

ABSTRACT
Candida albicans, a prevailing opportunistic fungal pathogen of humans, has a diploid genome containing three homologous FKS genes that are evolutionarily conserved. One of these, the essential gene FKS1, encodes the catalytic subunit of glucan synthase, which is the target of echinocandin drugs and also serves as a site of drug resistance. The other two glucan synthase-encoding genes, FKS2 and FKS3, are also expressed, but their roles in resistance are considered unimportant. However, we report here that expression of FKS1 is upregulated in strains lacking either FKS2 or FKS3. Furthermore, in contrast to what is observed in heterozygous FKS1 deletion strains, cells lacking FKS2 or FKS3 contain increased amounts of cell wall glucan, are more resistant to echinocandin drugs, and consistently are tolerant to cell wall-damaging agents. Our data indicate that C. albicans FKS2 and FKS3 can act as negative regulators of FKS1, thereby influencing echinocandin susceptibility.
KEYWORDS: Candida albicans, FKS genes, echinocandins, transcriptional regulation
INTRODUCTION
Fungal infections are currently a leading cause of the global burden of infectious diseases, surpassing the death toll from drug-resistant tuberculosis or malaria (1). Candida infections are responsible for most systemic invasive fungal infections, with mortality rates due to invasive candidiasis reaching 40 to 60% (2). Despite an increase in the diversity of Candida species isolated from clinical samples, Candida albicans, a unicellular budding fungus with a diploid genome that propagates as an opportunistic pathogen in normal human gut or genital microflora, is still a predominant cause of infections, causing up to 50% of candidemia (3, 4).
Members of the echinocandin class of antifungal drugs are currently recommended as front-line treatment for C. albicans infections, because of their low toxicities and high efficacies, especially against isolates resistant to azole drugs (5). Echinocandins are fungus-specific drugs that inhibit 1,3-β-glucan synthase and, ultimately, the synthesis of 1,3-β-glucan, the principal component of cell walls, to which all other main components (1,6-β-glucans, mannoproteins, and chitin) are cross-linked (6). Initially, levels of resistance to echinocandins were low. However, expanding use of echinocandins to prevent or to treat invasive candidiasis has led to an increase in the number of breakthrough infections due to resistant strains of C. albicans, as well as other resistant Candida species (7, 8). The only generally accepted mechanism of C. albicans echinocandin clinical resistance involves point mutations in the essential FKS1 (FK506 sensitivity, also referred to as GSC1; orf19.2929) gene, encoding a catalytic subunit of the 1,3-β-glucan synthase complex (9). Resistance mutations are clustered in two “hot spot” regions, HS1 and HS2, encompassing residues 641 to 649 and residues 1345 to 1365, respectively (8). These mutations decrease the sensitivity of glucan synthase to echinocandins by several orders of magnitude (10).
The genome of C. albicans contains two additional FKS genes that share considerable sequence identity with homologous FKS1, i.e., FKS2 (also referred to as GSL2; orf19.3269) and FKS3 (also referred to as GSL1; orf19.2495). The FKS genes consist of open reading frames of 5.7 kb (FKS1), 4.9 kb (FKS2), and 4.7 kb (FKS3) (http://www.candidagenome.org/cgi-bin/locus.pl?dbid=CAL0000176780, http://www.candidagenome.org/cgi-bin/locus.pl?dbid=CAL0000186782, and http://www.candidagenome.org/cgi-bin/locus.pl?dbid=CAL0000179710), which correspond to predicted proteins of 218 kDa, 189 kDa, and 182 kDa, respectively. According to Mio et al. (11), who used C. albicans strain CAI4, FKS1 and FKS3 produced transcripts of approximately 6 kb and 3.5 kb, respectively, whereas a FKS2 transcript could not be detected on the Northern blot. However, our transcriptional profiles of three strains (reference strain SC5314, popular laboratory strain 3153A, and JRCT1 [12]), determined using transcriptome sequencing (RNA sequencing), showed the following respective normalized fragments per kilobase million (FPKM) values for each of the three genes: 56, 58, and 73 FPKM for FKS1; 7, 7, and 9 FPKM for FKS2; and 3, 3, and 3 FPKM for FKS3 (E. Rustchenko, unpublished data). Fks1p, Fks2p, and Fks3p are predicted to be integral membrane proteins containing 16 (Fks1p and Fks2p) or 10 (Fks3p) transmembrane helices (11). The amino acid sequence of Fks1p is 52.3% identical to that of Fks2p, Fks1p is 46.1% identical to Fks3p, and Fks2p is 45.8% identical to Fks3p. These proteins also share conserved hot spot regions and identical catalytic domains of 46 residues for glucan synthase activity (13).
The role of C. albicans FKS1 as encoding a catalytic subunit of glucan synthase complex and its role in drug resistance are well established (11, 14), but remarkably little is known about the roles of FKS2 and FKS3 in echinocandin susceptibility. Furthermore, conservation of FKS genes and their structural similarities among paralogs in other Candida species and in other microbes, including the model yeast Saccharomyces cerevisiae, as well as in plants and protists (13), are indicative of important functions for all three genes.
While the physiological roles of FKS2 and FKS3 in C. albicans are not yet clear, there are indications that they may be involved in determining susceptibility to echinocandins. Levels of expression of C. albicans FKS2 and FKS3, relative to FKS1, are decreased in spontaneous mutants harboring echinocandin resistance mutations in FKS1 (relative to FKS1+ parental strains), and relative levels of expression of FKS genes can vary widely in clinical isolates harboring resistance mutations in FKS1 (14). Furthermore, levels of expression of C. albicans FKS genes were decreased in mutant strains that became tolerant to caspofungin in the absence of FKS1 mutations (15). In addition, S. cerevisiae FKS3 was reported to be a positive regulator of Rho1p, a regulatory subunit in the glucan synthase complex (16), and there is evidence that S. cerevisiae FKS2 negatively regulates susceptibility to caspofungin (17). It was also reported that deletion of S. cerevisiae FKS2 and FKS3 resulted in the formation of abnormal spore walls (16).
In this work, to better understand the roles of FKS2 and FKS3 in laboratory resistance to echinocandins, we prepared and characterized deletion mutants lacking both copies of either FKS2 or FKS3. In order to compare the properties of all three FKS genes, we also independently deleted one copy of the essential FKS1 gene. We found that deletion mutants lacking either FKS2 or FKS3 exhibited decreased susceptibility to echinocandins and increased expression of FKS1, which was accompanied, as expected, by increased amounts of cell wall 1,3-β-glucan. In contrast, heterozygous deletion of FKS1 resulted in enhanced echinocandin susceptibility, decreased levels of cell wall glucan, and increased levels of chitin in the cell wall. Our findings suggest that aspects of FKS2 and FKS3 functions in C. albicans may be quite distinct from the functions of FKS1 and that FKS2 and FKS3 negatively control FKS1.
RESULTS
Susceptibility to caspofungin and anidulafungin in deletion mutants.
We prepared independent deletion mutants either lacking one copy of the essential gene FKS1 or lacking one or both copies of FKS2 or FKS3 (Table 1), as described in Materials and Methods and illustrated in Fig. S1 in the supplemental material. We took care to prepare three separately derived mutants of each kind, i.e., fks1+/−, fks2+/− or fks2−/−, and fks3+/− or fks3−/− (Table 1). (Note: Gene names are indicated by italicized capital letters, but the deleted genes use italicized lowercase letters.) All deletion mutants were analyzed with an agar-based spot assay for susceptibility to caspofungin, and one representative deletion strain lacking one copy of the FKS1 gene or two copies of either FKS2 or FKS3 was also analyzed for susceptibility to anidulafungin.
TABLE 1.
C. albicans strains used in this study
| Strain | Genotypea | Source |
|---|---|---|
| CAF2-1 | URA3/ura3Δ::λimm434 | 45 |
| CAF4-2 | ura3Δ::λimm434/ura3Δ::λimm434 | 45 |
| NF133 (fks1−/+) | fks1Δ::URA3-FLP/FKS1 | This study |
| NF1-1-1 (fks1−/+) | fks1Δ::URA3-FLP/FKS1 | This study |
| NF1-4 (fks1−/+) | fks1Δ::URA3-FLP/FKS1 | This study |
| NF2-3 (fks2−/+) | fks2Δ::URA3-FLP/FKS2 | This study |
| NFK2-5-9 (fks2−/−) | fks2Δ::URA3-FLP/fks2Δ::NAT1-FLP | This study |
| NF2-2-3 (fks2−/+) | fks2Δ::URA3-FLP/FKS2 | This study |
| NFK2-2-12 (fks2−/−) | fks2Δ::URA3-FLP/fks2Δ::NAT1-FLP | This study |
| NF2-3-4 (fks2−/+) | fks2Δ::URA3-FLP/FKS2 | This study |
| NFK2-3-1 (fks2−/−) | fks2Δ::URA3-FLP/fks2Δ::NAT1-FLP | This study |
| NF3-1-1 (fks3−/+) | fks3Δ::URA3-FLP/FKS3 | This study |
| NFK3-8-1 (fks3−/−) | fks3Δ::URA3-FLP/fks3Δ::NAT1-FLP | This study |
| NF3-2-1 (fks3−/+) | fks3Δ::URA3-FLP/FKS3 | This study |
| NFK3-7-19 (fks3−/−) | fks3Δ::URA3-FLP/fks3Δ::NAT1-FLP | This study |
| NF3-3-1 (fks3−/+) | fks3Δ::URA3-FLP/FKS3 | This study |
| NFK3-9-55 (fks3−/−) | fks3Δ::URA3-FLP/fks3Δ::NAT1-FLP | This study |
| NUNU38 (fks2−/− fks3−/−) | fks2Δ::FRT/fks2Δ::FRT/fks3Δ::URA3-FLP/fks3Δ::NAT1-FLP | This study |
| NUNU85 (fks2−/− fks3−/−) | fks2Δ::FRT/fks2Δ::FRT/fks3Δ::URA3-FLP/fks3Δ::NAT1-FLP | This study |
URA3-FLP denotes the FRT-SAP2P–FLP-URA3-FRT cassette (URA3 flipper). NAT1-FLP denotes the FRT-SAP2P–FLP-NAT1-FRT cassette (NAT1 flipper).
In pilot experiments, we determined optimal sub-MIC concentrations that revealed in vitro growth differences, i.e., 80 ng/ml for caspofungin and 9 ng/ml for anidulafungin, based on the number of growing spots of 10-fold serial dilutions of cultures for deletion mutants and the parental strain CAF4-2 (Ura−), as well as the related Ura+ strain CAF2-1. For example, the absence of any detectable growth of fks1+/− cells in any of four spots on the plate with caspofungin versus CAF4-2 and CAF2-1 was considered to indicate suppressed growth of the deletion mutant, compared to that of the control strains (Fig. 1A). Also, the growth of CAF4-2 and CAF2-1 in the first spot on the left and the reduced growth of CAF2-1 in next spot versus fks2−/− cells growing in all four spots were considered to indicate suppressed growth of the control strains versus the deletion mutant (Fig. 1A).
FIG 1.

Analysis of caspofungin or anidulafungin susceptibility phenotype. (A) Spot assay showing comparative growth of representative deletion strains lacking one copy of the essential FKS1 gene or one or two copies of either FKS2 or FKS3, compared to the Ura− parental strain CAF4-2 and the isogenic Ura+ strain CAF2-1, on YPD medium and on YPD medium supplemented with caspofungin (CAS) or anidulafungin (ANI). Strains and their genotypes are indicated on the left. Cells (from left to right, 104, 103, 102, and 101 cells) were spotted on a plate and incubated for 3 days at 37°C. Note that one experiment, representative of three independent experiments, is shown; see Fig. S2A and B for the two other assays. (B and C) Standard broth microdilution assays showing comparative growth of the strains in panel A after 24 h of incubation in media containing different concentrations of caspofungin (B) or anidulafungin (C), as indicated. Note that one experiment, representative of three independent experiments, is shown; see Fig. S3A and B for the two other assays. OD600, optical density at 600 nm.
We found that deletion of one copy of the FKS1 gene led to nearly complete suppression of growth in all serial dilution spots in the presence of caspofungin, compared to the control strains, thus indicating increased caspofungin susceptibility (Fig. 1A). This phenotype in C. albicans is similar to the phenotype resulting from FKS1 deletion in S. cerevisiae (note that S. cerevisiae FKS1 is not essential) and in C. albicans (17–20) by other laboratories.
In contrast to the results of FKS1 deletion, strains lacking FKS2 or FKS3 exhibited better growth than control strains, indicating decreased caspofungin susceptibility (Fig. 1A). We present the relative growth of diverse deletion mutants in one representative experiment in Fig. 1A, and the results of two repetitions are shown in Fig. S2A and B. These results are in agreement with previous observations that deletion of S. cerevisiae FKS2 resulted in decreased susceptibility to echinocandins (17). Results of growth assays in the presence of the echinocandin drug anidulafungin were similar to those for caspofungin; growth of the mutant lacking one copy of FKS1 was greatly diminished, while growth of mutants lacking either FKS2 or FKS3 was enhanced (Fig. 1A).
The impact of FKS genes on echinocandin susceptibility was also tested with a standard broth microdilution assay (see Materials and Methods), using one representative deletion mutant of each kind, i.e., fks1+/−, fks2−/−, and fks3−/−. As expected, the concentration of caspofungin (31.25 ng/ml) or anidulafungin (0.24 ng/ml) that completely inhibited growth of the fks1+/− mutant was lower than that needed to completely inhibit the control strains CAF4-2 and CAF2-1 (62.5 ng/ml caspofungin and 0.49 ng/ml anidulafungin) (Fig. 1B and C). The fks1+/− mutant also showed less growth overall. In contrast, the fks2−/− and fks3−/− mutants showed more growth overall than the control strains. The growth of the fks3−/− mutant and the control strains was completely inhibited by the same amount of caspofungin (62.5 ng/ml), but a higher concentration of anidulafungin (0.98 ng/ml) was needed to completely inhibit the fks3−/− mutant, compared to that needed to inhibit the control strains (0.49 ng/ml) (Fig. 1B and C). The growth of the fks2−/− mutant was completely inhibited by higher concentrations of both caspofungin (125 ng/ml) and anidulafungin (0.98 ng/ml), compared to the control strains. The fks2−/− and fks3−/− mutants also showed higher MIC50 values for caspofungin (>40 ng/ml for fks3−/− and ∼50 ng/ml for fks2−/− versus ∼30 ng/ml for parental CAF4-2), as well as higher MIC50 values for anidulafungin (∼0.37 ng/ml for fks3−/− and 0.24 ng/ml for fks2−/− versus ∼0.18 ng/ml for parental CAF4-2). Two more repeats of this experiment are presented in Fig. S3A and B. Thus, results using the broth microdilution approach are consistent with those from the spotting assay, indicative of different effects of the deletion of FKS1, compared with FKS2 and FKS3, on susceptibility to echinocandin drugs.
Viability of homozygous double deletion mutants.
We also prepared two independent deletion mutants, NUNU39 and NUNU85, lacking both copies of FKS2 and FKS3 (fks2−/− fks3−/−) (Table 1). As shown in Fig. S5, these mutants were viable and their growth on yeast-peptone-dextrose (YPD) plates was the same as growth of the parental CAF4-2 strain or the control CAF2-1 strain.
Susceptibility to cell wall-damaging agents in deletion mutants.
To test the effects of deletion of the different FKS genes on the structure of the cell wall, deletion mutants were tested for the ability to grow in the presence of cell wall-damaging agents. These agents included Congo red, which binds to glucan, calcofluor white, which binds to chitin, and hygromycin B, which inhibits polypeptide synthesis and exposes defects of mannoprotein glycosylation. The test was repeated in three independent experiments, as exemplified by one experiment in Fig. 2 (see Fig. S4 for two other experiments). We found that deletion of one copy of FKS1 rendered cells more sensitive to all three agents. However, these cell wall stressors had no effect on the growth of mutants with homozygous deletions of FKS2 or FKS3. This suggests that the cell wall remains largely intact in mutants lacking FKS2 or FKS3, unlike the situation for the mutant lacking one copy of FKS1, which is consistent with FKS1 encoding the principal glucan synthase used in cells.
FIG 2.

Susceptibilities of representative FKS deletion mutants to cell wall-damaging agents, compared to the parental strain. The spot assay shows comparative growth of representative strains lacking one copy of the essential FKS1 gene or two copies of either FKS2 or FKS3, compared to the Ura− parental strain CAF4-2 and the isogenic Ura+ control strain CAF2-1, on control YPD medium and on YPD medium supplemented with Congo red, calcofluor white, or hygromycin B, as indicated. Concentrations of the cell wall-damaging agents are indicated. Strains are indicated on the left. For more details, see the legend to Fig. 1. Note that one of three independent repeats of the spot assay is shown here; see Fig. S4A and B for the two other assays.
Levels of 1,3-β-glucan and chitin in cell walls of deletion mutants.
The cell walls of the independent deletion mutants NF133 and NF1-1-1, lacking one copy of FKS1 (fks1+/−), contained significantly decreased amounts of 1,3-β-glucan (∼70% of the amounts in the parental CAF4-2 strain, as exemplified by NF133 in Fig. 3A; see Fig. S6A for NF1-1-1). The fks1+/− cells also contained ∼2-fold increased amounts of chitin in the cell wall (exemplified by NF133 in Fig. 3B; see Fig. S6B for NF1-1-1). Such remodeling can be interpreted as a compensatory change to fortify a weakened cell wall. In contrast, independent deletion of both copies of FKS2 (fks2−/−) in mutants NFK2-2-12 and NFK2-3-1 resulted in ∼15% increased amounts of glucan, and independent deletion of both copies of FKS3 (fks3−/−) in mutants NFK3-8-1 and NFK3-9-55 resulted in ∼20% increased amounts of glucan (Fig. 3A; also see Fig. S6A) but had no significant effect on the amount of chitin (Fig. 3B; also see Fig. S6B).
FIG 3.

Levels of cell wall glucan (A) or chitin (B) in representative deletion mutants. Mutant NF133 (fks1+/−), lacking one copy of FKS1, and mutants NFK2-2-12 (fks2−/−) and NFK3-8-1 (fks3−/−), lacking both copies of either FKS2 or FKS3, as indicated, are shown. Also shown are the Ura− parental strain CAF4-2 and the Ura+ control strain CAF2-1. Results from three independent experiments were averaged. The amount of glucan was calculated relative to that in the parental strain CAF4-2, which was considered 100%. The asterisks indicate P values of <0.05, as determined using Student's t test. See Fig. S6A and B for the same experiments conducted with a second set of deletion mutants.
Expression of FKS genes in deletion mutants.
We determined the expression of intact FKS genes in representative deletion mutants using semiquantitative reverse transcription (RT)-PCR (see Materials and Methods for growth of the strains and the RT-PCR method). Changes in expression of the gene of interest were calculated as the mutant/parent ratios of normalized expression values. In the mutant NF133, with only one copy of the essential FKS1 gene, the expected downregulation of FKS1 transcript levels (∼1.5-fold) occurred, with no effect on the expression of either FKS2 or FKS3 (Fig. 4A). In contrast, the lack of either FKS2 (mutant NFK2-2-12) (Fig. 4B) or FKS3 (mutant NFK3-8-1) (Fig. 4C) upregulated FKS1 expression ∼1.5-fold. We assume that the degree of FKS1 upregulation can vary in strains depending on the genetic background. The data presented in Fig. 4B and C indicate that there are differences between FKS2 and FKS3 regarding their effects on the expression of other FKS genes. The lack of FKS3 enhanced expression of both FKS1 and FKS2 ∼1.5-fold (Fig. 4C); however, the deletion of FKS2 reduced FKS3 expression ∼2-fold, while enhancing FKS1 expression ∼1.5-fold (Fig. 4B).
FIG 4.
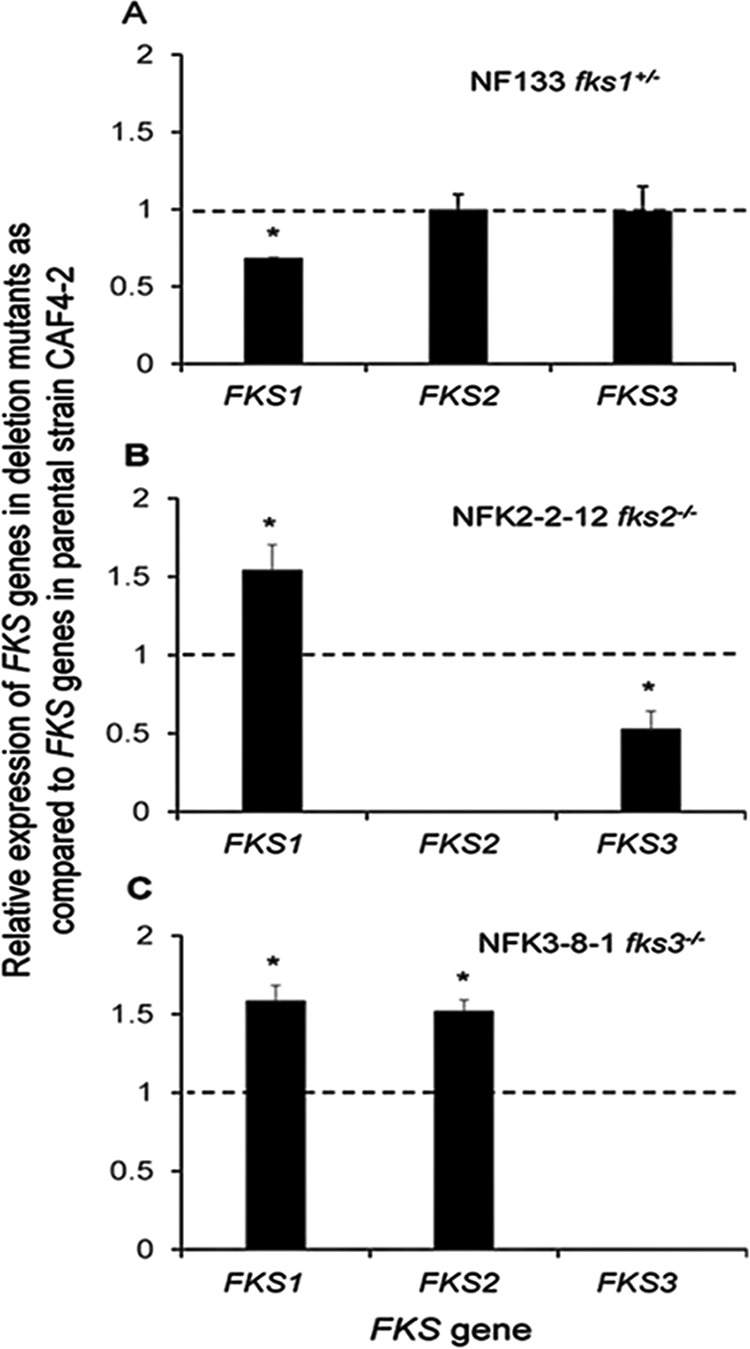
Changes in FKS1, FKS2, or FKS3 expression in mutants lacking one copy of FKS1 (A), both copies of FKS2 (B), or both copies of FKS3 (C). The names of the mutants are indicated at the top of each panel (also see Table 1). Expression changes were determined as the mutant/parent ratio for each gene, by semiquantitative RT-PCR analysis of three independent RNA preparations. The y axis shows the relative level of expression for each gene. The dashed lines indicate the expression level of each FKS gene in the parental strain CAF4-2. The asterisks indicate P values of <0.05, as determined by Student's t test. The absent bars indicate no expression of the lacking genes.
Inhibition of glucan synthase by caspofungin in deletion strains.
We assessed direct inhibition of glucan synthases by caspofungin by determining half-maximal inhibitory concentration (IC50) values for glucan synthase enzymes partially purified from strains lacking various FKS genes (one copy of FKS1 or both copies of FKS2 or FKS3) (Table 1). Caspofungin inhibition of glucan synthase for the control strain CAF4-2 and the aforementioned mutants could be fit to curves characterized by single inflection points, reflecting IC50s of 9.1, 3.6, 9.7, and 7.2 ng/ml, respectively (Fig. 5). These biochemical assays have inherent variability. The overall shape and inflection of the drug inhibition curves are key features. There was no statistical difference between the parental CAF4-2 strain and mutants lacking FKS2 or FKS3. The FKS1/fks1 heterozygote was more sensitive, which is consistent with previous work (19). Our data indicated that glucan synthases isolated from each of the strains were sensitive to caspofungin and the potency of the drug was approximately the same in all cases. Apparently, glucan synthase inhibition data confirm that FKS1 is still expressed and glucan synthase is assembled in deletion mutants.
FIG 5.

Inhibition of glucan synthases by caspofungin (CAS) in different deletion strains. Mutant NF133 (fks1+/−), lacking one copy of FKS1, and mutants NFK2-2-12 (fks2−/−) and NFK3-8-1 (fks3−/−), lacking both copies of either FKS2 or FKS3, as indicated, are shown. Also shown is the parental strain CAF4-2.
Germ tube formation in deletion strains.
We investigated whether lack of FKS genes affected the ability to form germ tubes. Cells of the parental strain CAF4-2, as well as the fks1+/−, fks2−/−, fks3−/−, and fks2−/− fks3−/− deletion mutants, were exposed to undiluted serum (see Materials and Methods), and then germinated cells were counted. Experiments were performed in triplicate; the numbers of germinated cells from the three replicates were averaged, and standard deviations were calculated. We found that approximately 88.00% ± 5.20% of cells of the parental strain CAF4-2 responded to serum exposure by germination. Deletion mutants showed a range of germinated cells, from 85.67% ± 4.04% to 94.33% ± 5.69%. Therefore, we concluded that lack of FKS genes does not affect germination and FKS genes are not responsible for germ tube formation.
DISCUSSION
While it was demonstrated previously that the clinical resistance of C. albicans to drugs of the echinocandin class depends on specific point mutations in FKS1 (8, 9), little is known about the roles of the related FKS2 and FKS3 genes in drug resistance. In this work, we present evidence that FKS2 and FKS3 negatively control laboratory susceptibility to echinocandins. We find that homozygous deletion of either FKS2 or FKS3 decreases susceptibility to echinocandins. These deletions result in increased expression of FKS1, accompanied by an increase in cell wall glucan, consistent with the known role of FKS1 as a catalytic subunit of glucan synthase, and no increased sensitivity to cell wall-damaging agents. In contrast, heterozygous deletion of the essential FKS1 gene results in increased susceptibility to echinocandins and to cell wall-disrupting treatments, most likely resulting from a decrease of cell wall glucan. Deletion of FKS1 also results in an increase in the cell wall chitin content, which may serve as compensation for the decrease in glucan content. Deletion of one copy of FKS1 had no effect on the expression of either FKS2 or FKS3. Assays of the effects of each of the FKS deletions on the ability of caspofungin to inhibit glucan synthase suggest that the assembled enzymes retain the same kinetic properties and that deletions do not affect echinocandin interactions with the cellular complements of glucan synthase.
Although previous reports indicated that increased chitin content correlates with decreased drug susceptibility, the increase in chitin content associated with heterozygous deletion of FKS1 is accompanied by increased drug susceptibility. This indicates that variations in cell wall chitin contents alone are not the only factor affecting echinocandin susceptibility.
Our data imply that C. albicans FKS2 and FKS3 do not play significant roles in cell wall biosynthesis in cells with the normal complement of Fks1p. The latter has a well-established role as a catalytic subunit of the 1,3-β-glucan synthase enzymatic complex, based on its high degree of sequence similarity to Fks1p from S. cerevisiae (which is also thought to be a catalytic subunit), on the reported decrease in glucan synthase activity resulting from decreased FKS1 gene dosage in C. albicans, and on the ability to immunoprecipitate glucan synthase activity using a monoclonal antibody raised against purified C. albicans Fks1p (11, 14). Thus, in contrast to FKS1, the FKS2 and FKS3 genes appear to play regulatory roles in cells. We also show here that there is mutual regulation between FKS2 and FKS3, which involves some complexity that requires future studies. It seems that FKS2 is a positive regulator of FKS3 expression, while FKS3 is a negative regulator of FKS2 expression.
As presented by Xie et al. (21), the transcription factor Cas5p plays a dual role by mediating cell cycle dynamics and cell wall stress responses in C. albicans. Homozygous deletion of CAS5 reduced resistance of a strain carrying the FKS1 F641S substitution, suggesting that Cas5p can influence FKS1-mediated echinocandin resistance (21). The nature of this interaction is unknown, and future studies will elucidate the relationship between FKS1, as well as genes such as CAS5, and FKS2 and FKS3.
It is of relevance to our data that the roles of FKS2 and FKS3 vary among different fungal species. For example, in S. cerevisiae, the most extensively studied system in this regard, FKS1 and FKS2 both appear to encode catalytic subunits of the glucan synthase complex, since residual glucan synthase activity is detected in strains completely lacking FKS1 and this residual activity can be immunodepleted with anti-Fks2p antibody (22, 23). Deletion of both FKS1 and FKS2 from S. cerevisiae is lethal, whereas, in contrast to the case for C. albicans (in which FKS1 is essential), S. cerevisiae strains containing single deletions of FKS1 and FKS2 are viable, indicating that there is redundancy in which the essential glucan synthase function can be encoded by either one (23). The functional redundancy of FKS1 and FKS2 is incomplete, however, as fks1Δ mutants grow slowly (24). Furthermore, in S. cerevisiae lacking FKS1, echinocandin resistance can arise through mutations in FKS2 corresponding to previously known resistance mutations in hot spot regions of FKS1 (25). Interestingly, deletion of S. cerevisiae FKS2 results in better growth in the presence of caspofungin (17). Although deletion of FKS3 results in formation of abnormal spore walls in S. cerevisiae (16), the exact function of FKS3 in this species remains unclear.
In the haploid species Candida glabrata, FKS1 and FKS2 are functionally redundant and, similar to the situation in S. cerevisiae, double deletions of fks1 and fks2 cannot be generated. C. glabrata strains with all three single-gene FKS deletions, as well as the double deletions fks1 fks3 and fks2 fks3, exhibit normal echinocandin susceptibilities. Echinocandin resistance mutations in C. glabrata occur in hot spot regions of either FKS1 or FKS2, but mutations of this type are more frequent in FKS2 (24).
The subunit composition of the glucan synthase complex remains poorly understood, beyond the determination that the complex includes at least a catalytic subunit encoded by an FKS gene and a regulatory subunit encoded by RHO1 (orf19.2843), which is a GTP-binding protein activating Fksp (26, 27). Thus, it has been proposed that the FKS genes encode alternative catalytic subunits of the glucan synthase complex (11; reviewed in reference 28).
In summary, our results raise three important points. (i) The roles of FKS2 and FKS3 in cells differ from the role of FKS1, raising questions about whether the functions of FKS2 and FKS3 depend on their catalytic activities, despite their similarities to FKS1. (ii) Deletion of FKS2 or FKS3 leads to an increase in transcript levels for FKS1, suggesting the possibility that FKS2 and FKS3 normally enhance echinocandin susceptibility by acting as negative regulators of FKS1, a possibility that is also supported by previous studies (10, 15). (iii) FKS2 and FKS3 in C. albicans are not completely functionally redundant, since individual deletion of the genes resulted in distinct phenotypes, related to their effects on expression of the remaining FKS genes. Future studies are needed to better understand the mechanisms of action of FKS2 and FKS3 and also to study the putative roles of FKS2 and FKS3 in clinical resistance.
MATERIALS AND METHODS
Strains, media, and growth conditions.
Strains used in this study are presented in Table 1. Cells were routinely maintained at 37°C. All strains were stored in 25% (vol/vol) glycerol solution at −70°C, to prevent induction of chromosome instability (29–32). Escherichia coli strain DH5α was used to propagate plasmids.
The preparation of YPD and synthetic dextrose media was described previously (33, 34). Uridine (50 μg/ml; Sigma, St. Louis, MO), nourseothricin (150 μg/ml; Gold Biotechnology, St. Louis, MO), caspofungin (80 ng/ml; Merck Sharp & Dohme, Kenilworth, NJ), or anidulafungin (9 ng/ml; Pfizer, New York, NY) was added when needed. Media were solidified with 2% (wt/vol) agar. Plasmids and primers used in this study are presented in Tables S1 and S2, respectively, in the supplemental material.
Gene deletions.
We performed deletions as described in reference 35. Briefly, recipient strain CAF4-2 was used to sequentially delete one or two copies of the entire open reading frame of each FKS gene. In order to diminish mutagenic effects of deletion cassette recycling (30, 36), we sequentially used two different deletion cassettes containing either URA3 or NAT1 marker. We independently generated strains lacking one copy of the essential FKS1 or both copies of FKS2 or FKS3. Each kind of deletion strain was obtained from at least three independent transformations, to ensure that the phenotype of interest was related to the deleted gene and not to a random mutation unintentionally generated by the mutagenic procedure (30).
The plasmid pSFU1 carrying the URA3 flipper (37) and pJK863 carrying the NAT1 flipper (38) were used for construction of deletion cassettes. Approximately 300 bp of flanking sequences of FKS genes were PCR amplified from the total genomic DNA of strain CAF4-2 and subcloned into either plasmid pSFU1 carrying the URA3 flipper or pJK863 carrying the NAT1 flipper, thus creating different deletion plasmids. The 5′ noncoding region (NCR) of the target gene was amplified with primers NF21 and NF22, which introduced KpnI and ApaI restriction sites (Table S1), and subsequently was subcloned in either the pSFU1 or pJK863 plasmid linearized with KpnI and ApaI restriction enzymes. The 3′-NCR sequence was amplified with primers NF23 and NF24. which introduced SacII and SacI restriction sites (Table S2), and subsequently was subcloned in either the pSFU1 or pJK863 plasmid linearized with SacII and SacI restriction enzymes. The deletion cassette was released by restriction digestion with KpnI and SacI enzymes and was used to transform CAF4-2 cells. Synthetic dextrose medium was used to select transformants carrying the URA3 flipper, and then YPD medium supplemented with 150 μg/ml nourseothricin was used to select transformants for putative null mutants carrying both the URA3 and NAT1 flippers. Transformants were randomly picked up and purified by subcloning on YPD agar plates. The proper integration of the deletion cassettes was confirmed with PCR using primers specific for junctions between the chromosome and the deletion cassette (Table S2).
Spot assay.
Cells from −70°C freezer stocks were streaked on YPD plates and incubated at 37°C until young colonies (∼2 × 105 cells per colony) appeared. Cells were collected, 10-fold dilutions were prepared, and 5 μl of each dilution was spotted on control YPD solid medium supplemented with 50 μg/ml uridine and either 80 ng/ml caspofungin or 9 ng/ml anidulafungin. After incubation, plates were photographed with a Molecular Imager Gel Doc XR+ system (Bio-Rad, Hercules, CA). Each assay was repeated three times.
Cell wall integrity.
Tests were performed by spotting serial dilutions of cells on plates (see above) supplemented with the following cell wall stressors: 100 μg/ml Congo red (MP Biomedicals, Solon, OH), 75 μg/ml calcofluor white (MP Biomedicals), or 200 μg/ml hygromycin B (Enzo Life Sciences, Farmingdale, NY). Plates lacking cell wall stressors were used as controls. After incubation, plates were photographed as described above. Each assay was repeated three times.
Broth microdilution assay.
We performed a broth microdilution test in accordance with the CLSI broth microdilution method for yeasts (39). An inoculum of 1 × 104 cells/ml of each strain was prepared in RPMI 1640 medium buffered to pH 7.0 with 0.165 M morpholinepropanesulfonic acid (MOPS). A series of 2-fold dilutions of the drug was prepared directly in 96-well, flat-bottom, polystyrene, microtiter plates. Subsequently, 100 μl of the cell suspension was added to each well to give a final concentration of 5 × 103 cells/ml in a total volume of 200 μl. The microtiter plates were incubated for 24 h at 35°C. Both negative (inoculum-free) and positive (drug-free) controls were included. Each strain was tested in duplicate on a microtiter plate. The turbidities were determined at 600 nm using a microplate reader (Spectra Max M5; Molecular Devices Corp.). Subsequently, values were normalized against the control value for parental CAF4-2 in the absence of drug and plotted as a curve. Each assay was repeated three times.
Determination of 1,3-β-glucan contents in cell walls.
Cells were cultured in YPD broth to the log phase, harvested, and washed twice with TE buffer (10 mM Tris-HCl, 1 mM EDTA [pH 8.0]). The same number of cells was prepared from each strain. The 1,3-β-glucan levels were determined by the aniline blue assay, as described previously (12, 35, 40). Briefly, 500 μl of cell suspension from each strain was resuspended with 100 μl of 6 M NaOH, followed by incubation at 80°C for 30 min. After glucan solubilization, 2.1 ml of aniline blue mixture (0.03% aniline blue, 0.18 M HCl, 0.49 M glycine-NaOH [pH 9.5]) was added to each sample, and the samples were incubated at 50°C for 30 min and then cooled to room temperature for 30 min. After incubation, fluorescence was measured in a black 96-well microplate using a fluorescence plate reader (Spectra Max M5; Molecular Devices Corp.), with an excitation wavelength of 400 nm, an emission wavelength of 460 nm, and a wavelength cutoff of 455 nm. Each assay was repeated three times.
Determination of chitin contents in cell walls.
The chitin contents were determined by measuring the absorbance of glucosamine released by acid hydrolysis of the purified cell wall, as described previously (12, 35). Briefly, ∼3 × 103 CFU were plated on YPD plates and incubated at 37°C. Young colonies of each strain (see above) were harvested from the surface of the plates with sterile distilled water and collected by centrifugation. Then, cells were disrupted with 0.5-mm glass beads (product no. 11079105; BioSpec Products, Bartlesville, OK) using a Mini-Beadbeater (BioSpec Products). The pellet was washed five times with 1 M NaCl, extracted with SDS-mercaptoethanol extraction buffer (50 mM Tris, 2% sodium dodecyl sulfate, 0.3 M mercaptoethanol, 1 mM EDTA [pH 8.0]) at 100°C for 10 min, and then washed three times with distilled water. Cells were dried with a SpeedVac concentrator (Phoenix Equipment, Rochester, NY) and weighed. Dry samples were suspended in 1 ml of 6 M HCl and boiled for 17 h. The acid was evaporated at 65°C. The hydrolyzed samples were resuspended in 1 ml of sterile distilled water. A 100-μl portion of each sample was mixed with 100 μl of 1.5 M Na2CO3 in 4% acetyl acetone. The mixture was boiled for 20 min, and then 700 μl of 96% ethanol and 100 μl of p-dimethylaminobenzaldehyde solution in a 1:1 mixture of ethyl alcohol and concentrated HCl were added, followed by 1 h of incubation at room temperature. Optical densities were read at 520 nm with a plate reader (Spectra Max M5; Molecular Devices Corp., Sunnyvale, CA). Glucosamine (Sigma-Aldrich, St. Louis, MO) was used as a standard for measurement of chitin contents. The final chitin level in each sample was calculated as a percentage of the cell wall dry weight. Each assay was repeated three times.
Germ tube formation.
Cells from −70°C freezer stocks were streaked on YPD plates and incubated at 37°C. Young colonies (∼2 × 105cells/colony) were collected, prepared as cell suspensions in YPD liquid medium at a concentration of 104 cells/ml, and cultured at 37°C. When the cell density reached ∼107 cells/ml, cells were harvested and suspended in heat-inactivated undiluted fetal bovine serum (Gibco, Gaithersburg, MD) to yield 105 cells/ml (41). Suspensions were incubated for 1.5 h at 37°C and then fixed with 4% paraformaldehyde in phosphate-buffered saline for 15 min at room temperature (42). Subsequently, 20 μl of cell suspension was transferred to a microscope slide, and a total of 200 cells were examined. Each strain was tested in triplicate.
PCR.
For cloning purposes, PCR was conducted using Ex Taq DNA polymerase (TaKaRa Biomedicals, Shiga, Japan), according to the manufacturer's instructions. Other PCRs were conducted using Dream Taq DNA polymerase (Thermo Scientific, Rockford, IL), according to the manufacturer's instructions. PCR products were electrophoresed on 1% agarose gels at 95 V for 45 min and stained with 1 μg/ml ethidium bromide for 10 min. Gel images were obtained using a Molecular Imager Gel Doc XR+ system (Bio-Rad).
Semiquantitative RT-PCR.
To determine the expression of FKS genes, we standardized the growth of C. albicans cells. Briefly, 100 ml of liquid synthetic medium in which glucose was substituted for sorbitol was seeded with ∼2 × 105 cells and incubated at 37°C for 22 to 24 h until the cell count reached ∼1 × 107 to 2 × 107 cells/ml. Three batches of total RNA were prepared from three independent cultures of each strain. RNA extraction and DNase I treatment were conducted as described previously (43). Reverse transcription was performed using a high-capacity cDNA reverse transcription kit (Applied Biosystems; Thermo Fisher Scientific, Waltham, MA), according to the manufacturer's instructions. PCRs were performed using Dream Taq DNA polymerase (Thermo Scientific, Rockford, IL), according to the manufacturer's instructions.
We performed a pilot PCR study with cDNA from the parental strain CAF4-2 to find appropriate internal control genes showing expression levels similar to those of each FKS gene. We choose ASN1 (orf19.198) for the control of FKS1 and APG7 (orf19.707) for the control of FKS2 and FKS3. The pilot PCR study showed that 23 to 28 amplification cycles produced amplicons with linear increases of DNA. For the final PCRs, approximately 50- to 200-ng (each) aliquots of cDNA for a gene of interest and a control gene were used as the templates, in order to amplify the two genes in the same tube for the aforementioned numbers of cycles. Each transcript was examined in three independent RNA batches. PCR products from several consecutive cycles in the exponential phase were electrophoresed on 1% agarose gels at 95 V for 45 min and stained with 1 μg/ml ethidium bromide for 10 min. The stained DNA bands were photographed using a Molecular Imager Gel Doc XR+ system (Bio-Rad), and band intensities were detected using Image Lab software (Bio-Rad). The gene of interest was normalized against the control gene by calculating the mean ratio of densitometry values. Each assay was repeated three times.
Measurement of inhibition of glucan synthase by caspofungin.
Strains were grown to the stationary phase in YPD broth at 37°C, in a shaking incubator (150 rotations per minute), and were collected by centrifugation. Cell disruption, membrane protein extraction, and partial 1,3-β-glucan synthase purification by-product entrapment were performed as described previously (10). Reactions were initiated by the addition of product-entrapped glucan synthase. Sensitivity to caspofungin was measured in a polymerization assay using a 96-well, 0.65-μm, MultiScreenHTS filtration system (Millipore Corp., Bedford, MA), in a final volume of 100 μl, as described previously (19). Serial dilutions of the drug (0.01 ng/ml to 10,000 ng/ml) were used as calibration standards. Caspofungin was dissolved in water. Inhibition profiles and IC50s were determined using a normalized-response (variable-slope) curve-fitting algorithm with GraphPad Prism 6.05 software (Prism Software, Irvine, CA). Each assay was repeated three times.
Miscellaneous.
Plasmid DNA purification from gels was performed with the QIAquick gel extraction kit (Qiagen, Valencia, CA). For transformation of C. albicans cells, the lithium acetate method was used, as described previously (44). Plates were photographed with a Molecular Imager Gel Doc XR+ system (Bio-Rad).
Supplementary Material
ACKNOWLEDGMENTS
We thank Robert Bambara and Mark Dumont for inspiring discussions and intellectual input. We also thank Mark Dumont for reading the manuscript. We thank Constantine G. Haidaris for helpful discussions.
This work was supported by National Institutes of Health grant AI110764 to E.R. and grant AI109025 to D.S.P.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02299-17.
REFERENCES
- 1.Li D, Calderone R. 2017. Exploiting mitochondria as targets for the development of new antifungals. Virulence 8:159–168. doi: 10.1080/21505594.2016.1188235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calandra T, Roberts JA, Antonelli M, Bassetti M, Vincent JL. 2016. Diagnosis and management of invasive candidiasis in the ICU: an updated approach to an old enemy. Crit Care 20:125. doi: 10.1186/s13054-016-1313-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guinea J. 2014. Global trends in the distribution of Candida species causing candidemia. Clin Microbiol Infect 20(Suppl 6):S5–S10. doi: 10.1111/1469-0691.12539. [DOI] [PubMed] [Google Scholar]
- 4.Lockhart S. 2014. Current epidemiology of Candida infection. Clin Microbiol Newsl 36:131–136. doi: 10.1016/j.clinmicnews.2014.08.001. [DOI] [Google Scholar]
- 5.Pappas PG, Kauffman CA, Andes DR, Clancy CJ, Marr KA, Ostrosky-Zeichner L, Reboli AC, Schuster MG, Vazquez JA, Walsh TJ, Zaoutis TE, Sobel JD. 2016. Clinical practice guideline for the management of candidiasis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis 62:e1–e50. doi: 10.1093/cid/civ1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klis FM, Mol P, Hellingwerf K, Brul S. 2002. Dynamics of cell wall structure in Saccharomyces cerevisiae. FEMS Microbiol Rev 26:239–256. doi: 10.1111/j.1574-6976.2002.tb00613.x. [DOI] [PubMed] [Google Scholar]
- 7.Arendrup MC, Perlin DS. 2014. Echinocandin resistance: an emerging clinical problem? Curr Opin Infect Dis 27:484–492. doi: 10.1097/QCO.0000000000000111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perlin DS. 2015. Mechanisms of echinocandin antifungal drug resistance. Ann N Y Acad Sci 1354:1–11. doi: 10.1111/nyas.12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perlin DS. 2011. Current perspectives on echinocandin class drugs. Future Microbiol 6:441–457. doi: 10.2217/fmb.11.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Effron G, Park S, Perlin DS. 2009. Correlating echinocandin MIC and kinetic inhibition of fks1 mutant glucan synthases for Candida albicans: implications for interpretive breakpoints. Antimicrob Agents Chemother 53:112–122. doi: 10.1128/AAC.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mio T, Adachi-Shimizu M, Tachibana Y, Tabuchi H, Inoue SB, Yabe T, Yamada-Okabe T, Arisawa M, Watanabe T, Yamada-Okabe H. 1997. Cloning of the Candida albicans homolog of Saccharomyces cerevisiae GSC1/FKS1 and its involvement in β-1,3-glucan synthesis. J Bacteriol 179:4096–4105. doi: 10.1128/jb.179.13.4096-4105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang F, Kravets A, Bethlendy G, Welle S, Rustchenko E. 2013. Chromosome 5 monosomy of Candida albicans controls susceptibility to various toxic agents, including major antifungals. Antimicrob Agents Chemother 57:5026–5036. doi: 10.1128/AAC.00516-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson ME, Edlind TD. 2012. Topological and mutational analysis of Saccharomyces cerevisiae Fks1. Eukaryot Cell 11:952–960. doi: 10.1128/EC.00082-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas CM, D'Ippolito JA, Shei GJ, Meinz M, Onishi J, Marrinan JA, Li W, Abruzzo GK, Flattery A, Bartizal K, Mitchell A, Kurtz MB. 1997. Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-β-d-glucan synthase inhibitors. Antimicrob Agents Chemother 41:2471–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang F, Zhang L, Wakabayashi H, Myers J, Jiang Y, Cao Y, Jimenez-Ortigosa C, Perlin DS, Rustchenko E. 2017. Tolerance to caspofungin in Candida albicans is associated with at least three distinctive mechanisms that govern expression of FKS genes and cell wall remodeling. Antimicrob Agents Chemother 61:e00071-17. doi: 10.1128/AAC.00071-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishihara S, Hirata A, Nogami S, Beauvais A, Latge JP, Ohya Y. 2007. Homologous subunits of 1,3-beta-glucan synthase are important for spore wall assembly in Saccharomyces cerevisiae. Eukaryot Cell 6:143–156. doi: 10.1128/EC.00200-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesage G, Sdicu AM, Ménard P, Shapiro J, Hussein S, Bussey H. 2004. Analysis of β-1,3-glucan assembly in Saccharomyces cerevisiae using a synthetic interaction network and altered sensitivity to caspofungin. Genetics 167:35–49. doi: 10.1534/genetics.167.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinoso-Martín C, Schüller C, Schuetzer-Muehlbauer M, Kuchler K. 2003. The yeast protein kinase C cell integrity pathway mediates tolerance to the antifungal drug caspofungin through activation of Slt2p mitogen-activated protein kinase signaling. Eukaryot Cell 2:1200–1210. doi: 10.1128/EC.2.6.1200-1210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park S, Kelly R, Kahn JN, Robles J, Hsu MJ, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, Perlin DS. 2005. Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49:3264–3273. doi: 10.1128/AAC.49.8.3264-3273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García R, Botet J, Rodríguez-Peña JM, Bermejo C, Ribas JC, Revuelta JL, Nombela C, Arroyo J. 2015. Genomic profiling of fungal cell wall-interfering compounds: identification of a common gene signature. BMC Genomics 16:683. doi: 10.1186/s12864-015-1879-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie JL, Qin L, Miao Z, Grys BT, Diaz JC, Ting K, Krieger JR, Tong J, Tan K, Leach MD, Ketela T, Moran MF, Krysan DJ, Boone C, Andrews BJ, Selmecki A, Ho Wong K, Robbins N, Cowen LE. 2017. The Candida albicans transcription factor Cas5 couples stress responses, drug resistance and cell cycle regulation. Nat Commun 8:499. doi: 10.1038/s41467-017-00547-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Douglas CM, Foor F, Marrinan JA, Morin N, Nielsen JB, Dahl AM, Mazur P, Baginsky W, Li W, el-Sherbeini M, Clemas JA, Mandala SM, Frommer BR, Kurtz MA. 1994. The Saccharomyces cerevisiae FKSI (ETGJ) gene encodes an integral membrane protein which is a subunit of 1,3-β-d-glucan synthase. Proc Natl Acad Sci USA 91:12907–12911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazur P, Morin N, Baginsky W, el-Sherbeini M, Clemas JA, Nielsen JB, Foor F. 1995. Differential expression and function of two homologous subunits of yeast 1,3-β-d-glucan synthase. Mol Cell Biol 15:5671–5681. doi: 10.1128/MCB.15.10.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, Edlind TD. 2012. Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother 56:6304–6309. doi: 10.1128/AAC.00813-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson ME, Katiyar SK, Edlind TD. 2011. New Fks hot spot for acquired echinocandin resistance in Saccharomyces cerevisiae and its contribution to intrinsic resistance of Scedosporium species. Antimicrob Agents Chemother 55:3774–3781. doi: 10.1128/AAC.01811-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Drgonová J, Drgon T, Tanaka K, Kollár R, Chen GC, Ford RA, Chan CS, Takai Y, Cabib E. 1996. Rho1p, a yeast protein at the interface between cell polarization and morphogenesis. Science 272:277–279. doi: 10.1126/science.272.5259.277. [DOI] [PubMed] [Google Scholar]
- 27.Qadota H, Python CP, Inoue SB, Arisawa M, Anraku Y, Zheng Y, Watanabe T, Levin DE, Ohya Y. 1996. Identification of yeast Rho1p GTPase as a regulatory subunit of 1,3-β-glucan synthase. Science 272:279–281. doi: 10.1126/science.272.5259.279. [DOI] [PubMed] [Google Scholar]
- 28.Alexander BD, Johnson MD, Pfeiffer CD, Jiménez-Ortigosa C, Catania J, Booker R, Castanheira M, Messer SA, Perlin DS, Pfaller MA. 2013. Increasing echinocandin resistance in Candida glabrata: clinical failure correlates with presence of FKS mutations and elevated minimum inhibitory concentrations. Clin Infect Dis 56:1724–1732. doi: 10.1093/cid/cit136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perepnikhatka V, Fischer FJ, Niimi M, Baker RA, Cannon RD, Wang YK, Sherman F, Rustchenko E. 1999. Specific chromosome alterations in fluconazole-resistant mutants of Candida albicans. J Bacteriol 181:4041–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahmad A, Kabir MA, Kravets A, Andaluz E, Larriba G, Rustchenko E. 2008. Chromosome instability and unusual features of some widely used strains of Candida albicans. Yeast 25:433–448. doi: 10.1002/yea.1597. [DOI] [PubMed] [Google Scholar]
- 31.Rustchenko-Bulgac EP. 1991. Variations of Candida albicans electrophoretic karyotypes. J Bacteriol 173:6586–6596. doi: 10.1128/jb.173.20.6586-6596.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang YK, Das B, Huber DH, Wellington M, Kabir MA, Sherman F, Rustchenko E. 2004. Role of the 14-3-3 protein in carbon metabolism of the pathogenic yeast Candida albicans. Yeast 21:685–702. doi: 10.1002/yea.1079. [DOI] [PubMed] [Google Scholar]
- 33.Rustchenko EP, Howard DH, Sherman F. 1994. Chromosomal alterations of Candida albicans are associated with the gain and loss of assimilating functions. J Bacteriol 176:3231–3241. doi: 10.1128/jb.176.11.3231-3241.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherman F. 2002. Getting started with yeast. Methods Enzymol 350:3–41. doi: 10.1016/S0076-6879(02)50954-X. [DOI] [PubMed] [Google Scholar]
- 35.Suwunnakorn S, Wakabayashi H, Rustchenko E. 2016. Chromosome 5 of human pathogen Candida albicans carries multiple genes for negative control of caspofungin and anidulafungin susceptibility. Antimicrob Agents Chemother 60:7457–7467. doi: 10.1128/AAC.01888-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wellington M, Rustchenko E. 2005. 5-Fluoro-orotic acid induces chromosome alterations in Candida albicans. Yeast 22:57–70. doi: 10.1002/yea.1191. [DOI] [PubMed] [Google Scholar]
- 37.Morschhäuser J, Michel S, Staib P. 1999. Sequential gene disruption in Candida albicans by FLP-mediated site-specific recombination. Mol Microbiol 32:547–556. doi: 10.1046/j.1365-2958.1999.01393.x. [DOI] [PubMed] [Google Scholar]
- 38.Shen J, Guo W, Köhler JR. 2005. CaNAT1, a heterologous dominant selectable marker for transformation of Candida albicans and other pathogenic Candida species. Infect Immun 73:1239–1242. doi: 10.1128/IAI.73.2.1239-1242.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard— 3rd ed M27-A3. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 40.Shedletzky E, Unger C, Delmer DP. 1997. A microtiter-based fluorescence assay for (1,3)-β-glucan synthases. Anal Biochem 249:88–93. doi: 10.1006/abio.1997.2162. [DOI] [PubMed] [Google Scholar]
- 41.Tsang PW, Fong WP, Samaranayake LP. 2017. Candida albicans orf19.3727 encodes phytase activity and is essential for human tissue damage. PLoS One 12:e0189219. doi: 10.1371/journal.pone.0189219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kretschmar M, Hube B, Bertsch T, Sanglard D, Merker R, Schröder M, Hof H, Nichterlein T. 1999. Germ tubes and proteinase activity contribute to virulence of Candida albicans in murine peritonitis. Infect Immun 67:6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilhelm BT, Marguerat S, Goodhead I, Bähler J. 2010. Defining transcribed regions using RNA-seq. Nat Protoc 5:255–266. doi: 10.1038/nprot.2009.229. [DOI] [PubMed] [Google Scholar]
- 44.Köhler JR, Fink GR. 1996. Candida albicans strains heterozygous and homozygous for mutations in mitogen-activated protein kinase signaling components have defects in hyphal development. Proc Natl Acad Sci U S A 93:13223–13228. doi: 10.1073/pnas.93.23.13223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fonzi WA, Irwine MY. 1993. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.