

Abstract
Herein we report the design, synthesis, X-ray structural, and biological studies of an exceptionally potent HIV-1 protease inhibitor, compound 5 ((3S,7aS,8S)-hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl ((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate). Using structure-based design, we incorporated an unprecedented 6-5-5-ring-fused crown-like tetrahydropyranofuran as the P2-ligand, a cyclopropylaminobenzothiazole as the P2′-ligand, and a 3,5-difluorophenylmethyl group as the P1-ligand. The resulting inhibitor 5 exhibited exceptional HIV-1 protease inhibitory and antiviral potency at the picomolar level. Furthermore, it displayed antiviral IC50 values in the picomolar range against a wide panel of highly multidrug-resistant HIV-1 variants. The inhibitor shows an extremely high genetic barrier against the emergence of drug-resistant variants. It also showed extremely potent inhibitory activity toward dimerization as well as favorable central nervous system penetration. We determined a high-resolution X-ray crystal structure of the complex between inhibitor 5 and HIV-1 protease, which provides molecular insight into the unprecedented activity profiles observed.
Keywords: antiviral agents, brain penetration, drug resistance, HIV-1 protease, structure-based design
Graphical abstract
Crystal clear: We report the structure-based design, synthesis, biological evaluation, and X-ray structural studies of a series of exceptionally potent HIV-1 protease inhibitors containing novel P1, P2, and P2′ ligands to interact with active site residues. Inhibitor 5 shows exceptional HIV-1 inhibitory and antiviral potency at the picomolar level. An X-ray structure of the inhibitor 5-bound HIV-1 protease complex provides molecular insight into the unprecedented activity profiles.
Introduction
The advent of combined antiretroviral therapy (cART) in the late 1990s is the most significant development in the treatment of patients with HIV-1 infection and AIDS.[1,2] The introduction of these drug regimens using a combination of reverse transcriptase and protease inhibitor drugs dramatically suppressed viral replication, decreased plasma HIV-1 viral loads, and improved CD4 cell counts in HIV-1-infected patients.[3,4] cART has significantly improved the life expectancy of patients who have access to these treatment regimens. There is currently no cure for HIV-1 infection or AIDS.[5] However, the mortality rates of HIV/AIDS patients are approaching those of the general population.[6,7] Despite these advances, the growing emergence of multidrug-resistant HIV-1 variants has raised serious concerns for the long-term viability of current treatment options.[8,9] Among cART, HIV-1 protease inhibitor (PI) drugs generally show a high genetic barrier against resistance. The latest approved PI, darunavir (DRV, 1, Figure 1), is the only PI drug recommended for first-line therapy.[10–12] It exhibits a high genetic barrier and it is the most widely prescribed PI for treatment-experienced and treatment-naïve patients with HIV-1 infection and AIDS.[13] This is possibly due to darunavir’s dual mechanism of action as 1) an inhibitor of catalytic HIV-1 protease, and 2) an inhibitor of the dimerization of protease monomers.[14,15] The current first-line cART with boosted PI-based and integrase-inhibitor-based regimens are effective against developing drug-resistant HIV-1 variants over an extended period of time. However, there are limitations, including side effects, toxicity, and rapid development of drug-resistant variants for some HIV/AIDS patient groups.[16,17] Furthermore, current cART regimens are ineffective for patients with HIV-associated neurocognitive disorders (HAND). A sub-therapeutic drug concentration in the central nervous system (CNS) may lead to the development of resistance and less virologic suppression in the CNS.[18–20]
Figure 1.
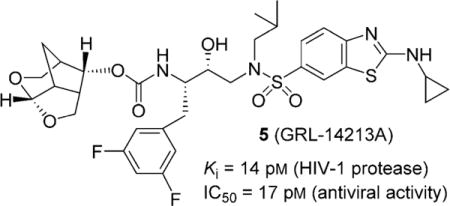
Structures of HIV-1 protease inhibitors 1–5.
In an effort to address the above issues, our PI design strategy is aimed at targeting the protein backbone to develop PIs that are very potent with greater specificity for HIV-1, show high genetic barrier to the emergence of multidrug-resistant HIV-1 variants, and are capable of penetrating the blood–brain barrier.[10, 21–23] Herein, we report a novel HIV-1 PI containing a 6-5-5-ring-fused crown-like tetrahydropyranofuran as the P2-ligand, an aminobenzothiazole as the P2′-ligand, and a two-fluorine containing phenylalanine side chain as the P1-ligand. The resulting PI exhibited unprecedented antiviral activity against a wide range of highly multidrug-resistant HIV-1 variants, and displayed high genetic barrier against the emergence of resistant variants. It also displayed very potent inhibition of dimerization of protease monomers and favorable CNS-penetrating capacity. We carried out X-ray structural studies of inhibitor-bound HIV-1 protease complex to gain structural insight.
Since our design and discovery of DRV, our continuing efforts focused on developing PIs that exhibit robust antiviral potency against a variety of existing multi-PI-resistant variants.[10, 24, 25] Our objectives are that these PIs need to significantly delay the emergence of HIV-1 variants resistant to the same PI, show better CNS penetration, and exhibit potent dimerization inhibition activity.[26] Toward these goals, we have reported a variety of novel PIs that meet many of these critical objectives. Recently, we reported the design and synthesis of a new class of PIs incorporating an unprecedented 6-5-5-ring-fused crown-like tetrahydropyranofuran (crn-THF) as the P2-ligand and cyclopropylamino-benzothiazole (Cp-Abt) as the P2′-ligand on an (R)-hydroxyethylsulfonamide isostere as the transition-state mimic.[23, 27] Inhibitor 3 showed superior antiviral activity and drug resistance over DRV (1). Our detailed structural analysis also provided molecular insight into its exceptional drug-resistance profiles. Based on this structural insight, we have further optimized ligand-binding site interactions in the active site of HIV-1 protease to improve the drug properties of inhibitor 5. Our overall design objectives were to preserve all critical hydrogen bonding interactions with the backbone atoms of the HIV-1 protease active site, while enhancing van der Waals interactions in the active site. In particular, we speculated that the incorporation of fluorine atoms in the P1-ligand would improve van der Waals contacts in the S1-subsite. Our hope is that these fluorine atoms in this PI would increase lipophilicity which would improve drug penetration in the CNS.[28, 29] Indeed, inhibitor 5, with a 3,5-difluorobenzyl moiety as the P1-ligand resulted in an exceptionally potent PI. This inhibitor exhibited unprecedented drug properties.[23] We have also prepared monofluoro derivatives and compared their antiviral activities against multidrug-resistant HIV-1 variants.
Synthesis of fluoroisosteres and inhibitors
Our synthesis of the 3,5-difluorobenzene-containing hydroxyethylamine sulfonamide isostere precursor in optically active form (compound 9) and its monofluoro counterparts 10 and 11 is shown in Scheme 1. Commercially available 1-bromo-3,5-difluorobenzene 6a was converted into the corresponding Grignard reagent. Reaction of the resulting 3,5-difluorophenyl-magnesium bromide with commercially available butadiene monoxide in the presence of a catalytic amount of CuCN at −78°C provided allylic alcohol 7a in 60% yield.[30] This was subjected to Sharpless asymmetric epoxidation[31, 32] using (−)-diethyl-D-tartrate to provide the corresponding epoxide in very good yield. Regioselective epoxide opening of the resulting epoxide with TMSN3 and Ti(OiPr)4 afforded azido diol 8a in 68% yield over two steps.[33] Reaction of this diol with 2-acetoxyisobutyryl chloride in CHCl3 at 0°C → 23°C provided the chloroacetate derivative, which was treated with NaOMe in THF at 23°C to afford azidooxirane 9 in 77% yield over two steps.[34] Monofluoro oxirane derivatives 10 and 11 were prepared by following the above sequence of reactions using commercially available fluorobromides (see the Experimental Section below).
Scheme 1.
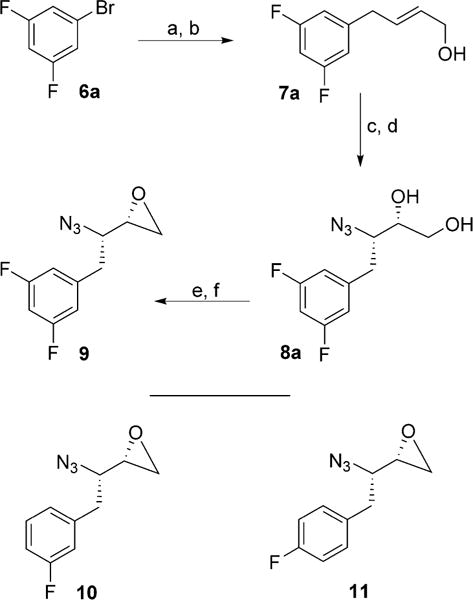
Synthesis of azidoepoxide 9: a) Mg, I2, THF, 23°C; b) butadiene-monoxide, CuCN (cat.), −78°C (60% over two steps); c) tBuOOH, (−)-DET, Ti(OiPr)4, CH2Cl2, −20°C; d) Ti(OiPr)4, TMSN3, PhH, (68% over two steps); e) AcOMe2CCOCl, CHCl3, 0°C→23°C; f) NaOMe, THF, 23°C, (77% over two steps).
Conversion of azidooxiranes into the corresponding fluoroisosteric amines 13 and 17 is shown in Scheme 2. Reaction of azidooxirane 9 with isobutylamine in isopropanol at 65°C for 12 h afforded amino alcohol 12a in excellent yield. Treatment of amino alcohol 12a with commercially available 4-nitrobenzenesulfonyl chloride in the presence of Et3N in CH2Cl2 at 23°C for 12 h provided the corresponding sulfonamide derivative in 67% yield from 9. Catalytic hydrogenation of the resulting azidosulfonamide derivative with Pd(OH)2 in EtOAc at 23°C for 12 h afforded diamine derivative 13 in 89% yield.[35] Amino alcohol 12a was converted into cyclopropylaminobenzothiazole derivatives as follows: treatment of amino alcohol 12a with sulfonyl chloride 14 in the presence of Et3N at 23°C for 12 h furnished sulfonamide derivative 15a in excellent yield.[27] This was subjected to Staudinger reduction[36] with Ph3P in aqueous THF at 23°C for 24 h to provide the corresponding amine. The resulting amine was reacted with (Boc)2O in the presence of NaHCO3 to provide Boc derivative 16a in very good yield. This was converted into aminobenzothiazole derivative 17 as follows: methyl sulfide was oxidized to its sulfone derivative with mCPBA in CH2Cl2 at 0°C→23°C for 12 h. Treatment of the resulting sulfone with cyclopropylamine in THF at 65°C for 12 h afforded the aminobenzothiazole derivative.[27] Removal of the Boc group by exposure to trifluoroacetic acid (TFA) in CH2Cl2 at 0°C→23°C for 1 h furnished difluoroisostere 17 in excellent yield. The synthesis of monofluoro isosteres 18 and 19 is shown in Scheme 3. As shown, reaction of oxiranes 10 and 11 with isobutylamine furnished amino alcohol derivatives 12b and 12c. These amino alcohols were reacted with sulfonyl chloride 14 to provide sulfonamide derivatives 15b and 15c in excellent yields. Reduction of azide with Ph3P followed by protection of the resulting amines gave Boc derivatives 16b and 16c. The methylbenzothiazole derivatives were converted into monofluorinated isosteric amines 18 and 19 by following the same sequence of reactions as for the difluorinated isostere 17.
Scheme 2.
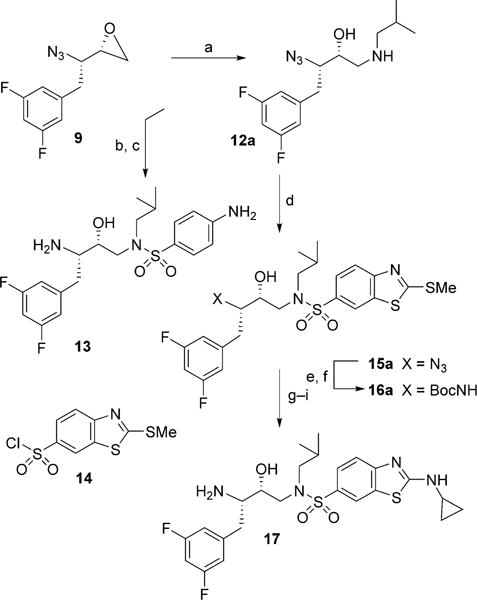
Synthesis of sulfonamide isosteres 13 and 17: a) iBuNH2, iPrOH, 65°C; b) 4-NO2-PhSO2Cl, Et3N, CH2Cl2, 23°C (67% over two steps); c) H2, Pd(OH)2-C, EtOAc, 23°C (89%); d) 14, CH2Cl2, Et3N, 23°C, (90% over two steps); e) Ph3P, THF/H2O (3:1), 23°C; f) Boc2O, THF/H2O (1:1), NaHCO3, 23°C, (87% over two steps); g) mCPBA, CH2Cl2, 23°C; h) cyclopropylamine, THF, 65°C, (91% over two steps); i) TFA, CH2Cl2, 0°C (99%).
Scheme 3.
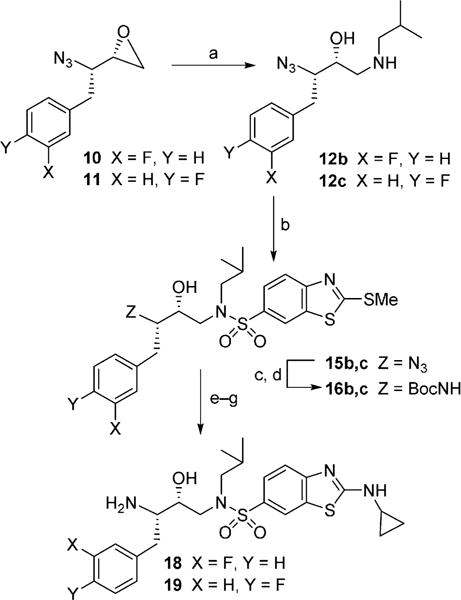
Synthesis of sulfonamide isosteres 18 and 19: a) iBuNH2, iPrOH, 65°C; b) 14, CH2Cl2, Et3N, 23°C, (87% over two steps for 15b and 95% over two steps for 15c); c) Ph3P, THF/H2O (3:1), 23°C; d) Boc2O, THF/H2O (1:1), NaHCO3, 23°C (83% over two steps for 16b and 70% over two steps for 16c); e) mCPBA, CH2Cl2, 23°C; f) cyclopropylamine, THF, 65°C, (97% over two steps for 18 and 98% over two steps for 19); g) TFA, CH2Cl2, 0°C.
Synthesis of inhibitors with fluoroisosteres is shown in Scheme 4. Synthesis of optically active crn-THF alcohol and its corresponding activated carbonate 20 was described recently.[27] Reaction of 4-aminobenzenesulfonamide derivative 13 with 3,5-difluorophenylmethyl as the P1-ligand and activated carbonate 20 in the presence of diisopropylethylamine (DIPEA) in CH3CN at 23°C for 72 h furnished inhibitor 4 in 88% yield. Similarly, reaction of activated carbonate 20 with fluoroisostere 17 in CH3CN under similar conditions afforded inhibitor 5 in 99% yield. Monofluoro derivatives 18 and 19 were reacted with activated carbonate 20 to provide inhibitors 21 and 22 in 90% and 94% yields, respectively.
Scheme 4.
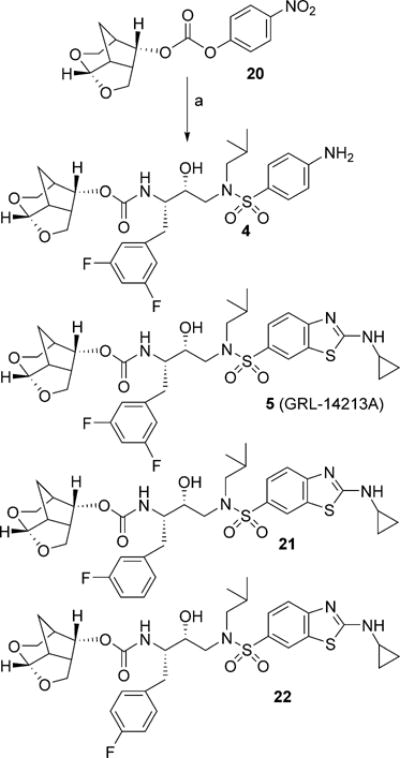
Synthesis of PIs: a) amines 13, 17–19 and carbonate 20, DIPEA, CH3CN, 23°C.
Results and Discussion
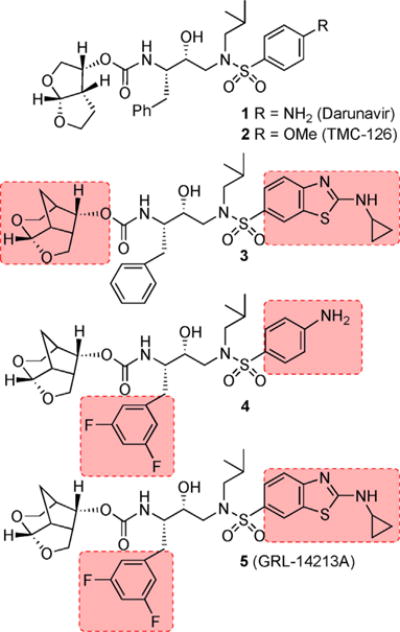
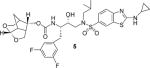
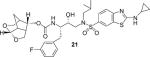
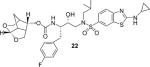
We recently reported a new class of crn-THF-derived inhibitors as represented in PI 3.[27] In this class of compounds we were able to demonstrate how the replacement of a bis-THF with a crn-THF P2-ligand led to enhanced van der Waals interactions in the S2 site. These increased hydrophobic interactions, in addition to the robust hydrogen bond interaction pattern of a cyclopropylaminobenzothiazole-based P2′-ligand, led to inhibitor 3, which exhibited unprecedented potency, particularly against multidrug-resistant HIV-1 strains. Based on our examination of the X-ray crystal structure of DRV and HIV-1 protease complex as well as our crn-THF-derived inhibitor 3-bound HIV-1 protease, we planned further modifications to improve ligand-binding site interactions.[27, 37] In particular, we speculated that the insertion of a fluorine atom on the P1-phenyl ring may lead to effective van der Waals interactions with Pro81′ and Val82′ residues in the active site. To test this hypothesis, we designed and synthesized inhibitors 4 and 5 with 3,5-difluoro derivatives. We also prepared inhibitors 21 and 22 with monofluorophenylmethyl derivatives as the P1-ligand. The structure and activity of these inhibitors are shown in Table 1. For determination of HIV-1 protease inhibitory activity, we used the assay protocol reported by Toth and Marshall.[38] As can be seen, inhibitor 4, with crn-THF as the P2-ligand and 2-fluorophenylmethyl as the P1-ligand, showed enzyme inhibitory potency with Ki value of 4 pm. Antiviral activity of this inhibitor was determined in MT-2 human-T-lymphoid cells exposed to HIVLAI.[17] Compound 4 displayed an antiviral IC50 value of 1.7 nm compared to IC50 values of 2.8 nm for DRV. Incorporation of cyclopropylaminobenzothiazole as the P2′-ligand resulted in inhibitor 5, with a Ki value of 14 pm. It showed antiviral IC50 value of 0.017 nm, a more than 150-fold potency enhancement over DRV and a 100-fold improvement over compound 4. The importance of difluoro substitution on the P1-ligand is clearly evident, as this inhibitor is over 15-fold more potent than inhibitor 3 with no fluorine atoms. Inhibitor 21, with 3-fluorophenylmethyl derivative as the P1-ligand, showed an HIV-1 protease inhibitory Ki value of 0.3 pm and an antiviral IC50 value of 0.055 nm, a significant improvement over p-aminosulfonamide derivative 4, but less potent than difluoro derivative 5. Antiviral IC50 values for inhibitors 5 and 21 were 0.017 ± 0.004 and 0.055 ± 0.0024 nm, respectively, in the same assay. The corresponding inhibitor 22, with a 4-fluorophenylmethyl as the P1-ligand, showed higher enzyme Ki and antiviral IC50 values than the 3-fluoro derivative 21. Overall, inhibitor 5, with 3,5-difluoro groups on the P1-ligand, displayed superior antiviral potency over sulfonamide derivative 4 and monofluoro derivatives 21 and 22.
Table 1.
Structures and activity of inhibitors 3–5 and 21–23.
Values are the mean of at least five data points; standard error in all cases was <7%. Darunavir Ki: 16 pM.
Antiviral IC50 values are the means of at least three experiments; standard error in all cases was <5%. Darunavir IC50: 1.6 nM.
As mentioned above, PI drugs, particularly DRV, are widely used for the treatment of naïve and experienced patients. Nonetheless, heavily ART-regimen-experienced HIV/AIDS patients are showing treatment-failure with currently approved PIs, including DRV.[13, 17] As a result, our inhibitor design is focused on developing PIs that can maintain robust antiviral activity against the existing multi-PI-resistant HIV-1 variants. Thus, we evaluated inhibitor 5 against DRV-resistant HIV-1 variants and compared antiviral IC50 values against approved PIs and inhibitors 3 and 4.
In these studies, MT-4 cells (1 × 104) were exposed to wild-type HIV-1, HIV-1NL4-3, and three highly DRV-resistant HIV-1 variants: HIVDRVRP20, HIVDRVRP30, and HIVDRVRP5.[17, 39] These were selected by propagating in the presence of increasing concentrations of DRV.[17] These resistant variants are highly resistant to all currently approved PIs, including DRV and nucleoside reverse transcriptase inhibitors such as tenofovir.[17, 23, 39] Antiviral IC50 values were determined using the p24 assay, and the results are listed in Table 2. As observed in our investigation, ATV failed to block the replication of these DRV-resistant HIV-1 variants. Inhibitor 4, containing crn-THF and difluorophenylmethyl as the P1-ligand and 4-aminobenzenesulfonamide as the P2′-ligand, showed similar antiviral activity against wild-type HIVNL4-3; however, its activity against DRV-resistant variants was significantly improved (Table 2). Inhibitor 3, containing crn-THF and Cp-Abt as the P2 and P2′-ligands and no fluorines at the P1-phenyl ring, blocked the replication of HIV-1NL4-3 14- to 23-fold better than DRV and inhibitor 4. Also, this inhibitor more effectively blocked the replication of all three highly DRV-resistant HIV-1 variants than inhibitor 4.
Table 2.
Antiviral activity of three novel compounds against highly DRV-resistant HIV-1 variants.
| Variant | IC50 [nM] ± SD (fold change)[a] | ||||
|---|---|---|---|---|---|
| ATV | DRV | 4 | 3 (GRL-12113A) | 5 (GRL-14213A) | |
| cHIVNL4-3WT | 5.2 ± 0.3 | 3.7 ± 0.7 | 5.9 ± 0.4 | 0.26 ± 0.03 | 0.016 ± 0.017 |
| HIVDRVRP20 | > 1000 (> 192) | 60 ± 6 (16) | 24 ± 9 (4) | 0.18 ± 0.1 (0.7) | 0.0016 ± 0.002 (0.1) |
| HIVDRVRP30 | > 1000 (> 192) | 134 ± 43 (36) | 11 ± 13 (1.9) | 1.4 ± 0.3 (5.3) | 0.015 ± 0.015 (1) |
| HIVDRVRP51 | > 1000 (> 192) | 4672 ± 1263 | 486 ± 52 (82) | 34 ± 10 (131) | 2.8 ± 0.2 (175) |
MT-4 cells (1 × 104) were exposed to 50 TCID50s of wild-type HIV-1NL4-3 or highly DRV-resistant HIV-1 variants (HIV-1DRVRP20, HIV-1DRVRP30, or HIV-1DRVRP51) and cultured in the presence of various concentrations of each compound, and IC50 values were determined by p24 assay. All assays were conducted in triplicate, and the data shown are mean values (± 1 standard deviation) derived from the results of two independent experiments. The amino acid substitutions identified in the protease of HIV-1DRVRP20 HIV-1DRVRP30, and HIV-1DRVRP51 relative to HIV-1NL4-3 include L10I/I15V/K20R/L24I/V32I/M36I/M46L/L63P/A71T/V82A/L89M, L10I/I15V/K20R/L24I/V32I/M36I/M46L/L63P/A71T/V82A/I84V/L89M, and L10I/I15V/K20R/L24I/V32I/L33F/M36I/M46L/I54M/L63P/A71T/V82I/I84V/L89M, respectively.
Inhibitor 5, resulting from the incorporation of two fluorine atoms on the P1-ligand of inhibitor 4, showed remarkable antiviral activity in the low picomolar range against two DRV-resistant HIV-1 variants (HIVDRVRP20 and HIVDRVRP30), and IC50 values were 1.6 and 15 pm, respectively. In contrast, DRV exhibited a 16-fold increase in its IC50 values (IC50=60 nm) relative to its IC50 value against HIV-1NL4-3. Interestingly, inhibitor 5 displayed over 10-fold improvement of its IC50 against HIVDRVRP20 compared with its IC50 against HIV-1NL4-3. Furthermore, inhibitor 5 exhibited similar IC50 values against HIVDRVRP30 (IC50 = 15 pm) relative to its IC50 value against HIV-1NL4-3. Against the most highly DRV-resistant HIV variants, HIVDRVRP51, inhibitor 5 showed 175-fold loss of antiviral activity (IC50=2.8 nm) compared with its IC50 value against wild-type virus. In comparison, DRV displayed >1000-fold increase in its IC50 value against HIVDRVRP51 (IC50 = 4672 nm). Inhibitor 5 also exerted very potent antiviral activity against the HIV-2 variants.[23] In addition, inhibitor 5 showed a much improved cytotoxicity profile, with a selectivity index (CC50/IC50) as high as 2473 0684 relative to DRV (41 562).[23]
We further investigated the ability of both inhibitors 3 and 5 against seven resistant variants selected in vitro against each of seven FDA-approved PIs. These drug-resistant HIV-1 variants were selected by propagating HIV-1NL4-3 or a mixture of 11 multidrug-resistant HIV-1 clinical isolates in the presence of increasing concentrations (up to 5 or 15 μm) of each FDA-approved PI in MT-4 cells.[17, 40] The results are listed in Table 3. ATV failed to block the replication of these variants effectively. DRV contrasted favorably compared with ATV and DRV variants, with IC50 values ranging from 6.1 to 275 nm, with fold changes of 1.6- to 74-fold. Inhibitor 3, without fluorines, exhibited very potent antiviral activity against all seven resistant HIV-1 variants, showing IC50 values ranging from 2.6 pm to 0.27 nm.[27] Inhibitor 5 exhibited unprecedented antiviral activity against all seven highly P1-resistant HIV-1 variants. This inhibitor showed antiviral IC50 values ranging from 2.4 fM (0.0000024 nm) to 0.02 nm against these drug-resistant HIV-1 variants. While inhibitor 3 very effectively blocked the replication of highly PI-resistant HIV-1 variants, the corresponding fluorinated inhibitor 5 has shown superior antiviral activity over inhibitor 3 without fluorine. Of particular note, inhibitor 5 has shown an extremely high genetic barrier to the emergence of HIV-1 variants resistant to this inhibitor.[23]
Table 3.
Antiviral activity of inhibitor 5 (GRL-14213A) against highly PI-resistant HIV-1 variants.
| Variant | Mean IC50 in nM ± SD (fold-change)[a] | |||
|---|---|---|---|---|
| ATV | DRV | 3 (GRL-12113A) | 5 (GRL-14313A) | |
| HIVNL4-3WT | 5.2 ± 0.3 | 3.7 ± 0.7 | 0.39 ± 0.06[b] | 0.016 ± 0.017 |
| HIVSQV-5 μM | 548 ± 177 (105) | 22 ± 6 (5.9) | 0.03 ± 0.01 (0.08)[b] | 0.00020 ± 0.00002 (0.01) |
| HIVIDV-5 μM | 59 ± 13 (11) | 42 ± 1 (11) | 0.012 ± 0.013 (0.03)[b] | 0.00025 ± 0.00035 (0.02) |
| HIVNFV-5 μM | 19 ± 1 (3.7) | 6.1 ± 0.8 (1.6) | 0.028 ± 0.02 (0.07)[b] | 0.00068 ± 0.00045 (0.04) |
| HIVAPV-5 μM | 2.4 ± 0.7 (0.5) | 43 ± 22 (12) | 0.27 ± 0.1 (0.7)[b] | 0.00001 ± 0.00001 (0.0006) |
| HIVLPV-5 μM | 36 ± 9 (7) | 275 ± 64 (74) | 0.0026 ± 0.0006 (0.007)[b] | 0.0000024 ± 0.0000016 (0.0002) |
| HIVATV-5 μM | > 1000 (> 192) | 26 ± 1 (7) | 0.19 ± 0.06 (0.5)[b] | 0.02 ± 0.01 (1.3) |
| HIVTPV-15 μM | > 1000 (> 192) | 41 ± 3 (11) | 0.057 ± 0.03 (0.15)[b] | 0.00037 ± 0.00007 (0.02) |
MT-4 cells (1 ×104) were exposed to 50 TCID50s of wild-type HIV-1NL4-3 or highly Pl-resistant HIV-1 variants (HIV-1SQV-5 μM, HIV-1IDV-5 μM, HIV-1NFV-5 μM, HIV-1APV-5 μM, HIV-1LPV-5 μM, HIV-1ATV-5 μM, or HIV-1TPV-5 μM) and cultured in the presence of various concentrations of each compound, and the IC50 values were determined by p24 assay. All assays were conducted in triplicate, and the data shown are mean values (± 1 standard deviation) derived from the results of two independent experiments. The amino acid substitutions identified in protease of HIV-1SQV-5 μM, HIV-1IDV-5 μM, HIV-1NFV-5 μM, HIV-1APV-5 μM, HIV-1LPV-5 μM, HIV-1ATV-5 μM, and HIV-1TPV-15 μM relative to HIV-1NL4-3 include L10I/N37D/G48V/I54V/L63P/G73C/I84V/L90M, L10F/L24I/M46I/I54V/L63P/A71V/G73S/V82T, L10F/K20T/D30N/M46I/A71V/T74S, L10F/M46I/I50V/I84V, L10F/V32I/M46I/I47A/A71V/I84V, L23I/E34Q/K43I/M46I/L50L/G51A/L63P/A71V/V82T, and L10I/L33I/M36I/M46I/I54VK55R/I62V/L63P/A71V/G73S/V82T/L90M/I93L, respectively.
Values are from reference [27].
The dimerization of HIV-1 protease subunits is an essential process for proteolytic activity. Therefore, disruption of the dimerization process can inhibit HIV-1 maturation. We previously reported that DRV potently inhibited the dimerization of HIV-1 protease monomer subunits.[39, 40] This was demonstrated by a fluorescence resonance energy transfer (FRET)-based HIV-1 expression assay.[15] The majority of other PIs do not show dimerization inhibitory activity. We examined the dimerization inhibition properties of inhibitor 5. As shown in Figure 2, we observed that in the absence of inhibitor, the mean CFPA/B ratio was 1.07, which indicates that protease dimerization clearly occurred. In the presence of 100 nm DRV, the ratio decreased to 0.89, demonstrating that DRV blocks dimerization of the protease subunits. However, inhibitor 5 was found to block the dimerization of protease subunits much more effectively at 0.1 nm, significantly lower than is the case for DRV. It appears that inhibitor 5 very potently inhibits dimerization by at least a factor of 1000-fold relative to DRV. These data demonstrate that inhibitor 5, like DRV, exhibits a dual mechanism of action on HIV-1 protease: 1) inhibition of dimerization, and 2) inhibition of proteolytic activity.
Figure 2.
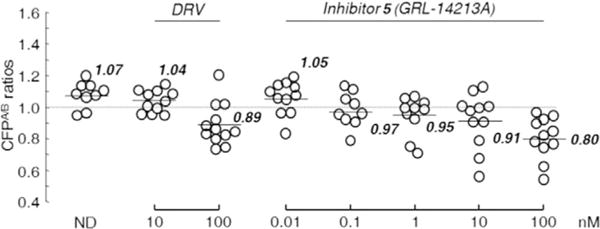
Inhibition of HIV-1 protease dimerization activity by GRL-14213A. COS-7 cells were exposed to various concentrations (0.01–100 nm) of GRL-142 or DRV and were subsequently co-transfected with two plasmids, pHIV-PRWT-CFP and pHIV-PRWT-YFP, respectively. After 72 h, cultured cells were examined in the FRET-based HIV-1 expression assay, and the CFPA/B ratios (y axis) were determined. The arithmetic mean values of the ratios obtained are shown as horizontal bars. A CFPA/B ratio >1 signifies protease dimerization, whereas a ratio <1 signifies disruption of protease dimerization. All experiments were conducted in a blind fashion. P values were determined using the Wilcoxon rank-sum test (JMP software, SAS, Cary, NC, USA) and were 0.3816 for the CFPA/B ratio in the absence of drug (CFPA/BNo-Drug) versus the CFPA/B ratio in the presence of 10 nm DRV (CFPA/B10-DRV), 0.004 for CFPA/BNo-Drug versus CFPA/B100-DRV, 0.8192 for CFPA/BNo-Drug versus CFPA/B0.01-GRL-142, 0.0459 for CFPA/BNo-Drug versus CFPA/B0.1-GRL-142, 0.0197 for CFPA/BNo-Drug versus CFPA/B1-GRL-142, 0.0248 for CFPA/BNo-Drug versus CFPA/B10-GRL-142, and 0.0003 for CFPA/BNo-Drug versus CFPA/B100-GRL-142.
We examined the CNS penetration of inhibitor 5 in rats and compared that with DRV.[23] Both inhibitor 5 and DRV were administered orally in two rats at a dose of 5 mgkg−1 with ritonavir (8.33 mgkg−1). The Cmax values were achieved at 360 min for inhibitor 5 and 90 min for DRV. The concentrations of inhibitor 5 in brain were 7.24 ± 9.65 and 32.6 ± 1.4 nm in 60 and 360 min, respectively. A brain concentration of 32.6 nm indicates an approximate 1880-fold greater drug level with respect to its IC50 value and is approximately 114-fold greater than its IC95 value for inhibitor 5.[23] These data strongly suggest that inhibitor 5 could block HIV-1 replication in the brain.
X-ray description
To gain molecular insight into the ligand-binding site interactions of inhibitor 5 (GRL-14213A), we co-crystallized this inhibitor with wild-type HIV-1 protease, and the X-ray structure was refined at a resolution of 1.67 Å. The structure was determined at a higher resolution than our recently reported structure with 2.0 Å resolution.[23] The complex crystallized in orthorhombic space group P21212 with one protease homodimer per asymmetric unit. The inhibitor binds in the active site in two alternate conformations related by 180° rotation with relative occupancy of 0.55/0.45. The overall dimer structure can be compared with the complex between HIV-1 protease and DRV[37] with a low RMSD of 0.2 Å for 198 equivalent Cα atoms. The largest deviation of 0.7 Å on comparison of the two structures occurs at residue Phe53′ in the flap region. Inhibitor 5 retains all the hydrogen bonds observed between DRV and the main-chain atoms of protease. Our previously reported inhibitor 5–HIV-1 protease complex was determined in P61 space group at 2 Å resolution, and the structure shows good similarity with an RMSD of 0.6 Å for 198 equivalent Cα atoms.[23] Large deviations of 1.8 Å and 1.9 Å occur at Gly40 and Pro44′, respectively, likely due to differences in the crystal packing contacts of the two structures. The key hydrogen bond interactions between inhibitor 5 and HIV-1 protease are shown in Figure 3. An overlay of X-ray structures of DRV-bound HIV-1 protease and inhibitor 5-bound HIV-1 protease is shown in Figure 4.
Figure 3.
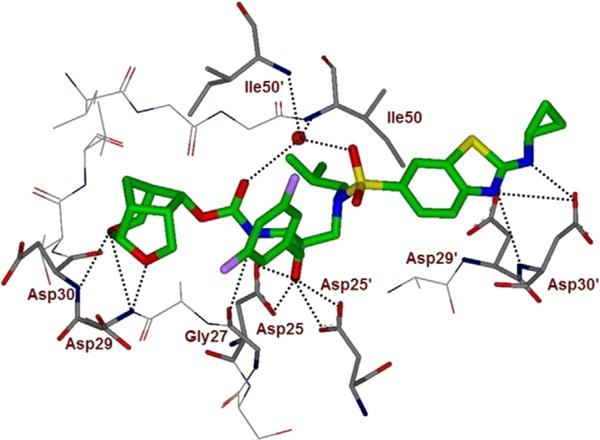
Inhibitor 5-bound X-ray structure of HIV-1 protease. The major orientation of the inhibitor is shown. The inhibitor carbon atoms are shown in green, water molecules are red spheres, and the hydrogen bonds are indicated by dotted lines (PDB ID: 6BZ2). Halogen interactions of the fluorine atoms are omitted for clarity.
Figure 4.
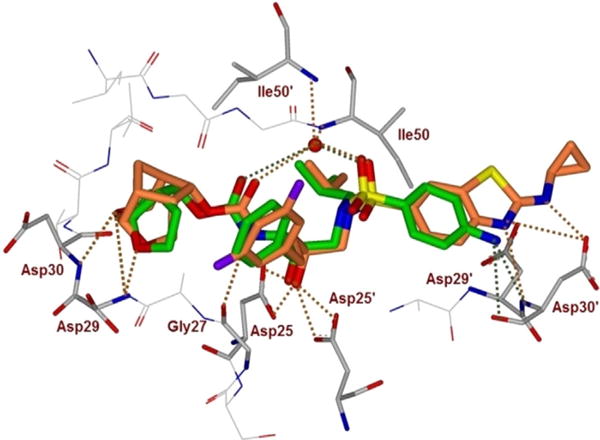
An overlay of the X-ray crystal structure of DRV-bound HIV-1 protease (green) with the X-ray structure of inhibitor 5 (orange)-bound HIV-1 protease.
In the present high-resolution structure, two fluorine atoms on the P1-ligand of inhibitor 5 form important halogen bond interactions with the protease. As shown in Figure 5, one of the fluorine atoms interacts with the tips of both flaps by forming a strong polar interaction (2.9 Å distance) with the main-chain NH group of Ile50 (C–F⋯H–N) and an orthogonal multipolar interaction (C–F⋯C–O) with the main-chain carbonyl group of Gly48 with an F⋯C distance of 2.8–2.9 Å.[41, 42] In addition, this fluorine forms hydrophobic interactions with Gly48, Ile50, and Pro81′. The second fluorine atom forms multipolar interactions with the guanidinium group of Arg8′, which is involved in a critical inter-subunit ion pair with Asp29. The second fluorine atom also has van der Waals contact with Val82′. The P2 group contains the crn-THF instead of bis-THF in DRV. Both oxygen atoms in the crn-THF form hydrogen bond interactions with the main-chain amides of Asp29 and Asp30, similar to those observed for DRV. However, unlike DRV, the crn-THF ligand has van der Waals interactions with all the side chain atoms of Ile47.
Figure 5.
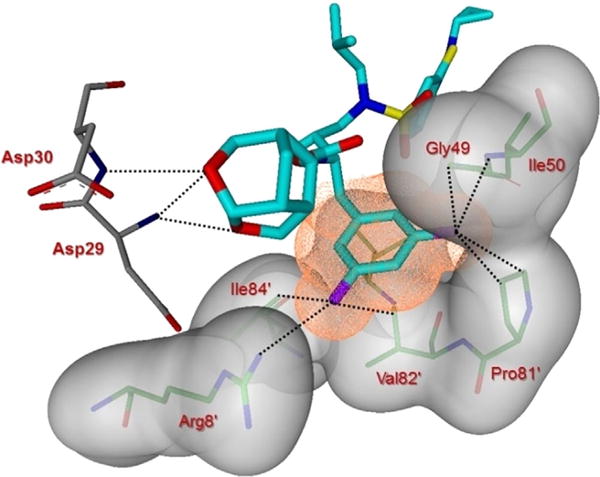
Side view of the S1 subsite. The protein surface is shown in transparent gray. The van der Waals surface for the fluorinated P1-ligand for inhibitor 5 is shown in orange wire mesh, and van der Waals contacts are shown by dotted lines.
The P2′ position of inhibitor 5 has a Cp-Abt moiety instead of aminobenzene in DRV. The extended P2′ moiety results in a deviation of ≈ 1 Å in position relative to P2′ of DRV as measured by the distance between the thiazole nitrogen atom of inhibitor 5 and the 4-aminobenzene nitrogen of DRV in both major and minor conformations (Figure 4). The thiazole nitrogen of 5 forms a hydrogen bond interaction with the main-chain amide of Asp30′, similar to the interaction of 4-aminobenzene of DRV. The P2′ amino group of inhibitor 5 has strong hydrogen bond interaction (2.5 Å length) with the side chain of Asp30′. Furthermore, the cyclopropyl group juts out and forms C–H⋯O interactions with the side chain of Asp29′. Thus, P1 and P2′ modifications in inhibitor 5 significantly contribute additional interactions with Arg 8′/8 and Asp29/29′, which stabilize the dimer of HIV-1 protease by a critical inter-subunit ion pair. In addition, the halogen bond interactions between the flap residues of protease and P1 fluorine of inhibitor 5, together with the van der Waals contacts between crn-THF and Ile47 increase the favorable inhibitor–HIV-1 protease interactions relative to the DRV complex. These new interactions are likely to explain the improved antiviral activity of inhibitor 5.
Conclusions
We have designed, synthesized, and evaluated fluorine-containing inhibitors with novel 6-5-5-ring-fused crown-like tetrahydropyranofuran as the P2-ligand, cyclopropyl aminobenzothiazole as the P2′-ligand, and 3,5-difluorophenylmethyl moiety as the P1-ligand. The corresponding fluorine-containing hydroxyethylamine sulfonamide isostere was synthesized using Sharpless asymmetric epoxidation followed by regioselective epoxide opening as the key steps. This synthetic route enabled us to synthesize both difluoro and monofluoro derivatives very efficiently. We have recently reported that crn-THF containing inhibitor 3 is a potent HIV-1 protease inhibitor with robust drug-resistance properties. We have specifically incorporated 3,5-difluorophenylmethyl group on this inhibitor to further modulate van der Waals interactions in the S1 sub-pocket of HIV-1 protease. Inhibitor 5 displayed enhanced enzyme inhibitory and antiviral potency over inhibitor 3 and DRV. In particular, inhibitor 5 exhibited unprecedented antiviral activity against a variety of highly multidrug-resistant HIV-1 variants, with antiviral activity ranging from picomolar to femtomolar levels. Inhibitor 5 displayed an extremely high genetic barrier to resistance. It also showed a dual mechanism of action, with potent dimerization inhibition of HIV-1 protease monomers as well as potent inhibition of proteolytic activity. In fact, inhibitor 5 showed nearly 1000-fold more potent dimerization inhibition activity than DRV. Furthermore, this compound showed much improved CNS penetration over DRV, and its brain level in rats strongly suggests that this inhibitor would block replication of HIV-1 effectively in the brain. Furthermore, it showed significantly higher selectivity (2473 684) than DRV (41 562). We obtained a high-resolution X-ray crystal structure of the inhibitor 5–HIV-1 protease complex. Inhibitor 5 forms extensive interactions in the HIV-1 protease active site. In particular, it forms a network of hydrogen bonds with the backbone atoms of HIV-1 protease. Our structural analysis provided the molecular basis for enhanced potency of inhibitor 5. Our present studies suggest that inhibitor 5 would be a favorable agent for development, and further studies are ongoing.
Experimental Section
General
All reactions were carried out under an argon atmosphere in either flame- or oven-dried (120°C) glassware. All reagents and chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted. Anhydrous solvents were obtained as follows: dichloromethane from calcium hydride, diethyl ether and tetrahydrofuran from sodium benzophenone, methanol and ethanol from activated magnesium under argon. All purification procedures were carried out with reagent-grade solvents (purchased form VWR) in air. TLC analysis was conducted using glass-backed thin-layer silica gel chromatography plates (60 Å, 250 μm thickness, F254 indicator). Column chromatography was performed using 230–400 mesh, 60 Å pore diameter silica gel. 1H and 13C NMR spectra were recorded at room temperature on Bruker ARX-400 and DRX-500 instruments. Chemical shifts (δ values) are reported in parts per million, and are referenced to the deuterated residual solvent peak. NMR data are reported as: δ value (chemical shift, J-value (Hz), integration, where s = singlet, d = doublet, t = triplet, q = quartet, brs = broad singlet). LRMS and HRMS spectra were recorded at the Purdue University Department of Chemistry Mass Spectrometry Center.
(E)-4-(3,5-Difluorophenyl)but-2-en-1-ol (7a)
To a suspension of magnesium turnings (450 mg, 18.54 mmol) in THF (15 mL) was added a solution of 1-bromo-3,5-difluorobenzene 6a (3.3 g, 17.11 mmol) in THF (3 mL). The mixture was heated at 65°C for 1 h and the solution was cooled to 23°C. The resulting Grignard solution was added dropwise to a mixture of CuCN (128 mg, 1.426 mmol) and butadiene monoxide (1.0 g, 14.26 mmol) in 80 mL of anhydrous THF at 78°C. The reaction mixture was stirred for 1 h at 78°C, after which it was quenched with 15 mL of saturated NH4Cl solution followed by 15 mL of NH4OH. The aqueous layer was extracted twice with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. The residue was purified by silica gel column chromatography (20% EtOAc in hexane) to give 7a (1.9 g, 60% yield over two steps). 1H NMR (400 MHz, CDCl3): δ=6.72–6.67 (m, 2H), 6.63 (tt, J=4.0, 8.0 Hz, 1H), 5.79 (dtt, J=1.2, 6.5, 15.5 Hz, 1H), 5.71 (dtt, J=1.2, 6.5, 16.0 Hz, 1H), 4.13 (dd, J=1.0, 5.5 Hz, 2H), 3.35 (d, J=6.5 Hz, 2H), 1.70 ppm (s, 1H); 13C NMR (100 MHz, CDCl3): δ=164.1 (d, J=13.0 Hz), 161.7 (d, J=13.0 Hz), 143.8 (t, J=9.0 Hz), 131.5, 129.3, 111.2 (d, J=7.0 Hz), 111.1 (d, J=6.0 Hz), 101.5 (t, J=25 Hz), 63.0, 38.1 ppm; LRMS-APCI (m/z): 167 [M–OH]+.
(2S,3S)-3-Azido-4-(3,5-difluorophenyl)butane-1,2-diol (8a)
Molecular sieves (500 mg) were flame-dried in a flask and then allowed to cool to 23°C. Under argon, dry dichloromethane (30 mL) and (−)-DET (439 mg, 2.13 mmol) were added and the suspension was cooled to −25°C. To this, Ti(OiPr)4 (318 μL, 1.06 mmol) and TBHP (4.3 mL, 5.5m solution in decane, 23.43 mmol) were added and the mixture was stirred at −25°C for 30 min. A solution of 7a (1.96 g, 10.65 mmol) in dry CH2Cl2 (20 mL) was added to the above mixture and run for 12 h at −25°C. To the reaction mixture H2O (10 mL) was added and stirred at 0°C for 30 min. A 10% aqueous NaOH solution was then added and the mixture was warmed to room temperature for 1 h. The product was extracted with CH2Cl2 (3×), dried with Na2SO4, filtered, and concentrated under reduced pressure. Purification by silica gel column chromatography (30–40% EtOAc in hexanes) afforded epoxy alcohol (1.77 g, 83%) as a liquid. Dry benzene (20 mL), Ti(OiPr)4 (1.52 mL, 5.10 mmol), and TMSN3 (1.35 mL, 10.20 mmol) were held at reflux for 5 h. A solution of above epoxy alcohol (680 mg, 3.40 mmol) in dry benzene (10 mL) was added and the solution was held at reflux for 30 min. After cooling to room temperature, 5% H2SO4 (8 mL) was added and the solution was stirred at room temperature for 1 h. The layers were separated and the aqueous portion was extracted with ethyl acetate (3×). The organic solution was dried with Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (50% EtOAc in hexanes) to afford 8a (558 mg, 68%). 1H NMR (400 MHz, CDCl3 + CD3OD): δ=6.78–6.72 (m, 2H), 6.63 (tt, J=2.0, 11.0 Hz, 1H), 3.69−3.57 (m, 2H), 3.56−3.48 (m, 2H), 3.02 (dd, J=3.0, 14.0 Hz, 1H), 2.66 ppm (dd, J=9.1, 14.0 Hz, 1H); 13C NMR (100 MHz, CDCl3 + CD3OD): δ=164 (d, J=13.0 Hz), 161.6 (d, J=13.0 Hz), 141.6, 112.1 (d, J=6.0 Hz), 112.0 (d, J=7.0 Hz), 102.0 (t, J=25 Hz), 72.9, 64.5, 62.9, 36.4 ppm; LRMS-APCI (m/z): 266 [M+Na]+, 216 [M−N2]+.
(S)-2-((S)-1-Azido-2-(3,5-difluorophenyl)ethyl)oxirane (9)
To a stirred solution of 8a (243 mg, 1.0 mmol) in chloroform (5 mL) was added 1-chlorocarbonyl-1-methylethyl acetate (215 μL, 1.50 mmol) at 0°C, and the solution was stirred at 0°C for 30 min. The reaction mixture was warmed to room temperature and stirred for 1 h. A saturated solution of NaHCO3 (aq) was added and the mixture stirred for 15 min. The crude chloroacetate was extracted with chloroform, dried with Na2SO4, filtering, and concentrated in vacuo. The residue was dissolved in dry THF (8 mL) and cooled to 0°C. Solid NaOMe (65 mg, 1.20 mmol) was added and the reaction mixture was stirred at room temperature for 2 h. The reaction was quenched with saturated NH4Cl(aq) and extracted with ethyl acetate. Drying over Na2SO4, filtered, and concentrating under reduced pressure afforded a residue which was purified by flash column chromatography (12% EtOAc in hexanes) to afford pure azido epoxide 9 as a liquid (174 mg, 77% yield over two steps). 1H NMR (400 MHz, CDCl3): δ=6.82−6.76 (m, 2H), 6.71 (tt, J=2.3, 11.3 Hz, 1H), 3.54 (quintet, J=4.3, 5.5 Hz, 1H), 3.05 (m, 1H), 2.96 (dd, J=4.0, 14.0 Hz, 1H), 2.85 (m, 1H), 2.82−2.73 ppm (m, 2H); 13C NMR (100 MHz, CDCl3): δ=164.1 (d, J=13.0 Hz), 161.6 (d, J=13.0 Hz), 140.3 (t, J=9.0 Hz), 112.2 (d, J=6.0 Hz), 112.1 (d, J=7.0 Hz), 102.5 (t, J=25.0 Hz), 63.1, 52.7, 45.2, 37.8 ppm; LRMS-APCI (m/z): 198 [M N2]+.
(E)-4-(3-Fluorophenyl)but-2-en-1-ol (7b)
The title compound 7b (942 mg, 67% yield over two steps) was obtained from 6b (1.47 g, 8.4 mmol) by following the procedure outlined for compound 7a. 1H NMR (500 MHz, CDCl3): δ=7.24 (m, 1H), 6.96 (d, J=7.6 Hz), 6.92−6.86 (m, 2H), 5.83 (m, 1H), 5.71 (m, 1H), 4.13 (dd, J=1.1, 5.6 Hz, 2H), 3.38 (d, J=6.5 Hz, 2H), 1.56 ppm (s, 1H).
(2S,3S)-3-Azido-4-(3-fluorophenyl)butane-1,2-diol (8b)
The title compound 8b (1.0 g, 78% yield over two steps) was obtained from 7b (940 mg, 5.66 mmol) by following the procedure outlined for compound 8a. 1H NMR (400 MHz, CDCl3): δ=7.29 (m, 1H), 7.05 (d, J=7.6 Hz, 1H), 7.02–6.93 (m, 2H), 3.86–3.73 (m, 2H), 3.72–3.62 (m, 2H), 3.07 (dd, J=3.4, 14.0 Hz, 1H), 2.79 (dd, J=8.8, 14.0 Hz, 1H), 2.49 ppm (brs, 2H).
(S)-2-((S)-1-Azido-2-(3-fluorophenyl)ethyl)oxirane (10)
The title compound 10 (265 mg, 69% yield over two steps) was obtained from 8b (417 mg, 1.85 mmol) by following the procedure outlined for compound 9. 1H NMR (400 MHz, CDCl3): δ=7.29 (m, 1H), 7.03 (d, J=7.7 Hz, 1H), 7.00−6.93 (m, 2H), 3.57 (quintet, J=4.2, 5.6 Hz, 1H), 3.06 (ddd, J=2.6, 3.7, 6.2 Hz, 1H), 2.99 (dd, J=4.2, 14.0 Hz, 1H), 2.87–2.76 ppm (m, 3H); 13C NMR (100 MHz, CDCl3): δ=164.1, 161.7, 139.2 (d, J=7.2 Hz), 130.1 (d, J=8.2 Hz), 125.1, 116.4 (d, J=21.0 Hz), 114.0 (d, J=20.8 Hz), 108.8, 63.5, 53.0, 45.3, 38.0 ppm.
(E)-4-(4-Fluorophenyl)but-2-en-1-ol (7c)
The title compound 7c (1.6 g, 61 %) was obtained from 6c (2.74 g, 15.68 mmol) by following the procedure outlined for compound 7a. 1H NMR (400 MHz, CDCl3): δ = 7.15−7.10 (m, 2H), 7.00−6.94 (m, 2H), 5.87−5.78 (m, 1 H), 5.73−5.64 (m, 1 H), 4.15−4.08 (m, 2 H), 3.35 (d, J = 6.4 Hz, 2 H), 1.51 ppm (brs, 1 H); 13C NMR (100 MHz, CDCl3): δ = 162.6, 160.1, 135.4 (d, J = 2.6 Hz), 131.2, 130.4, 129.8 (d, J = 7.7 Hz), 115.1(d, J = 21.0 Hz), 63.3, 37.6 ppm; LRMS-APCI (m/z): 149 [M–OH]+.
(2S,3S)-3-Azido-4-(4-fluorophenyl)butane-1,2-diol (8 c)
The title compound 8c (760 mg, 56% yield over two steps) was obtained from 7c (1.0 g, 6.02 mmol) by following the procedure outlined for compound 8a. 1H NMR (400MHz, CDCl3 + CD3OD): δ = 7.21−7.15 (m, 2 H), 6.98−6.92 (m, 2H), 3.73−3.58 (m, 2H), 3.57−3.52 (m, 2 H), 3.05−2.99 (m, 1 H), 2.68 ppm (dd, J = 9.1, 14.2 Hz, 1 H); 13C NMR (100 MHz, CDCl3 + CD3OD): δ = 162.9, 160.5, 133.2 (d, J = 2.8 Hz), 130.7 (d, J = 7.0 Hz), 115.2 (d, J = 21.0 Hz), 72.9, 65.2, 63.0, 35.9 ppm; LRMS-APCI (m/z): 248 [M + Na], 198 [M–N2]+.
(S)-2-((S)-1-Azido-2-(4-fluorophenyl)ethyl)oxirane (11)
The title compound 11 (700 mg, 76% over two steps) was obtained from 8c (1.0 g, 4.44 mmol) by following the procedure outlined for compound 9. 1H NMR (400 MHz, CDCl3): δ = 7.23−7.18 (m, 2H), 7.01 (tt, J = 2.1, 11.7 Hz, 1 H), 3.53 (ddd, J = 4.5, 5.5, 8.7 Hz, 1 H), 3.04 (ddd, J = 2.5, 3.8, 5.5 Hz, 1 H), 2.97 (dd, J = 4.5, 14.1 Hz, 1 H), 2.85−2.75 ppm (m, 3 H); 13C NMR (100 MHz, CDCl3): δ = 163.1, 160.6, 132.1, 130.7 (d, J = 8.0 Hz), 115.3 (d, J = 21.0 Hz), 63.6, 52.7, 45.1, 37.3 ppm; LRMS-APCI (m/z): 180 [M–N2]+.
(2R,3S)-3-Amino-4-(3,5-difluorophenyl)-1-(isobutylamino)butan-2-ol (12)
To a stirred solution of 9 (200 mg, 2.66 mmol) in isopropanol was added isobutylamine (0.27 mL, 2.66 mmol) at 23 °C under argon atmosphere. The reaction mixture was stirred at 65° C for 12 h. After this period, isopropanol was removed under reduced pressure. 1H NMR (400 MHz, CDCl3): δ = 6.83−6.77 (m, 2 H), 6.70 (tt, J = 2.3, 11.3 Hz, 1 H), 3.59−3.51 (m, 2H), 3.01 (dd, J = 3.2, 14.3 Hz, 1 H), 2.87 (dd, J = 3.0, 12.0 Hz, 1 H), 2.86 (brs, 1 H), 2.76−2.64 (m, 2 H), 2.45 (dd, J = 2.0, 6.9 Hz, 2 H), 1.74 (m, 1 H), 0.93 (d, J = 1.4 Hz, 3H), 0.92 ppm (d, J = 1.4 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ = 164.1 (d, J = 13.0 Hz), 161.6 (d, J = 13.0 Hz), 141.5 (t, J = 9.0 Hz), 112.2 (d, J = 6.0 Hz), 112.0 (d, J = 6.0 Hz), 102.2 (t, J = 25.0 Hz), 70.0, 66.2, 57.5, 50.8, 36.7, 28.2, 20.3 ppm; LRMS-APCI (m/z): 299 [M+ H]+.
4-Amino-N-((2R,3S)-3-amino-4-(3,5-difluorophenyl)-2-hydroxybutyl)-N-isobutylbenzenesulfonamide (13)
To a stirred solution of crude product 12a (331 mg) in dichloromethane was added 4-nitrobenzenesulfonyl chloride (246 mg, 1.11 mmol) and triethyl amine (0.5 mL, 3.33 mmol) at 23°C under argon atmosphere. The reaction mixture was stirred at 23°C for 12 h. Solvent was removed under reduced pressure and the crude product was purified by silica gel column chromatography (55% EtOAc in hexane) to give azide (348 mg, 67% over two steps). 1H NMR (400 MHz, CDCl3): δ= 8.39−8.35 (m, 2 H), 8.02−7.98 (m, 2 H), 6.81 (d, J = 7.2 Hz, 2 H), 6.70 (t, J = 8.9 Hz, 1 H), 3.76 (t, J = 6.8 Hz, 1 H), 3.57 (ddd, J = 9.4, 6.2, 4.1 Hz, 1 H), 3.32 (brs, 1 H), 3.29−3.20 (m, 2 H), 3.12−3.03 (m, 2 H), 2.94 (dd, J = 13.5, 6.9 Hz, 1 H), 2.77 (dd, J = 14.3, 9.2 Hz, 1 H), 1.86 (tt, J = 13.8, 6.9 Hz, 1 H), 0.90 (d, J = 6.6 Hz, 3 H), 0.87 ppm (d, J = 6.5 Hz, 3 H); 13C NMR (100 MHz, CDCl3): δ = 164.4 (d, J = 13.0 Hz), 161.9 (d, J = 13.0 Hz), 150.3, 144.4, 141.0 (t, J = 9.1 Hz), 128.6, 124.6, 114.3−110.5 (m), 102.6 (t, J = 25.2 Hz), 71.6, 66.0, 58.5, 52.6, 36.6, 27.1, 20.1, 19.8 ppm. To a stirred solution of above azide (338 mg, 0.7 mmol) in EtOAc (5 mL) was added 20% Pd(OH)2/C (80 mg). The resulting solution was stirred at 23°C under 1 atm H2 gas over 20 h. Upon completion, the mixture was filtered through a plug of Celite and solvents were removed under reduced pressure. The crude product was purified by silica gel column chromatography (5% NH3 in MeOH in CH2Cl2) to afford 13 (265 mg, 89% yield). 1H NMR (400 MHz, CDCl3): δ = 7.53 (d, J = 8.7 Hz, 2H), 6.76−6.71 (m, 2 H), 6.68−6.62 (m, 3 H), 4.26 (brs, 2 H), 3.68 (dt, J = 7.5, 4.8 Hz, 1 H), 3.19−3.12 (m, 2H), 3.03 (dt, J = 9.2, 4.3 Hz, 1 H), 2.99−2.91 (m, 2H), 2.80 (dd, J = 13.4, 6.8 Hz, 1 H), 2.47 (dd, J = 13.5, 9.7 Hz, 1 H), 1.85 (dt, J = 13.6, 6.8 Hz, 1 H), 0.88 ppm (dd, J = 14.4, 6.6 Hz, 6 H); 13C NMR (100 MHz, CDCl3): δ = 164.2 (d, J = 12.8 Hz), 161.7 (d, J = 12.9 Hz), 151.2, 143.1 (t, J = 9.0 Hz), 129.3, 125.6, 114.0, 112.5−110.9 (m), 101.8 (t, J = 25.3 Hz), 77.4, 73.0, 58.6, 55.3, 52.8, 38.8, 27.1, 20.1, 19.9 ppm; LRMS-ESI (m/z): 428.1 [M + H]+.
N-((2R,3S)-3-Azido-4-(3,5-difluorophenyl)-2-hydroxybutyl)-N-isobutyl-2-(methylthio)benzo[d]thiazole-6-sulfonamide (15a)
To a stirred solution of amine 12a in dichloromethane was added sulfonyl chloride 14 (248 mg, 0.89 mmol) and triethyl amine (0.37 mL, 2.66 mmol) at 23°C under argon atmosphere. The reaction mixture was stirred at 23°C for 12 h. Upon completion, solvent was removed under reduced pressure and the crude product was purified by silica gel column chromatography (15% EtOAc in hexane) to give 15a (433 mg, 90% over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.23−8.21 (m, 1 H), 7.89 (d, J = 8.6 Hz, 1 H), 7.80−7.76 (m, 1 H), 6.79 (d, J = 6.2 Hz, 2 H), 6.65 (t, J = 8.9 Hz, 1 H), 3.76 (d, J = 17.6 Hz, 2 H), 3.56 (ddd, J = 9.5, 6.1, 3.7 Hz, 1 H), 3.27−3.17 (m, 2 H), 3.05 (ddd, J = 21.8, 13.7, 5.8 Hz, 2 H), 2.89 (dd, J = 13.4, 6.8 Hz, 1 H), 2.76 (s, 3 H), 2.74−2.70 (m, 1 H), 1.88−1.78 (m, 1 H), 0.87 (d, J = 6.6 Hz, 3H), 0.84 ppm (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 173.6, 164.4 (d, J = 12.7 Hz), 161.9 (d, J = 12.9 Hz), 156.1, 141.3 (t, J = 9.1 Hz), 136.0, 133.7, 125.1, 121.9, 121.1, 112.6, 112.3, 102.6 (t, J = 25.3 Hz), 71.9, 66.0, 59.1, 53.2, 36.6, 27.3, 20.2, 19.9, 16.1 ppm; LRMS-ESI (m/z): 542.1 [M + H]+.
N-((2R,3S)-3-Azido-4-(3-fluorophenyl)-2-hydroxybutyl)-N-isobutyl-2-(methylthio)benzo[d]thiazole-6-sulfonamide (15 b)
The title compound (15 b) was obtained by following the procedure outlined for compound 15a, (129 mg, 87% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.24 (d, J = 1.7 Hz, 1 H), 7.94 (d, J = 8.6 Hz, 1 H), 7.81 (dd, J = 8.6, 1.8 Hz, 1 H), 7.30−7.24 (m, 1 H), 7.05 (d, J = 7.6Hz, 1 H), 7.01−6.92 (m, 2H), 3.78 (td, J = 6.2, 3.2 Hz, 1 H), 3.63−3.55 (m, 2 H), 3.28 (dd, J = 15.2, 9.1 Hz, 1 H), 3.17 (dd, J = 15.2, 2.5 Hz, 1 H), 3.11 (dd, J = 14.3, 3.6 Hz, 1 H), 3.08−3.04 (m, 1 H), 2.88 (dd, J = 13.4, 6.6 Hz, 1 H), 2.81 (s, 3 H), 2.79−2.75 (m, 1 H), 1.84 (tt, J = 13.3, 6.6 Hz, 1 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.87 ppm (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ = 173.5, 163.9, 161.9, 155.9, 139.8 (d, J = 7.2 Hz), 135.8, 133.6, 130.2 (d, J = 8.2 Hz), 125.3−125.0 (m), 121.8, 121.0, 116.3 (d, J = 21.2 Hz), 113.9 (d, J = 20.9 Hz), 71.8, 66.2, 58.9, 52.9, 36.6, 27.2, 20.2, 19.8, 16.1 ppm; LRMS-ESI (m/z): 546.2 [M + Na]+.
N-((2R,3S)-3-Azido-4-(4-fluorophenyl)-2-hydroxybutyl)-N-isobutyl-2-(methylthio)benzo[d]thiazole-6-sulfonamide (15 c)
The title compound (15c) was obtained by following the procedure outlined for compound 15 a, (171 mg, 93% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.24 (d, J = 1.5 Hz, 1 H), 7.94 (d, J = 8.6 Hz, 1 H), 7.81 (dd, J = 8.6, 1.7 Hz, 1 H), 7.23 (dd, J = 8.4, 5.5 Hz, 2 H), 7.00 (t, J = 8.6 Hz, 2 H), 3.79−3.72 (m, 1 H), 3.59−3.51 (m, 2 H), 3.27 (dd, J = 15.2, 9.1 Hz, 1 H), 3.16 (dd, J = 15.2, 2.3 Hz, 1 H), 3.12−3.04 (m, 2 H), 2.87 (dd, J = 13.4, 6.6 Hz, 1 H), 2.81 (s, 3 H), 2.77 (dd, J = 14.2, 9.2 Hz, 1 H), 1.83 (dp, J = 13.3, 6.6 Hz, 1 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.87 ppm (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ = 173.6, 162.9, 161.0, 156.0, 135.9, 133.6, 132.9 (d, J = 2.5 Hz), 131.0 (d, J = 7.8 Hz), 125.1, 121.8, 121.1, 115.5 (d, J = 21.2 Hz), 71.8, 66.6, 58.9, 53.0, 36.1, 27.2, 20.2, 19.8, 16.1 ppm; LRMS-ESI (m/z): 524.0 [M + H]+.
tert-Butyl ((2S,3R)-1-(3,5-difluorophenyl)-3-hydroxy-4-((N-isobutyl-2-(methylthio)benzo[d]thiazole)-6-sulfonamido)butan-2-yl)-carbamate (16a)
To a stirred solution of 15a (275 mg, 0.5 mmol) in THF/H2O (3:1 ratio, 4 mL) was added triphenylphosphine (160 mg, 0.61 mmol) at 23°C. The reaction mixture was stirred at 23°C for 24 h. After this period, to the reaction mixture Boc anhydride (133 mg, 0.61 mmol) and sodium bicarbonate (85 mg, 1 mmol) were added at 23°C. The reaction mixture was stirred at 23°C for 20 h. Upon completion, the mixture was concentrated under reduced pressure and diluted with EtOAc. The reaction mixture was washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (30% EtOAc in hexane) to give 16a (270 mg, 87% over two steps). 1H NMR (400 MHz, CDCl3): δ = 8.22 (d, J = 1.4 Hz, 1 H), 7.92 (d, J = 8.6 Hz, 1 H), 7.79 (dd, J = 8.6, 1.6 Hz, 1 H), 6.77 (d, J = 6.2 Hz, 2 H), 6.64 (tt, J = 9.2, 2.2 Hz, 1 H), 4.74 (d, J = 8.7 Hz, 1 H), 4.05 (s, 1 H), 3.84 (s, 1 H), 3.71 (tt, J = 9.4, 4.9 Hz, 1 H), 3.14 (qd, J = 15.1, 5.9 Hz, 2 H), 3.01−2.84 (m, 4 H), 2.81 (s, 3 H), 1.94−1.79 (m, 1 H), 1.34 (s, 9 H), 0.87 ppm (dd, J = 10.4, 6.6 Hz, 6 H); 13C NMR (100 MHz, CDCl3): δ = 173.5, 164.3 (d, J = 12.7 Hz), 161.8 (d, J = 13.2 Hz), 156.0 (d, J = 7.8 Hz), 142.3 (t, J = 8.9 Hz), 135.9, 133.9, 125.1, 121.8, 121.1, 112.6, 112.3, 108.8, 102.0 (t, J = 25.4 Hz), 80.2, 73.0, 58.9, 54.7, 53.8, 35.2, 28.3, 27.3, 20.2, 20.0, 16.1 ppm; LRMS-ESI (m/z): 638.1 [M+ Na]+.
tert-Butyl ((2S,3R)-1-(3-fluorophenyl)-3-hydroxy-4-((N-isobutyl-2-(methylthio)benzo[d]thiazole)-6-sulfonamido)butan-2-yl)carbamate (16 b)
The title compound (16b) was obtained by following the procedure outlined for compound 16a, (123 mg, 83% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.24−8.18 (m, 1 H), 7.90 (d, J = 8.6 Hz, 1 H), 7.77 (dd, J = 8.6, 1.7 Hz, 1 H), 7.21 (q, J = 7.8 Hz, 1 H), 7.00 (d, J = 7.6 Hz, 1 H), 6.94 (d, J = 9.8 Hz, 1 H), 6.88 (td, J = 8.4, 2.1 Hz, 1 H), 4.75 (d, J = 8.3 Hz, 1 H), 3.99 (s, 1 H), 3.83 (brs, 1 H), 3.73 (tt, J = 9.3, 5.1 Hz, 1 H), 3.13 (td, J = 16.3, 15.1, 9.7 Hz, 2 H), 3.04−2.93 (m, 2 H), 2.89 (dd, J = 13.4, 7.1 Hz, 2 H), 2.79 (s, 3 H), 1.86 (dp, J = 13.7, 6.7 Hz, 1 H), 1.32 (s, 9 H), 0.86 ppm (dd, J = 13.5, 6.6 Hz, 6 H); 13C NMR (125 MHz, CDCl3): δ = 173.3, 163.8, 161.9, 156.1, 155.9, 140.9−140.6 (m), 135.7, 133.9, 129.9 (d, J = 8.2 Hz), 125.2 (d, J = 18.0 Hz), 121.7, 121.0, 116.4 (d, J = 21.0 Hz), 113.3 (d, J = 20.9 Hz), 79.9, 72.9, 58.6, 54.7, 53.6, 35.2, 28.3, 27.2, 20.16, 20.0, 16.1 ppm; LRMS-ESI (m/z): 620.4 [M+ Na]+.
tert-Butyl ((2S,3R)-1-(4-fluorophenyl)-3-hydroxy-4-((N-isobutyl-2-(methylthio)benzo[d]thiazole)-6-sulfonamido)butan-2-yl)carbamate (16c)
The title compound (16c) was obtained by following the procedure outlined for compound 16a, (127 mg, 70% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.21 (s, 1 H), 7.91 (d, J = 8.6 Hz, 1 H), 7.77 (d, J = 8.6 Hz, 1 H), 7.19 (dd, J = 8.0, 5.6 Hz, 2 H), 6.96 (t, J = 8.6 Hz, 2 H), 4.68 (d, J = 8.6 Hz, 1 H), 3.98 (s, 1 H), 3.81 (brs, 1 H), 3.76−3.67 (m, 1 H), 3.18−3.09 (m, 2 H), 2.97 (t, J = 10.6 Hz, 2 H), 2.92−2.84 (m, 2 H), 2.81 (s, 3 H), 1.86 (dp, J = 13.4, 6.5 Hz, 1 H), 1.32 (s, 9 H), 0.87 ppm (dd, J = 13.9, 6.6 Hz, 6H); 13C NMR (125 MHz, CDCl3): δ = 173.4, 162.7, 160.7, 156.1, 156.0, 135.8, 133.7 (d, J = 31.9 Hz), 131.0 (d, J = 7.6 Hz), 125.1, 121.7, 121.0, 115.3 (d, J = 21.1 Hz), 79.9, 72.8, 58.7, 54.8, 53.7, 34.6, 28.3, 27.2, 20.2, 20.0, 16.1 ppm; LRMS-ESI (m/z): 598.2 [M + H]+.
N-((2R,3S)-3-Amino-4-(3,5-difluorophenyl)-2-hydroxybutyl)-2-(cyclopropylammo)-N-isobutylbenzo[d]thiazole-6-sulfonamide (17)
To a stirred solution of 16a (960 mg, 1.56 mmol) in dichloromethane (10 mL) was added mCPBA (807 mg, 4.68 mmol) at 0°C under argon atmosphere and the mixture was stirred at 23°C for 12 h. After this period, the reaction mixture was quenched by the addition of saturated aqueous Na2S2O3 (2 mL) and extracted with dichloromethane. The extracts were washed with saturated aqueous NaHCO3, dried (Na2SO4) and concentrated under reduced pressure. To the crude product in dry THF (10 mL) at 23°C under argon atmosphere was added cyclopropylamine (0.35 mL, 4.68 mmol) and the mixture was stirred at 65°C for 12 h. Upon completion, solvent was removed under reduced pressure and the crude product was purified by silica gel column chromatography (50% EtOAc in hexane) to give cyclopropylamine (880 mg, 91 % over two steps). 1H NMR (400 MHz, CDCl3 + CD3OD): δ = 7.93 (s, 1 H), 7.57−7.52 (m, 1 H), 7.38 (d, J = 8.5 Hz, 1 H), 6.63 (d, J = 6.6 Hz, 2 H), 6.49 (t, J = 9.0 Hz, 1 H), 5.47 (d, J = 9.4 Hz, 1 H), 3.68−3.60 (m, 1 H), 3.58−3.47 (m, 1 H), 3.21−3.10 (m, 1 H), 2.83 (dtd, J = 39.5, 15.1, 13.3, 5.5 Hz, 4 H), 2.61−2.50 (m, 2 H), 1.84−1.71 (m, 1 H), 1.18 (s, 9 H), 0.75 (dd, J = 9.7, 5.8 Hz, 8 H), 0.62−0.57 ppm (m, 2 H); 13C NMR (100 MHz, CDCl3 + CD3OD): δ = 173.3, 164.1 (d, J = 13.3 Hz), 161.7 (d, J = 12.5 Hz), 156.7, 155.5, 131.0, 130.4, 125.4, 120.9, 118.3, 112.4 (d, J = 24.1 Hz), 101.9 (d, J = 26.8 Hz), 79.9, 72.8, 60.6, 58.6, 54.5, 53.5, 35.4, 28.2, 27.1, 26.6, 20.0, 7.7 ppm; LRMS-ESI (m/z): 625.2 [M + H]+. To a stirred solution of above cyclopropylamine (870 mg, 1.39 mmol) in dichloromethane (15 mL) was added TFA (5 mL) at 0°C under argon atmosphere and the mixture was stirred at 23°C for 1 h. Upon completion, solvent was removed under reduced pressure to give 17 (730 mg) used for next coupling reaction without further purification. LRMS-ESI (m/z): 525.2 [M + H]+.
N-((2R,3S)-3-Amino-4-(3-fluorophenyl)-2-hydroxybutyl)-2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole-6-sulfonamide (18)
The cyclopropylamine was obtained from 16 b by following the procedure outlined for compound 17, (116 mg, 97% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.08 (s, 1 H), 7.68 (d, J = 8.2 Hz, 1 H), 7.54 (d, J = 8.4 Hz, 1 H), 7.25−7.18 (m, 1 H), 7.03 (d, J = 6.3 Hz, 1 H), 6.95 (d, J = 9.1 Hz, 1 H), 6.89 (t, J = 7.9 Hz, 1 H), 4.78 (d, J = 7.4 Hz, 1 H), 3.85 (s, 1 H), 3.76 (brs, 1 H), 3.20−3.08 (m, 2 H), 3.00 (dt, J = 22.3, 10.4 Hz, 2 H), 2.88 (dt, J = 18.6, 12.0 Hz, 2 H), 2.75 (s, 1 H), 1.93−1.80 (m, 1 H), 1.34 (s, 9 H), 0.91 (td, J = 17.8, 6.1 Hz, 8 H), 0.79 ppm (s, 2 H); 13C NMR (125 MHz, CDCl3): δ = 173.4, 163.9, 161.9, 156.1, 155.8, 140.8 (d, J = 6.8 Hz), 131.3, 130.4, 129.9 (d, J = 7.6 Hz), 125.4 (d, J = 20.3 Hz), 121.0, 118.5, 116.5 (d, J = 20.8 Hz), 113.4 (d, J = 20.9 Hz), 80.0, 72.9, 59.0, 54.7, 53.9, 35.2, 28.4, 27.4, 26.6, 20.3, 20.0, 8.0 ppm; LRMS-ESI (m/z): 629.5 [M + Na]+. The title compound (18) was obtained by following the de-Boc procedure outlined for compound 17, (97 mg) used for next coupling reaction without further purification. LRMS-ESI (m/z): 507.3 [M + H]+.
N-((2R,3S)-3-Amino-4-(4-fluorophenyl)-2-hydroxybutyl)-2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole-6-sulfonamide (19)
The cyclopropylamine was obtained form 16c by following the procedure outlined for compound 17, (125 mg, 98% yield over two steps). 1H NMR (500 MHz, CDCl3): δ = 8.08 (d, J = 1.5 Hz, 1 H), 7.68 (dd, J = 8.5, 1.8 Hz, 1 H), 7.56 (d, J = 8.5 Hz, 1 H), 7.24−7.18 (m, 2 H), 6.97 (t, J = 8.6 Hz, 2 H), 4.68 (d, J = 8.7 Hz, 1 H), 4.05 (s, 1 H), 3.82 (brs, 1 H), 3.77−3.69 (m, 1 H), 3.11 (d, J = 6.5 Hz, 2 H), 3.03−2.94 (m, 2 H), 2.92−2.81 (m, 2 H), 2.76 (tt, J = 6.7, 3.5 Hz, 1 H), 1.85 (dp, J = 12.9, 6.4 Hz, 1 H), 1.34 (s, 9 H), 0.97−0.93 (m, 2 H), 0.91 (d, J = 6.6 Hz, 3 H), 0.87 (d, J = 6.6 Hz, 3 H), 0.79 ppm (p, J = 5.0 Hz, 2 H); 13C NMR (125 MHz, CDCl3): δ = 173.2, 162.7, 160.8, 156.1, 155.8, 134.0−133.6 (m), 131.4, 131.1 (d, J = 7.7 Hz), 130.5, 125.5, 121.0, 118.7, 115.3 (d, J = 21.1 Hz), 79.9, 73.0, 59.0, 54.8, 54.0, 34.6, 28.4, 27.4, 26.8, 20.3, 20.0, 8.1 ppm; LRMS-ESI (m/z): 607.2 [M + H]+. The title compound (19) was obtained by following the de-Boc procedure outlined for compound 17, (166 mg) used for next coupling reaction without further purification. LRMS-ESI (m/z): 507.4 [M + H]+.
(3S,7 aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl((2S,3R)-4-((4-amino-N-isobutylphenyl)sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate (4)
To a stirred solution of activated alcohol 20 (12 mg, 0.037 mmol) and isostere 13 (19 mg, 0.045 mmol) in acetonitrile (2 mL) was added DIPEA (33 μL, 0.19 mmol) at 23°C under argon atmosphere. The reaction mixture was stirred at 23°C until completion. Upon completion, solvents were removed under reduced pressure and crude product was purified by silica gel column chromatography (55% EtOAc in hexane) to give inhibitor 4 (20 mg, 88% yield). 1H NMR (400 MHz, CDCl3): δ = 7.54 (d, J = 8.5 Hz, 2 H), 6.78 (d, J = 6.4 Hz, 2 H), 6.68 (d, J = 8.5 Hz, 2 H), 6.64 (dt, J = 9.2, 2.4 Hz, 1 H), 5.44 (d, J = 6.6 Hz, 1 H), 5.26 (d, J = 9.0 Hz, 1 H), 4.83 (dd, J = 8.9, 5.8 Hz, 1 H), 4.19 (brs, 2 H), 3.92−3.79 (m, 4 H), 3.62 (ddd, J = 19.0, 10.2, 7.2 Hz, 2 H), 3.05 (dt, J = 14.4, 4.8 Hz, 2 H), 2.97 (d, J = 16.3 Hz, 1 H), 2.94−2.89 (m, 1 H), 2.86−2.64 (m, 5 H), 2.38−2.32 (m, 1 H), 1.82 (d, J = 12.0 Hz, 2 H), 1.45 (dd, J = 12.0, 3.7 Hz, 1 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.88 ppm (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 164.3 (d, J = 12.9 Hz), 161.9 (d, J = 13.1 Hz), 155.7, 151.0, 142.2 (t, J = 9.0 Hz), 129.6, 126.1, 114.3, 113.1−111.8 (m), 104.5, 102.2 (t, J = 25.1 Hz), 75.2, 72.9, 68.5, 60.1, 59.1, 55.0, 53.8, 45.0, 42.1, 37.5, 35.3, 27.5, 23.7, 20.3, 20.1 ppm; LRMS-ESI (m/z): 610.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C29H38F2N3O7S, 610.2399; found 610.2394.
(3S,7 aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(3,5-difluorophenyl)-3-hydroxybutan-2-yl)carbamate (5)
Compound 20 (200 mg, 0.62 mmol) was treated with fluoro isostere 17 (392 mg, 0.75 mmol) by following the procedure outlined for inhibitor 4 to give inhibitor 5 (434 mg, 99% yield). 1H NMR (500 MHz, CDCl3): δ = 8.27 (s, 1 H), 8.10−8.07 (m, 1 H), 7.73−7.67 (m, 1 H), 7.52 (d, J = 8.5 Hz, 1 H), 6.76 (d, J = 6.3 Hz, 2 H), 6.61 (t, J = 8.9 Hz, 1 H), 5.84 (d, J = 9.2 Hz, 1 H), 5.41 (d, J = 6.7 Hz, 1 H), 4.82 (dd, J = 8.9, 5.8 Hz, 1 H), 4.16 (brs, 1 H), 3.98−3.92 (m, 1 H), 3.85 (dt, J = 12.6, 6.1 Hz, 3 H), 3.60 (dd, J = 9.0, 6.5 Hz, 1 H), 3.54 (dd, J = 11.1, 7.9 Hz, 1 H), 3.17–3.11 (m, 2H), 3.06 (dd, J = 14.0, 3.4 Hz, 1 H), 2.96 (dd, J = 13.3, 8.0 Hz, 1 H), 2.89 (dd, J = 13.4, 7.1 Hz, 1 H), 2.79 (dd, J = 13.9, 10.5 Hz, 1 H), 2.75−2.68 (m, 2 H), 2.67−2.61 (m, 1 H), 2.34−2.27 (m, 1 H), 1.86 (dp, J = 14.5, 6.9 Hz, 1 H), 1.78 (d, J = 11.9 Hz, 1 H), 1.43 (dd, J = 8.0, 4.0 Hz, 1 H), 0.93 (t, J = 6.7 Hz, 2 H), 0.87 (dd, J = 12.6, 7.1 Hz, 6 H), 0.79−0.75 ppm (m, 2H); 13C NMR (125 MHz, CDCl3): δ = 173.7, 163.9 (d, J = 12.7 Hz), 161.9 (d, J = 12.8 Hz), 155.7 (d, J = 18.0 Hz), 142.4 (t, J = 8.8 Hz), 131.1, 130.2, 125.5, 121.0, 118.2, 112.5−112.0 (m), 104.3, 102.0 (t, J = 25.1 Hz), 74.8, 72.9, 68.3, 59.9, 58.8, 55.1, 53.5, 44.9, 42.2, 37.4, 31.6, 27.3, 26.7, 23.6, 22.7, 20.2, 20.0, 7.8 ppm; LRMS-ESI (m/z): 707.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H41F2N4O7S2, 707.2385; found 707.2379.
(3S,7 aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(3-fluorophenyl)-3-hydroxybutan-2-yl)carbamate(21)
Compound 20 (7 mg, 0.022 mmol) was treated with fluoro isostere 18 (13 mg, 0.026 mmol) by following the procedure outlined for inhibitor 4 to give inhibitor 21 (13.5 mg, 90% yield). 1H NMR (500 MHz, CDCl3): δ = 8.09−8.07 (m, 1 H), 7.70−7.67 (m, 1 H), 7.56 (d, J = 8.5 Hz, 1 H), 7.24 (d, J = 7.8 Hz, 1 H), 7.06 (brs, 1 H), 7.04−7.00 (m, 1 H), 6.96 (d, J = 9.4 Hz, 1 H), 6.94−6.88 (m, 1 H), 5.42 (d, J = 6.6 Hz, 1 H), 5.23 (d, J = 8.7 Hz, 1 H), 4.81 (dd, J = 9.0, 5.8 Hz, 1 H), 3.94−3.85 (m, 4 H), 3.82−3.76 (m, 1 H), 3.59 (dd, J = 7.9, 4.8 Hz, 2 H), 3.17 (dd, J = 15.0, 8.5 Hz, 1 H), 3.10−2.98 (m, 3 H), 2.89−2.81 (m, 2 H), 2.78−2.69 (m, 2 H), 2.68−2.62 (m, 1 H), 2.33 (q, J = 4.6, 4.1 Hz, 1 H), 1.86 (dd, J = 13.3, 6.6 Hz, 1 H), 1.81 (d, J = 12.2 Hz, 1 H), 1.44 (dt, J = 12.0, 3.8 Hz, 1 H), 0.97−0.93 (m, 5 H), 0.88 (d, J = 6.5 Hz, 3 H), 0.82−0.78 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ = 173.1, 164.0, 162.0, 155.8 (d, J = 11.0 Hz), 140.5 (d, J = 7.1 Hz), 131.5, 130.4, 130.1 (d, J = 8.3 Hz), 125.4 (d, J = 33.7 Hz), 121.0, 118.8, 116.4 (dd, J = 20.9, 8.3 Hz), 113.7 (dd, J = 20.7, 8.2 Hz), 104.5, 75.2, 72.9, 68.5, 60.0, 59.1, 55.1, 53.9, 45.0, 42.1, 37.5, 35.2, 29.8, 27.5, 26.8, 23.7, 20.3, 20.0, 8.1 ppm; LRMS-ESI (m/z): 689.3 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2479; found 689.2474.
(3S,7 aS,8S)-Hexahydro-4H-3,5-methanofuro[2,3-b]pyran-8-yl((2S,3R)-4-((2-(cyclopropylamino)-N-isobutylbenzo[d]thiazole)-6-sulfonamido)-1-(4-fluorophenyl)-3-hydroxybutan-2-yl)carbamate (22)
Compound 20 (15 mg, 0.047 mmol) was treated with fluoro isostere 19 (28 mg, 0.056 mmol) by following the procedure outlined for inhibitor 4 to give inhibitor 22 (30 mg, 94% yield). 1H NMR (500 MHz, CDCl3): δ = 8.10−8.06 (m, 1 H), 7.68 (dd, J = 8.5, 1.7 Hz, 1 H), 7.55 (d, J = 8.5 Hz, 1 H), 7.45 (brs, 1 H), 7.20 (dd, J = 8.1, 5.5 Hz, 2 H), 6.97 (t, J = 8.6 Hz, 2 H), 5.42 (d, J = 6.7 Hz, 1 H), 5.32−5.27 (m, 1 H), 4.79 (dd, J = 8.9, 5.8 Hz, 1 H), 3.93−3.83 (m, 4H), 3.76 (d, J = 9.2 Hz, 1 H), 3.57 (dt, J = 9.1, 7.1 Hz, 2H), 3.17 (dd, J = 15.0, 8.6 Hz, 1 H), 3.07−2.97 (m, 3 H), 2.88−2.79 (m, 2 H), 2.75 (dq, J = 6.8, 3.4 Hz, 1 H), 2.73−2.67 (m, 1 H), 2.67−2.61 (m, 1 H), 2.35−2.28 (m, 1 H), 1.86 (dt, J = 14.3, 7.2 Hz, 1 H), 1.81 (d, J = 12.3 Hz, 1 H), 1.46−1.40 (m, 1 H), 0.97−0.94 (m, 2 H), 0.92 (d, J = 6.6 Hz, 3 H), 0.88 (d, J = 4.2 Hz, 3 H), 0.81−0.76 ppm (m, 2 H); 13C NMR (125 MHz, CDCl3): δ = 173.3, 162.8, 160.8, 155.8 (d, J = 9.2 Hz), 133.5, 131.4, 131.0 (d, J = 7.6 Hz), 130.4, 125.5, 121.0, 118.6, 115.5 (d, J = 21.1 Hz), 104.4, 75.1, 72.8, 68.4, 60.0, 59.0, 55.3, 53.9, 45.0, 42.1, 37.5, 31.7, 27.5, 26.8, 23.6, 22.8, 20.3, 20.0, 8.0 ppm; LRMS-ESI (m/z): 689.2 [M + H]+; HRMS-ESI (m/z): [M + H]+ calcd for C33H42FN4O7S2, 689.2479; found 689.2474.
X-ray crystal structure of the complex between HIV-1 protease and inhibitor 5
The HIV-1 protease was expressed and purified as described.[43] The protease-inhibitor complex was crystallized by the hanging drop vapor diffusion method with well solutions of 1.65 M NaCl, 0.1 M sodium acetate, pH 5.5. Diffraction data were collected on a single crystal cooled to 90 K at SER-CAT (22-ID beamline), Advanced Photon Source, Argonne National Lab (Chicago, IL, USA) with X-ray wavelength of 0.8 Å, and processed by HKL-2000[44] to give an Rmerge of 7.1%. The crystal structure was solved by PHASER[45] in CCP4i Suite[46–48] using one of the previously reported isomorphous structures[49] as the initial model, and refined by SHELX-97[50, 51] with 1.67 Å resolution diffraction data. PRODRG-2[52] was used to construct the inhibitor and the restraints for refinement. COOT[53, 54] was used for modification of the model. Alternative conformations were modeled, isotropic atomic displacement parameters (B factors) were applied for all atoms including solvent molecules. The final refined solvent structure comprised one Na+ ion, two Cl− ions, one acetate ion and 169 water molecules. The crystallographic statistics are listed in Table 1 (Supporting Information). The coordinates and structure factors of the protease complex with inhibitor 5 have been deposited in the RCSB Protein Data Bank[55] with code 6BZ2.
Supplementary Material
Acknowledgments
This research was supported by the US National Institutes of Health (Grant GM53386 to A.K.G., and Grant GM62920 to I.T.W.). This work was also supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health, and in part by a Grant-in-Aid for Scientific Research (Priority Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (Monbu Kagakusho), a Grant for Promotion of AIDS Research from the Ministry of Health, Welfare, and Labor of Japan, and the Grant to the Cooperative Research Project on Clinical and Epidemiological Studies of Emerging and Reemerging Infectious Diseases (Renkei Jigyo) of Monbu Kagakusho. The authors thank the Purdue University Center for Cancer Research, which supports the shared NMR and mass spectrometry facilities. We also thank the staff at the Southeast Regional Collaborative Access Team at the Advanced Photon Source, Argonne National Laboratory, for assistance during X-ray data collection.
Footnotes
Supporting information and the ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/cmdc.201700824.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Marcus JL, Chao CR, Leyden WA, Xu L, Quesenberry CP, Klein DB, Towner WJ, Horberg MA, Silverberg MJ. J Acquired Immune Defic Syndr. 2016;73:39–46. doi: 10.1097/QAI.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samji H, Cescon A, Hogg RS, Modur SP, Althoff KN, Buchacz K, Burchell AN, Cohen M, Gebo KA, Gill MJ, Justice A, Kirk G, Klein MB, Korthuis PT, Martin J, Napravnik S, Rourke SB, Sterling TR, Silverberg MJ, Deeks S, Jacobson LP, Bosch RJ, Kitahata MM, Goedert JJ, Moore R, Gange SJ. PLoS One. 2013;8:e81355. doi: 10.1371/journal.pone.0081355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh AK, Osswald HL, Prato G. J Med Chem. 2016;59:5172–5208. doi: 10.1021/acs.jmedchem.5b01697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montaner JSG, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon K, Harrigan PR, Hogg RS, Daly P, Kendall P. Lancet. 2010;376:532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deeks SG, Lewin SR, Ross AL, Ananworanich J, Benkirane M, Cannon P, Chomont N, Douek D, Lifson JD, Lo YR, Kuritzkes D, Margolis D, Mellors J, Persaud D, Tucker JD, Barre-Sinoussi F, Alter G, Auerbach J, Autran B, Barouch DH, Behrens G, Cavazzana M, Chen Z, Cohen ÉA, Corbelli GM, Eholié S, Eyal N, Fidler S, Garcia L, Grossman C, Henderson G, Henrich TJ, Jefferys R, Kiem HP, McCune J, Moodley K, Newman PA, Nijhuis M, Nsubuga MS, Ott M, Palmer S, Richman D, Saez-Cirion A, Sharp M, Siliciano J, Silvestri G, Singh J, Spire B, Taylor J, Tolstrup M, Valente S, van Lunzen J, Walensky R, Wilson I, Zack J. Nat Med. 2016;22:839–850. doi: 10.1038/nm.4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohse N, Hansen AB, Gerstoft J, Obel N. J Antimicrob Chemother. 2007;60:461–463. doi: 10.1093/jac/dkm241. [DOI] [PubMed] [Google Scholar]
- 7.Edmonds A, Yotebieng M, Lusiama J, Matumona Y, Kitetele F, Napravnik S, Cole SR, Van Rie A, Behets F. PLoS Med. 2011;8:e1001044. doi: 10.1371/journal.pmed.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents. 2016 Jul 14; https://aidsinfo.nih.gov/contentfiles/lvguidelines/adultandadolescentgl.pdf.
- 9.Hue S, Gifford RJ, Dunn D, Fernhill E, Pillay D. J Virol. 2009;83:2645–2654. doi: 10.1128/JVI.01556-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh AK, Anderson DD, Weber IT, Mitsuya H. Angew Chem Int Ed. 2012;51:1778–1802. doi: 10.1002/anie.201102762. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem. 2012;124:1812–1838. [Google Scholar]
- 11.a) Koh Y, Aoki M, Danish ML, Aoki-Ogata H, Amano M, Das D, Shafer RW, Ghosh AK, Mitsuya H. J Virol. 2011;85:10079–10089. doi: 10.1128/JVI.05121-11. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. Antimicrob Agents Chemother. 2003;47:3123–3129. doi: 10.1128/AAC.47.10.3123-3129.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Chapsal BD, Mitsuya H. Darunavir, a New PI with Dual Mechanism: From a Novel Drug Design Concept to New Hope against Drug-Resistant HIV. In: Ghosh AK, editor. Aspartic Acid Proteases as Therapeutic Targets, Methods and Principles in Medicinal Chemistry. Vol. 45. Wiley-VCH; Weinheim: 2010. pp. 205–243. [Google Scholar]
- 13.De Meyer S, Lathouwers E, Dierynck I, De Paepe E, Van Baelen B, Vangeneugden T, Spinosa-Guzman S, Lefebvre E, Picchio G, de Béthune MP. AIDS. 2009;23:1829–1840. doi: 10.1097/QAD.0b013e32832cbcec. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi H, Takamune N, Nirasawa T, Aoki M, Morishita Y, Das D, Koh Y, Ghosh AK, Misumi S, Mitsuya H. Proc Natl Acad Sci USA. 2014;111:12234–12239. doi: 10.1073/pnas.1400027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh Y, Matsumi S, Das D, Amano M, Davis DA, Li J, Leschenko S, Baldridge A, Shioda T, Yarchoan R, Ghosh AK, Mitsuya H. J Biol Chem. 2007;282:28709–28720. doi: 10.1074/jbc.M703938200. [DOI] [PubMed] [Google Scholar]
- 16.Clotet B, Feinberg J, van Lunzen J, Khuong-Josses MA, Antinori A, Dumitru I, Pokrovskiy V, Fehr J, Ortiz R, Saag M, Harris J, Brennan C, Fujiwara T, Min S. Lancet. 2014;383:2222–2231. doi: 10.1016/S0140-6736(14)60084-2. [DOI] [PubMed] [Google Scholar]
- 17.Koh Y, Amano M, Towata T, Danish M, Leshchenko-Yashchuk S, Das D, Nakayama M, Tojo Y, Ghosh AK, Mitsuya H. J Virol. 2010;84:11961–11969. doi: 10.1128/JVI.00967-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saylor D, Dickens AM, Sacktor N, Haughey N, Slusher B, Pletnikov M, Mankowski JL, Brown A, Volsky DJ, McArthur JC. Nat Rev Neurol. 2016;12:234–248. doi: 10.1038/nrneurol.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heaton RK, Clifford DB, Franklin DR, Woods SP, Ake C, Vaida F, Ellis RJ, Letendre SL, Marcotte TD, Atkinson JH, Rivera-Mindt M, Vigil OR, Taylor MJ, Collier AC, Marra CM, Gelman BB, McArthur JC, Morgello S, Simpson DM, McCutchan JA, Abramson I, Gamst A, Fennema-Notestine C, Jernigan TL, Wong J, Grant I. Neurology. 2010;75:2087–2096. doi: 10.1212/WNL.0b013e318200d727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tozzi V, Balestra P, Serraino D, Bellagamba R, Corpolongo A, Piselli P, Lorenzini P, Visco-Comandini U, Vlassi C, Quartuccio ME, Giulianelli M, Noto P, Galgani S, Ippolito G, Antinori A, Narciso P. AIDS Res Hum Retroviruses. 2005;21:706–713. doi: 10.1089/aid.2005.21.706. [DOI] [PubMed] [Google Scholar]
- 21.Ghosh AK, Chapsal BD, Weber IT, Mitsuya H. Acc Chem Res. 2008;41:78–86. doi: 10.1021/ar7001232. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh AK, Chapsal BD. Design of the Anti-HIV-1 Protease Inhibitor Darunavir. In: Ganellin CR, Jefferis R, Roberts S, editors. Introduction to Biological and Small Molecule Drug Research and Development: Theory and Case Studies. Elsevier; London: 2013. pp. 355–384. [Google Scholar]
- 23.Aoki M, Hayashi H, Rao KV, Das D, Higashi-Kuwata N, Bulut H, Aoki-Ogata H, Takamatsu Y, Yedidi RS, Davis DA, Hattori S-i, Nishida N, Hasegawa K, Takamune N, Nyalapatla PR, Osswald HL, Jono H, Saito H, Yarchoan R, Misumi S, Ghosh AK, Mitsuya H. eLife. 2017;6:e28020. doi: 10.7554/eLife.28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh AK, Chapsal BD, Baldridge A, Steffey MP, Walters DE, Koh Y, Amano M, Mitsuya H. J Med Chem. 2011;54:622–634. doi: 10.1021/jm1012787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghosh AK, Sridhar PR, Leshchenko S, Hussain AK, Li J, Kovalevsky AY, Walters DE, Wedekind J, Grum-Tokars V, Das D, Koh Y, Maeda K, Gatanaga H, Weber IT, Mitsuya H. J Med Chem. 2006;49:5252–5261. doi: 10.1021/jm060561m. [DOI] [PubMed] [Google Scholar]
- 26.Ghosh AK, Yashchuk S, Mizuno A, Chakraborty N, Agniswamy J, Wang Y-F, Aoki M, Salcedo Gomez PM, Amano M, Weber IT, Mitsuya H. ChemMedChem. 2015;10:107–115. doi: 10.1002/cmdc.201402358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh AK, Rao KV, Nyalapatla PR, Osswald HL, Martyr CD, Aoki M, Hayashi H, Agniswamy J, Wang YF, Bulut H, Das D, Weber IT, Mitsuya H. J Med Chem. 2017;60:4267–4278. doi: 10.1021/acs.jmedchem.7b00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park BK, Kitteringham NR, O’Neill PM. Annu Rev Pharmacol Toxicol. 2001;41:443–470. doi: 10.1146/annurev.pharmtox.41.1.443. [DOI] [PubMed] [Google Scholar]
- 29.Smart BE. J Fluorine Chem. 2001;109:3–11. [Google Scholar]
- 30.Ghosh AK, Thompson WJ, Holloway MK, McKee SP, Duong TT, Lee HY, Munson PM, Smith AM, Wai JM, Darke PL. J Med Chem. 1993;36:2300–2310. doi: 10.1021/jm00068a006. [DOI] [PubMed] [Google Scholar]
- 31.Katsuki T, Sharpless KB. J Am Chem Soc. 1980;102:5974–5976. [Google Scholar]
- 32.Gao Y, Hanson RM, Klunder JM, Ko SY, Masamue H, Sharpless KB. J Am Chem Soc. 1987;109:5765–5780. [Google Scholar]
- 33.Caron M, Carlier K, Sharpless KB. J Org Chem. 1988;53:5185–5187. [Google Scholar]
- 34.Ghosh AK, McKee SP, Lee HY, Thompson WT. J Chem Soc Chem Commun. 1992:273–274. doi: 10.1039/C39920000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghosh AK, Leshchenko S, Noetzel M. J Org Chem. 2004;69:7822–7829. doi: 10.1021/jo049156y. [DOI] [PubMed] [Google Scholar]
- 36.Gololobov YG, Zhmurova IN, Kasukhin LF. Tetrahedron. 1981;37:437–472. [Google Scholar]
- 37.Tie Y, Boross PI, Wang Y-F, Gaddis L, Hussian AK, Leshchenko S, Ghosh AK, Louis JM, Harrison RW, Weber IT. J Mol Biol. 2004;338:341–352. doi: 10.1016/j.jmb.2004.02.052. [DOI] [PubMed] [Google Scholar]
- 38.Toth MV, Marshall GR. Int J Pept Protein Res. 1990;36:544–550. doi: 10.1111/j.1399-3011.1990.tb00994.x. [DOI] [PubMed] [Google Scholar]
- 39.Aoki M, Hayashi H, Yedidi RS, Martyr CD, Takamatsu Y, Aoki-Ogata H, Nakamura T, Nakata H, Das D, Yamagata Y, Ghosh AK, Mitsuya H. J Virol. 2016;90:2180–2194. doi: 10.1128/JVI.01829-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aoki M, Danish ML, Aoki-Ogata H, Amano M, Ide K, Das D, Koh Y, Mitsuya H. J Virol. 2012;86:13384–13396. doi: 10.1128/JVI.07234-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zürcher M, Diederich F. J Org Chem. 2008;73:4345–4361. doi: 10.1021/jo800527n. [DOI] [PubMed] [Google Scholar]
- 42.Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 43.Mahalingam B, Louis JM, Hung J, Harrison RW, Weber IT. Proteins Struct Funct Bioinf. 2001;43:455–464. doi: 10.1002/prot.1057. [DOI] [PubMed] [Google Scholar]
- 44.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In: Carter CW Jr, Sweet RM, editors. Methods in Enzymology, 276: Macromolecular Crystallography, Part A. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Acta Crystallogr Sect D. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collaborative Computational Project, Number 4. Acta Crystallogr Sect D. 1994;50:760–763. [Google Scholar]
- 48.Potterton E, Briggs P, Turkenburg M, Dodson E. Acta Crystallogr Sect D. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 49.Shen CH, Wang YF, Kovalevsky AY, Harrison RW, Weber IT. FEBS J. 2010;277:3699–3714. doi: 10.1111/j.1742-4658.2010.07771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheldrick GM. Acta Crystallogr Sect A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 51.Sheldrick GM, Schneider TR. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 52.Schüttelkopf AW, van Aalten DMF. Acta Crystallogr Sect D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr Sect D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Emsley P, Cowtan K. Acta Crystallogr Sect D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 55.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.