

Abstract
Most ovarian cancer patients respond well to initial platinum-based chemotherapy. However, within a year, many patients experience disease recurrence with a platinum resistant phenotype that responds poorly to second line chemotherapies. As a result, new strategies to address platinum resistant ovarian cancer (PROC) are needed. Herein, we report that NP co-delivery of cisplatin (CP) and wortmannin (Wtmn), a DNA repair inhibitor, synergistically enhances chemoradiotherapy (CRT) and reverses CP resistance in PROC. We encapsulated this regimen in FDA approved poly(lactic-co-glycolic acid)-poly(ethylene glycol) (PLGA-PEG) NPs to reduce systemic side effects, enhance cellular CP uptake, improve Wtmn stability, and increase therapeutic efficacy. Treatment of platinum-sensitive ovarian cancer (PSOC) and PROC murine models with these dual-drug loaded NPs (DNPs) significantly reduced tumor burden versus treatment with combinations of free drugs or single-drug loaded NPs (SNPs). These results support further investigation of this NP-based, synergistic drug regimen as a means to combat PROC in the clinic.
Keywords: nanoparticle, combination therapy, platinum resistance, treatment synergy, ovarian cancer
Introduction
Ovarian cancer is the deadliest gynecological malignancy in the United States with over 15,000 deaths in 2012 [1]. For patients with advanced disease, treatment typically involves surgery followed by platinum-based chemotherapy. Initial ovarian cancer response rates to this regimen are high (>80 %), but many patients will relapse within a year [2]. Additionally, most of these patients’ recurrent disease will be resistant to further platinum based therapy and require second line chemotherapies, such as bevacizumab, for treatment [3–5]. However, clinical trials evaluating these second line chemotherapies in PROC have shown low response rates with poor efficacy. Therefore, new routes to overcome PROC are needed to improve patient outcomes.
Research has shown PROC stems from two primary mechanisms: (1) reduced CP uptake via downregulation of metal transport proteins and (2) enhanced DNA repair after CP induced damage [6–9]. Therefore, PROC treatment strategies must address these two mechanisms to enhance platinum-based therapies. As a solution for the latter issue, Wtmn, a phosphoinositide 3-kinase (PI3K) inhibitor, can block downstream DNA repair pathways and potently “re”-sensitize cancer cells to CRT [10,11]. However, free Wtmn shows poor in vivo stability and high hepatotoxicity when delivered systemically [12,13]. Moreover, Wtmn alone does not address the co-resistance mechanism (1) of poor CP uptake.
As a solution, we previously showed that NP delivery greatly improves the therapeutic efficacy of Wtmn by improving its solubility and protecting it’s bio-active furan moiety from degradation by extracellular amino acids [13–15]. Additionally, NP vehicles can improve the uptake of CP into resistant tumor cells, reduce systemic toxicities, and deliver a precise, synergistic ratio of both drugs to the tumor bed [6,16–21].
Therefore, we investigated biocompatible PLGA-PEG NPs as a vehicle to deliver both Wtmn and CP in a single formulation and reverse platinum resistance in PROC (Fig. 1). The DNP combination strongly and synergistically enhanced the cytotoxicity of CP based chemotherapy and radio-sensitized PROC cells. Mechanistic studies of Wtmn delivered with CPP by NP co-encapsulation demonstrates that Wtmn blocks DNA repair and enhances the synergistic cytotoxicity of the drug combination in PROC. In murine models of both PSOC and PROC, the DNPs greatly improved CRT versus a clinical dose of CP, combinations of free drugs, and mixtures of SNPs without increasing off-target toxicity.
Fig. 1.

Mechanism of action for Wtmn:CPP DNPs. PROC resists chemotherapy via reducing CP uptake and enhancing DNA repair pathways. NP mediated co-delivery of normally unstable Wtmn and CP synergistically enhances CP therapy by promoting CP uptake via alternative pathways and Wtmn inhibition of DNA repair mechanisms.
Materials and Methods
Materials
To improve CP encapsulation in the NPs, we synthesized and utilized a caprylic acid modified, Pt(IV) cisplatin prodrug (CPP) as previously described (Fig. S1) [22–26]. PEG-PLGA (3,600–30,000 MW, 50:50 LA:GA) and PLGA (50,000–75,000 MW, 85:15 LA:GA) were acquired from PolySciTech. Poly-(vinyl alcohol) (PVA) was acquired from Polysciences, Inc. Cell culture reagents were purchased from Gibco by Life Technologies. Anti-Cisplatin modified DNA antibody was purchased from Abcam. Anti-rat IgG-Alexafluor®555 antibodies were purchased from Cell Signaling. All other chemicals were acquired from Sigma Aldrich and used without further purification.
Cell Culture
The PSOC and PROC cell lines, A2780 and A2780cis, respectively, were obtained from American Type Culture Collection (ATCC). Cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin. CP (2 μM) was added to the A2780cis culture medium every other passage to maintain CP resistance. Before use in experiments, the A2780cis cells were passed twice to ensure no residual CP was bound to cells.
Animal Maintenance
Six to eight week old, female, nude mice (~20–30 g) were supplied by the University of North Carolina Animal Facility and maintained under pathogen-free conditions in the Center for Experimental Animals (AAALAC accredited animal facility). The experimental protocol was approved by the UNC Institutional Animal Care and Use committee and adhered to the NIH Guide for the Care and Use of Laboratory Animals (no. 86-23, revised 1985).
Synthesis of NPs
NPs were formed via nanoprecipitation. Stock solutions of formulation components were made prior to nanoprecipitation. Wtmn was dissolved in acetonitrile (ACN, 4 mg/mL), and CPP was dissolved in acetone (2 mg/ml). PLGA and mPEG-PLGA were dissolved in ACN (both at 20 mg/ml). 50 μL of each polymer stock was mixed with varying volumes of drug stock solutions to produce an organic phase with a range of drug feeding ratios (%FR) expressed as wt% versus polymer weight. This organic phase was then diluted with ACN to a final volume of 500 μL (2 mg/mL PLGA, 2 mg/mL PEG-PLGA). The NP suspension was then formed by dripping the organic solution drop-wise into an excess aqueous phase (0.1 v/v% poly-vinyl alcohol, 3 mL). The suspension was stirred for 3 h to allow for the organic phase to evaporate. The resulting NPs were concentrated and washed three times with deionized water by centrifugation using an Amicon Ultra-4 filter (MWCO-100 kDa) at 1,000 g for 15 min (EMD. Millipore, Billerica, MA, USA). The concentrated NP suspension was finally re-suspended in PBS (8 mg/mL, 250 μL).
Characterization of NPs
The size, polydispersity, and zeta (ζ)-potential of NPs (1 mg/mL) were determined by dynamic light scattering (DLS). Transmission electron microscopy (TEM) images were obtained with a JEOL JEM 1230 TEM. Prior to imaging, concentrated NP suspensions were diluted 100-fold with deionized water and then adsorbed onto a 400-mesh carbon filmed copper grid. The grid was negatively stained with potassium phosphotungstate (2 wt%, pH 7.0) for 1 min. Excess stain was removed using filter paper, and the grid was allowed to air dry before imaging.
Determination of Drug loading in NPs
NPs (40 μL, 8 mg/mL) were diluted with ACN (200 μL), vortexed, and then sonicated for 10 min to dissolve the NP structure. Treated samples (10 μL) were then analyzed with a Shimadzu SPD-M20A high performance liquid chromatography (HPLC) equipped with a diode array detector and a Chromolith Fast Gradient RP-18e 50 × 2mm column (EMD. Millipore, Billerica, MA, USA). Samples were eluted using a binary solvent system (A:B, A-Water, B-ACN) at a flow rate of 0.25 ml/min. The gradient followed: 0% to 100% B (0 to 20 min), 100% B (20 to 25 min), 50% B (25 to 30 min), 0% B (30 to 35 min). Wtmn and CPP elution were monitored at 262 nm (retention time 9.7 min) and 220 nm (retention time 13.3 min), respectively.
In vitro Drug Release
In vitro drug-release profiles of loaded NPs were determined under physiological sink conditions. NP suspensions (50 μL, 2.67 mg/mL) were loaded into Slide-A-Lyzer MINI dialysis microtubes (MWCO 20 kDa, Pierce, Rockford, IL, USA). Samples were then dialyzed against 4 L of PBS under gentle stirring at 37°C. NP samples (40 μL) were collected from the microtubes at the indicated times and the residual drugs’ concentrations were determined by HPLC as described above. Drug release was calculated by (1 − Ct/C0) × 100%, where C0 and Ct represent the retained drug concentration at time 0 and t, respectively.
DNA Repair Inhibition by Wtmn Formulations
Monitoring of γH2AX phosphorylation foci in the nucleus was used as an assay to determine the efficacy of DNA repair inhibition by various Wtmn formulations. Prolonged γH2AX phosphorylation post radiotherapy (XRT) indicates inhibition of DNA repair mechanisms and persistence of cytotoxic DNA double strand breaks [27–29].
A2780 cells were loaded in 6 well plates (250,000 cells/well) and incubated overnight. Plated cells were washed with PBS and then treated with an IC50 dose of a Wtmn formulation in media (the control arm received media only) for 2 h. Each well was then washed with PBS (x3) and media was added. After 45 min, the treated cells were irradiated (2Gy) and then incubated for an additional 3 or 24 h. The irradiated cells were fixed with neutral buffered formalin (10% v/v) for 15 min, washed with PBS (x2), and then stored at 4°C until staining.
For staining, fixed cells were washed with PBS (1% v/v Tween 20 [PBST]), permeabilized with Triton-X 100 (0.25% v/v in PBST) for 15 min, and washed again with PBST. Cells were blocked with BSA (5% w/v in PBST) for 1 h, stained with a primary phospho-γ-H2AX (Ser139) antibody (mouse, diluted 1:200 in 1% BSA in PBST) for 1 h, and then washed with PBST. The secondary, AF594 antibody (donkey anti-mouse, diluted 1:1000 in 1% w/v BSA PBST) was added to the cells for 30 min followed by washing with PBST (x3). Cover slips were mounted on glass slides with Prolong Diamond Anti-fade DAPI and stored at 4°C until imaged.
Z stack images of the samples were acquired on an Olympus BX61 microscope. Fiji software was used to create a maximal frontal projection of each set of images. Phosphorylated γH2AX foci were counted with Find Maxima on the AF594 red channel, and cells per image were counted using the DAPI blue channel. Representative images were de-convoluted using Autoquant.
In vitro Cytotoxicity
Cells (3,600 cells/well) were plated in a 96-well plate and allowed to recover overnight. Cells were then washed with PBS and treated with varying concentrations and ratios of free small-molecule drugs or drug loaded NPs at 37°C for 2 h. Cells were then washed with PBS three times and allowed to grow in complete culture media for an additional 72 h. After incubation, cell viability was analyzed by MTS assay (Promega) per the vendor’s instructions. Sample absorbance at 492 nm was recorded via a microtiter plate reader (Molecular Devices Corporation, California, USA). The IC50 values for each treatment was calculated by fitting the dose-dependent cell viabilities to a four-parameter logistic model using the MasterPlex 2010 software pack (MiraiBio Group, Hitachi Solutions America, Ltd.). Combination indexes (CI) were determined for each treatment arm using CompuSyn software.
Clonogenic Survival Assay [10]
Cells (600, 800, 1,200, 2,400 or 5,000 cells/well) were seeded in 6-well plates and incubated in complete cell culture medium for 2 h to allow for attachment. Cells were then washed with PBS and treated with various drug formulations at the IC20 dose for 2 h. Cells were washed again with PBS three times, and complete cell culture medium was added. Cells received an irradiation dose (X-RAD 320, Precision X-ray Inc., North Branford, CT, USA) according to the number of cells seeded: 0 Gy for 600 cells, 2 Gy for 800 cells, 4 Gy for 1,200 cells, 6 Gy for 2,400 cells, and 8 Gy for 5,000 cells. The irradiated cells were allowed to grow for 14 days to generate colonies. Colonies were washed with PBS, fixed, and then stained with a mixture of glutaraldehyde (6 v/v%) and crystal violet (0.5 w/v%) for 1 h. The cells were then gently rinsed with deionized water and dried at room temperature. Colonies with more than 50 cells were counted.
The plating efficiency (PE) was calculated via the number of colonies formed divided by the number of cells seeded. The surviving fraction (SF) of an irradiation dose was calculated by the treatment’s PE divided by the PE of non-irradiation treatment. D0 and N were acquired by fitting calculated SF to the single-hit multi-target model:
D represents the dose of irradiation. D0 and N, the average lethal dose parameters, were determined using GraphPad Prism version 6.01 for Windows (GraphPad Software, La Jolla California USA). The sensitization enhancement ratio (SER) was calculated using D0 of untreated cells divided by that of drug treated cells [30].
In Vivo Anticancer Efficacy
Low CP Dose Therapy
A murine ovarian cancer xenograft model was generated by injecting one million cells (0.2 ml of 50 v/v% RPMI-1640 medium and 50 v/v% Matrigel®) into the right flank. Tumors were allowed to grow to 80–150 mm3 before initiating treatment, and then mice were randomized into six groups (5 or 6 mice per group). Mice receiving drug therapy were treated with the same molar equivalent of free CP (0.15 mg/kg ~ 500 nmol/kg) or NP encapsulated CPP (0.30 mg/kg ~ 500 nmol/kg). Mice were treated once by tail vein injection on day 0 with either (1) PBS (200 μL), (2) mixture of free Wtmn (0.15 mg/kg) and CP (0.15 mg/kg), (3) PBS (200 μL), (4) mixture of free Wtmn (0.15 mg/kg) and CP (0.15 mg/kg), (5) a mixture of single drug loaded Wtmn (0.15 mg/kg) NPs and CPP (0.3 mg/kg) NPs (SNPs), or (6) DNPs (Wtmn 0.15 mg/kg and CPP 0.3 mg/kg). Mice in groups 3, 4, 5 and 6 additionally received 5 Gy of radiation on days 0, 1, and 2 (15 Gy total). Tumor length (L) and width (W) were measured for several days after treatment, and the tumor volume was calculated using: (W2 x L)/2, where L ≥ W. The body weight of the mice was also recorded. Mice were euthanized using CO2 inhalation method when tumor dimensions became larger than 2 cm in one direction.
Clinical Dose CP Therapy
Murine A2780cis xenografts were generated the same as above. The mice were then randomized into four arms (8 mice per group) and then treated with 200μl tail vein injection of either (1) PBS, (2) a mixture of free CP (0.15 mg/kg ~ 500 nmol/kg) and free WTMN (0.15 mg/kg ~ 350 nmol/kg), (3) a clinically comparable dose of free CP (2.5mg/kg ~ 8.33 μmol/kg) [31], or (4) DNPs co-loaded with CPP (0.3 mg/kg ~ 500 nmol/kg) and WTMN (0.15 mg/kg ~ 350 nmol/kg). All arms received 5 Gy of radiation on the same day as treatment, and another 5 Gy on each of the following two days (15 Gy total). Tumor sizes were first measured on day 0 after treatment, and all subsequent measurements were normalized based on the initial size. Mice were euthanized using CO2 inhalation method when tumor dimensions became larger than 2 cm in one direction.
Toxicity Determination
Murine A2780cis xenografts were generated the same as above. The mice were randomized into three groups (5 mice per group) and then treated with either (1) a mixture of free CP (0.15 mg/kg ~ 500 nmol) and free Wtmn (0.15 mg/kg ~ 350 nmol/kg), (2) a mixture of single drug loaded Wtmn (0.15 mg/kg) and single drug loaded CPP (0.3 mg/kg) SNPs, or (3) DNPs (Wtmn 0.15 mg/kg ~ 350 nmol/kg and CPP 0.3 mg/kg ~ 500 nmol/kg). All arms received 5 Gy of radiation on the same day as treatment, and another 5 Gy on each of the following two days (15 Gy total). Three days after treatment whole blood was drawn via sub-mandibular bleed.
For hematological toxicity, 500 μL of whole blood was stored in a heparin treated tube at 4°C before analyzing. For hepatotoxicity and nephrotoxicity, whole blood was centrifuged at 4,000 rpm for 10 min to separate and collect the plasma and then stored at −20 °C before analyzing. The whole-blood and the isolated plasma was analyzed by the Department of Pathology & Laboratory Medicine, Animal Histopathology & Laboratory Medicine Core, University of North Carolina, for blood cell counts, serum aspartate aminotransferase (AST), Blood Urea Nitrogen (BUN), and Creatinine (Crea) levels [14].
Immunofluorescent Tumor Section Imaging
Tumors were excised from one euthanized mouse in each arm on day 3 after initial treatment. The tissue was washed with PBS, fixed in formalin (10 v/v%) for 24 h, dehydrated, and then embedded in paraffin. Blank paraffin slides were hydrated and then microwaved twice for 10 min in sodium citrate solution (0.01 μmol/L, pH 6.0). The slides were cooled to room temperature and washed with PBS. The slides were then treated with a PBS solution containing hydrogen peroxide (3 v/v%) and Triton X-100 (0.5 w/v%) and subsequently washed three times with PBS.
Tumor sections were mounted and then blocked with bovine serum albumin (BSA, 10 w/v%) for 1 h. The slides were treated with 1:400 PBS diluted anti-cisplatin modified DNA antibody (Abcam, ab103261) overnight at 4°C. Secondary antibody was then added with Anti-rat IgG (H+L) (Alexa Fluor® 555 Conjugate, Cell Signaling, 4417S). After 1 h, slides were washed with PBS (0.2% Tween 20) and sections were treated with DAPI (1 μg/ml, Thermo Fisher Scientific, D1306) for 20 min. Slides were washed with PBS and then sealed with Fluoromount™ aqueous mounting medium (Sigma-Aldrich, F4680-25ML) and a cover glass. Immunofluorescence images were obtained via a laser scanning confocal microscope (ZEISS, LSM 700).
The extent of cisplatin-DNA complexing within the nucleus was quantified using Fiji (ImageJ) by calculating the mean fluorescence intensity of the cisplatin-DNA complex channel divided by that of the DAPI channel in identified nuclear regions. Data were presented using the mean of each group minus that of the PBS group.
Statistical Analysis
All experiments were performed at least three times, and expressed as mean ± SD for in vitro or mean ± SEM for in vivo studies. Unless otherwise noted, statistical differences/significance were determined using two-tailed Student’s t-test. The exceptions were the statistical significance of γH2AX foci differences was determined using a Tukey test, and the in vivo efficacy study employed a single-tailed Student’s t-test.
Results
Drug Loading and Release Efficacy
Free CP loads poorly into the hydrophobic core of PLGA-PEG NPs, so we utilized a CPP to enhance CP’s hydrophobicity and loading (Fig. S1) [22–26]. We tested the ability of PLGA-PEG NPs to load both drugs at a range of feeding ratios (%FR, wt% versus polymer) during nanoprecipitation. For SNPs, the hydrophobic CPP loaded the most efficiently with a maximum loading of 9.94 ± 2.07 wt% (25 %FR, Fig. S2, Table S1). Wtmn loaded at much lower levels with a high 25 %FR yielding 1.59 ± 0.70 wt% of loaded drug. Both drugs showed decreased encapsulation efficiencies (%EE) as drug %FR increased with maximum values of 12.50 ± 2.46% and 40.03 ± 6.00% for Wtmn and CPP, respectively.
We next investigated whether Wtmn and CPP affected each other’s loading when co-encapsulated in DNPs (Fig. 2, Table S2). Since CPP loaded at higher levels than Wtmn, we maintained Wtmn’s %FR at a high 25 wt% and varied the CPP %FR from 2.5 to 15 wt%. Wtmn had limited effect on CPP loading as CPP loaded at relatively the same level in DNPs as the SNPs (SNP-2.75±0.08% versus DNP-2.72±0.29% at 5% FR CPP). On the other hand, CPP had a varying regulatory effect on Wtmn loading. At low CPP %FRs (< 12.5 %), CPP increased the amount of loaded Wtmn by as much as 87% (2.98 ± 0.20 wt%, 12.30 ± 0.85 %EE) versus Wtmn SNPs (1.59 ± 0.70 wt%, 4.33 ± 1.45 %EE). However, at CPP %FRs ≥12.5 %, Wtmn loading dropped (0.78 ± 0.41 wt%, 3.18 ± 1.66 %EE). Despite this loading variability, we were able to synthesize nanoformulations with a dynamic range of loaded Wtmn:CPP molar ratios (2:1 to 1:5), which allowed for screening of the most synergistic ratios.
Fig. 2.

Encapsulation efficiency (%EE) and drug loading (wt%) of a) CPP or b) Wtmn in DNPs. The Wtmn %FR was held constant at 25 wt%, while the CPP %FR was varied. A CPP %FR of 7.5 wt% produced the loaded 1:1.4 Wtmn:CPP molar ratios used in all further studies.
We characterized the nanoformulations physical properties by TEM and DLS analysis (Fig. S3, Table S3–S4). Except at the highest %FR for CPP, the Wtmn SNPs typically skewed larger (~115 nm) than their CPP SNP counterparts (< 100 nm). The synthesized particles also generally showed low polydispersity values (~0.1), but some CPP SNPs became more polydisperse (~0.20) when CPP %FR exceeded 20%. TEM analysis of particle morphology confirmed the DLS size determination by showing spherical particles that range from 80 – 200 nm (Fig. S4). All NPs had negative zeta potentials (ζ) in the range of −15 to −40 mV, but DNPs (~ −18 mV) typically exhibited lower ζ values than SNPs (> − 20 mV).
We also determined the drug release kinetics of both the SNPs and DNPs under physiological sink conditions (Fig. S5). For Wtmn, its release half-life (t1/2) was approximately the same in both cases (t1/2 ~ 3.3 hours for SNPs to t1/2 ~ 2.8 DNPs). In contrast, CPP release slowed slightly when co-encapsulated from t1/2 ~ 3.1 to t1/2 ~ 4.8 hours.
NP In Vitro Activity and Cytotoxicity
We investigated whether Wtmn maintained its function to inhibit DNA repair upon NP encapsulation. A2780 cells were treated with an IC50 dose of the various Wtmn formulations (no Wtmn, free Wtmn, Wtmn SNP, or DNP) prior to XRT. Cells were then monitored for changes in γH2AX foci, which serve as a marker for DNA double strand breaks and inhibition of DNA repair mechanisms [27–29]. Control cells that received no Wtmn showed a statistically significant reduction in γH2AX foci (~40% fewer foci) after 24 hours indicating ongoing repair of the XRT induced DNA damage (Fig. S6–S7). However, all Wtmn formulations showed γH2AX foci levels remained elevated between 3 and 24 hours. For example, both Wtmn NP formulations statistically showed the same high foci count between the two time points and significantly higher levels of foci than the control at 24 h. These results confirm that Wtmn still propagates to the nucleus and maintains its DNA repair inhibitory function even upon NP encapsulation.
Next, we examined the cytotoxicity of various free drug and NP combinations against PSOC (A2780) and PROC (A2780cis) cell lines. PROC showed substantial resistance to both chemotherapies versus the PSOC cell line with a ~4.8 fold higher required CP and Wtmn monotherapeutic dose (Fig. 3a, Table S5). However, when free CP and Wtmn were combined, they lowered the required drug dose by as much as 2.7 fold in A2780cis and displayed potent synergism with a combination index (CI) of 0.272 (1:1 Wtmn:CP, Fa=0.5, Fig. S8, Table S6). In contrast, the free Wtmn and CP combination only mildly enhanced cytotoxicity in PSOC A2780 with a 13% IC50 decrease and an additive CI of 0.903 (1:4 Wtmn:CP, Fig. S9, Table S7).
Fig 3.

a) In Vitro cytotoxicity of formulations without radiation and b) clonogenic survival curves with radiation treatment. In b), cells were treated with the IC20 dose of their respective drug combination 2 h prior to radiation treatment. (* p ≤ .05, ** p ≤ .01, *** p ≤ .001, n.s. = not significant)
We also evaluated the differences in potency when therapies and their combinations were delivered in NP form (Fig. 3a, Table S5). SNPs given as monotherapies had some effect on the Wtmn and CP toxicity. Wtmn toxicity in A2780cis increased 2.1 fold versus the free drug, and CP toxicity increased 3.3 and 1.9 fold for A2780 and A2780cis, respectively. Encouragingly, DNPs (1:1.4 Wtmn:CPP) strongly and synergistically enhanced CP and Wtmn efficacy in A2780cis with IC50 dose decreases of ~21 fold (CI ~ 0.04) versus CPP SNPs given alone. Yet, no significant difference existed between the cytotoxicity of mixtures of SNPs or DNPs in vitro. In the PSOC A2780 cells, the required drug doses of mixed SNPs or DNP were nearly identical as CPP NPs given alone. Indeed, the two drugs had only an additive effect in A2780 cells (CI ~ 0.95 – 1.2, Fig. S7, Table S7). Due to the significantly enhanced cytotoxicity to PROC and excellent %EE of drugs at the 1:1.4 Wtmn:CPP ratio, we chose to use this formulation for all further experimentation.
Since CP and Wtmn are known radiosensitizers, we examined their ability to sensitize cells to XRT with the clonogenic survival assay (Fig. 3b) [14,30]. Cells were monitored and compared for reductions in colony formation when cultured with each formulations’ pre-determined IC20 dose 2 h prior to irradiation. The SER, a measure of radiosensitizer potency, was determined for all treatment arms in the A2780cis cell line. Both mixed SNPs and DNPs (1:1.4 Wtmn:CPP) showed statistically significant increases in SER (1.20 ± 0.24 and 1.29 ± 0.17, respectively) versus PBS indicating their potential to sensitize PROC to radiotherapy. However, the free drug combination’s SER (0.98 ± 0.24) did not statistically differ from treatment with just PBS (1.00 ± 0.11).
NPs’ In Vivo Efficacy
We tested our DNPs in vivo to determine if they improve the therapeutic efficacy of this synergistic drug combination (Fig. 4, Fig. S10). Once ovarian cancer xenografts became palpable, we administered drug formulations at sub-therapeutic levels (5% of CP’s and 20% of Wtmn’s maximum tolerated dose [MTD]). Although higher dosing levels that could completely eliminate tumor burden were possible, this lower dose therapy was initially chosen to allow for more facile monitoring of tumor growth differences between treatment arms.
Fig. 4.
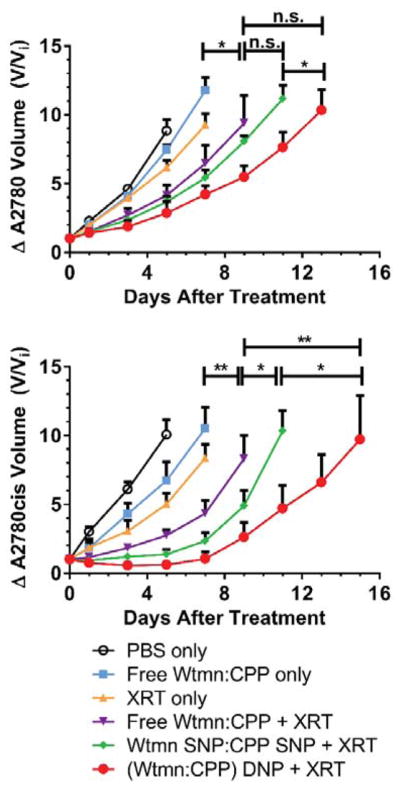
In vivo efficacy of NPs in a) A2780 and b) A2780cis models. Mice were treated with low dose therapy (5% CP MTD and 20 % Wtmn MTD) once tumors became palpable. Data are represented as the change in tumor volume (V/Vi) after treatment (* p ≤ .05, ** p ≤ .01, n.s. = not significant)
Administering CRT with fractionated XRT and free Wtmn:CP (1:1.4) significantly blunted tumor growth in both models versus either XRT or the free drug combination alone. This effect was further enhanced when the drug combination was loaded into NPs and delivered as mixed SNPs. More importantly, the DNPs outperformed all treatment arms in both models. The DNPs were particularly more potent in the PROC model than mixed SNPs (p ≤ 0.05 versus Mixed NPs, p ≤ 0.01 versus Free Drugs + XRT).
We also examined the efficacy of the DNPs versus a much higher, clinically relevant dose of CP (2.5 mg/kg, 8.3 μmol/kg) in A2780cis xenografts (Fig. S11) [31]. Despite the DNPs containing a significantly lower concentration of platinum (0.3 mg/kg, 500 nmol/kg), the DNPs reduced tumor growth rates significantly versus both the high dose CP and the control receiving XRT alone (both p < .001).
Aside from accumulating in tumors via passive targeting, PLGA-PEG NPs accumulate in the liver and kidneys [14,15,32–34]. Furthermore, Wtmn exhibits high hepatotoxicity, which initially led to its abandonment as a potential therapeutic [12,13]. Therefore, we examined serum hepatic, renal, and hematological toxicity markers to determine if the NP co-encapsulation increased off-target side effects in these organs (Table S8). Although the NPs increased the treatment’s potency, the NP formulations did not increase serum liver and kidney toxicity markers above normal levels (Table S8) or cause dramatic shifts in mouse bodyweight (Fig. S12). However, as with most platinum-based chemotherapies, mice did expectedly show leukopenia (Table S8).
We also examined representative tumor sections for increased CP nuclear localization via immunofluorescence to qualitatively corroborate the improvements in tumor drug uptake and treatment efficacy (Fig. 5, S13) [35]. Mice given XRT concurrently with free drugs showed enhanced CP-DNA binding versus mice receiving only the free drug. Mice given CPP SNP form showed greater localization of CP adducts in the nucleus versus either free drug arm. The DNP treatment showed the greatest nuclear accumulation of CP.
Fig. 5.
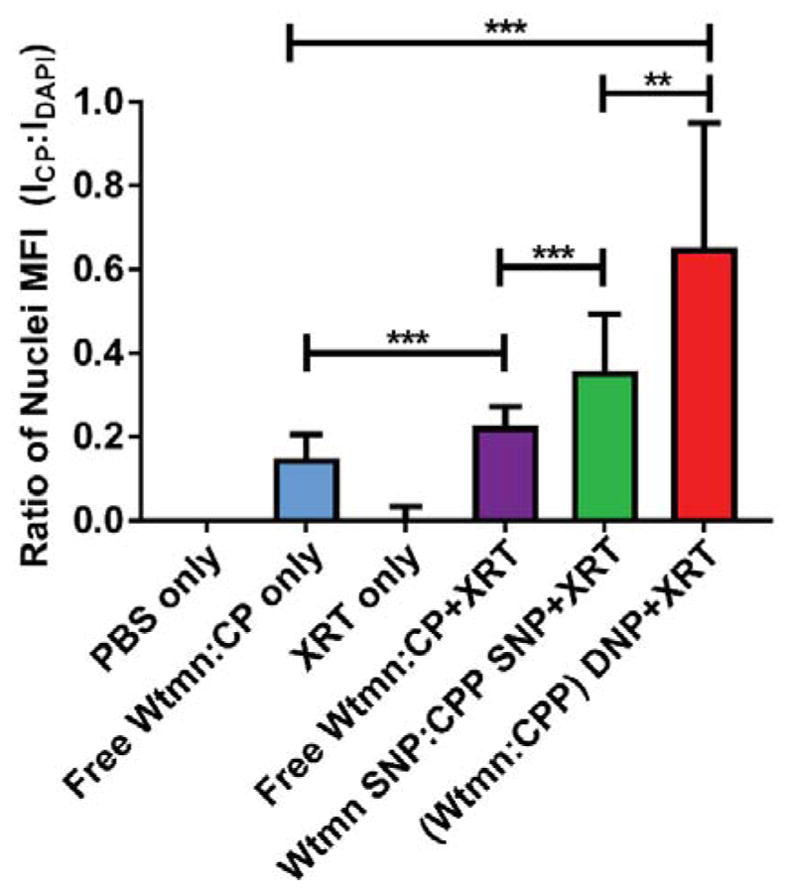
Fluorescent detection of CP nuclear localization in A2780cis tumor sections. The data are represented as the ratio of the mean fluorescent intensity (MFI) between CP (ICP) and DAPI (IDAPI) fluorescence in identified nuclear regions. (* p ≤ .05, ** p ≤ .01, *** p ≤ .001, n.s. = not significant)
Discussion
Platinum based chemotherapy is part of the standard treatment regimen for ovarian cancer [3,5]. Generally, initial response rates to treatment are high, but a significant proportion of ovarian cancer patients will experience recurrence with PROC [2]. This significantly complicates further treatment and greatly increases patient mortality. Therefore, we became interested in new strategies to address PROC. As a solution, Wtmn can potently inhibit the DNA repair pathways that are involved in CP resistance and, therefore, synergistically enhance CP’s cytotoxicity [10,11]. Unfortunately, most of its analogues were abandoned as therapeutics due to its poor in vivo stability, low solubility, and hepatotoxicity [12,13]. We have previously shown that NP delivery of Wtmn alleviates these difficulties by increasing its in vivo solubility, stability, and therapeutic index [13–15]. Additionally, NP vehicles can enhance CP uptake in resistant cells (mechanism #1), limit systemic exposure, and improve its pharmacokinetic profile [6,34,36,37]. However, most importantly, NP co-encapsulation allows for the delivery of drugs in a precise, therapeutically synergistic ratio [6,16–21]. Indeed, co-encapsulation of other therapeutic combinations into a single nanoformulation has proved a powerful strategy to enhance treatment synergy [17,19,20]. Therefore, we reasoned NP co-encapsulation of Wtmn and CP could improve PROC treatment by accurately co-delivering a powerfully synergistic CRT regimen (Fig. 1).
Drug Loading and Release
For this study, we chose to use biocompatible PLGA-PEG based NPs due to their potential for rapid translation into the clinic [38]. Since active CP shows poor loading within hydrophobic PLGA, a fatty acid modified CPP (Fig. S1) was used to allow for facile loading. This octanoic acid modified CPP has been previously shown to easily permeate the cell membrane and release active CP upon reduction by intracellular reductants (e.g., glutathione, ascorbate) [22–26]. Conveniently, cancer cells often have elevated levels of cytosolic glutathione, which can promote CP formation from CPP.
As expected, using this prodrug strategy provided high CPP loading wt% and %EEs when nanoprecipitated at a range of %FR (Fig. 2, S2, Table S1–S2). CPP loaded more efficiently than Wtmn in both the SNP and DNPs. Therefore, higher CPP:Wtmn ratios could easily be generated when co-loading both into a single nanoformulation. In mice, this trend corresponds nicely with greater NP delivery of CP, since higher CP doses (~ 6 mg/kg) are tolerated better than Wtmn (~ 0.7 mg/kg) [13]. However, a high CPP:Wtmn ratio may not necessarily provide the most potent or synergistic formulation (vide infra). In DNPs, CPP actually boosted the loading efficiency of Wtmn versus Wtmn SNPs provided the CPP’s %FR was ≤ 7.5 wt%. Wtmn loading was nearly two-fold higher when co-loaded with a CPP %FR of 7.5 wt%. This potentially provides a useful strategy to boost drug loading efficiency. As the CPP %FR increased past this level, Wtmn loading returned to levels observed in Wtmn SNPs. This increased loading effect could stem from CPP increasing the hydrophobicity of the NP core and, thus, further increasing hydrophobic Wtmn retention. However, at high CPP %FR (> 7.5 wt%), the PLGA core likely becomes saturated with CPP blocking further Wtmn loading. Regardless of this loading variability, we were able to formulate DNPs containing a wide range of Wtmn:CPP molar ratios (2:1 to 1:5) to screen for potentially synergistic treatment ratios.
Despite the drugs interacting during loading, the release of Wtmn and CPP only slowed slightly when co-encapsulated versus the SNPs, and both therapeutics had fully released by 24 hours. This suggests that the SNPs and DNPs would have similar release rates in vivo. albeit, at faster rates than in vitro [37].
Formulation Synergy and Radiosensitization
We determined dose response curves for our various nanoformulations and compared their IC50 values to free drug monotherapies and combinations (Fig. 3, Table S5). For both cell lines, encapsulation of CP in a NP improved the therapeutic response versus the free drug. Previous studies have shown that this effect arises from NPs’ ability to bypass defective CP transporting proteins and deliver CP via a proposed endosomolytic or passive diffusion pathway [6]. However, this effect was modest indicating that reduced CP uptake may play only a minor role in platinum resistance for the A2780 cell lines.
PSOC cells were largely unaffected by any combination of Wtmn and CPP when compared to treating with only CP or CPP SNP. This likely indicates these cells do not significantly upregulate DNA repair to blunt CP action. In contrast, NP delivery of Wtmn and CPP combinations in PROC cells (A2780cis) produced profound enhancements in cytotoxicity (22-fold) and displayed strong treatment synergy (e.g., CI ~0.04, Fa = 0.5, 1:1.4 Wtmn:CPP, Fig. S6, Table S6). These synergistic enhancements suggest that Wtmn effectively blocks A2780cis’ primary CP resistance mechanism of enhanced DNA repair. This observation is further corroborated by the ability of Wtmn containing NPs to blunt DNA repair mechanisms as evidenced by the prolonged elevation of phosphorylated γH2AX foci levels in XRT treated cells (Fig. S6–S7) [27–29]. However, no statistically significant differences existed in vitro between delivering the combination as a mixture of SNPs or together in a DNP. This results from limited barriers to delivery of a precise therapeutic ratio that would be present in vivo [15].
Free drug combinations also showed synergy in A2780cis, but they required an order of magnitude higher dose than the NP arms to elicit the same effect. This difference is likely due to free Wtmn’s poor stability in the extracellular milieu, which also contributed to its abandonment for clinical use. Furthermore, the improved uptake and cytotoxicity of particulate CP versus free CP likely provides an additional synergistic effect that makes the NP drug combination significantly more potent.
In addition to being synergistic, CP and Wtmn radiosensitize cancer cells by inhibiting the mechanisms that repair DNA double strand breaks. Mixtures of SNPs and DNPs both enhanced XRT slightly versus the free drug arms (Fig. 3b). This could result from improved delivery of both components into the cell imparted by the NP vehicles. Although the SER in A2780cis cell lines was modest, employing XRT in the treatment regimen could have other positive effects on treatment efficacy. For example, XRT can remodel the tumor microenvironment (TME) making it more responsive to chemotherapy [39].
Toxicity and In Vivo Efficacy
Wtmn’s hepatotoxicity concerns have greatly limited its application as a chemotherapeutic. However, the NP vehicles exhibited low off-target kidney and liver cytotoxicity (Table S8). Mice dosed with free Wtmn also exhibited low toxicity. However, this is likely due to the low free Wtmn dose (~20% MTD) employed which failed to elicit a comparable therapeutic response. Treatment did lead to a decrease in white blood cell counts, which is common for platinum-based chemotherapies. This condition is often addressed easily in the clinic with stimulating cytokine administration [40].
We initially examined the efficacy of our treatment regimens in both A2780 and A2780cis xenograft murine models (Fig. 4, Fig. S10). In both models, combining XRT and free drug Wtmn:CPP significantly improved treatment versus either XRT or chemotherapy given alone. Therefore, despite only the modest SER improvements in vitro, combined Wtmn:CPP CRT can enhance this regimen for ovarian cancer in vivo. As stated above, RT can improve chemotherapy by other complex mechanisms aside from direct tumor cell killing, such as remodeling the TME or recruiting immune cells that potentiate chemotherapies’ effects. The increased accumulation of CP observed in the nuclei of XRT treated tumors versus the free drug combination further indicates radiation might enhance CP function in resistant cells (Fig. 5, Fig. S13).
DNPs significantly outperformed all treatment arms in both models at a low, sub-therapeutic dose (~20% Wtmn MTD, ~5% CP MTD). Additionally, immunofluorescent imaging of tumor sections showed treatment with DNPs greatly increased the amount of CP-DNA adducts within the nucleus. This improved efficacy versus the free drug + XRT arm could stem from the improved uptake of CPP into tumor cells via alternative pathways and the improved pharmacokinetic profile NP vehicles impart on Wtmn and CPP [41].
However, the causative factors for the efficacy differences between SNP and DNP arms are not as easy to determine. The in vitro release data suggests that the in vivo drug release rates should be relatively the same between SNPs and DNPs, albeit faster. Therefore, it appears unlikely that differences in the drugs’ circulation times led to differences in their efficacy. Instead, the DNPs may improve the therapeutic profile due to their ability to deliver the precise Wtmn:CPP ratio (1:1.4) that produces the greatest synergy [17]. Differences in properties between the SNPs when co-administered could lead to unequal accumulation of one SNP over the other within the tumor bed (Fig. S3–S4, Table S3–4). This would shift the tumor observed dosing ratio. For example, CPP NPs skewed smaller than Wtmn NPs and had more negative zeta potentials. This could lead to differences in their tumor accumulation [15]. In contrast, DNPs would accumulate the precise 1:1.4 ratio, since both drugs are ensconced inside and have similar drug release profiles.
The DNPs given at the same low dose of CPP as above (0.3 mg/kg CPP, 500 nmol) also significantly outperformed (p < .001) a higher, clinical dose of CP (2.5 mg/kg, 8.3 μmol/kg) (Fig. S11) [31]. Generally, CRT with CP at this high dose is administered no more than once every 4 weeks, but the low dose DNP’s superior performance suggests possible alternative routes to improve CP administration. In this work, the in vivo efficacy studies utilized a single, low dose of chemotherapy to allow for facile monitoring of tumor growth differences. Due to the low toxicity and the improved performance of the DNP formulations, it might be possible to administer more regular chemotherapy doses to improve the current regimen. However, further dosing optimization studies were outside the scope of this initial pre-clinical study and ongoing work within our lab seeks to address these possibilities.
Conclusion
Overcoming PROC remains a pressing challenge in oncology clinics. We have demonstrated a CP delivery strategy that takes a two-pronged approach to overcome this resistance by utilizing FDA approved PLGA-PEG NPs that improve CP uptake and deliver Wtmn, a DNA repair inhibitor. Delivery of this combination synergistically reverses CP resistance and improves CP’s therapeutic efficacy in both PSOC and PROC murine models. As platinum resistance is present in other cancers, it is our hope that this work will inspire future investigations of this NP based regimen for not only the treatment of PROC but other cancers as well.
Supplementary Material
Acknowledgments
We would like to thank Colin O’banion for his help analyzing fluorescent images. This work was supported by National Institutes of Health/National Cancer Institute (R01CA17848-01, R21 CA182322, U54CA198999 for Carolina Center of Cancer Nanotechnology Excellence (CCNE)-Nano Approaches to Modulate Host Cell Response for Cancer Therapy. Zachary Rodgers was supported by the Carolina Cancer Nanotechnology T32 Training Program (C-CNTP, NIH- 1T32CA196589)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tapia G, Diaz-Padill I. Ovarian Cancer - A Clin Transl Updat. InTech; 2013. Molecular Mechanisms of Platinum Resistance in Ovarian Cancer. [DOI] [Google Scholar]
- 2.Davis A, Tinker AV, Friedlander M. “Platinum resistant” ovarian cancer: What is it, who to treat and how to measure benefit? Gynecol Oncol. 2014;133:624–631. doi: 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- 3.Markman M. New, expanded, and modified use of approved antineoplastic agents in ovarian cancer. Oncologist. 2007;12:186–90. doi: 10.1634/theoncologist.12-2-186. [DOI] [PubMed] [Google Scholar]
- 4.Markman M. Second-Line Treatment of Ovarian Cancer. Oncologist. 2000;5:26–35. doi: 10.1634/theoncologist.5-1-26. [DOI] [PubMed] [Google Scholar]
- 5.Luvero D, Milani A, Ledermann JA. Treatment options in recurrent ovarian cancer: latest evidence and clinical potential. Ther Adv Med Oncol. 2014;6:229–239. doi: 10.1177/1758834014544121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Min Y, Mao CQ, Chen S, Ma G, Wang J, Liu Y. Combating the Drug Resistance of Cisplatin Using a Platinum Prodrug Based Delivery System. Angew Chemie Int Ed. 2012;51:6742–6747. doi: 10.1002/anie.201201562. [DOI] [PubMed] [Google Scholar]
- 7.Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin Resistance: A Cellular Self-Defense Mechanism Resulting from Multiple Epigenetic and Genetic Changes. Pharmacol Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parker RJ, Eastman A, Bostick-Bruton F, Reed E. Acquired cisplatin resistance in human ovarian cancer cells is associated with enhanced repair of cisplatin-DNA lesions and reduced drug accumulation. J Clin Invest. 1991;87:772–777. doi: 10.1172/JCI115080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 10.Hashimoto M, Rao S, Tokuno O, Yamamoto KI, Takata M, Takeda S, Utsumi H. DNA-PK: the major target for wortmannin-mediated radiosensitization by the inhibition of DSB repair via NHEJ pathway. J Radiat Res. 2003;44:151–9. doi: 10.1269/jrr.44.151. http://www.ncbi.nlm.nih.gov/pubmed/13678345. [DOI] [PubMed] [Google Scholar]
- 11.Okayasu R, Suetomi K, Ullrich RL. Wortmannin inhibits repair of DNA double-strand breaks in irradiated normal human cells. Radiat Res. 1998;149:440–5. http://www.ncbi.nlm.nih.gov/pubmed/9588354. [PubMed] [Google Scholar]
- 12.Wipf P, Halter RJ. Chemistry and biology of wortmannin. Org Biomol Chem. 2005;3:2053. doi: 10.1039/b504418a. [DOI] [PubMed] [Google Scholar]
- 13.Karve S, Werner ME, Sukumar R, Cummings ND, Copp JA, Wang EC, Li C, Sethi M, Chen RC, Pacold ME, Wang AZ. Revival of the abandoned therapeutic wortmannin by nanoparticle drug delivery. Proc Natl Acad Sci. 2012;109:8230–8235. doi: 10.1073/pnas.1120508109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Au KM, Min Y, Tian X, Zhang L, Perello V, Caster JM, Wang AZ. Improving Cancer Chemoradiotherapy Treatment by Dual Controlled Release of Wortmannin and Docetaxel in Polymeric Nanoparticles. ACS Nano. 2015;9:8976–8996. doi: 10.1021/acsnano.5b02913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caster JM, Yu SK, Patel AN, Newman NJ, Lee ZJ, Warner SB, Wagner KT, Roche KC, Tian X, Min Y, Wang AZ. Effect of particle size on the biodistribution, toxicity, and efficacy of drug-loaded polymeric nanoparticles in chemoradiotherapy, Nanomedicine Nanotechnology. Biol Med. 2017 doi: 10.1016/j.nano.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107:17939–44. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CMJ, Zhang L. Nanoparticle-based combination therapy toward overcoming drug resistance in cancer. Biochem Pharmacol. 2012;83:1104–1111. doi: 10.1016/j.bcp.2012.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Miao L, Guo S, Zhang J, Kim WY, Huang L. Nanoparticles with Precise Ratiometric Co-Loading and Co-Delivery of Gemcitabine Monophosphate and Cisplatin for Treatment of Bladder Cancer. Adv Funct Mater. 2014;24:6601–6611. doi: 10.1002/adfm.201401076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lancet JE, Cortes JE, Hogge DE, Tallman MS, Kovacsovics TJ, Damon LE, Komrokji R, Solomon SR, Kolitz JE, Cooper M, Yeager AM, Louie AC, Feldman EJ. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123:3239–3246. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raut L. Novel formulation of cytarabine and daunorubicin: A new hope in AML treatment. South Asian J Cancer. 2015;4:38. doi: 10.4103/2278-330X.149950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Min Y, Caster JM, Eblan MJ, Wang AZ. Clinical Translation of Nanomedicine. Chem Rev. 2015;115:11147–11190. doi: 10.1021/acs.chemrev.5b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci. 2013;110:18638–18643. doi: 10.1073/pnas.1303958110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnstone TC, Lippard SJ. The effect of ligand lipophilicity on the nanoparticle encapsulation of Pt(IV) prodrugs. Inorg Chem. 2013;52:9915–9920. doi: 10.1021/ic4010642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao H, Qi R, Liu S, Hu X, Duan T, Zheng Y, Huang Y, Jing X. Biodegradable polymer – cisplatin(IV) conjugate as a pro-drug of cisplatin(II) Biomaterials. 2011;32:7732–7739. doi: 10.1016/j.biomaterials.2011.06.072. [DOI] [PubMed] [Google Scholar]
- 25.Xiao H, Song H, Yang Q, Cai H, Qi R, Yan L, Liu S, Zheng Y, Huang Y, Liu T, Jing X. A prodrug strategy to deliver cisplatin(IV) and paclitaxel in nanomicelles to improve efficacy and tolerance. Biomaterials. 2012;33:6507–6519. doi: 10.1016/j.biomaterials.2012.05.049. [DOI] [PubMed] [Google Scholar]
- 26.Xiao H, Song H, Zhang Y, Qi R, Wang R, Xie Z, Huang Y, Li Y, Wu Y, Jing X. The use of polymeric platinum(IV) prodrugs to deliver multinuclear platinum(II) drugs with reduced systemic toxicity and enhanced antitumor efficacy. Biomaterials. 2012;33:8657–8669. doi: 10.1016/j.biomaterials.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 27.Kuo LJ, Yang LX. Gamma-H2AX - a novel biomarker for DNA double-strand breaks. In Vivo. n.d;22:305–9. http://www.ncbi.nlm.nih.gov/pubmed/18610740. [PubMed] [Google Scholar]
- 28.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–68. doi: 10.1074/jbc.273.10.5858. http://www.ncbi.nlm.nih.gov/pubmed/9488723. [DOI] [PubMed] [Google Scholar]
- 29.Mah LJ, El-Osta A, Karagiannis TC. γH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia. 2010;24:679–686. doi: 10.1038/leu.2010.6. [DOI] [PubMed] [Google Scholar]
- 30.Radiobiology for the Radiologist. Radiology. (5) 2002;224:512–512. doi: 10.1148/radiol.2242022530. [DOI] [Google Scholar]
- 31.FDA. Cisplatin Injection. 2015:1–11. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/018057s083lbl.pdf.
- 32.Semete B, Booysen L, Kalombo L, Ramalapa B, Hayeshi R, Swai HS. Effects of protein binding on the biodistribution of PEGylated PLGA nanoparticles post oral administration. Int J Pharm. 2012;424:115–120. doi: 10.1016/j.ijpharm.2011.12.043. [DOI] [PubMed] [Google Scholar]
- 33.Rafiei P, Haddadi A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int J Nanomedicine. 2017;12:935–947. doi: 10.2147/IJN.S121881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M, Panagi Z, Avgoustakis K, Reineke J. Physiologically based pharmacokinetic modeling of PLGA nanoparticles with varied mPEG content. Int J Nanomedicine. 2012;7:1345–1356. doi: 10.2147/IJN.S23758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tian J, Min Y, Rodgers Z, Au KM, Hagan CT, Zhang M, Roche K, Yang F, Wagner K, Wang AZ. Co-delivery of paclitaxel and cisplatin with biocompatible PLGA-PEG nanoparticles enhances chemoradiotherapy in non-small cell lung cancer models. J Mater Chem B. 2017;5:6049–6057. doi: 10.1039/C7TB01370A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Avgoustakis K. PLGA–mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79:123–135. doi: 10.1016/S0168-3659(01)00530-2. [DOI] [PubMed] [Google Scholar]
- 37.Avgoustakis K. Pegylated poly(lactide) and poly(lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Deliv. 2004;1:321–33. doi: 10.2174/1567201043334605. [DOI] [PubMed] [Google Scholar]
- 38.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Préat V. PLGA-based nanoparticles: An overview of biomedical applications. J Control Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 39.Kershaw MH, Devaud C, John LB, Westwood JA, Darcy PK. Enhancing immunotherapy using chemotherapy and radiation to modify the tumor microenvironment. Oncoimmunology. 2013;2:e25962. doi: 10.4161/onci.25962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Confer DL, Miller JP. Long-term safety of filgrastim (rhG-CSF) administration. Br J Haematol. 2007;137:77–78. doi: 10.1111/j.1365-2141.2007.06524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105:17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.