30.Hohenester E, Tisi D, Talts JF, Timpl R. The crystal structure of a laminin G-like module reveals the molecular basis of alpha-dystroglycan binding to laminins, perlecan, and agrin. Mol Cell. 1999;4(5):783–792. doi: 10.1016/s1097-2765(00)80388-3. [DOI] [PubMed] [Google Scholar]
- 31.Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD. Self-assembly of laminin isoforms. J Biol Chem. 1997;272(50):31525–31532. doi: 10.1074/jbc.272.50.31525. [DOI] [PubMed] [Google Scholar]
- 32.Yurchenco PD, Cheng YS. Self-assembly and calcium-binding sites in laminin. A three-arm interaction model. J Biol Chem. 1993;268:17286–17299. [PubMed] [Google Scholar]
- 33.McKee KK, Harrison D, Capizzi S, Yurchenco PD. Role of laminin terminal globular domains in basement membrane assembly. J Biol Chem. 2007;282(29):21437–21447. doi: 10.1074/jbc.M702963200. [DOI] [PubMed] [Google Scholar]
- 34.McKee KK, Capizzi S, Yurchenco PD. Scaffold-forming and adhesive contributions of synthetic laminin-binding proteins to basement membrane assembly. J Biol Chem. 2009;284(13):8984–8994. doi: 10.1074/jbc.M809719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carafoli F, Hussain SA, Hohenester E. Crystal structures of the network-forming short-arm tips of the laminin beta1 and gamma1 chains. PLoS One. 2012;7(7):e42473. doi: 10.1371/journal.pone.0042473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hussain SA, Carafoli F, Hohenester E. Determinants of laminin polymerization revealed by the structure of the alpha5 chain amino-terminal region. EMBO Rep. 2011;12(3):276–282. doi: 10.1038/embor.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavassini BF, Carboni N, Nielsen JE, Danielsen ER, Thomsen C, Svenstrup K, Bello L, Maioli MA, Marrosu G, Ticca AF, Mura M, Marrosu MG, Soraru G, Angelini C, Vissing J, Pegoraro E. Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve. 2011;44(5):703–709. doi: 10.1002/mus.22132. [DOI] [PubMed] [Google Scholar]
- 38.Di Blasi C, Piga D, Brioschi P, Moroni I, Pini A, Ruggieri A, Zanotti S, Uziel G, Jarre L, Della Giustina E, Scuderi C, Jonsrud C, Mantegazza R, Morandi L, Mora M. LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch Neurol. 2005;62(10):1582–1586. doi: 10.1001/archneur.62.10.1582. [DOI] [PubMed] [Google Scholar]
- 39.Patton BL, Wang B, Tarumi YS, Seburn KL, Burgess RW. A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J Cell Sci. 2008;121(Pt. 10):1593–1604. doi: 10.1242/jcs.015354. [DOI] [PubMed] [Google Scholar]
- 40.Hollfelder D, Frasch M, Reim I. Distinct functions of the laminin beta LN domain and collagen IV during cardiac extracellular matrix formation and stabilization of alary muscle attachments revealed by EMS mutagenesis in Drosophila. BMC Dev Biol. 2014;14:26. doi: 10.1186/1471-213X-14-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Blahova K, Bockenhauer D, Cheong HI, Maruniak-Chudek I, Cochat P, Dotsch J, Gajjar P, Hennekam RC, Janssen F, Kagan M, Kariminejad A, Kemper MJ, Koenig J, Kogan J, Kroes HY, Kuwertz-Broking E, Lewanda AF, Medeira A, Muscheites J, Niaudet P, Pierson M, Saggar A, Seaver L, Suri M, Tsygin A, Wuhl E, Zurowska A, Uebe S, Hildebrandt F, Antignac C, Zenker M. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat. 2010;31(9):992–1002. doi: 10.1002/humu.21304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colognato H, Yurchenco PD. The laminin alpha2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers. Curr Biol. 1999;9(22):1327–1330. doi: 10.1016/s0960-9822(00)80056-1. [DOI] [PubMed] [Google Scholar]
- 43.Yurchenco PD, McKee KK, Reinhard JR, Ruegg MA. Laminin-deficient muscular dystrophy: molecular pathogenesis and structural repair strategies. Matrix Biol. 2017 Nov 27; doi: 10.1016/j.matbio.2017.11.009. https://doi.org/10.1016/j.matbio.2017.11.009 (Epub ahead of print, pii: S0945-053X1730333-5) [DOI] [PMC free article] [PubMed]
- 44.Mohney BG, Pulido JS, Lindor NM, Hogan MC, Consugar MB, Peters J, Pankratz VS, Nasr SH, Smith SJ, Gloor J, Kubly V, Spencer D, Nielson R, Puffenberger EG, Strauss KA, Morton DH, Eldahdah L, Harris PC. A novel mutation of LAMB2 in a multigenerational mennonite family reveals a new phenotypic variant of Pierson syndrome. Ophthalmology. 2011;118(6):1137–1144. doi: 10.1016/j.ophtha.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mayer U, Nischt R, Poschl E, Mann K, Fukuda K, Gerl M, Yamada Y, Timpl R. A single EGF-like motif of laminin is responsible for high affinity nidogen binding. EMBO J. 1993;12:1879–1885. doi: 10.1002/j.1460-2075.1993.tb05836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stetefeld J, Mayer U, Timpl R, Huber R. Crystal structure of three consecutive laminin-type epidermal growth factor-like (LE) modules of laminin gamma1 chain harboring the nidogen binding site. J Mol Biol. 1996;257(3):644–657. doi: 10.1006/jmbi.1996.0191. [DOI] [PubMed] [Google Scholar]
- 47.Hopf M, Gohring W, Ries A, Timpl R, Hohenester E. Crystal structure and mutational analysis of a perlecan-binding fragment of nidogen-1. Nat Struct Biol. 2001;8(7):634–640. doi: 10.1038/89683. [DOI] [PubMed] [Google Scholar]
- 48.Reinhardt D, Mann K, Nischt R, Fox JW, Chu ML, Krieg T, Timpl R. Mapping of nidogen binding sites for collagen type IV, heparan sulfate proteoglycan, and zinc. J Biol Chem. 1993;268:10881–10887. [PubMed] [Google Scholar]
- 49.Funk SD, Bayer RH, Malone AF, McKee KK, Yurchenco PD, Miner JH. Pathogenicity of a human laminin beta2 mutation revealed in models of Alport syndrome. J Am Soc Nephrol. 2017 Dec 20; doi: 10.1681/ASN.2017090997. https://doi.org/10.1681/ASN.2017090997 (Epub ahead of print, pii: ASN.2017090997) [DOI] [PMC free article] [PubMed]
- 50.Lehnhardt A, Lama A, Amann K, Matejas V, Zenker M, Kemper MJ. Pierson syndrome in an adolescent girl with nephrotic range proteinuria but a normal GFR. Pediatr Nephrol. 2012 May;27(5):865–868. doi: 10.1007/s00467-011-2088-2. https://doi.org/10.1007/s00467-011-2088-2 (Epub 2012 Jan 8) [DOI] [PubMed] [Google Scholar]
- 51.Purvis A, Hohenester E. Laminin network formation studied by reconstitution of ternary nodes in solution. J Biol Chem. 2012;287(53):44270–44277. doi: 10.1074/jbc.M112.418426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen YM, Kikkawa Y, Miner JH. A missense LAMB2 mutation causes congenital nephrotic syndrome by impairing laminin secretion. J Am Soc Nephrol. 2011;22(5):849–858. doi: 10.1681/ASN.2010060632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen YM, Zhou Y, Go G, Marmerstein JT, Kikkawa Y, Miner JH. Laminin beta2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J Am Soc Nephrol. 2013;24(8):1223–1233. doi: 10.1681/ASN.2012121149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D. Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation. J Cell Biol. 1999;144(1):151–160. doi: 10.1083/jcb.144.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H. Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity. J Biol Chem. 1985;260:7636–7644. [PubMed] [Google Scholar]
- 56.Engel J, Odermatt E, Engel A, Madri JA, Furthmayr H, Rohde H, Timpl R. Shapes, domain organizations and flexibility of laminin and fibronectin, two multifunctional proteins of the extracellular matrix. J Mol Biol. 1981;150:97–120. doi: 10.1016/0022-2836(81)90326-0. [DOI] [PubMed] [Google Scholar]
- 57.McKee KK, Crosson SC, Meinen S, Reinhard JR, Ruegg MA, Yurchenco PD. Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J Clin Invest. 2017;127(3):1075–1089. doi: 10.1172/JCI90854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reinhard JR, Lin S, McKee KK, Meinen S, Crosson SC, Sury M, Hobbs S, Maier G, Yurchenco PD, Ruegg MA. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci Transl Med. 2017;9(396) doi: 10.1126/scitranslmed.aal4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Webb B, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2016;86:291–2937. doi: 10.1002/cpps.20. [DOI] [PubMed] [Google Scholar]
- 60.Reuten R, Patel TR, McDougall M, Rama N, Nikodemus D, Gibert B, Delcros JG, Prein C, Meier M, Metzger S, Zhou Z, Kaltenberg J, McKee KK, Bald T, Tuting T, Zigrino P, Djonov V, Bloch W, Clausen-Schaumann H, Poschl E, Yurchenco PD, Ehrbar M, Mehlen P, Stetefeld J, Koch M. Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes. Nat Commun. 2016;7:13515. doi: 10.1038/ncomms13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grandin M, Meier M, Delcros JG, Nikodemus D, Reuten R, Patel TR, Goldschneider D, Orriss G, Krahn N, Boussouar A, Abes R, Dean Y, Neves D, Bernet A, Depil S, Schneiders F, Poole K, Dante R, Koch M, Mehlen P, Stetefeld J. Structural decoding of the Netrin-1/UNC5 interaction and its therapeutical implications in cancers. Cancer Cell. 2016;29(2):173–185. doi: 10.1016/j.ccell.2016.01.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.