

ABSTRACT
Understanding bacterial pathogenesis requires adequate genetic tools to assess the role of individual virulence determinants by mutagenesis and complementation assays, as well as for homologous and heterologous expression of cloned genes. Our knowledge of Acinetobacter baumannii pathogenesis has so far been limited by the scarcity of genetic tools to manipulate multidrug-resistant (MDR) epidemic strains, which are responsible for most infections. Here, we report on the construction of new multipurpose shuttle plasmids, namely, pVRL1 and pVRL2, which can efficiently replicate in Acinetobacter spp. and in Escherichia coli. The pVRL1 plasmid has been constructed by combining (i) the cryptic plasmid pWH1277 from Acinetobacter calcoaceticus, which provides an origin of replication for Acinetobacter spp.; (ii) a ColE1-like origin of replication; (iii) the gentamicin or zeocin resistance cassette for antibiotic selection; and (iv) a multilinker containing several unique restriction sites. Modification of pVRL1 led to the generation of the pVRL2 plasmid, which allows arabinose-inducible gene transcription with an undetectable basal expression level of cloned genes under uninduced conditions and a high dynamic range of responsiveness to the inducer. Both pVRL1 and pVRL2 can easily be selected in MDR A. baumannii, have a narrow host range and a high copy number, are stably maintained in Acinetobacter spp., and appear to be compatible with indigenous plasmids carried by epidemic strains. Plasmid maintenance is guaranteed by the presence of a toxin-antitoxin system, providing more insights into the mechanism of plasmid stability in Acinetobacter spp.
KEYWORDS: Acinetobacter, arabinose, cloning, expression, gentamicin, inducible, plasmid, vector, zeocin
INTRODUCTION
Members of the genus Acinetobacter are ubiquitous microorganisms distributed in many environments. Some species are most often found in hospitals and are implicated in human disease (e.g., Acinetobacter baumannii, Acinetobacter nosocomialis, Acinetobacter pittii, Acinetobacter seifertii, Acinetobacter dijkshoorniae), while other species are environmental saprophytes and are seldom associated with infection (e. g., Acinetobacter calcoaceticus and Acinetobacter baylyi) (1). A. baumannii is the most dreaded species, with increasing reports of health care-related outbreaks and dissemination of multidrug-resistant (MDR) and extremely drug-resistant (XDR) strains being made worldwide (2, 3, 4). While A. baumannii is rapidly evolving toward pan-resistance, the traits responsible for its success as a human pathogen are not yet fully understood (5). Since MDR or XDR A. baumannii strains are almost invariably isolated from human infection, mining of their genomes by bioinformatic, omic, and reverse genetic approaches represents a valuable strategy to gain more insight into A. baumannii virulence. Unfortunately, the scarcity of genetic tools for the genetic manipulation of highly resistant A. baumannii is a shortcoming that limits genome-scale studies. To date, the Escherichia coli-Acinetobacter species shuttle-vector pWH1266 (6) and its derivatives (7, 8, 9) have often been used as cloning vehicles in A. baumannii. The pWH1266 vector was obtained by combining the pWH1277 natural cryptic plasmid of A. calcoaceticus BD413 with the E. coli pBR322 vector (6). Although pWH1266 has been widely used in A. baumannii, it has some limitations, including the following: (i) it contains inappropriate markers for selection in MDR A. baumannii strains (namely, blaTEM-1 and tetA genes conferring resistance to β-lactams and tetracycline [Tc], respectively), (ii) screening of recombinant clones is time-consuming, since it is based on marker inactivation, (iii) it has few unique cloning sites, (iv) its copy number is unknown, and (v) it has uncharacterized stability mechanisms.
Here, we illustrate the generation and the functional characterization of new E. coli-Acinetobacter species shuttle plasmids suited for gene cloning and expression in MDR and XDR A. baumannii. These plasmid vectors, referred to as the pVRL series, contain (i) two origins of replication, one from E. coli pBluescript II (pBS) and the other from the A. calcoaceticus BD413 plasmid pWH1277; (ii) antibiotic resistance markers suitable for selection in MDR or XDR clinical strains; and (iii) a complete multilinker with several unique restriction sites, allowing (iv) blue/white screening of recombinant clones and (v) tightly controlled expression of cloned genes under the control of the araC-PBAD regulatory element. The molecular mechanisms governing pVRL stability and maintenance in Acinetobacter spp. have also been elucidated.
RESULTS
Construction of pVRL vectors.
The pVRL shuttle vectors were assembled by combining plasmid fragments with PCR products (Table 1; see also Table S1 in the supplemental material). Briefly, pVRL1 was constructed by ligating three DNA fragments: (i) the cryptic plasmid pWH1277, which provides an origin of replication for Acinetobacter spp.; (ii) the aacC1 gene for gentamicin (Gm) resistance; and (iii) the pBS fragment encompassing the ColE1-like origin of replication for E. coli and the lacZα gene (10), including the multiple-cloning site (MCS). The 4,693-bp fragment corresponding to the entire sequence of the pWH1277 cryptic plasmid was obtained by PvuII digestion of pWH1266 (Fig. S1A) (6) and cloned into the unique SmaI site of pBS to generate the pBSoriAb vector (Fig. S1B). The replication of pBSoriAb in A. baumannii ATCC 19606T was verified by electrotransformation and selection on Luria-Bertani agar (LA) supplemented with carbenicillin (Cb) at 250 μg/ml. Transformation with the pBS vector was used as a negative control. The 1,071-bp fragment containing the aacC1 gene, encoding Gm N(3′)-acetyltransferase 1 (11), was obtained by PCR with primers aacC1 FW and aacC1 RV, using pEX19Gm (12) as the template (Fig. S1C). The 1,529-bp fragment encompassing the ColE1-like origin of replication (ori), the MCS, and the lacZα fragment from pBS was amplified by PCR using primers MCS ori ColE1-like FW and MCS ori ColE1-like RV (Fig. S1D). All three fragments were directionally ligated and introduced into E. coli DH5α by transformation to obtain pVRL1 (Fig. 1A and S1E). pVRL1 was entirely sequenced by primer walking to verify that no mutations were introduced during the cloning process. Being a pBS derivative, universal sequencing synthetic oligonucleotides (i.e., the T3, T7, M13 Fw, M13 Rv, pBS SK, and pBS KS primers) can anneal on pVRL1 regions flanking the MCS. Moreover, pVRL1 has a polylinker with 16 unique restriction sites located within the lacZα gene, thus allowing blue/white screening for selection of the recombinant clones in E. coli DH5α (Fig. S2). The appropriate combination of cohesive compatible ends employed during pVRL1 construction (i.e., BclI-BamHI and PstI-NsiI) generated spurious hexameric sequences, avoiding the loss of unique restriction sites within the polylinker.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristicsa | Source and/or reference |
|---|---|---|
| Strains | ||
| A. baumannii | ||
| ATCC 19606T | Type strain | 50 |
| ACICU | MDR clinical isolate, prototype of international clonal lineage II | 17 |
| AYE | MDR clinical isolate, prototype of international clonal lineage I | 18 |
| ATCC 19606T trpE19 | Transposon insertion derivative of A. baumannii ATCC 19606T; tryptophan auxotrophic mutant | 7 |
| A. baylyi BD413 trpE27 | Naturally transformable; tryptophan auxotrophic mutant | 51, 52 |
| E. coli | ||
| DH5α | recA1 endA1 hsdR17 supE44 thi-1 gyrA96 relA1 Δ(lacZYA-argF)U169 [ϕ80dlacZΔM15] F− Nalr | 42 |
| MC4100 | araD139 rpsL150 relA1 flbB5301 deoC1 pstF25 rbsR Δ(lacZYA-argF)U169 Strr | 53 |
| MG1655 | K-12 F− λ− ilvG− rfb-50 rph-1; type strain | 54 |
| A. pittii UKK_0145 | Member of the ACB complex | H. Seifert collection |
| A. nosocomialis UKK_0361 | Member of the ACB complex | H. Seifert collection |
| A. dijkshoorniae Scope 271 | Member of the ACB complex | H. Seifert collection (55) |
| A. seifertii HS A23-2 | Member of the ACB complex | H. Seifert collection (56) |
| Plasmids | ||
| pBluescript-II KS | Standard cloning vector; source of ColE1 origin of replication and MCS (GenBank accession no. X52327.1); Apr | 10 |
| pEX19Gm | Cloning vector, source of aacC1 cassette (GenBank accession no. KM887142.1); Gmr | 12 |
| pWH1266 | A. baumannii shuttle vector, source of oriAb; Apr Tcr | 6 |
| pME6032 | IPTG-inducible broad-host-range shuttle expression vector for genetic complementation; Tcr | 46 |
| miniCTX1-araC-PBAD | Vector carrying the araC-PBAD regulatory region with an altered RBS for stringent arabinose-dependent control in Pseudomonas aeruginosa; Tcr | 14 |
| miniTn7-gfp | Vector used as source of GFP-coding sequence; Apr Gmr | 44 |
| miniCTXmCherry | Vector used as source of mCherry-coding sequence; Tcr | 57 |
| pCR-Blunt II-TOPO | Cloning vector, source of the ble cassette; Zeor Kmr | Thermo Fisher |
| pBSoriAb | Vector carrying the origin of replication for Acinetobacter spp., derived from pWH1266; Apr | This work |
| pVRL1 | E. coli-Acinetobacter species shuttle vector for general cloning purposes; Gmr | This work |
| pVRL1a | pVRL1 with rrnB transcriptional terminators; Gmr | This work |
| pVRL1b | pVRL1a with a deletion in the CAP binding site and lac promoter (Plac); Gmr | This work |
| pVRL2 | pVRL1b carrying the araC-PBAD arabinose-inducible expression cassette; Gmr | This work |
| pVRL1trpEwt | trpE gene with its endogenous promoter cloned into pVRL1; Gmr | This work |
| pVRL2trpEPL | trpE promoterless gene cloned into pVRL2; Gmr | This work |
| pVRL2lacZ | lacZ promoterless gene cloned into pVRL2; Gmr | This work |
| pVRL2tetA | tetA promoterless gene cloned into pVRL2; Gmr Tcr | This work |
| pVRL2gfp | gfp promoterless gene cloned into pVRL2; Gmr | This work |
| pVRL2mCherry | mCherry promoterless gene cloned into pVRL2; Gmr | This work |
| pVRL1ΔTA | pVRL1 carrying a deletion in the TA system; Gmr | This work |
| pVRL1ΔpaaA2-like | pVRL1 carrying a deletion in the antitoxin (paaA2-like) protein; Gmr | This work |
| pME6032paaA2-like | pME6032 containing the promoterless paaA2-like antitoxin gene; Tcr | This work |
| pVRL1Z | pVRL1-derived vector containing the ble cassette; Zeor | This work |
| pVRL2Z | pVRL2-derived vector containing the ble cassette; Zeor | This work |
Nalr, nalidixic acid resistance; Strr, streptomycin resistance; ilvG−, frameshift mutation in the ilvG gene that knocks out acetohydroxy acid synthase II; Apr, ampicillin resistance; Gmr, gentamicin resistance; Tcr, tetracycline resistance; Zeor, zeocin resistance; Kmr, kanamycin resistance; ACB complex, Acinetobacter calcoaceticus-Acinetobacter baumannii complex.
FIG 1.

Physical and functional map of the pVRL1 and pVRL2 plasmid vectors. Relevant features of pVRL1 (A) and pVRL2 (B) are annotated on plasmid maps in different colors. The rrnB term (red box) denotes the Rho-independent transcriptional terminators of the rrnB ribosomal gene. The araC gene (dark green) and PBAD promoter (brown) were cloned upstream of the MCS. Other features of the plasmid are indicated in different colors: the Gm resistance cassette (green), the nickase-like gene (cyan), oriAb (violet), the parE2-like toxin gene (light purple), the paaA2-like antitoxin gene (orange), the ColE1-like origin of replication (ori; yellow), the lac promoter (Plac; pink), and the lacZα fragment in pVRL1 and ΔlacZα in pVRL2 (gray). Annealing regions for universal sequencing primers, such as M13 Fw, M13 Rv, pBS SK, pBS KS, T3, and T7, are not shown. (C) Detail of the PBAD promoter sequence preceding the RBS and the first restriction site (XhoI) of MCS. The Shine-Dalgarno sequence (SD) is composed of a noncanonical pentamer (CTTCT), and it is located 4 nt upstream of the XhoI restriction site. Unique cutter restriction enzymes are indicated. All genes are reported in scale over the total length of the vector. Images were obtained by the use of SnapGene software (from GSL Biotech).
With the aim of constructing an expression vector for MDR A. baumannii, the pVRL1 plasmid was used as a scaffold to generate an arabinose-inducible expression system (Fig. S3A). A 353-bp fragment containing the three head-to-tail rrnB ribosomal gene transcriptional terminators (rrnB term) (13) was amplified from the pEX19Gm plasmid, using primers rrnB term FW and rrnB term RV, and ligated to NcoI-digested pVRL1, to obtain pVRL1a (Fig. S3B). The DNA region encompassing the CAP binding site, the lac promoter (Plac), and the first 24 codons of lacZα was deleted from pVRL1a using a Q5 site-directed mutagenesis kit with primers ΔlacP FW and ΔlacP RV, generating pVRL1b (Fig. S2C). Deletion of the region encompassing the lac promoter and the first 24 codons of the lacZα gene was necessary to eliminate the transcriptional readthrough of RNA polymerase from the Plac promoter and the formation of a chimeric protein composed of LacZα and the cloned insert. Subsequently, pVRL1b was digested at the NsiI (introduced with primer ΔlacP RV) and XhoI sites, to insert the arabinose-inducible araC-PBAD expression cassette amplified from miniCTX1-araC-PBADtolB (14), using primers araC-PBAD FW and araC-PBAD RV. The resulting plasmid, named pVRL2 (Fig. 1B and S3D), is an E. coli-Acinetobacter species shuttle vector for gene expression under the control of the PBAD promoter and the AraC regulator of the ara operon. In pVRL2, 5 nucleotides separate the PBAD noncanonical ribosome binding site (RBS) from the first available cloning site of the polylinker (XhoI; Fig. 1C), to ensure the low translation efficiency of the transcript in favor of tight regulation upon PBAD induction with arabinose (15).
Plasmids pVRL1 and pVRL2 were further modified by replacing the aacC1 gene with the ble gene, conferring resistance to zeocin (Zeo). The 780-bp region corresponding to the aacC1 gene was removed from pVRL1 and pVRL2 by PCR, using primers pVRL1ΔaacC1 FW and pVRL1ΔaacC1 RV and primers pVRL2ΔaacC1 FW and pVRL2ΔaacC1 RV (Table S1), respectively, to obtain the linearized markerless plasmids pVRL1ΔaacC1 and pVRL2ΔaacC1. The ble gene was generated from pCR-Blunt II-TOPO (Table 1) by PCR using primers Zeo 1 FW and Zeo 1 RV and primers Zeo 2 FW and Zeo 2 RV (Table S1). The resulting 568-bp and 569-bp amplicons were cloned into pVRL1ΔaacC1 and pVRL2ΔaacC1 at NsiI and ApaI/KpnI restriction sites, respectively. The resulting plasmids carrying the ble gene for Zeo resistance were named pVRL1Z and pVRL2Z (Fig. 1).
Complementation analysis of the trpE mutation in A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27.
To assess the potential of pVRL1 as a cloning vehicle, the A. baumannii trpE gene with its endogenous (wild-type [wt]) promoter (trpEwt) was cloned into pVRL1, and the resulting plasmid, pVRL1trpEwt, was used to complement tryptophan auxotrophy in A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27 mutants. Both auxotrophs complemented with the pVRL1trpEwt plasmid were able to grow on Vogel-Bonner minimal agar plates containing succinate as the carbon source (VBS agar plates), while auxotrophs carrying the empty vector pVRL1 did not (Fig. 2A). As expected, the growth of A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27 carrying pVRL1 was restored in tryptophan-supplemented VBS agar plates (Fig. 2A). These data demonstrate that pVRL1 can be used for genetic complementation in Acinetobacter spp.
FIG 2.

Complementation analysis of the trpE mutation in A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27. (A) A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27 auxotrophic mutants were naturally transformed and electrotransformed, respectively, with either pVRL1trpEwt or the corresponding empty plasmid, pVRL1. Bacteria were grown on VBS agar plates for 24 h at 37°C with or without 1 mM tryptophan (trp), as indicated. (B) A. baumannii ATCC 19606T trpE19 and A. baylyi BD413 trpE27 were transformed with either pVRL2trpEPL or the corresponding empty plasmid, pVRL2. Bacteria were grown on VBS agar plates for 24 h at 37°C in the presence of increasing concentrations of arabinose, as shown at the top.
To investigate the gene expression driven by the araC-PBAD element in Acinetobacter spp., the promoterless trpE gene (trpEPL) was cloned into pVRL2 to generate plasmid pVRL2trpEPL. No growth of A. baumannii ATCC 19606T trpE19(pVRL2trpEPL) or A. baylyi BD413 trpE27(pVRL2trpEPL) was observed on VBS agar plates without tryptophan, as was also observed for A. baumannii ATCC 19606T trpE19(pVRL2) and A. baylyi BD413 trpE27(pVRL2), used as negative controls (Fig. 2B). Supplementation of the medium with increasing concentrations of arabinose (from 0.5% to 2%) resulted in the increased growth of both A. baumannii ATCC 19606T trpE19(pVRL2trpEPL) and A. baylyi BD413 trpE27(pVRL2trpEPL) on VBS without tryptophan, whereas no growth was observed for auxotrophs carrying the empty vector, irrespective of the presence of arabinose (Fig. 2B). These data indicate that pVRL2 is suitable for inducible gene expression in Acinetobacter spp.
Expression analysis of the lacZ (β-galactosidase), tetA (tetracycline resistance), and gfp (green fluorescent protein [GFP]) genes reveals tight regulation of the araC-PBAD system in pVRL2.
To investigate more in depth the gene expression control of the araC-PBAD system in A. baumannii, the lacZ, tetA, and gfp genes were independently cloned into pVRL2, resulting in the pVRL2lacZ, pVRL2tetA, and pVRL2gfp recombinant plasmids, respectively. These plasmids were introduced into A. baumannii ATCC 19606T. Initially, the dose-response effect of arabinose induction on lacZ expression was visualized by growing A. baumannii ATCC 19606T(pVRL2lacZ) in Luria-Bertani broth (LB) supplemented with the β-galactosidase chromogenic substrate X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside). The level of production of the blue compound (i.e., 5,5′-dibromo-4,4′-dichloro-indigo) generated by X-Gal hydrolysis paralleled the arabinose concentration (from 0.125% to 2%) (Fig. 3A). Time course measurements of the β-galactosidase activity expressed by A. baumannii ATCC 19606T(pVRL2lacZ) upon induction with increasing arabinose concentrations (from 0.06% to 8%) showed a dose-dependent response at all the tested time points (Fig. 3B). Increasing arabinose concentrations caused a moderate decrease in bacterial growth during the first 3 h postinduction (Fig. 3B), likely due to a metabolic burden resulting from lacZ overexpression. Maximum β-galactosidase expression was obtained upon 24 h of induction with 2% to 8% arabinose (Fig. 3B).
FIG 3.

Arabinose-inducible expression of the genes under the control of the araC-PBAD promoter in pVRL2 plasmid. (A) Visualization of arabinose-induced β-galactosidase expression in A. baumannii ATCC 19606T(pVRL2lacZ) grown for 24 h at 37°C in LB supplemented with the chromogenic substrate X-Gal and different arabinose concentrations. (B) Expression of β-galactosidase in A. baumannii ATCC 19606T carrying pVRL2lacZ. Cells were grown in LB medium to an OD600 of 0.6 and then challenged with different concentrations of arabinose, as indicated. Expression of the reporter lacZ gene (black bars) and growth (white circles) were measured after 1, 3, and 24 h postinduction. Data are the means ± SDs from three independent experiments. (C) Expression levels of the Tc resistance gene (tetA) were determined as the Tc MIC of A. baumannii ATCC 19606T carrying pVRL2tetA (black circles) grown in the presence of different arabinose concentrations (0%, 0.5%, 2%). A. baumannii ATCC 19606T carrying the pVRL2 empty vector (black triangles) was used as a control. (D) Arabinose-induced tetA expression in A. baumannii ATCC 19606T(pVRL2tetA), as visualized by the disk diffusion assay. Bacteria were grown for 24 h at 37°C on LA without and with arabinose (2%). (E) Confocal microscopy observation of bacterium-associated fluorescence (GFP) upon induction of A. baumannii ATCC 19606T(pVRL2gfp) with different arabinose concentrations (0%, 0.5%, 1%, 2%, 4%) for 6 h at 37°C. (Top) Representative details of images from Fig. S4 in the supplemental material are shown. Magnifications, ×6,300. (Bottom) The relative fluorescence values (fluorescence/OD600) of the bacterial suspensions were determined with a luminescence spectrophotometer (model LS-50B; PerkinElmer) at excitation and emission wavelengths of 475 nm and 515 nm, respectively. A. baumannii ATCC 19606T(pVRL2) was used as a control.
To gain more insight into the regulation of the pVRL2 araC-PBAD system in A. baumannii and to rule out relaxed transcription from the PBAD promoter, the recombinant pVRL2tetA plasmid was used. In this construct, controlled expression of tetA upon arabinose induction would confer resistance to tetracycline (Tc). Tc resistance was evaluated by both the broth microdilution method (Fig. 3C) and the disk diffusion assay (Fig. 3D). The MIC of Tc in the absence of arabinose was 2 μg/ml for both A. baumannii ATCC 19606T(pVRL2tetA) and A. baumannii ATCC 19606T(pVRL2). Like what was observed for β-galactosidase expression, arabinose induced the expression of tetA from pVRL2tetA in A. baumannii ATCC 19606T in a dose-dependent manner (Fig. 3C). In fact, induction with 0.5% and 2% arabinose resulted in, respectively, 2- and 8-fold increases in the Tc MIC (4 and 16 μg/ml, respectively) in the microdilution assay. A. baumannii ATCC 19606T(pVRL2), used as a control, showed the same Tc susceptibility profile in the presence and in the absence of arabinose. Likewise, the disk diffusion assay showed that arabinose addition markedly reduced the growth inhibition halo caused by Tc (30 μg) in A. baumannii ATCC 19606T(pVRL2tetA) compared with that in the uninduced control and A. baumannii ATCC 19606T(pVRL2) under both induced and uninduced conditions (Fig. 3D).
To use a more sensitive gene expression probe, the gfp gene was cloned under PBAD control in pVRL2, and GFP levels were quantified by combining confocal microscopy and fluorometric analyses (Fig. 3E and S4). Bacterium-associated fluorescence became evident upon addition of 0.5% arabinose, although not all cells appeared fluorescent. In the presence of 1% arabinose, all cells became fluorescent, and this was concomitant with a 2-fold increase in fluorescence relative to that obtained with the lowest arabinose concentration used (0.5%). In line with previous expression profiles, arabinose induced the expression of gfp in a dose-dependent manner, reaching a plateau in the presence of 2% to 4% arabinose. To rule out any possible fluorescent noise of A. baumannii cells, A. baumannii ATCC 19606T(pVRL2) was used as a negative control, and no autofluorescence was detected by either confocal microscopy or fluorimetry (Fig. 3E and S4).
Overall, no gene expression was detectable in the absence of arabinose, whereas increased reporter gene expression paralleled the increase in arabinose (inducer) concentrations. Taken together, these results indicate that gene expression under the control of the araC-PBAD system in pVRL2 is tightly regulated in A. baumannii.
Gentamicin resistance gene aacC1 is a suitable selectable marker in XDR A. baumannii strains.
On the basis of the results generated from routine antimicrobial susceptibility testing, A. baumannii ACICU is an XDR strain resistant to the majority of antibiotics (16). The presence of the aacA4 cassette in the chromosomal AbaR2 region of A. baumannii ACICU likely confers Gm resistance (17). We wondered whether the pVRL1 and pVRL2 vectors could be introduced and selected in A. baumannii strains, like ACICU, which are classified as Gm resistant by standard antibiotic susceptibility tests, upon selection with Gm at concentrations higher than the reported MIC (4 μg/ml) (16). Initially, liquid and agar dilution methods were used to determine the MIC of Gm in all the host strains employed in this study (Table 2; Fig. S5). These experiments revealed that E. coli DH5α and A. baylyi BD413 trpE27 showed the same sensitivity to Gm, with a MIC of 1 μg/ml in LB and a MIC of 2 μg/ml on LA, while the corresponding strains harboring either pVRL plasmid showed Gm resistance up to 128 μg/ml and 64 μg/ml, respectively. A. baumannii ATCC 19606T and A. baumannii ACICU showed MICs of 16 μg/ml and 64 μg/ml, respectively, both in liquid LB and on solid LB (Table 2; Fig. S5). The level of Gm resistance was considerably increased in both strains transformed with either pVRL1 or pVRL2, attaining MIC values of >512 μg/ml (Table 2; Fig. S5). Overall, these data demonstrate that the pVRL plasmids can also successfully be selected in XDR strains classified as clinically resistant to Gm, such as ACICU. However, some strains, like AYE (18), show exceedingly high levels of resistance to Gm (MIC > 512 μg/ml) so that pVRL1 and pVRL2 transformants cannot be selected using this marker. However, replacement of aacC1 with ble in pVRL1Z and pVRL2Z allowed AYE transformants to be selected on low-salt Luria-Bertani agar (LA) plates supplemented with 250 μg/ml Zeo (Table 2). Indeed, all A. baumannii strains used in this work showed Zeo MICs of ≤32 μg/ml, which increased to >512 μg/ml after transformation with the pVRL1Z and pVRL2Z plasmids (data not shown). Other members of the A. calcoaceticus-A. baumannii (ACB) complex could be selected on low-salt LA plates supplemented with 250 μg/ml Zeo upon transformation with pVRL1Z (Table 2).
TABLE 2.
MIC of gentamicin in LB medium for strains carrying pVRL plasmids, transformation efficiency, and gentamicin or zeocin concentrations required for pVRL selection
| Strain | Gm MICa (μg/ml) with no plasmid | pVRL1 |
pVRL2 |
Gm (Zeo) concn (μg/ml) required for selectiona | ||
|---|---|---|---|---|---|---|
| Gm MICa (μg/ml) | Transformation efficiencyb (no. of CFU/μg DNA) | Gm MICa (μg/ml) | Transformation efficiencyb (no. of CFU/μg DNA) | |||
| E. coli DH5α | 1 | 128 | (1.3 ± 0.3) × 105 | 128 | (1.3 ± 0.2) × 105 | 10 (25) |
| A. baumannii ATCC 19606T | 16 | >512 | (6.5 ± 1.1) × 102 | >512 | (7.7 ± 0.9) × 101 | 100 (250) |
| A. baumannii ACICU | 64 | >512 | (1.8 ± 0.3) × 105 | >512 | (1.9 ± 0.2) × 105 | 200 (250) |
| A. baumannii AYE | >512 | NDc | ND | ND | ND | ND (250) |
| A. baylyi BD413 trpE27 | 1 | 64 | (9.5 ± 0.4) × 103 | 64 | (2.2 ± 0.5) × 104 | 10 (ND) |
| A. pittii UKK_0145 | 4 | >512 | (2.5 ± 0.2) × 105 | >512 | (6.1 ± 1.1) × 105 | 50 (250) |
| A. nosocomialis UKK_0361 | 4 | 256 | (6.6 ± 0.3) × 103 | 256 | (7.9 ± 0.7) × 103 | 50 (250) |
| A. dijkshoorniae Scope 271 | 2 | 256 | (6.7 ± 0.2) × 102 | 256 | (3.1 ± 0.6) × 102 | 50 (250) |
| A. seifertii HS A23-2 | 8 | 128 | (3.7 ± 0.7) × 100 | 128 | (8.9 ± 0.7) × 100 | 50 (250) |
The MIC for Gm was determined by the broth microdilution assay as described in Materials and Methods.
The transformation efficiency is expressed as the mean ± standard deviation from three independent experiments. E. coli chemically competent cells were transformed by heat shock as described by Sambrook et al. (42); A. baumannii electrocompetent cells were transformed by electroporation as described by Yildirim et al. (20); A. baylyi naturally competent cells were transformed as described by Renda et al. (43).
ND, not determined.
Host range, transformation efficiency, copy number, and stability of pVRL1 and pVRL2 plasmids.
Having a ColE1-derived origin of replication, pVRL plasmids can replicate in Enterobacteriaceae (19). The second origin is from pWH1277, a plasmid from A. calcoaceticus (6). Repeated attempts to transform various Pseudomonas spp. with pVRL plasmids were unsuccessful, suggesting that replication from the pWH1277 origin is genus specific and not supported in other members of the Pseudomonadales order (data not shown).
To evaluate the transformation efficiency of pVRL1 and pVRL2, both plasmids were extracted and purified from E. coli DH5α and A. baumannii ATCC 19606T using a commercial system, and plasmid yields were calculated as the number of micrograms of DNA per milliliter of culture (Table 3). Plasmid yields were high both in E. coli DH5α (i.e., 1.21 ± 0.03 and 1.04 ± 0.03 for pVRL1 and pVRL2, respectively) and in A. baumannii ATCC 19606T (i.e., 0.70 ± 0.09 and 0.57 ± 0.10 for pVRL1 and pVRL2, respectively). Interestingly, yields were higher for pVRL1 than for pVRL2 in both species, plausibly due to the smaller size of pVRL1 (7,276 bp) than pVRL2 (8,722 bp).
TABLE 3.
Stability, yields, and copy number of pVRL plasmids
| Strain (plasmid) | Stability (NGm/N0) | Plasmid yield (μg DNA/ml culture)a | PCN/chromosome |
|---|---|---|---|
| E. coli DH5α(pVRL1) | 0.86 ± 0.18 | 1.21 ± 0.03 | 70.1 ± 5.2 |
| E. coli DH5α(pVRL2) | 1.00 ± 0.33 | 1.04 ± 0.03 | NDb |
| E. coli DH5α(pVRL1ΔTA) | 0.10 ± 0.02 | ND | ND |
| A. baumannii ATCC 19606T(pVRL1) | 0.92 ± 0.29 | 0.70 ± 0.09 | 56.8 ± 5.1 |
| A. baumannii ATCC 19606T(pVRL2) | 0.85 ± 0.16 | 0.57 ± 0.10 | ND |
| A. baumannii ATCC 19606T(pVRL1ΔTA) | 0.57 ± 0.20 | ND | ND |
| A. baumannii ACICU(pVRL1) | 0.93 ± 0.34 | ND | ND |
| A. baumannii ACICU(pVRL2) | 0.72 ± 0.15 | ND | ND |
| A. baylyi BD413 trpE27(pVRL1) | 1.01 ± 0.20 | ND | ND |
| A. baylyi BD413 trpE27(pVRL2) | 0.99 ± 0.17 | ND | ND |
Plasmid DNA was extracted from 3 ml of a 16-h bacterial culture, diluted to an OD600 of 1, and then purified using a Wizard Plus SV Minipreps DNA purification system (Promega).
ND, not determined.
The transformation efficiency in E. coli DH5α and various Acinetobacter spp., also including the MDR A. baumannii strains ACICU and AYE, was determined for the pVRL vectors (Table 2). In E. coli DH5α, the efficiency was 1.3 × 105 CFU/μg DNA for both pVRL1 and pVRL2. Similarly high values were observed for A. baumannii ACICU, while the efficiency was ca. 1,000-fold lower in A. baumannii ATCC 19606T. The transformation efficiency of pVRL1Z and pVRL2Z was 4.2 × 103 and 3.9 × 103 CFU/μg, respectively, for A. baumannii AYE upon selection on low-salt LA plates supplemented with 250 μg/ml Zeo (data not shown). Strain-to-strain variation in transformation efficiency is quite common in A. baumannii, especially among clinical isolates (20). Also, naturally competent A. baylyi BD413 trpE27 and species belonging to the ACB complex could be transformed with either pVRL plasmid, showing variable transformation efficiencies and different increases in the Gm susceptibilities of the transformants (Table 2).
Moreover, the plasmid copy number (PCN) per chromosome unit was assessed by means of quantitative PCR analysis (21), as described in Materials and Methods. The PCN of pVRL1 in E. coli DH5α and A. baumannii ATCC 19606T were 70.1 ± 5.2 and 56.8 ± 5.1 copies per chromosome unit, respectively (Table 3). Thus, pVRL vectors can be classified as high-copy-number plasmids according to the conventional definitions (22).
Finally, the stability of pVRL1 and pVRL2 in E. coli and A. baumannii strains was assessed. Bacteria were grown in liquid medium for 24 h without antibiotic selection and then plated on LA with or without Gm. Plasmid stability was expressed as the NGm/N0 ratio, where NGm and N0 are the numbers of CFU obtained when the bacteria were grown on LA supplemented and not supplemented with Gm, respectively. Both pVRL1 and pVRL2 were maintained for 24 h in all the tested strains, with NGm/N0 values ranging from 0.72 ± 0.15 to 1.01 ± 0.20 (Table 3).
Since pVRL plasmids are maintained in the absence of antibiotic selection, we reasoned that either a high copy number or genetic elements involved in plasmid maintenance, or both, could account for plasmid stability. To investigate this issue, the entire sequence of the pWH1277-derived fragment was analyzed. Analysis with a combination of the BLASTX and ORFfinder programs identified at least six putative open reading frames (ORFs; ORF-1 to ORF-6) flanking the origin of replication for Acinetobacter spp. (Fig. 4). Four ORFs are likely involved in plasmid replication and mobilization, since they encode two putative MobA/MobL proteins (ORF-1 and ORF-5), one putative rolling-circle-replication (RCR) protein (ORF-4), and a putative nickase (ORF-6). Notably, ORF-2 and ORF-3 code for a putative antitoxin and a putative toxin, respectively, possibly constituting a toxin-antitoxin (TA) system involved in plasmid maintenance.
FIG 4.

Physical map of pWH1277 and functional predictions of putative gene products. The 4,693-bp linear map of pHW1277 containing at least six putative ORFs (colored arrows), predicted with ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/). The origin of replication for A. baumannii (oriAb), which was completely sequenced by Hunger et al. (6), includes the A+T-rich regions (yellow) and the palindromic element (fluorescent green). Predicted promoters (black arrows) and bidirectional Rho-independent transcriptional terminators (Ω) are indicated. Unique cutter restriction enzymes are highlighted in bold. All genes are reported in scale over the total length of the vector. The image was obtained by the use of SnapGene software (from GSL Biotech). aa, number of amino acids.
In particular, BLASTX analysis predicted that the 270-nucleotide (nt) ORF-3 codes for a protein sharing 100% sequence identity with a ParE toxin of the type II TA system of Gammaproteobacteria (GenBank accession no. WP005166293.1). Coherently, homology modeling with both I-TASSER and SWISS-MODEL software generated very high template modelling (TM) and Qualitative Model Energy ANalysis (QMEAN) scores (0.969 and −1.48, respectively; Table S2) for the structure superimposition of the protein encoded by ORF-3 to the ParE2 toxin from E. coli O157 cocrystallized with its cognate antitoxin PaaA2 (ParE-associated antitoxin 2; PDB accession no. 5CW7) (23) (Fig. 5A).
FIG 5.
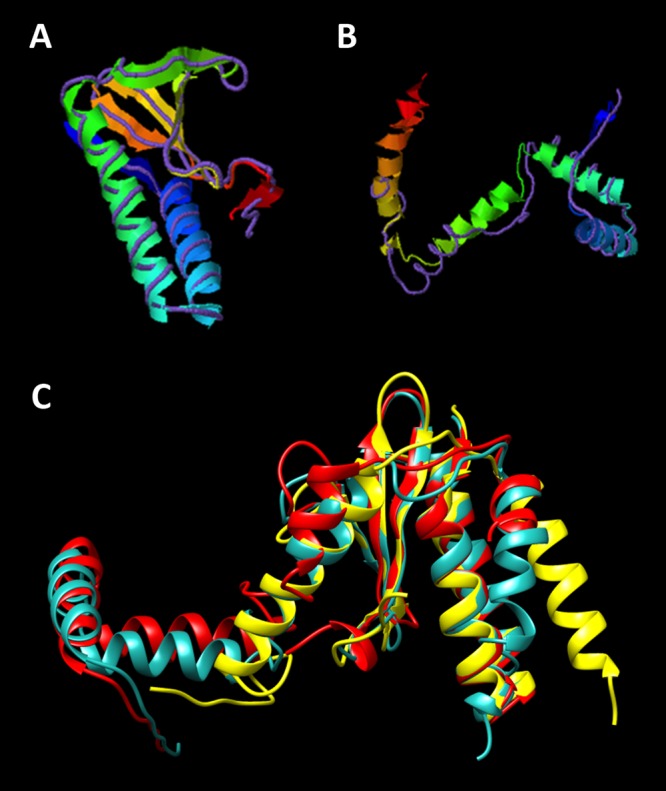
Homology modeling of the ParE2-like toxin and the PaaA2-like antitoxin from pWH1277, and superimposition of the ParE2-PaaA2-like TA complex on the E. coli ParE2-PaaA2 TA crystal structure. The query structure is shown with ribbon diagrams, while the structural analog is displayed as a backbone trace. (A) ParE2-like toxin; (B) PaaA2-like antitoxin. The imperfect superimposition on the backbone trace of the PaaA2 antitoxin (B) is probably due to the intrinsically disordered structure of the toxin-binding domain, which provides a certain degree of overall flexibility needed for the interaction with the toxin, a feature in common with several type II antitoxins (23). Only the first-ranked model predicted by I-TASSER (47) for each query is shown. (C) Superimposition within the crystal structure of ParE2-PaaA2 (yellow; PDB accession no. 5CW7) (23), I-TASSER (cyan), and SWISS-MODEL (red) homology modeling prediction for the pWH1277 toxin and antitoxin. The image was obtained using UCSF Chimera software (available at http://www.cgl.ucsf.edu/chimera) (49).
The 294-nt ORF-2 was identified by analysis with the BLASTX and ORFfinder programs to be a putative transcriptional regulator (GenBank accession no. WP005166297.1), while it was modeled by I-TASSER and SWISS-MODEL software on the YafQ protein (Fig. 5B; Table S2), the antitoxin of the E. coli DinJ-YafQ TA system (PDB accession no. 4ML0) (24). Although sequence homology and structure-based modeling provided apparently diverging functional predictions, they actually revealed two different roles of the same protein, consistent with the notion that some members of the type II TA family act as transcriptional regulators of the TA module itself (25). Of note, neither I-TASSER and SWISS-MODEL provided high TM and QMEAN scores for this prediction (0.571 and −2.43, respectively; Table S2), probably because antiantitoxin proteins show considerable structural flexibility that limits the superimposition of the ORF-2 product on the template structure. Nevertheless, a significant similarity of the ORF-3/ORF-2-encoded putative TA system was observed upon superimposition of both the I-TASSER- and SWISS-MODEL-derived complexes onto the ParE2-PaaA2 crystal structure complex (PDB accession no. 5CW7) (23) (Fig. 5C). The ParE2 toxin, like the unrelated F-encoded toxin CcdB and quinolones, binds the gyrase enzyme and stalls the DNA gyrase cleavage complex (26). Given the structural similarity between the pWH1277 TA system and the ParE2-PaaA2 complex, we hypothesized that the predicted pWH1277-encoded toxin could function as a gyrase inhibitor, ensuring plasmid stability by killing plasmid-free daughter cells. Taken together, these results suggest that the cryptic natural plasmid pWH1277 contains a TA system composed of a ParE2-like toxin (ORF-3), which could interact with gyrase and inhibit its activity, and a PaaA2-like antitoxin (ORF-2), which could neutralize the ParE2-like toxin activity and probably ensures plasmid stability and maintenance in daughter cells (27).
A ParE2-PaaA2-like TA system is implicated in pVRL plasmid stability.
To demonstrate the involvement of the predicted TA system in pVRL plasmid stability, a pVRL1 derivative deleted of the 544-bp region encompassing the putative ParE2-like toxin and PaaA2-like antitoxin system (pVRL1ΔTA) was generated. The stability values (NGm/N0 ratios) of pVRL1 and pVRL1ΔTA in E. coli DH5α after 24 h of growth in LB without antibiotic selection were 0.86 ± 0.18 and 0.10 ± 0.02, respectively, while in A. baumannii ATCC 19606T the TA deletion decreased plasmid stability from 0.92 ± 0.29 to 0.57 ± 0.20 (Table 3). These results indicate that the ParE2-PaaA2-like TA system plays a key role in the maintenance of pVRL plasmids. To support this result and provide evidence of the toxicity of the ParE2-like toxin, a pVRL1 variant carrying a 274-bp deletion of the entire paaA2-like antitoxin gene was generated (pVRL1ΔpaaA2-like) and introduced into E. coli DH5α cells harboring a pME6032-derived plasmid that allows isopropyl-β-d-1-thiogalactopyranoside (IPTG)-inducible expression of the paaA2-like antitoxin gene (pME6032paaA2-like). In the resulting strain, the paaA2-like antitoxin gene should be expressed only upon IPTG induction, while the parE2-like toxin should be constitutively expressed from its indigenous σ70-dependent promoter (Fig. 6A). Assuming that the ParE2-like protein is a toxin and the PaaA2-like protein is the cognate antitoxin, cells should be viable only upon expression of the paaA2-like antitoxin gene, i.e., in the presence of IPTG. In fact, the pME6032paaA2-like plasmid allowed bacterial growth on LA only in the presence of IPTG (Fig. 6B, inset). Consistent with this observation, analysis of the growth profiles in LB revealed that IPTG concentrations of ≥0.12 mM stimulated bacterial growth after a 10-h lag phase. Lower IPTG concentrations (<0.06 mM) did not allow bacterial growth, probably because, under these conditions, the expression level of the paaA2-like antitoxin gene from pME6032paaA2-like is insufficient to neutralize the parE2-like toxin gene product. Supplementation of LB with an intermediate IPTG concentration (0.06 mM) allowed moderate bacterial growth (Fig. 6B). Interestingly, filamenting cells (up to 30 μm in length) from cultures exposed to 0.06 mM and 2 mM IPTG were observed, corroborating the inhibitory effect of the ParE2-like toxin on gyrase activity during E. coli cell division (Fig. S6A). To further investigate this issue, both E. coli DH5α and A. baumannii ATCC 19606T harboring pVRL1 or pVRL1ΔTA were imaged by laser scanning confocal microscopy (Fig. S6B). In line with the previous observation, the presence of the TA system in pVRL1 altered the E. coli cell morphology, resulting in filamentous bacteria, without causing major changes to the bacterial growth profile or yields (Fig. S6B and C). Conversely, the pVRL1 variant lacking the TA system (i.e., pVRL1ΔTA) did not affect E. coli cell morphology (Fig. S6B). Notably, the pVRL1-encoded TA system had no apparent effect on the cell morphology or growth kinetics of A. baumannii ATCC 19606T (Fig. S6B and C).
FIG 6.

Inducible expression of the paaA2-like antitoxin protects from the lethal effect of the parE2-like toxin. (A) Schematic illustration of the experimental model. The IPTG-inducible expression of the paaA2-like antitoxin gene provided in trans from pME6032 allows the growth of E. coli DH5α(pVRL1ΔpaaA2-like) expressing the parE2-like toxin gene. (B) Growth was monitored in LB under appropriate antibiotic selection. To induce the expression of the paaA2-like antitoxin gene from the IPTG-inducible Ptac promoter, the media were supplemented with different IPTG concentrations, as indicated. OD600 values are the means ± SDs from three independent experiments. (Inset) Bacterial growth was also assessed after 24 h at 37°C on LA with or without 1 mM IPTG.
DISCUSSION
The main aim of this work was the generation of new user-friendly, multipurpose shuttle vectors that replicate in both E. coli and Acinetobacter spp. and that are suitable for use in MDR and XDR A. baumannii strains. The pVRL vectors described here were derived from pWH1277, a cryptic natural plasmid of A. calcoaceticus BD413 carrying a functional origin of replication for Acinetobacter spp. (6). Although the 1,337-bp region encompassing the minimal origin of replication region for A. calcoaceticus (oriAb) was sequenced a long time ago (6), the mechanisms responsible for plasmid replication and maintenance have not been investigated so far. In silico analysis of pWH1277 identified at least six putative gene products (ORF-1 to ORF-6; Fig. 4). Among them, two ORFs (ORF-2 and ORF-3) were modeled using a combination of I-TASSER and SWISS MODEL software, generating three-dimensional models of the putative toxin- and antitoxin-like gene products (Fig. 5; see Table S2 in the supplemental material).
On the basis of sequence homology predictions, the 1,337-bp region encompassing the pWH1277 origin of replication for Acinetobacter spp. shows no significant similarity with sequences deposited in GenBank (except those derived from pWH1277), and pWH1277 does not belong to any known incompatibility group (28). The pVRL vectors can be maintained together with indigenous plasmids, which are very common in MDR and XDR A. baumannii strains, as is the case for ACICU and AYE (17, 18). In fact, we did not observe the exclusion of indigenous plasmids carried by A. baumannii ACICU (i.e., pACICU1 and pACICU2 [17]) and AYE (i.e., p1ABAYE, p2ABAYE p3ABAYE, and p4ABAYE [18]) upon transformation with pVRL plasmids and extensive subculturing of transformants (for ca. 40 generations) in the absence of antibiotic selection (data not shown).
The two predicted genes encoding the TA module are similar to the genes encoding the E. coli ParE2-PaaA2 TA system, as observed by superimposition of our models on the crystal structure of ParE2-PaaA2 (Fig. 5C). The presence of a helix-turn-helix (HTH) motif in the antitoxin could suggest its role in the regulation of gene expression, since HTH motifs represent a widespread DNA-binding domain in transcriptional regulators (29). This is in line with the functional predictions obtained with the BLASTX and ORFfinder programs, which, in the first instance, categorized the paaA2-like antitoxin gene as a transcriptional regulator. Indeed, type II TA systems are able to negatively regulate their own expression at the level of transcription, with the antitoxin module directly repressing the promoter of the TA operon, hence generating a negative-feedback loop required for fine expression control of the TA system (25).
The entire sequence of pWH1277 was used in combination with the Gm resistance cassette (i.e., aacC1), the ColE1-like origin of replication, and the MCS from pBS to generate pVRL1 (Fig. 1A). Being the product of the valuable implementation of genetic tools available for cloning and gene manipulation in A. baumannii, the pVRL E. coli-Acinetobacter species shuttle vectors generated in this work should facilitate the cloning, sequencing, and expression of genes. In fact, pVRL1 represents a useful tool for cloning and allows the rapid screening of cloned inserts, since it includes (i) 16 unique restriction sites in the polylinker, (ii) blue/white screening for the convenient selection of E. coli colonies containing recombinant plasmids (Fig. S2), and (iii) the presence of a region which could be used for direct sequencing of cloned inserts using commercially available primers (i.e., T3, T7, M13 Fw, M13 Rv, pBS SK, and pBS KS). Moreover, the pVRL1 plasmid overcomes some of the limitations of pWH1266, such as the presence of two selective markers (i.e., the blaTEM-1 and tetA genes) conferring resistance to β-lactams or tetracycline, two classes of antibiotics unsuitable for plasmid selection in A. baumannii (30). It should also be taken into account that blaTEM-1 confers resistance to sulbactam (31), which is still a viable therapeutic option, eventually in combination with other antibiotics, for the treatment of MDR A. baumannii infections (32, 33, 34, 35), raising concern about the use of the blaTEM-1 marker in A. baumannii.
Using pVRL1 as a scaffold, the pVRL2 plasmid was constructed with the aim of generating a useful tool for controlled gene expression in Acinetobacter spp. (Fig. 1B). Both pVRL1 and pVRL2 were successfully employed for the direct cloning of PCR products, upon digestion with suitable restriction enzymes, and for the complementation of tryptophan auxotrophy in A. baylyi BD413 trpE27 and A. baumannii ATCC 19606T trpE19 (Fig. 2). Remarkably, we recently managed to clone a large insert (up to 10 kb, corresponding to the A. baumannii ACICU gene cluster from the ACICU_00873 to ACICU_00880 genes) directly into pVRL1 (data not shown), supporting the potential of this plasmid in large-scale genome studies. Moreover, pVRL2 carries a noncanonical RBS sequence, 5′-CTTCT-3 (derived from miniCTX1-araC-PBAD tolB), instead of the more common 5′-AGGAG-3′ sequence (Fig. 1C) (14). Modification of the RBS sequence and AraC-dependent promoter repression reduce the basal expression level of the cloned insert in the absence of the inducer (i.e., without arabinose) (15), hence silencing the effect of cloned genes. This is a valuable property for cloning antibiotic resistance and toxin genes. Expression studies of the tetA gene cloned in pVRL2 confirmed the tight control of transcription from the araC-PBAD promoter in A. baumannii. Indeed, the same level of Tc resistance was observed in A. baumannii carrying either the empty pVRL2 vector or the recombinant pVRL2tetA plasmid without arabinose induction, whereas Tc resistance increased 8-fold upon 2% arabinose induction in A. baumannii ATCC 19606T(pVRL2tetA) (Fig. 3C). The expression of the lacZ reporter gene under araC-PBAD control was quantified using the β-galactosidase assay (36) (Fig. 3B). We demonstrated that the presence of arabinose was able to induce the expression of lacZ from pVRL2lacZ in A. baumannii ATCC 19606T with a dose-response and time-dependent expression pattern. We further exploited the tight regulation of the araC-PBAD system in pVRL2 using the recombinant plasmid pVRL2gfp. We observed high levels of fluorescence emitted by cells in the presence of arabinose (1% to 4%), while no detectable fluorescence was measured in the absence of arabinose (Fig. 3E). It should also be pointed out, however, that gene expression in A. baumannii is affected by the codon usage of the cloned gene, since no expression of the mCherry red fluorescent protein was observed by A. baumannii cells transformed with the recombinant plasmid pVRL2mCherry, though mCherry expression was evident in E. coli(pVRL2mCherry) (Fig. S7A). The lack of mCherry expression in A. baumannii(pVRL2mCherry) could be due to the high content of rare codons for A. baumannii in the mCherry sequence compared with that in the lacZ and gfp sequences (Fig. S7B).
Furthermore, pVRL1 and pVRL2 can be purified with high yields from both E. coli and A. baumannii, consistent with the high PCN estimated in both species (Table 3). In line with a previous study, the electrotransformation efficiency was highly variable, depending on the A. baumannii recipient strain (Table 2) (20).
A major shortcoming of plasmid-based tools for genetic complementation is the need for antibiotic selection to ensure plasmid maintenance in the cells. Here, we determined the stability of pVRL1 and pVRL2 in E. coli DH5α and Acinetobacter spp., including the MDR A. baumannii ACICU strain, in the absence of Gm. In vitro assays revealed that both plasmids are very stable over time (up to 24 h) even without antibiotic selection, due to the presence of the ParE2-PaaA2-like TA system, responsible for maintenance and segregational stability. In fact, deletion of this TA system in pVRL1 caused a drastic decrease in plasmid stability, especially in E. coli (Table 3), in the absence of antibiotic selection. Of note, E. coli cells harboring pVRL plasmids showed changes in morphology, as inferred by the observation of filamenting cells upon transformation with these plasmids (Fig. S6 and S7). Many microorganisms adopt a filamentous shape caused by cell division arrest, concomitant with the continuous cell volume growth, in response to a variety of stressful environments, including treatment with fluoroquinolones (37, 38). Upon exposure to subinhibitory ciprofloxacin concentrations, E. coli cells adopt a filamenting phenotype very similar to the one observed under our conditions (37). The ParE2 protein, like quinolones and all members of the CcdB family of toxins, inhibits gyrase activity by stabilizing the DNA gyrase cleavage complex (26). We speculate that filamentation is probably due to the activity of the parE2-like toxin expressed by pVRL plasmids. We further demonstrated that E. coli cells harboring a pVRL1 variant lacking the PaaA2-like antitoxin (pVRL1ΔpaaA2-like) were unable to grow (Fig. 6B), confirming the poisoning effect of the ParE2-like toxin on bacterial vitality when it is not neutralized by its cognate antitoxin.
We were able to select the MDR A. baumannii ACICU strain transformed with pVRL plasmids on Gm, although this strain is reported to be clinically resistant to Gm (16). Standard clinical definitions and classifications of drug sensitivity for bacteria are based on achievable nontoxic levels of antibiotics in the human body. However, when assessing the in vitro use of antibiotics as selectable markers, these definitions can be reconsidered, because it is possible to achieve significantly higher drug concentrations in vitro than in vivo. Here, we demonstrated that Gm concentrations that cannot be reached in vivo can be used in vitro (up to 200 μg/ml) for selection of A. baumannii MDR clinical strains, like ACICU, whose MIC for Gm is 64 μg/ml in LB and LA media. Recent studies have shown that other nonclinically relevant antimicrobials, such as tellurite and Zeo, can be used for in vitro selection (39, 40). Zeo is not intended for clinical use, though it has already been employed as a selectable marker for XDR A. baumannii strains (40). In this study, replacement of the aacC1 cassette of pVRL1 and pVRL2 with the Zeo resistance gene (i.e., ble) led to the generation of pVRL1Z and pVLR2Z, respectively (Table 1). The latter vectors allow plasmid selection in A. baumannii strains that are extremely resistant to Gm (e.g., AYE; Table 2). Moreover, replacement of the aacC1 cassettes of pVRL plasmids with the ble gene should prevent homologous recombination with the chromosomal aacC1 gene in a RecA-proficient background.
Furthermore, the pVRL plasmids share a narrow host range, since they replicate in different strains belonging to the ACB complex (Table 2) but they do not replicate in Pseudomonas spp. This characteristic prevents the possible spread of resistance genes to neighbor species, rendering pVRL vectors safe tools for manipulation of class 2 biological agents.
In conclusion, pVRL plasmids represent promising tools for gene cloning and expression in A. baumannii, and we expect that they will be useful for future genetic studies in MDR and XDR A. baumannii strains.
MATERIALS AND METHODS
Bacterial strains and culture media.
The bacterial strains used in this study are listed in Table 1. A. baumannii and E. coli were routinely grown in Luria-Bertani broth (LB) or on Luria-Bertani agar (LA) plates at 37°C, while A. baylyi BD413 trpE27, A. pittii UKK_0145, A. nosocomialis UKK_0361, A. dijkshoorniae Scope 271, and A. seifertii HS A23-2 were grown in tryptic soy broth (TSB) or on tryptic soy broth agar (TSA) plates at 37°C. Vogel-Bonner minimal medium (41) agar plates supplemented with 20 mM sodium succinate as the carbon source (VBS agar plates) were also used. When required, VBS agar plates were supplemented with 1 mM tryptophan (Trp). Carbenicillin (Cb), gentamicin (Gm), tetracycline (Tc), and zeocin (Zeo) were added when needed. The Cb concentration used for A. baumannii ATCC 19606T was 250 μg/ml. The Gm concentration used for E. coli and A. baylyi BD413 trpE27 was 10 μg/ml, while 100 μg/ml and 200 μg/ml were used for A. baumannii ATCC 19606T and A. baumannii ACICU, respectively. The Tc concentration used for E. coli was 12.5 μg/ml. The Zeo concentrations used for E. coli and A. baumannii were 25 μg/ml and 250 μg/ml, respectively. Zeo selection was performed on low-salt LA (40).
DNA manipulation.
Chromosomal DNA was isolated using a QIAamp DNA minikit (Qiagen), and plasmid DNA was purified from bacterial cultures using a Wizard Plus SV Minipreps DNA purification system (Promega Corporation), according to the manufacturer's instructions. PCRs were performed using Thermo Scientific Phusion High-Fidelity DNA polymerase and the primers listed in Table S1 in the supplemental material. FastDigest restriction enzymes were purchased from Thermo Fisher Scientific. DNA sequencing was performed using an ABI 3730 sequencer (service by Bio-Fab Research, Rome, Italy) and the primers listed in Table S1.
Plasmid construction.
Plasmid construction details are provided in Results. All plasmids used in this study are listed in Table 1.
Preparation of A. baumannii electrocompetent cells.
Competent A. baumannii cells were prepared for electrotransformation as previously described (20) with minor modifications. Briefly, bacteria were grown in 3 ml of LB for 18 h. Then, bacterial cultures were diluted 1:100 into 50 ml of prewarmed LB and the cells were grown for 24 h at 37°C with vigorous shaking. Cells were harvested by centrifugation (3,000 × g for 15 min), washed twice with 25 ml of 10% glycerol at room temperature, and suspended in 1.5 ml of 10% glycerol. Fifty-microliter aliquots of competent cells were stored at −80°C until they were used. Electroporation was performed using 300 ng/μl of plasmid in 0.2-cm electroporation cuvettes (Gene Pulser; Bio-Rad). After pulsing (2.5 kV/cm, 200 Ω, 25 μF), cells were immediately recovered in 1 ml of prewarmed SOC medium (2% tryptone, 0.5% yeast extract, 10 mM NaCl, 2.5 mM KCl, 10 mM MgCl2, 10 mM MgSO4, 20 mM glucose) and incubated at 37°C for 1.5 h. Transformants were selected on LA or low-salt LA with the appropriate antibiotic concentration. Competent E. coli cells were prepared by the calcium chloride method (42).
Cloning, transformation, and complementation of the trpE gene.
The primers and restriction enzymes used for cloning of the trpE gene (GenBank accession no. HMPREF0010_01955) are listed in Table S1. The location of the A. baumannii ATCC 19606T trpE σ70-dependent promoter was predicted using the BPROM program (SoftBerry). The promoterless trpE gene (trpEPL) or the trpE gene including its endogenous promoter (trpEwt) was amplified by PCR from the A. baumannii ATCC 19606T genome. The 1,791-bp (trpEwt)- and 1,513-bp (trpEPL)-derived amplicons were digested with XhoI and PstI for cloning in the corresponding sites of pVRL1 and pVRL2, respectively. The resulting plasmids, termed pVRL1trpEwt and pVRL2trpEPL, were introduced into E. coli DH5α, and transformants were selected on LA supplemented with 10 μg/ml Gm and 40 μg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) for blue/white screening (Fig. S2). After plasmid extraction, the naturally competent A. baylyi BD413 trpE27 and the transposon insertion derivative of A. baumannii ATCC 19606T trpE19 were transformed with each plasmid. Transformation of A. baylyi was performed as previously described (43). Briefly, 70 μl of an A. baylyi BD413 trpE27 bacterial culture grown 18 h in TSB was combined with 1 ml of fresh TSB and 130 ng of each plasmid DNA. The bacteria were incubated at 37°C for 3 h and plated onto TSA supplemented with 10 μg/ml Gm. Complementation of the trpE mutation was investigated using VBS agar plates with or without the addition of 1 mM tryptophan.
Cloning of lacZ and β-galactosidase activity assay.
The lacZ gene (GenBank accession no. EG10527) was amplified from E. coli MG1655 genomic DNA. The primers and restriction enzymes used for cloning of the lacZ gene are listed in Table S1. The resulting 3,094-bp amplicon was digested using the XhoI and PstI restriction enzymes and ligated to the corresponding sites of pVRL2. The resulting plasmid (pVRL2lacZ) was used to transform A. baumannii ATCC 19606T. Transformants were incubated at 37°C under vigorous shaking until the culture reached an optical density at 600 nm (OD600) of ca. 0.6. Then, cells were challenged with different concentrations of arabinose (from 0.06% to 8%). The β-galactosidase assay was performed at 1 h, 3 h, and 24 h postinduction (36).
Cloning of the tetA gene and tetracycline susceptibility testing.
The tetracycline resistance (tetA) gene was amplified from pBR322, using the primers listed in Table S1. The resulting 1,223-bp amplicon was digested with XhoI and PstI for directional cloning into the corresponding sites of pVRL2. The resulting plasmid (pVRL2tetA) was introduced into A. baumannii ATCC 19606T. Bacteria were grown for 18 h in LB supplemented with 2% arabinose and then diluted 1:1,000 in cation-adjusted Mueller-Hinton (MH) broth. The Tc MIC was determined by microdilution in the absence or presence of arabinose (0.5% or 2%). The disk diffusion assay was performed using as the inoculum a bacterial culture grown for 18 h in LB supplemented with 2% arabinose. Cells were washed, diluted in saline to an OD600 of 0.1, and then seeded onto LA plates without or with arabinose (0.5% or 2%). The sensitivity to Tc was evaluated as the zone of growth inhibition around a commercial disk containing 30 μg Tc.
Cloning of fluorescent reporter genes, cell imaging by laser scanning confocal microscopy, and fluorescence measurements.
The green fluorescent protein (GFP) gene was excised from miniTn7-gfp (44) by XbaI and NotI digestion and then directionally cloned into the corresponding sites of pVRL2. The red fluorescent protein mCherry was amplified from miniCTXmCherry (Table 1) using the primers listed in Table S1, and the 708-bp amplicon was digested with XhoI and EcoRI for directional cloning into the corresponding sites of pVRL2. The resulting plasmids (pVRL2gfp and pVRL2mCherry) were independently introduced into A. baumannii ATCC 19606T. For GFP detection, bacterial cells from 18-h cultures in LB were diluted 1:20 in LB supplemented or not with arabinose (0.5%, 1%, 2%, and 4%). The presence of bacterium-associated GFP fluorescence was evaluated after 6 h at 37°C by laser scanning confocal microscopy (Leica SP5; 63× oil immersion objective). In parallel, bacteria from cultures exposed to the different arabinose concentrations were washed with saline and diluted to an OD600 of 0.25, and GFP fluorescence was measured in a fluorescence/luminescence spectrophotometer (model LS-50B; PerkinElmer) with excitation and emission wavelengths of 475 nm and 515 nm, respectively. The excitation and emission wavelengths for mCherry were 587 nm and 610 nm, respectively.
Transformation efficiency and in vitro stability of pVRL plasmids.
The transformation efficiency was determined using 100 ng and 300 ng of each plasmid DNA for calcium-competent or electrocompetent E. coli and A. baumannii cells, respectively. Naturally competent A. baylyi BD413 trpE27 cells were also used. Transformants were plated on LA or TSA supplemented with 10 μg/ml of Gm for E. coli DH5α and A. baylyi BD413 trpE27, while 100 μg/ml and 200 μg/ml of Gm were used for A. baumannii ATCC 19606T and A. baumannii ACICU, respectively. The number of CFU grown on the transformation plates was calculated. Transformation efficiency was expressed as the number of CFU per microgram of plasmid DNA. To establish plasmid stability determinants, pVRL1 carrying a complete deletion of the TA system (544 bp) was generated, using primers ΔTA FW and ΔTA RV (Table S1) and a Q5 site-directed mutagenesis kit (New England BioLabs). The resulting plasmid (pVRL1ΔTA) was introduced into E. coli DH5α and A. baumannii ATCC 19606T for the stability assay, and the bacterial strains were grown for 18 h in LB or TSB with the appropriate Gm concentration, diluted 1,000-fold in LB or TSB without antibiotic, and cultured for an additional 24 h at 37°C (stationary phase). Bacterial colony counts were determined on LA or TSA (N0) and on LA or TSA supplemented with Gm at the appropriate concentrations (NGm). Plasmid stability is defined by the ratio NGm/N0 (6).
Determination of plasmid copy number (PCN).
The copy number of pVRL1 in A. baumannii ATCC 19606T and E. coli DH5α was determined by real-time quantitative PCR (RT-qPCR), as previously described (21), with minor modifications. Three primer pairs for the amplification of the Gm resistance gene (aacC1) and of the d-1-deoxyxylulose 5-phosphate synthase gene (dxs) of A. baumannii ATCC 19606T (GenBank accession no. HMPREF0010_02600) or E. coli DH5α (GenBank accession no. G6237) were designed (Table S1). The aacC1 gene is present in a single copy in the pVRL1 plasmid, while it is absent in the A. baumannii ATCC 19606T and E. coli DH5α chromosomes, and dxs is a single-copy gene in both A. baumannii ATCC 19606T and E. coli DH5α. Consequently, RT-qPCR quantification of aacC1 and dxs in samples containing both pVRL1 and the A. baumannii ATCC 19606T or E. coli DH5α chromosome relative to that in samples containing known amounts of pVRL1 or A. baumannii ATCC 19606T or E. coli DH5α chromosomal DNA alone makes it possible to extrapolate the copy number of pVRL1 in these bacteria. The total DNA from A. baumannii ATCC 19606T and E. coli DH5α harboring or not harboring pVRL1 (i.e., both the plasmid and chromosome or the chromosome alone, respectively) was purified using a QIAamp DNA minikit (Qiagen) following the manufacturer's instructions. The pVRL1 plasmid was purified from A. baumannii ATCC 19606T and E. coli DH5α cells with a PureYield plasmid miniprep system (Promega) following the manufacturer's instructions. The amount of DNA in each sample was quantified by spectrophotometric analysis and normalized to a concentration of 25 ng/μl. RT-qPCR was performed using an AriaMX real-time PCR system (Agilent) with software accompanying the system (version 1.0). Reactions were performed in a 20-μl total volume containing 1× iTaq Universal SYBR green supermix (Bio-Rad), 0.4 μM each primer, and 2 μl of diluted template DNA (range of final DNA amount, 0.05 ng to 50 ng per sample). For detection of chromosome- or plasmid-specific amplicons, separate reaction mixtures were prepared for each template DNA concentration. The thermal cycling protocol was as follows: initial denaturation of 4 min at 95°C, followed by 40 cycles of 15 s at 95°C and 45 s at 60°C. Cycle threshold (CT) values were determined after automatic adjustment of the baseline and manual adjustment of the fluorescence threshold. Standard curves (R2 ≥ 0.99) for five independent samples containing A. baumannii ATCC 19606T or E. coli DH5α chromosomal DNA (chromosome) or pVRL1 (plasmid) alone were generated by placing the log value of the amount of template DNA (determined according to the dilution) on the x axis and the average CT value on the y axis. These standard curves were used to extrapolate the copy number of pVRL1 in samples containing both chromosomal and plasmid DNA.
Gentamicin susceptibility testing.
The MIC of Gm was determined using both the broth microdilution method and the agar dilution method (45). First, bacteria were inoculated at ca. 5 × 105 CFU/ml in a 96-well microtiter plate containing 200 μl of TSB or LB per well. The Gm concentrations tested ranged from 0 μg/ml to 512 μg/ml, depending on the strain. The MICs were determined to be the lowest concentration of Gm that inhibited visible growth after 16 to 20 h of incubation at 37°C. Similarly, LA or TSA plates were prepared by adding increasing concentrations of Gm (0 μg/ml to 512 μg/ml) in a 20-ml medium volume. In this case, the bacterial concentrations were adjusted in order to obtain ca. 5 × 104 cells per spot. Growth was assessed after incubation at 37°C for 16 to 20 h. The MIC (expressed in micrograms per milliliter) was defined as the lowest concentration of the antimicrobial agent that prevented the visible growth of a microorganism.
Construction of pVRL1ΔpaaA2-like and pME6032paaA2-like plasmids.
The primers and restriction enzymes used for generation of pVRL1ΔpaaA2-like and pME6032paaA2-like are listed in Table S1. The deletion of paaA2-like (294 bp) from pVRL1 was obtained by use of a Q5 site-directed mutagenesis kit (New England BioLabs) following the manufacturer's instructions with ΔpaaA2-like FW and ΔTA RV primers. The resulting plasmid, termed pVRL1ΔpaaA2-like, was introduced into E. coli DH5α, and transformants were selected on LA supplemented with 10 μg/ml Gm. The promoterless paaA2-like gene was amplified by PCR from the pVRL1 plasmid with primers paaA2-like FW and paaA2-like RV. The 317-bp derived amplicon was digested at the EcoRI and SacI restriction sites and cloned into the corresponding sites of the pME6032 vector (46). The derived plasmid, termed pME6032paaA2-like, was introduced into E. coli DH5α carrying pVRL1ΔpaaA2-like, and transformants were selected on LA supplemented with 12.5 μg/ml Tc, 10 μg/ml Gm, and 1 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG). The growth assay was performed in a 96-well microtiter plate (BD Falcon) using a Tecan Spark 10M microtiter reader. Briefly, a 16-h bacterial culture of E. coli DH5α(pVRL1ΔpaaA2-like, pME6032paaA2-like) was diluted 1,000-fold in a 200-μl final volume of LB supplemented with the appropriate antibiotic concentration and different IPTG concentrations (no IPTG or 0.01 mM to 2 mM IPTG). The bacterial cultures were incubated at 37°C, and the optical density at 600 nm (OD600) was measured every 2 h for 24 h. Bacteria were also spotted on LA supplemented or not supplemented with 1 mM IPTG, and growth was observed after 24 h of incubation at 37°C. Finally, bacteria grown in the presence of different IPTG concentrations (0.06 mM and 2 mM) were visualized by laser scanning confocal microscopy (Leica SP5; 63× oil immersion objective).
Homology searches and protein modeling.
The complete sequence of the A. calcoaceticus-derived portion of pWH1266 was submitted to analysis with BLASTX (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) software for interrogation of the nonredundant NCBI protein database, in order to detect the presence of any homologous sequence. All homology results were confirmed using I-TASSER (47) and SWISS-MODEL prediction software (48), to assign putative protein structures and functions. Match marker analyses and superimposition of proteins were performed using UCSF Chimera software (49).
Accession number(s).
The full-length pVRL1 and pVRL2 sequences have been deposited in the GenBank database under accession numbers MG462882 and MG551985, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We thank Fabio Polticelli for help and assistance with bioinformatics analysis. We also thank Harald Seifert for providing A. baylyi BD413 trpE27, A. pittii UKK_0145, A. nosocomialis UKK_0361, A. dijkshoorniae Scope 271, and A. seifertii HS A23-2, Paul G. Higgins for providing the pWH1266 plasmid, and Luis A. Actis for providing A. baumannii ATCC 19606T trpE19.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02480-17.
REFERENCES
- 1.Turton JF, Shah J, Ozongwu C, Pike R. 2010. Incidence of Acinetobacter species other than A. baumannii among clinical isolates of Acinetobacter: evidence for emerging species. J Clin Microbiol 48:1445–1449. doi: 10.1128/JCM.02467-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. 2010. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. doi: 10.1371/journal.pone.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wong D, Nielsen TB, Bonomo RA, Pantapalangkoor P, Luna B, Spellberg B. 2017. Clinical and pathophysiological overview of Acinetobacter infections: a century of challenges. Clin Microbiol Rev 30:409–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antunes LC, Visca P, Towner KJ. 2014. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis 71:292–301. doi: 10.1111/2049-632X.12125. [DOI] [PubMed] [Google Scholar]
- 6.Hunger M, Schmucker R, Kishan V, Hillen W. 1990. Analysis and nucleotide sequence of an origin of DNA replication in Acinetobacter calcoaceticus and its use for Escherichia coli shuttle plasmids. Gene 87:45–51. doi: 10.1016/0378-1119(90)90494-C. [DOI] [PubMed] [Google Scholar]
- 7.Dorsey CW, Tomaras AP, Actis LA. 2002. Genetic and phenotypic analysis of Acinetobacter baumannii insertion derivatives generated with a transposome system. Appl Environ Microbiol 68:6353–6360. doi: 10.1128/AEM.68.12.6353-6360.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aranda J, Poza M, Pardo BG, Rumbo S, Rumbo C, Parreira JR, Rodríguez-Velo P, Bou G. 2010. A rapid and simple method for constructing stable mutants of Acinetobacter baumannii. BMC Microbiol 10:279. doi: 10.1186/1471-2180-10-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richie DL, Takeoka KT, Bojkovic J, Metzger LE IV, Rath CM, Sawyer WS, Wei JR, Dean CR. 2016. Toxic accumulation of LPS pathway intermediates underlies the requirement of LpxH for growth of Acinetobacter baumannii ATCC 19606. PLoS One 11:e0160918. doi: 10.1371/journal.pone.0160918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alting-Mees MA, Short JM. 1989. pBluescript II: gene mapping vectors. Nucleic Acids Res 22:9494. doi: 10.1093/nar/17.22.9494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wohlleben W, Arnold W, Bissonnette L, Pelletier A, Tanguay A, Roy PH, Gamboa GC, Barry GF, Aubert E, Davies J. 1989. On the evolution of Tn21-like multiresistance transposons: sequence analysis of the gene (aacC1) for gentamicin acetyltransferase-3-I(AAC(3)-I), another member of the Tn21-based expression cassette. Mol Gen Genet 217:202–208. doi: 10.1007/BF02464882. [DOI] [PubMed] [Google Scholar]
- 12.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 13.Orosz A, Boros I, Venetianer P. 1991. Analysis of the complex transcription termination region of the Escherichia coli rrnB gene. Eur J Biochem 201:653–659. doi: 10.1111/j.1432-1033.1991.tb16326.x. [DOI] [PubMed] [Google Scholar]
- 14.Lo Sciuto A, Fernández-Piñar R, Bertuccini L, Iosi F, Superti F, Imperi F. 2014. The periplasmic protein TolB as a potential drug target in Pseudomonas aeruginosa. PLoS One 9:e103784. doi: 10.1371/journal.pone.0103784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makrides SC. 1996. Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60:512–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Longo B, Pantosti A, Luzzi I, Tarasi A, Di Sora F, Gallo S, Placanica P, Monaco M, Dionisi AM, Volpe I, Montella F, Cassone A, Rezza G. 2007. Molecular findings and antibiotic-resistance in an outbreak of Acinetobacter baumannii in an intensive care unit. Ann Ist Super Sanita 43:83–88. [PubMed] [Google Scholar]
- 17.Iacono M, Villa L, Fortini D, Bordoni R, Imperi F, Bonnal RJ, Sicheritz-Ponten T, De Bellis G, Visca P, Cassone A, Carattoli A. 2008. Whole-genome pyrosequencing of an epidemic multidrug-resistant Acinetobacter baumannii strain belonging to the European clone II group. Antimicrob Agents Chemother 52:2616–2625. doi: 10.1128/AAC.01643-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fournier PE, Vallenet D, Barbe V, Audic S, Ogata H, Poirel L, Richet H, Robert C, Mangenot S, Abergel C, Nordmann P, Weissenbach J, Raoult D, Claverie JM. 2006. Comparative genomics of multidrug resistance in Acinetobacter baumannii. PLoS Genet 2:e7. doi: 10.1371/journal.pgen.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kües U, Stahl U. 1989. Replication of plasmids in gram-negative bacteria. Microbiol Rev 53:491–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yildirim S, Thompson MG, Jacobs AC, Zurawski DV, Kirkup BC. 2016. Evaluation of parameters for high efficiency transformation of Acinetobacter baumannii. Sci Rep 6:22110. doi: 10.1038/srep22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee C, Kim J, Shin SG, Hwang S. 2006. Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J Biotechnol 123:273–280. doi: 10.1016/j.jbiotec.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Mayer MP. 1995. A new set of useful cloning and expression vectors derived from pBlueScript. Gene 163:41–46. doi: 10.1016/0378-1119(95)00389-N. [DOI] [PubMed] [Google Scholar]
- 23.Sterckx YG, Jove T, Shkumatov AV, Garcia-Pino A, Geerts L, De Kerpel M, Lah J, De Greve H, Van Melderen L, Loris R. 2016. A unique hetero-hexadecameric architecture displayed by the Escherichia coli O157 PaaA2-ParE2 antitoxin-toxin complex. J Mol Biol 428:1589–1603. doi: 10.1016/j.jmb.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 24.Liang Y, Gao Z, Wang F, Zhang Y, Dong Y, Liu Q. 2014. Structural and functional characterization of Escherichia coli toxin-antitoxin complex DinJ-YafQ. J Biol Chem 289:21191–21202. doi: 10.1074/jbc.M114.559773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Unterholzner SJ, Poppenberger B, Rozhon W. 2013. Toxin-antitoxin systems: biology, identification, and application. Mob Genet Elements 3:e26219. doi: 10.4161/mge.26219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan J, Sterckx Y, Mitchenall LA, Maxwell A, Loris R, Waldor MK. 2010. Vibrio cholerae ParE2 poisons DNA gyrase via a mechanism distinct from other gyrase inhibitors. J Biol Chem 285:40397–40408. doi: 10.1074/jbc.M110.138776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goeders N, Van Melderen L. 2014. Toxin-antitoxin systems as multilevel interaction systems. Toxins 6:304–324. doi: 10.3390/toxins6010304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carattoli A, Bertini A, Villa L, Falbo V, Hopkins KL, Threlfall EJ. 2005. Identification of plasmids by PCR-based replicon typing. J Microbiol Methods 63:219–228. doi: 10.1016/j.mimet.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Brennan RG, Matthews BW. 1989. The helix-turn-helix DNA binding motif. J Biol Chem 264:1903–1906. [PubMed] [Google Scholar]
- 30.Perez F, Hujer AM, Hujer KM, Decker BK, Rather PN, Bonomo RA. 2007. Global challenge of multidrug-resistant Acinetobacter baumannii. Antimicrob Agents Chemother 51:3471–3484. doi: 10.1128/AAC.01464-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krizova L, Poirel L, Nordmann P, Nemec A. 2013. TEM-1 β-lactamase as a source of resistance to sulbactam in clinical strains of Acinetobacter baumannii. J Antimicrob Chemother 68:2786–2791. doi: 10.1093/jac/dkt275. [DOI] [PubMed] [Google Scholar]
- 32.Levin AS, Levy CE, Manrique AE, Medeiros EA, Costa SF. 2003. Severe nosocomial infections with imipenem-resistant Acinetobacter baumannii treated with ampicillin/sulbactam. Int J Antimicrob Agents 21:58–62. doi: 10.1016/S0924-8579(02)00276-5. [DOI] [PubMed] [Google Scholar]
- 33.Kempf M, Djouhri-Bouktab L, Brunel JM, Raoult D, Rolain JM. 2012. Synergistic activity of sulbactam combined with colistin against colistin-resistant Acinetobacter baumannii. Int J Antimicrob Agents 39:180–181. doi: 10.1016/j.ijantimicag.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Pei G, Mao Y, Sun Y. 2012. In vitro activity of minocycline alone and in combination with cefoperazone-sulbactam against carbapenem-resistant Acinetobacter baumannii. Microb Drug Resist 18:574–577. doi: 10.1089/mdr.2012.0076. [DOI] [PubMed] [Google Scholar]
- 35.Kalin G, Alp E, Akin A, Coskun R, Doganay M. 2014. Comparison of colistin and colistin/sulbactam for the treatment of multidrug resistant Acinetobacter baumannii ventilator-associated pneumonia. Infection 42:37–42. doi: 10.1007/s15010-013-0495-y. [DOI] [PubMed] [Google Scholar]
- 36.Miller JH. 1972. Experiments in molecular genetics, p 352–355. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 37.Mason DJ, Power EG, Talsania H, Phillips I, Gant VA. 1995. Antibacterial action of ciprofloxacin. Antimicrob Agents Chemother 39:2752–2758. doi: 10.1128/AAC.39.12.2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bos J, Zhang Q, Vyawahare S, Rogers E, Rosenberg SM, Austin RH. 2015. Emergence of antibiotic resistance from multinucleated bacterial filaments. Proc Natl Acad Sci U S A 112:178–183. doi: 10.1073/pnas.1420702111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin IM, Richmond GE, Sen P, Koh TH, Piddock LJ, Chua KL. 2013. A method for generating marker-less gene deletions in multidrug-resistant Acinetobacter baumannii. BMC Microbiol 13:158. doi: 10.1186/1471-2180-13-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luna BM, Ulhaq A, Yan J, Pantapalangkoor P, Nielsen TB, Davies BW, Actis LA, Spellberg B. 2017. Selectable markers for use in genetic manipulation of extensively drug-resistant (XDR) Acinetobacter baumannii HUMC1. mSphere 2(2):e00140-17. doi: 10.1128/mSphere.00140-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogel HJ, Bonner DM. 1956. Acetylornithinase of Escherichia coli: partial purification and some properties. J Biol Chem 218:97–106. [PubMed] [Google Scholar]
- 42.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 43.Renda BA, Chan C, Parent KN, Barrick JE. 2016. Emergence of a competence-reducing filamentous phage from the genome of Acinetobacter baylyi ADP1. J Bacteriol 198:3209–3219. doi: 10.1128/JB.00424-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambertsen L, Sternberg C, Molin S. 2004. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ Microbiol 6:726–732. doi: 10.1111/j.1462-2920.2004.00605.x. [DOI] [PubMed] [Google Scholar]
- 45.Wiegand I, Hilpert K, Hancock RE. 2008. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3:163–175. doi: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 46.Heeb S, Itoh Y, Nishijyo T, Schnider U, Keel C, Wade J, Walsh U, O'Gara F, Haas D. 2000. Small, stable shuttle vectors based on the minimal pVS1 replicon for use in gram-negative, plant-associated bacteria. Mol Plant Microbe Interact 13:232–237. doi: 10.1094/MPMI.2000.13.2.232. [DOI] [PubMed] [Google Scholar]
- 47.Zhang Y. 2008. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics 9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42(Web Server issue):W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 50.Janssen P, Maquelin K, Coopman R, Tjernberg I, Bouvet P, Kersters K, Dijkshoorn L. 1997. Discrimination of Acinetobacter genomic species by AFLP fingerprinting. Int J Syst Bacteriol 47:1179–1187. doi: 10.1099/00207713-47-4-1179. [DOI] [PubMed] [Google Scholar]
- 51.Juni E, Janik A. 1969. Transformation of Acinetobacter calco-aceticus (Bacterium anitratum). J Bacteriol 98:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaneechoutte M, Young DM, Ornston LN, De Baere T, Nemec A, Van Der Reijden T, Carr E, Tjernberg I, Dijkshoorn L. 2006. Naturally transformable Acinetobacter sp. strain ADP1 belongs to the newly described species Acinetobacter baylyi. Appl Environ Microbiol 72:932–936. doi: 10.1128/AEM.72.1.932-936.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Casadaban MJ, Cohen SN. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol 138:179–207. doi: 10.1016/0022-2836(80)90283-1. [DOI] [PubMed] [Google Scholar]
- 54.Bachman BJ. 1996. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12, p 2460–2488. In Neidhardt FC, Ingraham JL, Low KB, Magasanik B, Schaechter M, Umbarger HE (ed), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed, vol 2 ASM Press, Washington, DC. [Google Scholar]
- 55.Cosgaya C, Marí-Almirall M, Van Assche A, Fernández-Orth D, Mosqueda N, Telli M, Huys G, Higgins PG, Seifert H, Lievens B, Roca I, Vila J. 2016. Acinetobacter dijkshoorniae sp. nov., a member of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex mainly recovered from clinical samples in different countries. Int J Syst Evol Microbiol 66:4105–4111. doi: 10.1099/ijsem.0.001318. [DOI] [PubMed] [Google Scholar]
- 56.Nemec A, Krizova L, Maixnerova M, Sedo O, Brisse S, Higgins PG. 2015. Acinetobacter seifertii sp. nov., a member of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex isolated from human clinical specimens. Int J Syst Evol Microbiol 65:934–942. doi: 10.1099/ijs.0.000043. [DOI] [PubMed] [Google Scholar]
- 57.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, Facchini M, Pasquali P, Bragonzi A, Visca P. 2016. Role of iron uptake systems in Pseudomonas aeruginosa virulence and airway infection. Infect Immun 84:2324–2335. doi: 10.1128/IAI.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.