

Abstract
4-Anilinoquinolines were identified as potent and narrow spectrum inhibitors of the cyclin G associated kinase (GAK), an important regulator of viral and bacterial entry into host cells. Optimization of the 4-anilino group and the 6,7-quinoline substituents produced GAK inhibitors with nanomolar activity, over 50,000-fold selectivity relative to other members of the numb-associated kinase (NAK) sub-family, and narrow activity spectra in the broader kinome. These compounds may be useful tools to explore the therapeutic potential of GAK in prevention of a broad range of infectious and systemic diseases.
Keywords: Cyclin G Associated Kinase (GAK), 4-Anilinoquinolines, 4-Anilinoquinazolines, Numb-Associated Kinase (NAK) family, Chemogenomic
1. Introduction
Cyclin G associated kinase (GAK) is a ubiquitously expressed 160 kDa serine/threonine kinase involved in membrane trafficking[1]. GAK was initially identified as a protein that associated with cell cycle regulator cyclin G[1b]. GAK knockdown by siRNA initiated cell-cycle arrest at the metaphase, demonstrating the requirement of GAK for normal mitotic progression[2]. GAK is a member of the Numb-Associated Kinase (NAK) family, which includes AAK1 (adaptor-associated kinase), STK16/MPSK1 (serine/threonine kinase 16/myristoylated and palmitoylated serine/threonine kinase 1), and BMP2K/BIKE (BMP-2 inducible kinase)[3].
GAK and AAK1 are host cell kinases that regulate clathrin-mediated endocytosis, a critical regulatory process by which oligomeric clathrin and adaptor protein complexes facilitate entry of macromolecules, proteins and nutrients into cells[4]. GAK and AAK1 recruit clathrin and adaptor protein complexes to the cell membrane in part through phosphorylation of T156 in the μ2 subunit of adaptor protein-2 (AP-2). The clathrin/AP-2 complex facilitates vesicle assembly and efficient internalization of cargo proteins. In addition, GAK also regulates recycling of clathrin back to the cell surface, while AAK1 mediates the rapid recycling of receptors back to the plasma membrane. Through their involvement in clathrin-mediated endocytosis, GAK and AAK1 regulate epidermal growth factor receptor (EGFR) internalization, thereby promoting EGF uptake and EGFR signaling[5].
Clathrin-mediated endocytosis is a common mechanism by which viruses, toxins, and bacteria enter their host cells[6]. Several viruses that add significant global disease burden, such as influenza, HCV, dengue, Hantaan virus, and Junin arenavirus, use this mechanism to infect cells[6]. HIV, Ebola and Zika have also been characterized as entering the cell via this uptake pathway[7]. In addition, the endocytosis of anthrax and diphtheria toxins is mediated by clathrin[8]. Inhibition of NAK family kinases has been proposed as a new approach to development of host-centered anti-viral drugs[9]. A notable advantage of this approach is that these host molecular targets may be less prone to viral and bacterial resistance mechanisms.
GAK participates in a variety of other important biological processes through its roles in cell proliferation and receptor trafficking. GAK is a prognostic marker in advanced diseases, including hormone refractory prostate cancer[10]. GAK interacts directly with the androgen receptor (AR) and sensitizes it to low levels of androgens; the expression of GAK increases upon prolonged androgen treatment and during the progression of cells to hormone independence[10–11]. GAK is over-expressed in osteosarcoma cell lines and tissues where it contributes to proliferation and survival[11b]. Genome-wide association studies (GWAS) have identified single nucleotide polymorphisms in the GAK gene associated with susceptibility to Parkinson’s disease[12]. Additional studies support a functional role for GAK in the pathology of Parkinson’s disease—for example, enhanced toxicity was observed upon siRNA knockdown of GAK in HEK293 cells overexpressing α-synuclein, a major constituent of Lewy bodies, the protein aggregates in nerve cells of patients with Parkinson’s disease and other forms of dementia[13].
Prior reports have disclosed potent GAK inhibitors that lack selectivity over other protein kinases (Figure 1), such as the pyridinylimidazole p38 inhibitor SB203580 and the c-Jun N-terminal kinase inhibitor SP600125[14]. Intriguingly, the clinically-approved EGFR inhibitors gefitinib, erlotinib and Bcr-Abl and Src inhibitors dasatinib and bosutinib show off target activity on GAK, with potency in the low nanomolar range[15]. It is not known whether the clinical efficacy or adverse events observed with these kinase inhibitor drugs are connected to their inhibition of GAK activity. These drugs were originally prepared to target EGFR and Bcr-Abl/Src, and only later was GAK identified as a collateral kinase target. The utility of these inhibitors as tools to study the biology of GAK is severely limited. Notably, it has been proposed that GAK inhibition causes clinical toxicity due to pulmonary alveolar dysfunction, but this controversial hypothesis has not been addressed with a selective small molecule GAK inhibitor[16].
Figure 1.
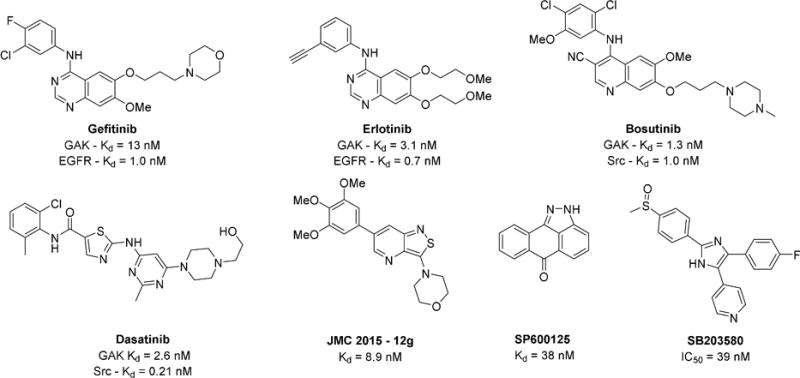
Known compounds with reported GAK activity
Recently isothiazolo[5,4-b]pyridines have been described as first generation chemical probes for GAK[9a]. Analog 12g was identified as a potent narrow spectrum GAK inhibitor with only a limited number of kinase off-targets including KIT, PDGFRB, FLT3, and MEK5. The availability of chemical probes with improved selectivity for GAK or a different spectrum of off-targets would be useful in target validation studies. Here we report the synthesis and characterization of 4-anilinoquinolines and 4-anilinoquinazolines as potent narrow spectrum GAK inhibitors. Several of these compounds have potential for development into high quality chemical probes for the study of GAK biology.
2. Results
2.1 Initial screening
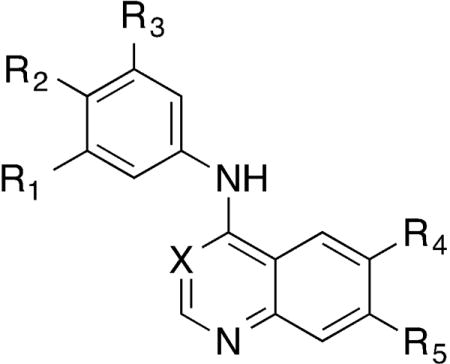
The chemogenomic sets PKIS and PKIS2 contain hundreds of biologically and chemically diverse kinase inhibitors with potent activity across the protein kinome[17]. Through the screening of PKIS2 we identified several 4-anilinoquinolines and 4-anilinoquinazolines with nanomolar activity on GAK and varying degrees of selectivity with respect to other kinases (Table 1A and Figure 2). 4-Anilinoquinoline 1 had a Kd = 31 nM against GAK and showed potent activity on only one other kinase, ADCK3 (Kd = 220 nM). The analog 2, containing a 4-pyridyl substituent at the 7-positon of the quinoline, retained GAK activity (84% I at 1 μM) and also inhibited eighteen other kinases. The 4-anilinoquinazoline 3 showed a Kd = 15 nM on GAK with activity on only three additional protein kinases, EGFR (Kd = 0.32 nM), ERBB2 (Kd = 85 nM), and BUB1 (Kd = 130 nM). We then used a panel of binding assays to determine selectivity across the NAK family and were encouraged by the results (Table 1B): 1 and 2 were potent and over 1000-fold selective, while compound 3 had weaker potency and lower selectivity. Our interest in the quinoline/quinazoline scaffold was heightened by the reported GAK activity of gefitinib, erlotinib, and bosutinib (Figure 1)[18].
Table 1A.
Chemical starting points identified by screening PKIS2 Screening.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | R3 | R4 | R5 | GAK Kd (μM) |
Kinase off targetsa |
| 1 | CH | OMe | OMe | OMe | CF3 | H | 31 | 1 |
| 2 | CH | OMe | OMe | OMe | H | 4-pyridyl | (85% at 1 μM) | 18 |
| 3 | N | Br | H | H | OMe | OMe | 29 | 3 |
Number of kinases with over 95% binding at 1 μM in a DiscoverX KINOMEscan® experiment.
Figure 2.
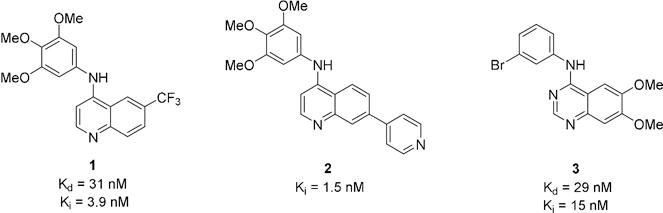
PKIS/PKIS2 quinoline and quinazoline hit compounds and GAK activities
Table 1B.
Chemical starting points profiled in the NAK family assay panel
| Cmpd | Binding displacement assay Ki (μM) | NAK selectivityb | |||
|---|---|---|---|---|---|
| GAK | AAK1 | BMP2K | STK16 | ||
| 1 | 0.0039 | 54 | >100 | 17 | 4300 |
| 2 | 0.0015 | 2.8 | 8.6 | >10 | 1900 |
| 3 | 0.015 | 3.1 | 6.6 | >10 | 210 |
Selectivity against the nearest NAK family member
2.2 Chemistry
To further explore the structural requirements for GAK activity, we synthesized analogs of 1 and 3 as outlined in Scheme 1. 4-Anilinoquinolines (1, 4–12, 16–56) were prepared by heating the corresponding 4-chloroquinoline and substituted aniline in ethanol at reflux for 18 hours. Products were isolated by direct crystallization from the crude reaction mixture or following chromatographic purification. The 4-anilinoquinazolines (13 & 57) were prepared by heating the corresponding 4-chloroquinazoline and substituted aniline in ethanol (13) or n-butanol (57) at reflux for 18 hours. The lower reactivity of the 4-chloroquinazoline required heating at the higher temperature, and these reactions were generally not as clean, requiring chromatographic purification to isolate the products. Several of the less nucleophilic anilines required an alternative method to be employed in which the aniline was added through a Buchwald-Hartwig coupling reaction to produce the corresponding 4-anilinoquinazolines 14 and 15[19].
Scheme 1.
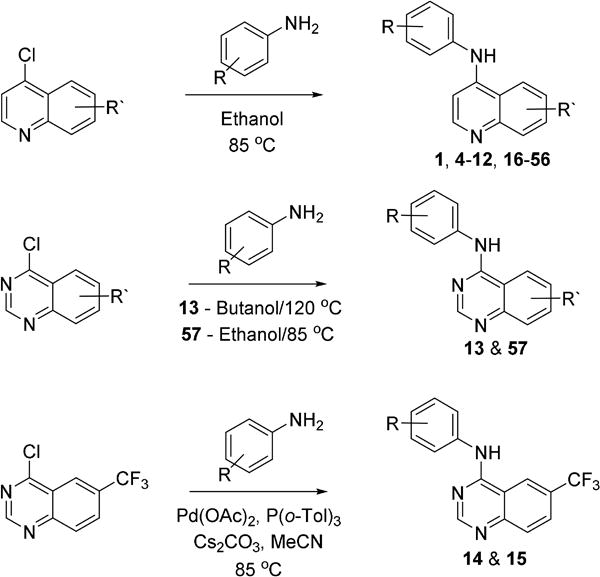
Preparation of quinoline and quinazoline based GAK inhibitors
2.3 NAK family screening and determination of cell potency
The final compounds were initially screened at 10 μM for their ability to affect the melting temperature of the GAK protein using a differential scanning fluorimetry (DSF) assay. The change in denaturation temperature of kinase protein (ΔTm) in the presence and absence of a small molecule inhibitor was previously established as predictive of compound affinity [20]. The compounds were next screened for activity on the kinase domains of all four members of the NAK sub-family (GAK, AAK1, BMP2K, and STK16) using a Time-Resolved Fluorescence Energy Transfer (TR-FRET) binding displacement assay in a 16-point dose response format to determine the inhibition constant (Ki).
The most active analogs were then tested in a GAK nanoBRET cellular target engagement assay[21]. Briefly an N-terminal fusion of GAK and Nano Luciferase, a small and extremely bright luciferase, was transiently expressed in HEK293 cells. Addition of Nano Luciferase enzyme substrate and a cell permeable tracer, comprised of a red shifted dye tethered to a fragment shown to bind the ATP site of GAK, led to a bioluminescence resonance energy transfer (BRET) signal. Small molecule GAK inhibitors competed away the tracer from the fusion protein, leading to the observable loss of BRET signal and forming the basis of the nanoBRET assay. The assay was conducted in a 8-point dose response format to determine the cellular binding potency IC50[22].
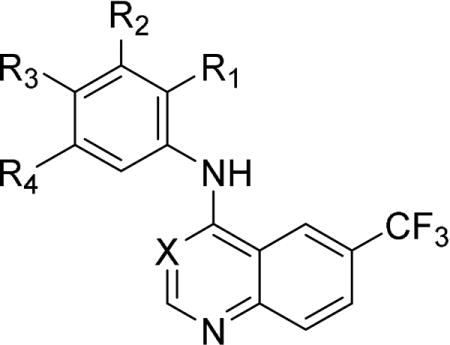
Because 4-anilinoquinolines generally have significantly lower EGFR activity than their corresponding 4-anilinoquinazolines[23], we decided to focus initially on the former series. The 4-anilinoquinoline 1 had ΔTm = 6.3°C in the GAK DSF assay (Table 2). In the GAK binding displacement assay 1 demonstrated Ki = 3.9 nM with only weak activity on other members of the NAK family and a selectivity index, defined as the ratio of GAK Ki and the Ki of nearest NAK family member, over 4000 (Table 2). The first series of analogs explored deletion of one or more of the methoxy substituents. Removal of either the 4-methoxy (4) or 3-methoxy (5) group led to a 10-fold drop in potency against GAK and a slight increase in activity on other NAK family members. Modulating the position of the methoxy groups to 2,4-dimethoxy (6) or 2,5-dimethoxy (7) positions led to a decrease in GAK activity. The effect of substituting the aniline with a single methoxy group were more dramatic. Incorporating a single 4-methoxy group, as in 8, resulted in a 100-fold drop in activity. However, analog 9 with a single 3-methoxy group retained potent GAK activity, demonstrating Ki = 5.7 nM and a NAK family selectivity index >2500. The 2-methoxy substituted 10 was an order of magnitude less potent than the parent compound 1 and maintained moderate NAK family selectivity. Two analogs were prepared in which the alkoxy substituents were bridged by a methylene (11) and an ethylene unit (12). However, both compounds showed a decrease in GAK activity compared to the corresponding 3,4-dimethoxy analog 6.
Table 2.
Small modifications to the hit compound - 1
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | R3 | R4 | GAK DSF ΔTm (°C)a |
Binding displacement assay Ki (μM) | NAK selectivityb | |||
| GAK | AAK1 | BMP2K | STK16 | ||||||||
| 1 | CH | H | OMe | OMe | OMe | 6.3 | 0.0039 | 54 | >100 | 17 | 4300 |
| 4 | CH | H | OMe | H | OMe | 4.3 | 0.054 | 34 | 85 | 52 | 640 |
| 5 | CH | H | OMe | OMe | H | 3.4 | 0.058 | 24 | 44 | 24 | 420 |
| 6 | CH | OMe | H | OMe | H | 1.8 | 0.86 | 71 | >100 | 70 | 81 |
| 7 | CH | OMe | H | H | OMe | 2.6 | 0.14 | 80 | >100 | 58 | 420 |
| 8 | CH | H | H | OMe | H | 1.7 | 0.73 | 56 | 75 | 19 | 26 |
| 9 | CH | H | OMe | H | H | 5.8 | 0.0057 | 14 | 23 | 18 | 2500 |
| 10 | CH | OMe | H | H | H | 3.0 | 0.044 | 4.8 | 11 | 29 | 110 |
| 11 | CH | H | OCH2O | H | 3.2 | 0.29 | 30 | 60 | 25 | 85 | |
| 12 | CH | H | OCH2CH2O | H | 1.5 | 0.88 | 44 | 56 | 13 | 14 | |
| 13 | N | H | OMe | OMe | OMe | 2.4 | 0.037 | 7.8 | 18 | 2.7 | 73 |
| 14 | N | H | OMe | H | OMe | 0.7 | 0.31 | >100 | >100 | 8.0 | 26 |
| 15 | N | H | H | OMe | H | 0.3 | 1.8 | 11 | >100 | 2.7 | 1.5 |
Average of 4 experiments
Selectivity against the nearest NAK family member
Switching to the 4-anilinoquinazoline core, the effect of variation in methoxy substitution was explored in three analogs (Table 2). The rank order activity of the mono-, di-, and trimethoxy 4-anilinoquinazolines 13–15 paralleled that of the corresponding 4-anilinoquinoline analogs 1, 4, and 5, but the quinazolines showed a 10-fold decrease in GAK potency compared with the quinolines. The 3,4,5-trimethoxyanilino analog 13 was the most potent on GAK but showed a greatly reduced NAK family selectivity. We also note that the changes in the substitution of the aniline and 6- and 7-quinazoline position drastically decreased EGFR activity (for example, 13 - EGFR Kd = 6.1 μM (n=2), GAK Kd = 190 nM (n=2) (Figure S12)) relative to quinazoline 3 in the same assay system (Table 1). The lower potency and selectivity of the 4-anilinoquinazoline core led us to focus our efforts on the 4-anilinoquinolines.
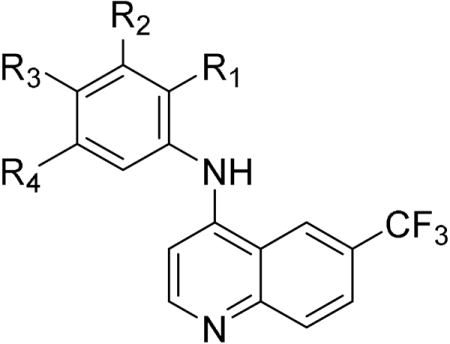
The next series of 4-anilinoquinoline analogs explored the variation in electronic properties and steric bulk of the aniline substituents (Table 3). Replacing the methoxy groups with fluorine yielded several potent compounds. Mono-, di-, and trifluoro analogs 16–20 showed potent activity on GAK but with reduced NAK family selectivity. Analogs with chlorine substitution (21–24) also retained potent GAK activity with the exception of the 2-chloro derivative 24. The bromo- and iodo-substituted analogs 27–30 were less potent. Non-halogen electron withdrawing substituents were generally poorly tolerated: cyano (31–33), trifluromethyl (34), and methylsulphonyl (37–38) analogs showed reduced activity. The bulky 4-tert-butoxy and 4-methylsulphonylmethyl analogs 39 and 40 had weaker activity. The two remaining compounds with potent GAK activity were the 3-acetylene (35) and the 3-acetyl (36), however both showed only modest NAK family selectivity.
Table 3.
Modifications to the aniline segment of compound - 1
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | R4 | GAK DSF ΔTm (°C)am |
Binding displacement assay Ki (μM) | NAK selectivityb | |||
| GAK | AAK1 | BMP2K | STK16 | |||||||
| 16 | H | F | F | F | 3.7 | 0.047 | 9.5 | >100 | >100 | 200 |
| 17 | H | F | F | H | 3.2 | 0.051 | 9.6 | >100 | 22 | 190 |
| 18 | H | H | F | H | 5.6 | 0.036 | 8.7 | 15 | 19 | 240 |
| 19 | H | F | H | H | 4.3 | 0.048 | 8.4 | 14 | 17 | 180 |
| 20 | F | H | H | H | 3.4 | 0.086 | 14 | 26 | 6.1 | 71 |
| 21 | H | F | Cl | H | 4.5 | 0.056 | 20 | 61 | 39 | 360 |
| 22 | H | F | H | Cl | 4.2 | 0.071 | 28 | >100 | 58 | 390 |
| 23 | H | Cl | Cl | H | 5.1 | 0.054 | 33 | >100 | >100 | 390 |
| 24 | H | H | Cl | H | 4.3 | 0.052 | 21 | 64 | 20 | 390 |
| 25 | H | Cl | H | H | 4.3 | 0.050 | 17 | 29 | 26 | 350 |
| 26 | Cl | H | H | H | 2.8 | 0.14 | 13 | 38 | 7.4 | 55 |
| 27 | H | H | Br | H | 3.6 | 0.13 | 48 | >100 | 32 | 250 |
| 28 | H | Br | H | H | 3.8 | 0.096 | 24 | >100 | 33 | 250 |
| 29 | Br | H | H | H | 1.8 | 0.29 | 18 | 40 | 10 | 34 |
| 30 | H | I | H | H | 2.6 | 0.21 | 35 | 34 | 50 | 160 |
| 31 | H | H | CN | H | 1.7 | 0.32 | 1.4 | 7.6 | 8.7 | 4.3 |
| 32 | H | CN | H | H | 1.1 | 0.46 | 19 | 61 | 9.8 | 22 |
| 33 | CN | H | H | H | 0.6 | 0.85 | 17 | 25 | 7.9 | 9.3 |
| 34 | H | CF3 | H | H | 1.3 | 1.2 | >100 | >100 | 52 | 42 |
| 35 | H | C≡C | H | H | 3.7 | 0.087 | 7.1 | 26 | 16 | 82 |
| 36 | H | Ac | H | H | 3.6 | 0.029 | 9.1 | 13 | 4.7 | 160 |
| 37 | H | SO2Me | H | H | 0.3 | 0.97 | 10 | 15 | 1.4 | 1.4 |
| 38 | H | H | SO2Me | H | 0.2 | 2.5 | 14 | 18 | 4.5 | 1.8 |
| 39 | H | H | OtBu | H | 0.3 | 5.3 | 4.8 | 18 | 35 | 0.9 |
| 40 | H | H | CH2SO2CH3 | H | 0.5 | 2.4 | 4.0 | 14 | 6.7 | 1.7 |
Average of 4 experiments
Selectivity relative to the nearest NAK family member
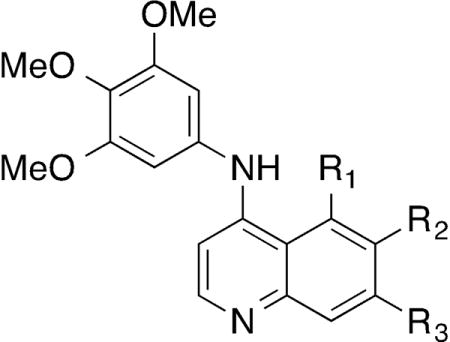
Having explored the steric and electronic requirements of the aniline group, our focus turned to the quinoline core (Table 4). The effect of varying the 6- and 7-substituent on the quinoline core was explored within the 3,4,5-trimethoxyaniline series. Removal of the trifluromethyl group (41) resulted in a 10-fold drop in NAK family potency and a 4-fold decrease in cellular potency relative to 1, although selectivity for GAK was retained. The 6-fluoro (42) and 7-fluoro (43) substituted analogs demonstrated potent GAK activity and >10,000-fold NAK family selectivity but 42 was twice as potent in cells. In contrast, the 5,7-difluoro compound 44 showed a significant drop in GAK activity compared with the 7-fluoro compound 43 and weak cellular potency by nanoBRET. Compounds with 6-tert-butyl (45), 6-cyano (46), and 6-sulfonylmethyl (47) substituents demonstrated potent GAK and NAK family selectivity >1000-fold, but while 45 and 46 were equipotent in the nanoBRET (IC50 approx. 350 nM), 47 showed a 10-fold drop in potency on GAK in cells. While 6-methoxy substitution (48) resulted in a slight decrease in GAK activity, 6,7-dimethoxy substitution (49) yielded a dramatic increase in GAK activity and NAK family selectivity. Compound 49 showed >8°C stabilization of GAK protein in the thermal denaturation assay and Ki = 0.54 nM in the TR-FRET assay with NAK family selectivity >50,000 and with a nanoBRET in cell GAK potency of IC50 = 56 nM. The 14–17-fold cellular potency increase for 49 over the single methoxy substitutions (48 & 50) constituted more than an additive effect. Finally, the 7-substituted analogs 50–52 showed single digit nanomolar GAK potency and NAK family selectivity of approximately 10,000-fold with comparable cellular GAK potency.
Table 4.
Modifications to the quinoline core of compound - 1
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | R1 | R2 | R3 | GAK DSF ΔTm (°C)a |
Binding displacement assay Ki (μM) | NAK selectivityb | NanoBRETc | |||
| GAK | AAK1 | BMP2K | STK16 | GAK IC50 (nM) | ||||||
| 1 | H | CF3 | H | 6.3 | 0.0039 | 54 | >100 | 17 | 4300 | 180 |
| 41 | H | H | H | 4.4 | 0.040 | >100 | >100 | >100 | >2500 | 780 |
| 42 | H | F | H | 5.9 | 0.0057 | 83 | >100 | >100 | 14000 | 270 |
| 43 | H | H | F | 5.8 | 0.0024 | 44 | >100 | >100 | 18000 | 510 |
| 44 | F | H | F | 3.1 | 0.14 | >100 | >100 | >100 | >720 | 4400 |
| 45 | H | tBu | H | 5.2 | 0.0081 | >100 | >100 | 68 | 8400 | 330 |
| 46 | H | CN | H | 4.4 | 0.0015 | 16 | 16 | 2.5 | 11000 | 350 |
| 47 | H | SO2Me | H | 3.3 | 0.018 | 73 | >100 | 61 | 3400 | 3100 |
| 48 | H | OMe | H | 6.0 | 0.013 | >100 | >100 | >100 | >7700 | 280 |
| 49 | H | OMe | OMe | 8.8 | 0.00054 | 28 | 63 | >100 | 51000 | 25 |
| 50 | H | H | OMe | 6.1 | 0.0026 | 58 | >100 | >100 | 22000 | 440 |
| 51 | H | H | CF3 | 5.6 | 0.0062 | 90 | >100 | >100 | 15000 | 520 |
| 52 | H | H | CN | 5.3 | 0.0027 | 67 | >100 | 25 | 9300 | 340 |
Average of 4 experiments
Selectivity relative to the nearest NAK family member
Average of 2 experiments
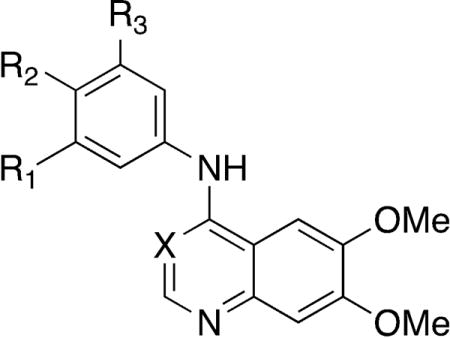
A final series of analogs explored the effect of matching the potent 6,7-dimethoxy core with a range of 4-anilino substituents (Table 5). The 3-methoxy analog 9 retained the potency of the trimethoxy analog 1 in the original series (Table 2). When 3-methoxyaniline was incorporated into the 6,7-dimethoxyquinoline core (53), there was a 4-fold reduction in GAK activity compared to the trimethoxy analog 49 and a reduction of 20-fold in cells. The 3-bromo (54) and 3-acetylene (55) substituted 4-anilino-6,7-dimethoxyquinolines showed similar drops in GAK potency and decreases in NAK family selectivity, but while 55 showed the same drop in cells as 53, 54 showed only a modest decrease of just under 3-fold. The 3-acetyl substituent (56) led to a 100-fold loss of GAK activity relative to 49, and this compound was equipotent across the NAK family. Finally, the 6,7-dimethoxyquinazoline 57, incorporating a 3,4,5-trimethoxyaniline, showed a two-fold loss in potency while maintaining a high selectivity relative to other NAK family members and better cellular potency than 53 with only a 13-fold drop relative to 49.
Table 5.
Small library of potentially optimized analogs of 49
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmpd | X | R1 | R2 | R3 | GAK DSF ΔTm (°C)a |
Binding displacement assay Ki (μM) | NAK selectivityb | NanoBRETc | |||
| GAK | AAK1 | BMP2K | STK16 | GAK IC50 (nM) | |||||||
| 49 | CH | OMe | OMe | OMe | 8.8 | 0.00054 | 28 | 63 | >100 | 51000 | 25 |
| 53 | CH | OMe | H | H | 7.7 | 0.0025 | 8.1 | 22 | >100 | 3300 | 520 |
| 54 | CH | Br | H | H | 5.1 | 0.0094 | 2.5 | >100 | 39 | 270 | 76 |
| 55 | CH | C≡C | H | H | 5.5 | 0.0035 | 0.9 | 2.4 | 19 | 260 | 500 |
| 56 | CH | Ac | H | H | 1.3 | 0.40 | 0.26 | 0.68 | 1.5 | – | – |
| 57 | N | OMe | OMe | OMe | 5.6 | 0.0014 | 7.4 | 19 | 35 | 5300 | 350 |
Average of 4 experiments
Selectivity relative to the nearest NAK family member
Average of 2 experiments
To rationalize the pivotal activity of the methoxy group on the aniline ring, we used molecular modelling to analyse the effect of inhibitor binding to the GAK catalytic site. Analogs 1, 5, 9, and 49 were docked into the active site of the GAK x-ray structure, and a hydration site analysis was performed with WaterMap using multiple orientations and conformations of each compound (Figure 3)[9a, 24]. These calculations identified a network of water molecules that could occupy the active site. Docking of the 4-aminoquinolines into the site resulted in potential displacement of one specific water molecule that could contribute up to 6.4 kcal/mol in binding energy (Figure 3). The trimethoxy substituted anilines (49 and 1) were predicted to displace this water molecule most effectively (Figure 3A/3B and 3C respectively), whereas other methoxy substitution patterns such as the 3,4-dimethoxy (5) were only partially able to displace the water molecule (Figure 3D/3E). The water site analysis also explained why the meta-methoxy analog 9 was more potent than the ortho- and para-analogs 8 and 10. 9 could fully displace the high-energy water molecule (Figure 3F), whereas 8 and 10 were not able to do so.
Figure 3.
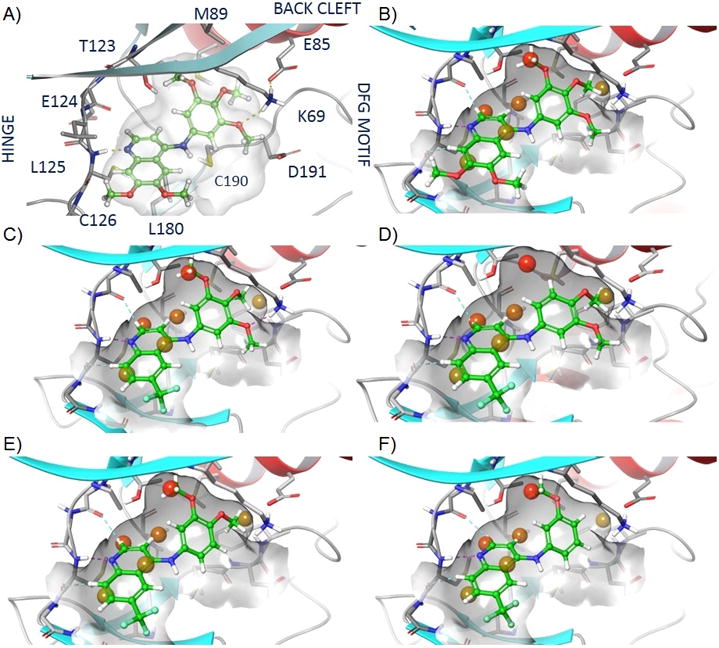
Active site WaterMap of compounds in GAK: A - Visualised GAK active site interaction of 49, B - WaterMap of 49, C - WaterMap of 1, D/E - WaterMap of 5 favoured (D) and unfavoured (E), F - WaterMap of 9
We measured GAK affinity of the 4-anilinoquinoline analogs with a wide range of ΔTm values and affinity over several orders of magnitude in the TR-FRET binding assay. The two binding assays were highly correlated, as shown by a plot of ΔTm versus −log Ki which has R = 0.86 (Figure 4).
Figure 4.
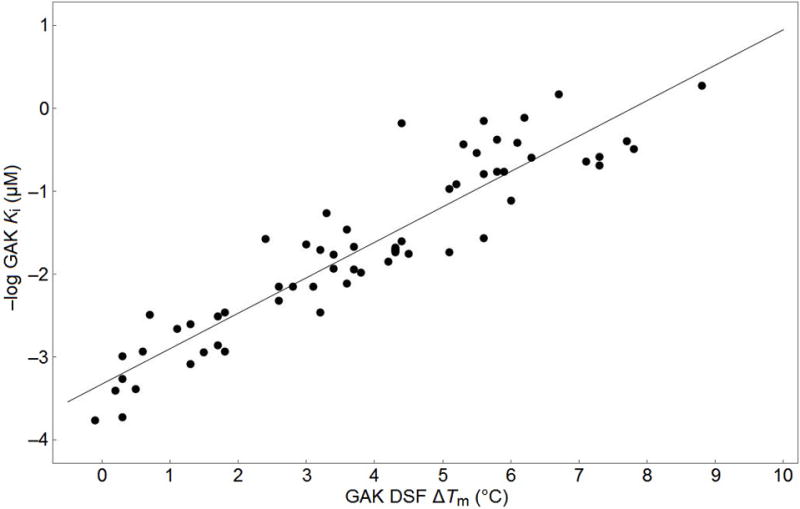
Correlation of GAK DSF ∆Tm vs-Log GAK Ki
2.4 Kinome selectivity evaluation
Having obtained 49, a compound with sub-nanomolar GAK potency and favourable NAK family selectivity, we next evaluated its selectivity in the context of the broader kinome present in cell lysate using multiplexed inhibitor beads coupled with mass spectrometry (MIB/MS)[25]. This methodology involves quantitation of kinases bound to immobilized inhibitors in the presence and absence of soluble inhibitors and allows for assessment of inhibitor binding across a large percentage of the expressed, functional kinome in a single MS run. GAK was the only kinase depleted by 1 in a dose dependent manner, suggesting a very narrow specificity for the compound at the concentrations tested (Figure 5A). 49 was used at lower concentrations (300 pM – 10 nM) in this assay and showed considerably greater GAK potency, depleting it by almost 10-fold at the highest concentration (Figure 5B). All four of the NAK family kinases (AAK1, STK16, BMP2K, and GAK) were profiled by this analysis, and both 1 and 49 showed exquisite selectivity for GAK. Among the >240 protein kinases profiled, these results indicate narrow selectivity for both GAK and RIPK2 of 1 and 49 (Figure 5A–D).
Figure 5.
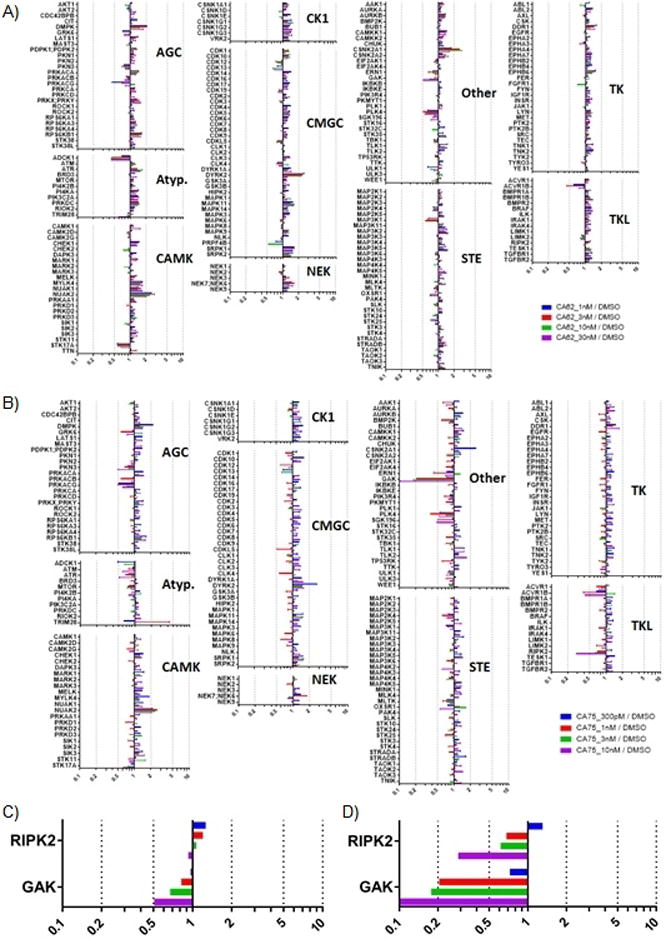
A - Compound 1 is highly selective for GAK. SUM159 cell lysates were incubated with DMSO or the indicated concentration of 1 for 30 minutes on ice. Kinases were then affinity purified using multiplexed inhibitor beads (MIBs) and analyzed by mass spectrometry. Kinase abundance was quantified label free using MaxQuant software. Bars represent the ratio of label free quantification values for the indicated kinase in lysate treated with drug over DMSO control lysates. A dose dependent decrease in kinase ratio indicates 1 binding. B - Compound 49 is highly selective for GAK, under the same conditions as 1. C - GAK and RIPK2 enrichment from experiment with 1. D - GAK and RIPK2 enrichment from experiment with 49.
3. Discussion
Chemical tractability is a hallmark of the protein kinases. Over 30 drugs have been approved that target the ATP binding site, predominantly in the field of oncology. However, most of these drugs were developed as multi-kinase inhibitors that leverage the conservation of the ATP pocket across the enzymes to increase their clinical efficacy. Development of kinase inhibitors for treatment of chronic diseases, such as inflammation and neurodegeneration, will require drugs with greatly improved selectivity profiles[26]. New chemical approaches and molecular insights into the development of highly selective kinase inhibitors that target the conserved ATP-binding site are urgently needed. Binding assays are the most accurate and robust method to measure potency and selectivity of ATP-competitive kinase inhibitors[27]. Binding assays available at commercial vendors are routinely used to profile kinase inhibitors for their selectivity across the human kinome[28]. Moreover, ligand binding displacement assays provide an accepted for direct measurement of kinase inhibition in drug optimization of ATP-binding site inhibitors[29]. This point is particularly relevant for neglected kinases such as GAK where there are currently no robust and validated enzyme activity assays. Here, we have uncovered a series of 4-anilinoquinolines that show high affinity for GAK as measured in two orthogonal biochemical assays (DSF and TR-FRET), which were highly correlated (Figure 4). Some of these compounds demonstrated remarkable binding selectivity for GAK over hundreds of other kinases and even for other members of the closely related NAK sub-family. Using medicinal chemistry and molecular modelling, we have delineated the structural features of these molecules that contribute to the binding selectivity within the NAK sub-family.
Common structural elements of kinase ligand interactions have been systematically described in the KLIFS database[30]. Briefly, kinase domains have C- and N-terminal domains bridged by a hinge region. ATP-competitive inhibitors generally make one to three hydrogen bonding interactions with the backbone residues in the hinge. The ATP binding site can be divided into a front cleft projecting towards solvent and a back cleft that has some degree of conformational plasticity, due in part to the disposition of a conserved loop containing an Asp-Phe-Gly (“DFG”) motif. The ease of access to the back cleft is largely determined by the size of the amino acid directly preceding the hinge, which is known as the “gatekeeper” residue.
Compounds containing quinoline and quinazoline ring systems have been the focus of prior kinase drug discovery campaigns, which in some cases have led to approved medicines[31]. Structural studies using x-ray crystallography have been reported for several quinoline and quinazoline-based kinase inhibitors in complex with their target kinases[32]. The dominant binding mode of these quinolines and quinazolines shows a hydrogen bond in the hinge region between N1 of the ligand and the third residue from the gatekeeper (GK + 3: Cys in GAK, BMP2K, and AAK1 and Phe in STK16) (Figure 3A). The aniline group at the 4-position of the quinazoline or quinoline ring system projects into the back-cleft pocket of GAK that is partially defined by the αC helix and the DFG motif. Finally, substituents at the 6- and 7-position of the quinoline are directed toward the surface of the protein. Changes in the aniline head group and on the quinoline core are known to affect kinase potency and selectivity profiles in other, structurally-related inhibitor series[33], as these parts of the ligand project into a pocket lined with varied functionality or having a range of conformational plasticity[30b].
We observed that GAK potency and NAK family selectivity was affected by a) the hinge interacting moiety (quinoline vs. quinazoline), b) the pendant aniline, and c) substituents at the quinoline 6- and 7-positions. The quinoline 1 was an order of magnitude more potent on GAK than the quinazoline 13. The hydrogen that projects from C3 of the quinoline restricts the torsion angle defined with the aniline to a much greater degree than the quinazoline N3, from near planar for quinazoline and up to 60° for quinoline. Quinoline 1 may be more conformationally restricted than 13, a proposition supported by gas phase OPLS3 torsional scanning (Figure S1) and Density Function Theory (DFT) optimised (B3LYP/6-31G**) force field calculations (Table S1). These results are consistent with small molecule x-ray structures determined for 1, 9, 13, 17, 37, 48, and 49 (Figure S2 and Tables S2 and S3). Thus, the restricted conformation of the quinoline relative to the quinazoline may favourably pre-organize 1 for binding to GAK (Figure 6).
Figure 6.
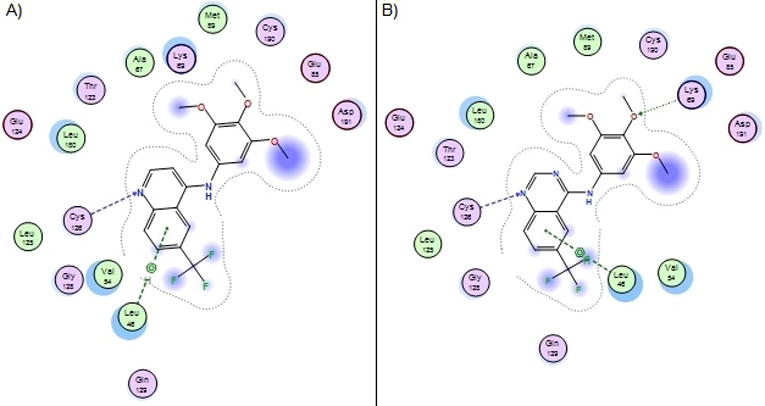
Comparison of GAK binding of compound 1 (A) and 13 (B). The quinoline scaffold is a tighter binder then the quinazoline despite the possibility for an additional hinge binding interaction.
WaterMap analysis of the binding site suggested that a coordinated water network in the protein pocket spans the region proximal the aniline and the 6-position of the ring system. We propose that compounds able to displace a poorly coordinated water molecule in this system will have increased potency due to entropic and enthalpic contributions to the free energy of binding (Figures 3 and S3). Compounds 1 and 16 are calculated to have their methoxy or fluoro substituents directly occupying the same space as the high-energy water molecule. On the other hand, 6 and 20 were comparatively weaker binders and are predicted to be unable to displace the water molecule. These models suggest that a water network within the GAK active site plays a critical role in defining the relative affinity of quinoline ligands. Extension of this model to other NAK family members or more distantly related kinases may lead to computational models to predict selectivity.
Variation of the substitution at the 6- and 7-positions was observed to have a large effect on GAK affinity. Comparing compounds in the 3,4,5-trimethoxyaniline series, the compound with the weakest affinity was the unsubstituted quinoline 41. The 6- and 7-positions are oriented towards solvent and may provide an opportunity to improve molecular properties. Indeed, the approved medicines gefitinib and erlotinib both incorporate water solubilizing groups at the 6- and 7-positions of their quinazoline cores (Figures S4–S7).
As a result of screening the four NAK family kinases in parallel, we also identified quinolines with interesting dual activity profiles (Figure S8). Most analogs had GAK affinity that was >100-fold higher than for the other NAK family kinases. However, there were some exceptions. 39, which has a 4-tert-butoxy aniline substituent, had affinity for all four NAK family kinases within a range of 8-fold. Compound 37 with a 3-SO2Me aniline substituent had a notable potency boost on STK16, to the extent that its affinity was within 2-fold of GAK. These bulky substituents are likely to bind into the back cleft of the active site. In addition, it was also observed that a number of compounds including 36 and 56 have pan-NAK activity (Figure S9). Further exploration of this pocket may yield quinolines with selectivity for other members of the NAK family.
The measurement of binding affinity across the NAK family kinases in parallel allowed us to explore the determinants of selectivity within four close phylogenetic relatives. Inhibitors of the NAK family kinases have a high likelihood of showing low selectivity across the family based on high identity of their primary amino acid sequence and the presence of structural features which are distinct from those observed in other kinases. Specifically, the four NAK family kinases have a large α-helical insert positioned C-terminal to the activation segment, in proximity to the ATP binding site[34]. Notably, GAK does have one fewer amino acid residue in the hinge region, which distinguishes it from the other NAK family kinases. Prior results with 144 clinically-used kinase inhibitors revealed few compounds selective for GAK across the NAK family. It is therefore remarkable that several of the 4-aminoquinolines bound to GAK with over 10,000-fold higher affinity compared to the other NAK family kinases.
In summary, the 4-anilinoquinoline 49 was identified as a narrow spectrum GAK inhibitor with low nanomolar potency in cells. 49 has remarkable selectivity within the NAK sub-family, over 50,000-fold as assessed by a ligand binding displacement assay. In a broader evaluation of selectivity, 49 showed binding to only GAK and RIPK2 in an unbiased MIB/MS experiment in cell lysate. Our results demonstrate that quinoline-based ATP-competitive kinase inhibitors can be designed with exquisite selectivity. More importantly, the 4-anilinoquinolines series exemplified by 1 and 49 has the potential to yield high quality chemical probes for use in the elucidation of GAK function in cells and perhaps in vivo.
4. Experimental
4.1 Chemistry
4.1.1 General
All reactions were performed using flame-dried round-bottomed flasks or reaction vessels unless otherwise stated. Where appropriate, reactions were carried out under an inert atmosphere of nitrogen with dry solvents, unless otherwise stated. Dry acetonitrile (MeCN) was obtained directly from manufacturer and used without further purification. Yields refer to chromatographically and spectroscopically pure isolated yields. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Reactions were monitored by thin-layer chromatography carried out on 0.25 mm E. Merck silica gel plates (60F-254) using ultraviolet light as visualizing agent. NMR spectra were recorded on a Varian Inova 400 or Inova 500 spectrometer and were calibrated using residual undeuterated solvent as an internal reference (CDCl3: 1H NMR = 7.26, 13C NMR = 77.16). The following abbreviations or combinations thereof were used to explain the multiplicities observed: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad. Liquid Chromatography (LC) and High Resolution Mass Spectra (HRMS) were recorded on a ThermoFisher hybrid LTQ FT (ICR 7T). The University of Southampton (Southampton, UK) small molecule x-ray facility collected and analysed all X-ray diffraction data.
4.1.2 General procedure for the synthesis of 4-anilinoquin(azo)olines
4.1.2.1 Method A
Quinoline (200 mg, 0.86 mmol) and aniline (0.95 mmol) were suspended in ethanol (10 mL) and refluxed at 90°C for 18 hours. The product was collected by filtration and washed with water (2 × 10 mL) and ether (3 × 10 mL) to yield the product as a free following solid.
4.1.2.2 Method B
Quinoline/Quinazoline (200 mg, 0.86 mmol) and aniline (0.95 mmol) were suspended in ethanol (10 mL) and refluxed at 90°C for 18 hours. The crude mixture was purified by flash chromatography ethyl acetate:hexane followed by 1–5% methanol:ethyl acetate and solvent removed under reduced pressure to yield the product as a free following solid.
4.1.2.3 Method C
Quinazoline (200 mg, 0.86 mmol) and aniline (0.95 mmol) were suspended in n-butanol (10 mL) and refluxed at 120°C for 18 hours. The crude mixture was purified by flash chromatography 20–100% ethyl acetate:hexane followed by 1–5% methanol:ethyl acetate and solvent removed under reduced pressure to yield the product as a free following solid.
4.1.2.4 Method D
Quinazoline (200 mg, 0.86 mmol) and aniline (0.95 mmol) palladium acetate (40 mg, 0.17 mmol), tri(o-tolyl)phosphine (55 mg, 0.18 mmol) and caesium carbonate (380 mg, 2.0 mmol) were all suspended in acetonitrile 10 mL and degassed for 5 minutes. The mixture was refluxed at 85°C for 18 hours. The crude mixture was passed through a plug of diatomaceous earth 545 before been purified by flash chromatography 20–100% ethyl acetate:hexane followed by 1–5% methanol:ethyl acetate and solvent removed under reduced pressure to yield the product as a free following solid.
4.1.3 Compound characterization
4.1.3.1 6-(Trifluoromethyl)-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (1)
1 was prepared by method B to afford a mustard solid (219 mg, 67%); m.p. 128–130°C; 1H NMR (400 MHz, DMSO-d6) δ 11.23 (s, 1H), 9.30 (s, 1H), 8.57 (d, J = 7.0 Hz, 1H), 8.29 (Br s, 2H), 6.96 (d, J = 7.0 Hz, 1H), 6.83 (s, 2H), 3.81 (s, 6H), 3.73 (s, 3H): 13C NMR (101 MHz, DMSO-d6) δ 155.3, 153.7 (s, 2C), 144.6, 140.7, 136.7, 132.6, 129.1, 126.6 (d, J = 32.9 Hz, CF3), 125.2, 122.5 (d, J = 5.1 Hz, 2C), 116.6, 103.1 (s, 2C), 101.5, 60.2, 56.2 (s, 2C); δ HRMS-ESI (m/z): [M+H]+ calcd for C19H17F3N2O3 - 379.1270; found 379.1261; LC - Tr = 3.86 min, purity > 95%.
4.1.3.2 N-(3,5-Dimethoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (4)
4 was prepared by method B to afford a tan solid (47 mg, 16%); m.p. >200°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 8.99 (s, 1H), 8.58 (d, J = 5.6 Hz, 1H), 8.09 (d, J = 8.8 Hz, 1H), 7.98 (dd, J = 8.9, 1.7 Hz, 1H), 7.10 (d, J = 5.6 Hz, 1H), 6.58 (d, J = 2.2 Hz, 2H), 6.37 (t, J = 2.2 Hz, 1H), 3.76 (s, 6H); 13C NMR (100MHz, DMSO-d6) 161.1, 153.7, 149.1, 141.5, 139.5, 135.6, 133.5, 128.3, 125.3, 124.8, 124.5, 121.4, 118.6, 103.1 (s, 2C), 101.0, 55.3 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H15F3N2O2 - 349.1164; found 349.1149; LC - Tr = 4.22 min, purity > 95%.
4.1.3.3 N-(3,4-Dimethoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (5)
5 was prepared by method B to afford a yellow solid (165 mg, 55%); m.p. 235–240°C; 1H NMR (400 MHz, DMSO-d6) δ 11.25 (s, 1H), 9.32 (s, 1H), 8.54 (d, J = 6.9 Hz, 1H), 8.28 (s, 2H), 7.14 (d, J = 8.5 Hz, 1H), 7.09 (s, 1H), 7.02 (d, J = 8.3 Hz, 1H), 6.81 (d, J = 6.9 Hz, 1H), 3.80 (d, J = 18.3 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 155.6, 149.6, 148.2, 144.3, 140.6, 129.5, 129.1 (d, J = 3.4 Hz), 126.6 (q, J = 32.7 Hz, CF3), 125.2, 122.6 – 122.1 (m, 2C), 117.6, 116.5, 112.4, 109.6, 101.0, 55.8 (s, 2C). HRMS-ESI (m/z): [M+H]+ calcd for C18H15F3N2O2 - 349.1164; found 349.1153; Tr = 3.85 min, purity > 95%.
4.1.3.4 N-(2,4-Dimethoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (6)
6 was prepared by method B to afford a mustard solid (202 mg, 67%); m.p. 93–95°C; 1H NMR (400 MHz, DMSO-d6) δ 9.30 (s, 1H), 8.48 (d, J = 6.6 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.17 (dd, J = 8.9, 1.4 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 6.80 (d, J = 2.5 Hz, 1H), 6.67 (dd, J = 8.6, 2.6 Hz, 1H), 6.31 (d, J = 6.6 Hz, 1H), 3.83 (s, 3H), 3.77 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.2, 155.4, 155.0, 145.7, 142.5, 128.8, 128.0 (d, J = 2.9 Hz), 126.1 (q, J = 32.7 Hz, CF3), 124.0, 122.6, 122.3 (d, J = 4.2 Hz), 118.0, 116.6, 105.6, 101.4, 99.8, 55.83, 55.61; HRMS-ESI (m/z): [M+H]+ calcd for C18H15F3N2O2 - 349.1164; found 349.1152; Tr = 4.14 min, purity > 95%.
4.1.3.5 N-(2,5-Dimethoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (7)
7 was prepared by method B to afford a mustard solid (204 mg, 68%); m.p. 116–118°C; 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H), 7.72 (d, J = 6.7 Hz, 1H), 7.42 – 7.36 (m, 2H), 6.44 (d, J = 8.7 Hz, 1H), 6.31 – 6.25 (m, 2H), 5.80 (d, J = 6.7 Hz, 1H), 3.04 (d, J = 4.1 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 155.4 (d, J = 96.8 Hz), 149.5, 146.6, 143.4, 129.1 (d, J = 3.1 Hz), 128.3 (q, J = 33.0 Hz, CF3), 126.7, 126.2, 124.7, 122.6 (d, J = 4.3 Hz), 117.9, 114.9, 114.5, 114.2, 103.0, 56.5, 56.2; HRMS-ESI (m/z): [M+H]+ calcd for C18H15F3N2O2 - 349.1164; found 349.1152; Tr = 4.05 min, purity > 95%.
4.1.3.6 N-(4-Methoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (8)
8 was prepared by method B to afford a green solid (190 mg, 68%); m.p. >220°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 11.27 (s, 1H), 9.33 (d, J = 1.7 Hz, 1H), 8.54 (d, J = 7.0 Hz, 1H), 8.28 (d, J = 1.1 Hz, 2H), 7.41 (d, J = 8.9 Hz, 2H), 7.14 (d, J = 8.9 Hz, 2H), 6.72 (d, J = 7.0 Hz, 1H), 3.83 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 158.5, 155.6, 144.3, 140.6, 129.4, 129.1 (d, J = 3.4 Hz), 127.0 (s, 2C), 126.5 (q, J = 33.0 Hz, CF3), 125.2, 122.4, 122.3, 116.6, 115.2 (s, 2C), 100.7, 55.5; HRMS-ESI (m/z): [M+H]+ calcd for C17H13F3N2O - 319.1058; found 319.1049; Tr = 4.04 min, purity > 95%.
4.1.3.7 N-(3-Methoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (9)
9 was prepared by method B to afford a light yellow solid (258 mg, 94%); m.p. 135–137°C; 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.34 (s, 1H), 8.58 (d, J = 6.8 Hz, 1H), 8.28 (q, J = 8.9 Hz, 2H), 7.48 (t, J = 8.3 Hz, 1H), 7.07 (s, 2H), 6.96 (dd, J = 26.3, 7.3 Hz, 2H), 3.81 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.6, 154.8, 145.4, 141.6, 138.6, 131.0, 129.1 (d, J = 3.4 Hz), 126.7 (q, J = 32.8 Hz, CF3), 125.4, 123.2, 122.7 (d, J = 3.9 Hz), 117.2, 117.2, 113.2, 110.9, 101.6, 55.6; HRMS-ESI (m/z): [M+H]+ calcd for C17H13F3N2O - 319.1058; found 319.1048; Tr = 4.08 min, purity >95%.
4.1.3.8 N-(2-Methoxyphenyl)-6-(trifluoromethyl)quinolin-4-amine (10)
10 was prepared by method B to afford a dark green solid (104 mg, 38%); m.p. 85–87°C; 1H NMR (400 MHz, DMSO-d6) δ 9.35 (s, 1H), 8.52 (d, J = 6.7 Hz, 1H), 8.29 (d, J = 8.9 Hz, 1H), 8.22 (dd, J = 8.9, 1.7 Hz, 1H), 7.46 (ddd, J = 8.3, 7.5, 1.7 Hz, 1H), 7.41 (dd, J = 7.8, 1.6 Hz, 1H), 7.28 (dd, J = 8.4, 1.1 Hz, 1H), 7.12 (td, J = 7.6, 1.2 Hz, 1H), 6.36 (d, J = 6.7 Hz, 1H), 3.79 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.6, 154.2, 142.0, 129.4, 128.3 (d, J = 2.9 Hz), 127.9, 126.3 (d, J = 32.7 Hz, CF3), 125.3, 125.1, 123.6, 122.6, 122.4 (d, J = 4.2 Hz), 121.2, 116.6, 112.9, 101.7, 55.7; HRMS-ESI (m/z): [M+H]+ calcd for C17H13F3N2O - 319.1058; found 319.1049; Tr = 4.00 min, purity > 95%.
4.1.3.9 N-(Benzo[d][1,3]dioxol-5-yl)-6-(trifluoromethyl)quinolin-4-amine (11)
11 was prepared by method B to afford a dark green solid (126 mg, 44%); m.p. 177–179°C; 1H NMR (400 MHz, DMSO-d6) δ 11.24 (s, 1H), 9.32 (s, 1H), 8.56 (d, J = 6.9 Hz, 1H), 8.28 (s, 2H), 7.14 – 7.05 (m, 2H), 6.95 (dd, J = 8.2, 2.0 Hz, 1H), 6.78 (d, J = 6.9 Hz, 1H), 6.14 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 155.6, 148.3, 146.7, 144.4, 140.7, 130.6, 129.0 (d, J = 2.7 Hz), 126.5 (q, J = 32.8 Hz, CF3), 125.2, 122.5 (d, J = 3.2 Hz, 2C), 119.2, 116.6, 109.0, 106.8, 101.9, 101.1; HRMS-ESI (m/z): [M+H]+ calcd for C17H11F3N2O2 - 333.0851; found 333.0840; Tr = 3.90 min, purity > 95%.
4.1.3.10 N-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-6-(trifluoromethyl)quinolin-4-amine (12)
12 was prepared by method B to afford a mustard solid (185 mg, 62%); m.p. 225–227°C; 1H NMR (400 MHz, DMSO-d6) δ 11.33 (s, 1H), 9.36 (s, 1H), 8.55 (d, J = 7.0 Hz, 1H), 8.33 – 8.24 (m, 2H), 7.05 (d, J = 8.5 Hz, 1H), 7.01 (d, J = 2.4 Hz, 1H), 6.94 (dd, J = 8.5, 2.5 Hz, 1H), 6.78 (d, J = 7.0 Hz, 1H), 4.31 (s, 4H); 13C NMR (101 MHz, DMSO-d6) δ 155.6, 144.1, 144.0, 142.9, 129.9, 129.1 (d, J = 2.8 Hz), 127.9, 126.6 (q, J = 32.8 Hz, CF3), 122.6 (d, J = 4.5 Hz), 122.4, 122.2, 118.5, 118.1, 116.6, 114.4, 100.9, 64.1 (s, 2C). HRMS-ESI (m/z): [M+H]+ calcd for C18H13F3N2O2 - 347.1007; found 347.0997; Tr = 4.05 min, purity > 95%.
4.1.3.11 6-(Trifluoromethyl)-N-(3,4,5-trimethoxyphenyl)quinazolin-4-amine (13)
13 was prepared by method B to afford a yellow solid (137 mg, 42%); m.p. 225–227°C; 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 9.26 (s, 1H), 8.94 (s, 1H), 8.33 (dd, J = 8.8, 1.6 Hz, 1H), 8.07 (d, J = 8.7 Hz, 1H), 7.14 (s, 2H), 3.81 (s, 6H). 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.7, 152.7 (s, 2C), 135.9, 134.5, 132.6, 131.0, 130.9, 129.4, 127.8, 126.5, 122.9, 122.3, 113.8, 102.3 (s, 2C), 60.2, 56.1 (s, 2C). HRMS-ESI (m/z): [M+H]+ calcd for C18H13F3N2O2 - 380.1222; found 380.1210; Tr = 4.38 min, purity > 95%.
4.1.3.12 N-(3,5-Dimethoxyphenyl)-6-(trifluoromethyl)quinazolin-4-amine (14)
14 was prepared by method D to afford a light yellow solid (114 mg, 38%); m.p. 250–255°C; 1H NMR (400 MHz, DMSO-d6) δ 11.51 (s, 1H), 9.32 (s, 1H), 8.98 (s, 1H), 8.34 (d, J = 10.3 Hz, 1H), 8.11 (d, J = 8.7 Hz, 1H), 7.01 (s, 2H), 6.50 (t, J = 2.2 Hz, 1H), 3.79 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 160.9 (s, 2C), 160.0, 153.9, 138.8-131.5 (m, 1C, CF3), 128.4, 128.0, 125.4, 123.5-123.4 (m, 1C), 122.7, 120.5, 114.2, 103.2 (s, 2C), 98.6, 55.9 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H14F3N3O2 - 350.1116; found 350.1110; Tr = 4.84 min, purity > 95%.
4.1.3.13 N-(4-Methoxyphenyl)-6-(trifluoromethyl)quinazolin-4-amine (15)
15 was prepared by method D to afford a light yellow solid (121 mg, 44%); m.p. 255–260°C; 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 9.37 (s, 1H), 8.93 (s, 1H), 8.36 (dd, J = 8.8, 1.6 Hz, 1H), 8.12 (d, J = 8.7 Hz, 1H), 7.77 – 7.51 (m, 2H), 7.19 – 6.95 (m, 2H), 3.81 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 159.4, 157.9, 155.8, 152.8, 131.4, 129.2 (d, J = 3.7 Hz), 128.1, 126.1 (s, 2C), 123.3-123.1 (m, 1C), 122.2, 121.9, 114.0, 113.6 (s, 2C), 55.4; HRMS-ESI (m/z): [M+H]+ calcd for C16H12F3N3O - 320.1011; found 320.1004; Tr = 4.08 min, purity > 95%.
4.1.3.14 6-(Trifluoromethyl)-N-(3,4,5-trifluorophenyl)quinolin-4-amine (16)
16 was prepared by method B to afford a tan solid (142 mg, 48%); m.p. 210–212°C; 1H NMR (400 MHz, DMSO-d6) δ 9.32 (s, 1H), 8.65 (d, J = 5.2 Hz, 1H), 8.24 (dd, J = 39.6, 8.5 Hz, 2H), 7.70 – 7.37 (m, 2H), 7.09 (d, J = 5.6 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 153.0, 151.9 (dd, J = 10.3, 4.7 Hz), 149.4 (dd, J = 9.8, 5.5 Hz), 147.0, 143.1, 138.2 (t, J = 14.7 Hz), 135.7 (t, J = 15.5 Hz), 134.7 (td, J = 10.8, 3.5 Hz), 128.1 – 127.9 (m, 1C), 126.4 (q, J = 32.7 Hz, CF3), 125.2, 124.6, 122.5, 117.6, 109.5 (d, J = 23.1 Hz), 102.5; HRMS-ESI (m/z): [M+H]+ calcd for C16H8N2F6 - 343.0670; found 343.0653; Full Scan at Tr = 4.24 min, purity > 95%.
4.1.3.15 N-(3,5-Difluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (17)
17 was prepared by method B to afford an off white solid (169 mg, 64%); m.p. 240–245°C; 1H NMR (400 MHz, DMSO-d6) δ 11.64 (s, 1H), 9.42 (s, 1H), 8.69 (d, J = 7.0 Hz, 1H), 8.37 (d, J = 9.0 Hz, 1H), 8.33 (dd, J = 9.0, 1.6 Hz, 1H), 7.52 – 7.22 (m, 3H), 7.15 (d, J = 7.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 164.1 (d, J = 15.0 Hz), 161.7 (d, J = 15.1 Hz), 155.1, 144.7, 140.2, 139.8 (t, J = 13.1 Hz), 130.3 – 128.7 (m, 1C), 127.0 (q, J = 32.9 Hz), 125.0, 122.8 (q, J = 3.7 Hz), 122.3, 122.2, 117.0, 109.6 – 107.9 (m, 1C), 103.2, 103.0 (t, J = 26.0 Hz); HRMS-ESI (m/z): [M+H]+ calcd for C16H9N2F5 - 325.0764; 325.07487; Tr = 4.07 min, purity > 95%.
4.1.3.16 N-(4-Fluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (18)
18 was prepared by method B to afford an off white solid (169 mg, 64%); m.p. 226–228°C; 1H NMR (400 MHz, DMSO-d6) δ 11.57 (s, 1H), 9.48 (s, 1H), 8.57 (d, J = 6.9 Hz, 1H), 8.36 (d, J = 8.9 Hz, 1H), 8.28 (dd, J = 8.9, 1.4 Hz, 1H), 7.68 – 7.48 (m, 2H), 7.48 – 7.31 (m, 2H), 6.78 (d, J = 6.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 162.0, 159.6, 155.4, 144.5, 140.8, 133.4, 129.0 (d, J = 3.1 Hz), 127.8 (d, J = 8.7 Hz), 126.6 (q, J = 32.8 Hz), 125.2, 122.9 (d, J = 4.2 Hz), 122.4, 116.8 (d, J = 8.7 Hz), 116.8, 100.9; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F4 - 307.0858; found 307.0844; Tr = 3.86 min, purity > 95%.
4.1.3.17 N-(3-Fluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (19)
19 was prepared by method B to afford a light yellow solid (204 mg, 77%); m.p. 193–195°C; 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.37 (s, 1H), 8.62 (d, J = 6.6 Hz, 1H), 8.32 (d, J = 8.8 Hz, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.59 (q, J = 7.9 Hz, 1H), 7.38 (dd, J = 15.9, 9.0 Hz, 2H), 7.23 (td, J = 8.5, 2.0 Hz, 1H), 7.01 (d, J = 6.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 163.8, 161.3, 153.9, 145.9, 142.1, 139.4 (d, J = 10.3 Hz), 131.5 (d, J = 9.4 Hz), 128.5 (d, J = 3.0 Hz), 126.5 (q, J = 32.7 Hz, CF3), 123.6, 122.9 – 122.0 (m, 1C), 120.7 (d, J = 2.8 Hz), 117.3, 113.6 (d, J = 20.9 Hz), 111.8 (d, J = 23.8 Hz), 101.8; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F4 307.0858; found 307.0844; Tr = 3.95 min, purity > 95%.
4.1.3.18 N-(2-Fluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (20)
20 was prepared by method B to afford a light yellow solid (135 mg, 51%); m.p. 108–110°C; 1H NMR (400 MHz, DMSO-d6) δ 9.37 (s, 1H), 8.61 (d, J = 6.4 Hz, 1H), 8.35 – 8.16 (m, 2H), 7.58 (t, J = 7.8 Hz, 1H), 7.54 – 7.44 (m, 2H), 7.39 (dt, J = 8.2, 4.7 Hz, 1H), 6.54 (dd, J = 6.3, 2.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 157.9, 155.4, 154.0, 146.5, 142.7, 129.4 (d, J = 7.8 Hz), 128.5, 128.1 – 128.1 (m, 1C), 126.4 (q, J = 32.8 Hz), 125.7 (d, J = 3.6 Hz), 125.3, 125.0 (d, J = 12.1 Hz), 124.3, 122.6 – 122.4 (m, 1C), 117.2 – 116.6 (m, 1C), 101.7; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F4 - 307.0858; found 307.0844; Tr = 3.81 min, purity > 95%.
4.1.3.19 N-(4-Chloro-3-fluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (21)
21 was prepared by method B to afford a bright yellow solid (200 mg, 68%); m.p. 112–114°C; 1H NMR (400 MHz, DMSO-d6) δ 1 6 H NMR (400 MHz, DMSO-d6) δ 9.26 (s, 1H), 8.63 (d, J = 5.8 Hz, 1H), 8.19 (dd, J = 44.3, 8.6 Hz, 2H), 7.69 (t, J = 8.4 Hz, 1H), 7.64 – 7.53 (m, 1H), 7.37 (d, J = 8.2 Hz, 1H), 7.10 (d, J = 5.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 158.6, 156.2, 151.7, 148.2 (d, J = 1.6 Hz), 144.8 (d, J = 1.6 Hz), 139.5 (d, J = 9.3 Hz), 131.3, 127.3, 125.1 (q, J = 33.3 Hz, CF3), 125.9, 122.3 (q, J = 3.7 Hz), 120.7 (d, J = 2.9 Hz), 118.0, 115.7 (d, J = 17.6 Hz), 112.0 (d, J = 23.4 Hz), 102.7; HRMS-ESI (m/z): [M+H]+ calcd for C16H9N2F4Cl - 341.0469; found 341.0454; Tr = 4.32 min, purity > 95%.
4.1.3.20 N-(3-Chloro-5-fluorophenyl)-6-(trifluoromethyl)quinolin-4-amine (22)
22 was prepared by method B to afford a tan solid (185 mg, 63%); m.p. 162–164°C; 1H NMR (400 MHz, DMSO-d6) δ 9.19 (s, 1H), 8.66 (s, 1H), 8.18 (dd, J = 34.8, 7.1 Hz, 2H), 7.41 (s, 1H), 7.33 (dd, J = 22.7, 8.5 Hz, 2H), 7.15 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 163.8, 161.3, 151.7, 148.0, 144.2, 141.5 (d, J = 11.8 Hz), 134.7 (d, J = 12.9 Hz), 127.5, 126.1 (q, J = 32.4 Hz), 125.5, 122.1 (d, J = 3.9 Hz), 119.4 - 119.3 (m, 1C), 118.0, 112.9 (d, J = 25.2 Hz), 109.3 (d, J = 24.1 Hz), 103.1; HRMS-ESI (m/z): [M+H]+ calcd for C16H9N2F4Cl - 341.0469; found 341.0458; Tr = 4.40 min, purity > 95%.
4.1.3.21 N-(3,4-Dichlorophenyl)-6-(trifluoromethyl)quinolin-4-amine (23)
23 was prepared by method B to afford a tan solid (207 mg, 67%); m.p. 145–147°C; 1H NMR (400 MHz, DMSO-d6) δ 1H NMR (400 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.71 – 8.51 (m, 1H), 8.15 (dd, J = 42.3, 8.2 Hz, 2H), 7.77 – 7.66 (m, 2H), 7.48 (d, J = 7.4 Hz, 1H), 7.06 (d, J = 4.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 151.3, 148.6, 145.2, 139.2, 131.7, 131.3, 127.1 – 126.7 (m, 1C), 127.0, 126.3, 125.8 (q, J = 32.3 Hz), 125.1, 123.5, 122.7, 122.1 (d, J = 3.9 Hz), 118.1, 102.6; HRMS-ESI (m/z): [M+H]+ calcd for C16H9N2F3Cl2 - 357.0173; found 357.0163; Tr = 4.71 min, purity > 95%.
4.1.3.22 N-(4-Chlorophenyl)-6-(trifluoromethyl)quinolin-4-amine (24)
24 was prepared by method B to afford a light yellow solid (181 mg, 65%); m.p. 111–114°C; 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.58 (d, J = 6.3 Hz, 1H), 8.24 (d, J = 8.8 Hz, 1H), 8.14 (dd, J = 8.9, 1.6 Hz, 1H), 7.59 – 7.48 (m, 4H), 6.94 (d, J = 6.3 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 152.4, 147.9, 144.6, 137.4, 129.9, 129.6 (s, 2C), 128.2 (d, J = 2.5 Hz), 127.5 – 127.0 (m, 1C), 126.4 – 125.3 (m, 3C), 122.7, 122.3 (d, J = 4.4 Hz), 117.8, 101.8; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Cl - 323.0563; found 323.0553; Tr = 4.30 min, purity > 95%.
4.1.3.23 N-(3-Chlorophenyl)-6-(trifluoromethyl)quinolin-4-amine (25)
25 was prepared by method B to afford a light yellow solid (201 mg, 72%); m.p. 187–189°C; 1H NMR (400 MHz, DMSO-d6) δ 11.40 (s, 1H), 9.35 (s, 1H), 8.62 (d, J = 6.6 Hz, 1H), 8.41 – 8.19 (m, 2H), 7.61 (s, 1H), 7.57 (d, J = 7.9 Hz, 1H), 7.47 (dd, J = 14.7, 7.8 Hz, 2H), 6.98 (d, J = 6.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 154.0, 145.8, 141.9, 139.1, 133.9, 131.5, 128.6 (d, J = 3.1 Hz), 126.8, 126.5 (q, J = 32.3Hz, CF3), 125.2, 124.6, 123.5, 122.8 – 122.3 (m, 1C), 122.5, 117.2, 101.7; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Cl - 323.0563; found 323.0553; Tr = 4.25 min, purity > 95%.
4.1.3.24 N-(2-Chlorophenyl)-6-(trifluoromethyl)quinolin-4-amine (26)
26 was prepared by method B to afford a light yellow solid (187 mg, 67%); m.p. 89–91°C; 1H NMR (400 MHz, DMSO-d6) δ 9.42 (s, 1H), 8.61 (s, 1H), 8.26 (dd, J = 36.5, 7.3 Hz, 2H), 7.84 – 7.34 (m, 4H), 6.96 (s, 1H); 13C NMR (101 MHz,, DMSO-d6) δ 154.5, 146.2, 142.3, 139.6, 134.3, 131.9, 129.1 – 128.9 (m, 1C), 127.2, 126.9 (q, J = 32.9 Hz, CF3), 125.6, 125.0, 123.8, 123.4 – 123.0 (m, 1C), 122.9, 117.7, 102.2; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Cl - 323.0563; found 323.0553; Tr = 4.04 min, purity > 95%.
4.1.3.25 N-(4-Bromophenyl)-6-(trifluoromethyl)quinolin-4-amine (27)
27 was prepared by method B to afford a yellow solid (225 mg, 71%); m.p. 118–120°C; 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.58 (d, J = 4.7 Hz, 1H), 8.19 (dd, J = 36.2, 8.4 Hz, 2H), 7.56 (dd, J = 100.3, 7.7 Hz, 4H), 6.95 (d, J = 5.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 152.5, 147.5, 144.0, 137.6, 132.6, 127.6 – 127.4 (m, 1C), 126.3, 126.0 (d, J = 32.6 Hz, CF3), 125.3, 125.2, 122.6, 122.2 (d, J = 4.1 Hz), 118.3, 117.6, 101.8; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Br - 367.0058; found 367.0046; Tr = 4.37 min, purity > 95%.
4.1.3.26 N-(3-Bromophenyl)-6-(trifluoromethyl)quinolin-4-amine (28)
28 was prepared by method B to afford a yellow solid (251 mg, 79%); m.p. 156–158°C; 1H NMR (500 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.61 (d, J = 6.1 Hz, 1H), 8.27 – 8.17 (m, 2H), 7.70 (s, 1H), 7.51 (q, J = 11.9, 9.9 Hz, 3H), 6.99 (d, J = 6.1 Hz, 1H); 13C NMR (126 MHz, DMSO-d6) δ 153.1, 147.0, 143.2, 139.8, 131.7, 129.2, 128.2 – 127.7 (m, 1C), 127.0, 126.3 (q, J = 32.6 Hz, CF3), 125.0, 124.6, 123.3, 122.9, 122.4 – 122.2 (m, 1C), 117.5, 101.9; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Br - 367.0058; found 367.0046; Tr = 4.39 min, purity > 95%.
4.1.3.27 N-(2-Bromophenyl)-6-(trifluoromethyl)quinolin-4-amine (29)
29 was prepared by method B to afford a yellow solid (219 mg, 69%); m.p. 88–90°C; 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.46 (d, J = 5.8 Hz, 1H), 8.14 – 7.98 (m, 2H), 7.83 (dd, J = 8.1, 1.2 Hz, 1H), 7.62 – 7.45 (m, 2H), 7.34 (td, J = 7.6, 1.8 Hz, 1H), 6.18 (d, J = 5.8 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 151.8, 150.3, 147.6, 138.5, 134.2, 129.8, 129.3, 128.4, 126.4 – 126.1 (m, 1C), 126.1, 125.6 (q, J = 32.2 Hz), 123.4, 122.2 – 121.7 (m, 2C), 118.3, 102.5; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3Br - 367.0058; found 367.0046; Tr = 4.10 min, purity > 95%.
4.1.3.28 N-(3-Iodophenyl)-6-(trifluoromethyl)quinolin-4-amine (30)
30 was prepared by method B to afford a yellow solid (240 mg, 67%); m.p. 135–137°C; 1H NMR (400 MHz, DMSO-d6) δ 9.22 (s, 1H), 8.58 (d, J = 6.2 Hz, 1H), 8.23 – 8.08 (m, 2H), 7.81 (s, 1H), 7.57 (dd, J = 60.3, 8.3 Hz, 2H), 7.28 (t, J = 7.9 Hz, 1H), 6.95 (d, J = 6.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 152.2, 147.8, 144.3, 139.9, 134.6, 132.3, 131.5, 127.4 (d, J = 3.0 Hz), 126.0 (q, J = 32.5 Hz, CF3), 125.5, 123.3, 122.7, 122.2 (q, J = 4.1 Hz), 117.7, 102.0, 95.3; HRMS-ESI (m/z): [M+H]+ calcd for C16H10N2F3I - 414.9919; found 414.9902; Tr = 4.55 min, purity > 95%.
4.1.3.29 4-((6-(Trifluoromethyl)quinolin-4-yl)amino)benzonitrile (31)
31 was prepared by method B to afford a yellow solid (251 mg, 79%); m.p. 235–240°C; 1H NMR (400 MHz, DMSO-d6) δ 11.76 (s, 1H), 9.45 (s, 1H), 8.71 (d, J = 6.9 Hz, 1H), 8.40 – 8.30 (m, 2H), 8.07 – 7.72 (m, 4H), 7.16 (d, J = 6.9 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 154.7, 144.8, 141.8, 140.4, 134.0 (s, 2C), 129.4 (d, J = 3.1 Hz), 127.0 (q, J = 32.9 Hz), 125.2 (s, 2C), 122.9 (q, J = 4.0 Hz), 122.3, 122.2, 118.5, 117.4, 109.1, 102.2; HRMS-ESI (m/z): [M+H]+ calcd for C17H10N3F3-314.0905; found 314.0892; Tr = 3.59 min, purity > 95%.
4.1.3.30 3-((6-(Trifluoromethyl)quinolin-4-yl)amino)benzonitrile (32)
32 was prepared by method B to afford an off-white solid (251 mg, 79%); m.p. >270°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 9.28 (s, 1H), 8.63 (d, J = 6.2 Hz, 1H), 8.30 – 8.15 (m, 2H), 7.95 (s, 1H), 7.83 (d, J = 7.7 Hz, 1H), 7.80 – 7.66 (m, 2H), 7.06 (d, J = 6.1 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 152.2, 147.8, 144.2, 139.6, 131.1, 129.4, 128.7, 127.7 – 127.3 (m, 1C) 127.0, 126.1 (q, J = 32.6 Hz), 125.5, 125.4, 122.6, 122.3 (d, J = 4.1 Hz), 117.9, 112.5, 102.3; HRMS-ESI (m/z): [M+H]+ calcd for C17H10N3F3 - 314.0905; found 314.0892; Tr = 3.57 min, purity > 95%.
4.1.3.31 2-((6-(Trifluoromethyl)quinolin-4-yl)amino)benzonitrile (33)
33 was prepared by method B to afford a yellow solid (184 mg, 68%); m.p. >270°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.30 (s, 1H), 7.95 (s, 2H), 7.88 (d, J = 7.8 Hz, 1H), 7.74 (t, J = 8.3 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.35 (t, J = 7.9 Hz, 1H), 6.37 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 13C NMR (101 MHz, DMSO-d6) δ 154.2, 150.8, 147.0, 140.3, 135.1, 134.4, 131.3, 129.3, 129.0, 126.3, 123.6, 121.7, 117.9, 112.5, 108.0, 103.7, 102.9; HRMS-ESI (m/z): [M+H]+ calcd for C17H10N3F3 - 314.0905; found 314.0893; Tr = 3.31 min, purity > 95%.
4.1.3.32 6-(Trifluoromethyl)-N-(3-(trifluoromethyl)phenyl)quinolin-4-amine (34)
34 was prepared by method B to afford a yellow solid (120 mg, 39%); m.p. 118–120°C; 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.62 (s, 1H), 8.14 (dd, J = 41.7, 7.8 Hz, 2H), 7.78 (s, 2H), 7.71 (s, 1H), 7.59 (d, J = 6.2 Hz, 1H), 7.09 – 6.99 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 151.1, 149.1, 145.7, 139.9, 130.8, 130.3 (q, J = 31.9 Hz, CF3), 127.0, 126.8 (s, 2C), 125.7 (q, J = 32.4 Hz), 125.3 (d, J = 23.7 Hz), 122.6 (d, J = 23.6 Hz), 122.0 – 121.1 (m, 2C), 119.8 (d, J = 3.5 Hz), 118.2, 102.3; HRMS-ESI (m/z): [M+H]+ calcd for C17H9N2F6 - 356.0748; found 357.0814; Tr = 4.60 min, purity > 95%.
4.1.3.33 N-(3-Ethynylphenyl)-6-(trifluoromethyl)quinolin-4-amine (35)
35 was prepared by method B to afford a mustard solid (159 mg, 59%); m.p. 149–151°C; 1H NMR (500 MHz, DMSO-d6) δ 9.26 (s, 1H), 8.59 (d, J = 6.4 Hz, 1H), 8.25 – 8.14 (m, 2H), 7.58 – 7.41 (m, 4H), 6.94 (d, J = 6.4 Hz, 1H), 4.31 (s, 1H); 13C NMR (126 MHz, DMSO-d6) δ 153.0, 147.3, 143.7, 138.5, 130.3, 129.6, 127.9 – 127.6 (m, 1C), 127.3, 126.2 (d, J = 32.6 Hz), 125.1, 125.0, 125.0, 123.2, 122.9, 122.3 (d, J = 4.1 Hz), 117.6, 101.8, 82.7, 81.8; HRMS-ESI (m/z): [M+H]+ calcd for C18H11N2F3 - 313.0953; found 313.0942; Tr = 4.17 min, purity > 95%.
4.1.3.34 1-(3-((6-(Trifluoromethyl)quinolin-4-yl)amino)phenyl)ethan-1-one (36)
36 was prepared by method B to afford a tan solid (185 mg, 65%); m.p. 228–230°C; 1H NMR (400 MHz, DMSO-d6) δ 9.39 (s, 1H), 8.59 (d, J = 6.4 Hz, 1H), 8.33 – 8.18 (m, 2H), 8.03 (s, 1H), 7.94 (d, J = 7.5 Hz, 1H), 7.77 (d, J = 7.7 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 6.95 (d, J = 6.5 Hz, 1H), 2.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.4, 153.7, 146.3, 142.7, 138.3, 130.3, 129.1, 128.3 – 127.9 (m, 1C), 127.0 – 125.8 (m, 2C), 125.3, 124.1, 124.0, 122.6, 122.8 – 122.3 (m, 1C), 117.4, 101.5, 26.9; HRMS-ESI (m/z): [M+H]+ calcd for C18H13N2OF3 - 331.1058; found 331.1047; Tr = 3.66 min, purity > 95%.
4.1.3.35 N-(3-(Methylsulfonyl)phenyl)-6-(trifluoromethyl)quinolin-4-amine (37)
37 was prepared by method B to afford a yellow solid (269 mg, 85%); m.p. >270°C; 1H NMR (400 MHz, DMSO-d6) δ 11.63 (s, 1H), 9.40 (s, 1H), 8.68 (d, J = 7.0 Hz, 1H), 8.40 – 8.31 (m, 2H), 8.07 (t, J = 1.7 Hz, 1H), 7.98 (dt, J = 7.4, 1.5 Hz, 1H), 7.94 – 7.84 (m, 2H), 7.06 (d, J = 7.0 Hz, 1H), 3.32 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 155.1, 144.7, 142.4, 140.3, 138.0, 131.3, 129.9, 129.4 (d, J = 3.1 Hz), 127.0 (q, J = 32.9 Hz), 125.6, 125.1, 123.3, 122.7 (q, J = 4.0 Hz), 122.4, 122.2, 117.1, 101.4, 43.4; HRMS-ESI (m/z): [M+H]+ calcd for C17H13N2O2SF3 - 367.0728; found 367.0716; Tr = 3.27 min, purity >95%.
4.1.3.36 N-(4-(Methylsulfonyl)phenyl)-6-(trifluoromethyl)quinolin-4-amine (38)
38 was prepared by method A to afford a tan solid (285 mg, 90%); m.p. >270°C; 1H NMR (400 MHz, DMSO-d6) δ 11.75 (s, 1H), 9.47 (s, 1H), 8.70 (d, J = 7.0 Hz, 1H), 8.35 (q, J = 8.2, 7.4 Hz, 2H), 7.96 (dd, J = 119.9, 8.7 Hz, 4H), 7.15 (d, J = 7.0 Hz, 1H), 3.30 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.8, 144.7, 141.9, 140.4, 138.8, 129.4 (d, J = 3.1 Hz), 128.9 (s, 2C), 127.0 (q, J = 32.9 Hz), 125.2 (s, 2C), 122.9 (q, J = 4.3, 3.8 Hz), 122.4 122.2, 117.3, 102.0, 43.5; HRMS-ESI (m/z): [M+H]+ calcd for C17H13N2O2SF3 - 367.0728; found 367.0710; Tr = 3.29 min, purity > 95%.
4.1.3.37 N-(4-(tert-Butoxy)phenyl)-6-(trifluoromethyl)quinolin-4-amine (39)
39 was prepared by method B to afford a tan solid (233 mg, 75%); m.p. 180–182°C; 1H NMR (400 MHz, DMSO-d6) δ 10.95 (s, 1H), 9.31 (s, 1H), 8.53 (d, J = 6.5 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 8.16 (dd, J = 8.9, 1.4 Hz, 1H), 7.37 (d, J = 8.7 Hz, 2H), 7.12 (d, J = 8.7 Hz, 2H), 6.81 (d, J = 6.5 Hz, 1H), 1.34 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 153.6, 146.6, 143.3, 132.6, 127.9 – 127.6 (m, 1C), 126.0 (q, J = 32.6 Hz, CF3), 125.7, 125.4, 124.6, 124.5, 122.6, 122.3 (q, J = 4.0 Hz), 117.2, 101.1, 78.4, 28.5 (s, 3C, tBu); HRMS-ESI (m/z): [M+H]+ calcd for C20H19N2OF3 - 361.1528; found 361.1515; Tr = 4.86 min, purity > 95%.
4.1.3.38 N-(4-((Methylsulfonyl)methyl)phenyl)-6-(trifluoromethyl)quinolin-4-amine (40)
40 was prepared by method B to afford a tan solid (210 mg, 64%); m.p. >250°C; 1H NMR (400 MHz, DMSO-d6) δ 10.78 (s, 1H), 9.22 (s, 1H), 8.60 (d, J = 6.5 Hz, 1H), 8.26 – 8.14 (m, 2H), 7.65 – 7.43 (m, 4H), 6.99 (d, J = 6.5 Hz, 1H), 4.57 (s, 2H), 2.96 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 152.9, 147.3, 138.2, 132.4 (s, 2C), 127.8 - 127.7 (m, 1C), 127.2, 126.1 (d, J = 32.5 Hz), 125.4, 125.1-124.9 (m, 1C), 124.2 (s, 2C), 122.7, 122.1 (d, J = 3.7 Hz), 117.6, 101.8, 58.9, 48.6; HRMS-ESI (m/z): [M+H]+ calcd for C18H15N2O2SF3 - 381.0885; found 381.0874; Tr = 3.37 min, purity > 95%.
4.1.3.39 N-(3,4,5-Trimethoxyphenyl)quinolin-4-amine (41)
41 was prepared by method B to afford a tan solid (288 mg, 76%); m.p. 224–226°C; 1H NMR (400 MHz, DMSO-d6) δ 10.84 (s, 1H), 8.82 (d, J = 8.4 Hz, 1H), 8.47 (d, J = 6.8 Hz, 1H), 8.11 (d, J = 8.0 Hz, 1H), 7.98 (t, J = 8.0 Hz, 1H), 7.76 (t, J = 8.1 Hz, 1H), 6.88 (d, J = 6.8 Hz, 1H), 6.82 (s, 2H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.5, 153.6 (s, 2C), 143.3, 139.3, 136.3, 133.3 (s, 2C), 133.2, 126.7, 123.6, 121.1, 117.3, 103.1, 100.4, 60.2, 56.1 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H18N2O3 - 311.1396; found 311.1387; Tr = 3.23 min, purity > 95%.
4.1.3.40 6-Fluoro-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (42)
42 was prepared by method A to afford a light greed solid (288 mg, 76%); m.p. 92–94°C; 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H), 8.72 (dd, J = 10.5, 2.7 Hz, 1H), 8.50 (d, J = 6.9 Hz, 1H), 8.20 (dd, J = 9.3, 5.1 Hz, 1H), 7.98 (ddd, J = 9.3, 8.0, 2.7 Hz, 1H), 6.92 (d, J = 6.9 Hz, 1H), 6.82 (s, 2H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, dmso) δ 161.0, 158.5, 154.7, 153.7 (s, 2C), 142.6, 136.6, 135.5, 132.7, 123.5-123.1 (m, 1C), 118.2 (d, J = 9.5 Hz), 108.4 (d, J = 25.1 Hz), 103.1 (s, 2C), 100.3, 60.2, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H17N2O3F - 329.1301; found 329.1287; Tr = 3.32 min, purity > 95%.
4.1.3.41 7-Fluoro-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (43)
43 was prepared by method a to afford a light green solid (206 mg, 64%); m.p. 228–230°C; 1H NMR (400 MHz, DMSO-d6) δ 11.17 (s, 1H), 8.97 (dd, J = 9.4, 5.6 Hz, 1H), 8.48 (d, J = 7.0 Hz, 1H), 7.91 (dd, J = 9.5, 2.6 Hz, 1H), 7.74 (ddd, J = 9.4, 8.4, 2.6 Hz, 1H), 6.84 (d, J = 7.0 Hz, 1H), 6.83 (s, 2H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 165.5, 163.0, 155.1, 153.6, 143.3, 140.1, 140.0, 136.6, 132.7, 127.5 (d, J = 10.6 Hz), 116.5 (d, J = 24.4 Hz), 114.2, 105.1 (d, J = 25.1 Hz), 103.3 (s, 2C), 100.4, 60.2, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H17N2O3F - 329.1301; found 329.1289; Tr = 3.14 min, purity > 95%.
4.1.3.42 5,7-Difluoro-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (44)
44 was prepared by method B to afford a bright yellow solid (191 mg, 55%); m.p. >200°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.38 (d, J = 5.9 Hz, 1H), 7.61 (d, J = 9.5 Hz, 1H), 7.58 – 7.50 (m, 1H), 6.74 (s, 3H), 3.74 (s, 6H), 3.66 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.5 (d, J = 15.8 Hz), 161.0 (d, J = 15.4 Hz), 158.5 (d, J = 14.6 Hz), 153.5, 151.5 – 150.5 (m, 1C), 149.2 – 147.3 (m, 1C), 146.1 (d, J = 12.6 Hz), 135.8, 134.1, 106.2 (d, J = 9.5 Hz), 105.4 (dd, J = 22.0, 4.1 Hz), 102.9, 102.6 – 101.0 (m, 1C), 60.2, 56.1 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C18H16F2N2O3 - 347.1207; found 347.1195; Tr = 3.19 min, purity > 95%.
4.1.3.43 6-(tert-Butyl)-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (45)
45 was prepared by method B to afford a grey solid (224 mg, 67%); m.p. 119–121°C; 1H NMR (400 MHz, DMSO-d6) δ 8.66 (s, 1H), 8.42 (d, J = 6.6 Hz, 1H), 8.10 – 7.98 (m, 2H), 6.82 (d, J = 3.0 Hz, 3H), 3.80 (s, 6H), 3.72 (s, 3H), 1.43 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 153.9, 153.6 (s, 2C), 149.6, 143.2, 136.1, 133.7, 131.4, 121.5, 118.9, 117.2, 103.1 (s, 2C), 100.5, 60.2, 56.1 (s, 2C), 35.4, 31.2 (s, 3C); HRMS-ESI (m/z): [M+H]+ calcd for C22H25N2O3 - 367.2022; found 367.2009; Tr = 4.59 min, purity > 95%.
4.1.3.44 4-((3,4,5-Trimethoxyphenyl)amino)quinoline-6-carbonitrile (46)
46 was prepared by method A to afford a yellow solid (245 mg, 69%); m.p. 245–250°C; 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.49 (d, J = 1.3 Hz, 1H), 8.55 (d, J = 7.1 Hz, 1H), 8.33 (dd, J = 8.8, 1.5 Hz, 1H), 8.24 (d, J = 8.8 Hz, 1H), 6.98 (d, J = 7.1 Hz, 1H), 6.83 (s, 2H), 3.80 (s, 6H), 3.73 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.9, 153.7 (s, 2C), 144.4, 140.4, 136.7, 134.7, 132.4, 130.6, 121.9, 117.9, 116.9, 109.0, 103.0 (s, 2C), 101.8, 60.2, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C19H17N3O3 - 336.1348; found 336.1334; Tr = 3.11 min, purity > 95%.
4.1.3.45 6-(Methylsulfonyl)-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (47)
47 was prepared by method B to afford a dark yellow solid (231 mg, 72%); m.p. 94–96°C; 1H NMR (400 MHz, DMSO-d6) δ 11.38 (s, 1H), 9.54 (s, 1H), 8.55 (d, J = 6.2 Hz, 1H), 8.47 – 8.16 (m, 2H), 6.99 (d, J = 6.1 Hz, 1H), 6.83 (s, 2H), 3.76 (d, J = 31.3 Hz, 9H), 3.44 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 155.3, 154.0 (s, 2C), 145.8, 142.4, 138.7, 136.8, 133.4 (s, 2C), 130.0, 125.2, 117.4, 103.1 (s, 2C), 102.3, 60.6, 56.6 (s, 2C), 44.0; HRMS-ESI (m/z): [M+H]+ calcd for C19H20N2O5S - 389.1171; found 389.1156; Tr = 2.98 min, purity > 95%.
4.1.3.46 6-Methoxy-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (48)
48 was prepared by method A to afford a light green solid (313 mg, 89%); m.p. 133–135°C; 1H NMR (400 MHz, DMSO-d6) δ 10.75 (s, 1H), 8.40 (d, J = 6.9 Hz, 1H), 8.19 (d, J = 2.6 Hz, 1H), 8.03 (d, J = 9.2 Hz, 1H), 7.67 (dd, J = 9.2, 2.6 Hz, 1H), 6.86 (d, J = 6.9 Hz, 1H), 6.82 (s, 2H), 3.99 (s, 3H), 3.81 (s, 3H), 3.73 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 157.9, 154.0, 153.7 (s, 2C), 140.7, 136.4, 133.0 (s, 2C), 125.3, 122.1, 118.3 (s, 2C), 103.3, 102.7, 100.0, 60.2, 56.5, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C19H19N2O4 - 340.1423; found 341.1486; Tr = 3.55 min, purity > 95%.
4.1.3.47 6,7-Dimethoxy-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (49)
49 was prepared by method A to afford a light green solid (285 mg, 86%); m.p. 171–173°C; 1H NMR (400 MHz, DMSO-d6) δ 10.56 (s, 1H), 8.32 (d, J = 6.9 Hz, 1H), 8.11 (s, 1H), 7.44 (s, 1H), 6.92 – 6.70 (m, 3H), 3.98 (d, J = 8.9 Hz, 6H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.5, 153.6 (s, 2C), 153.3, 149.4, 140.0, 136.3, 135.4, 133.2 (s, 2C), 111.5 (s, 2C), 103.3 (s, 2C), 99.6, 60.2, 56.7, 56.2, 56.1 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C20H22N2O5 - 371.1607; found 371.1589; Tr = 3.60 min, purity > 95%.
4.1.3.48 7-Methoxy-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (50)
50 was prepared by method A to afford an off white solid (278 mg, 79%); m.p. 217–219°C; 1H NMR (400 MHz, DMSO-d6) δ 10.81 (s, 1H), 8.71 (d, J = 9.4 Hz, 1H), 8.38 (d, J = 7.0 Hz, 1H), 7.46 (d, J = 2.5 Hz, 1H), 7.41 (dd, J = 9.3, 2.5 Hz, 1H), 6.80 (s, 2H), 6.76 (d, J = 7.0 Hz, 1H), 3.96 (s, 3H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 163.4, 155.1, 154.0 (s, 2C), 141.1, 136.9, 133.4 (s, 2C), 125.9, 118.5, 111.7, 103.8 (s, 2C), 100.5, 100.0, 60.6, 56.6 (s, 2C), 56.4; HRMS-ESI (m/z): [M+H]+ calcd for C19H19N2O4 - 340.1423; found 341.1485; Tr = 3.41 min, purity > 95%.
4.1.3.49 7-(Trifluoromethyl)-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (51)
51 was prepared by method A to afford a bright yellow solid (239 mg, 73%); m.p. 260–265°C; 1H NMR (400 MHz, DMSO-d6) δ 11.12 (s, 1H), 8.99 (d, J = 8.9 Hz, 1H), 8.62 (d, J = 7.0 Hz, 1H), 8.45 (s, 1H), 8.14 (dd, J = 8.9, 1.5 Hz, 1H), 6.99 (d, J = 7.0 Hz, 1H), 6.83 (s, 2H), 3.81 (s, 6H), 3.73 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.9, 153.7 (s, 2C), 144.4, 138.3 (d, J = 31.7 Hz, CF3), 136.7, 132.6, 125.8, 124.6, 122.3 (d, J = 3.5 Hz), 119.3, 118.3, 105.5, 103.1 (s, 2C), 101.7, 60.2, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C19H17F3N2O3 - 379.1270; found 379.1253; Tr = 3.81 min, purity > 95%.
4.1.3.50 4-((3,4,5-Trimethoxyphenyl)amino)quinoline-7-carbonitrile (52)
52 was prepared by method A to afford a yellow solid (224 mg, 63%); m.p. >255°C (decomp.); 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 9.55 (s, 1H), 8.54 (d, J = 7.1 Hz, 1H), 8.32 (dd, J = 8.8, 1.5 Hz, 1H), 8.25 (d, J = 8.8 Hz, 1H), 6.97 (d, J = 7.1 Hz, 1H), 6.84 (s, 2H), 3.80 (s, 6H), 3.72 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 154.9, 153.6 (s, 2C), 144.2, 140.3, 136.7, 134.7, 132.4, 130.7, 121.7, 117.9, 116.8, 108.9, 103.0 (s, 2C), 101.7, 60.2, 56.2 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C19H17N3O3 - 336.1348; found 336.1333; Tr = 3.04 min, purity > 95%.
4.1.3.51 N-(3-Bromophenyl)-6,7-dimethoxyquinolin-4-amine (53)
53 was prepared by method B to afford a colourless solid (183 mg, 57%); m.p. 170–171°C; 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.31 (d, J = 5.3 Hz, 1H), 7.69 (s, 1H), 7.50 (t, J = 1.9 Hz, 1H), 7.35 (ddd, J = 8.1, 2.1, 1.1 Hz, 1H), 7.28 (t, J = 7.9 Hz, 1H), 7.23 (s, 1H), 7.19 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H), 6.91 (d, J = 5.3 Hz, 1H), 3.89 (d, J = 15.2 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 151.8, 148.4, 148.1, 145.9, 145.4, 143.3, 131.1, 125.1, 123.4, 122.0, 119.6, 114.6, 108.1, 102.2, 101.4, 56.0, 55.5; HRMS-ESI (m/z): [M+H]+ calcd for C17H15N2O2Br - 359.0395; found 359.0380; Tr = 3.99 min, purity > 95%.
4.1.3.52 6,7-Dimethoxy-N-(3-methoxyphenyl)quinolin-4-amine (54)
54 was prepared by method B to afford a colourless solid (180 mg, 65%); m.p. 178–180°C; 1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 8.31 (d, J = 6.4 Hz, 1H), 8.08 (s, 1H), 7.43 (s, 1H), 7.40 (t, J = 8.4 Hz, 1H), 7.06 – 6.98 (m, 2H), 6.87 (ddd, J = 8.4, 2.4, 0.9 Hz, 1H), 6.81 (d, J = 6.4 Hz, 1H), 3.99 (s, 3H), 3.94 (s, 3H), 3.79 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 160.2, 153.6, 150.9, 149.0, 142.3, 139.9, 138.6, 130.4, 116.2, 112.5, 111.3, 109.9, 102.4, 102.4, 100.0, 56.6, 55.9, 55.3; HRMS-ESI (m/z): [M+H]+ calcd for C18H18N2O3 - 311.1396; found 311.1382; Tr = 3.65 min, purity > 95%.
4.1.3.53 N-(3-Ethynylphenyl)-6,7-dimethoxyquinolin-4-amine (55)
55 was prepared by method A to afford a dark colourless solid (193 mg, 71%); m.p. 232–234°C; 1H NMR (400 MHz, DMSO-d6) δ 10.94 (s, 1H), 8.35 (d, J = 6.9 Hz, 1H), 8.25 (s, 1H), 7.60 (q, J = 1.4 Hz, 1H), 7.58 – 7.52 (m, 2H), 7.51 – 7.43 (m, 2H), 6.76 (d, J = 6.9 Hz, 1H), 4.34 (s, 1H), 3.99 (d, J = 19.4 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 154.6, 152.9, 149.4, 139.8, 138.1, 135.3, 130.2, 130.0, 128.1, 125.9, 123.2, 111.9, 103.0, 99.7, 99.31, 82.6, 81.9, 56.9, 56.1; HRMS-ESI (m/z): [M+H]+ calcd for C19H16N2O2 - 305.1290; found 305.1278; Tr = 3.75 min, purity >95%.
4.1.3.54 1-(3-((6,7-dimethoxyquinolin-4-yl)amino)phenyl)ethan-1-one (56)
56 was prepared by method A to afford a colourless solid (193 mg, 67%); m.p. 238–240°C; 1H NMR (400 MHz, DMSO-d6) δ 10.96 (s, 1H), 8.35 (d, J = 6.9 Hz, 1H), 8.24 (s, 1H), 8.02 (s, 1H), 7.95 (d, J = 7.7 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.50 (s, 1H), 6.78 (d, J = 6.9 Hz, 1H), 3.99 (d, J = 19.6 Hz, 6H), 2.62 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 197.8, 155.1, 153.3, 149.9, 140.6, 138.7, 130.7, 130.1, 124.9, 112.4, 103.2, 100.5, 99.7, 57.2, 56.6, 27.4; HRMS-ESI (m/z): [M+H]+ calcd for C19H18N2O3 - 323.1396; found 323.1383; Tr = 2.81 min, purity > 95%.
4.1.3.55 6,7-Dimethoxy-N-(3,4,5-trimethoxyphenyl)quinolin-4-amine (57)
57 was prepared by method C to afford a bright yellow solid (261 mg, 79%) m.p. 230–232°C; 1H NMR (400 MHz, DMSO-d6) δ 11.41 (s, 1H), 8.81 (s, 1H), 8.35 (s, 1H), 7.37 (s, 1H), 7.09 (s, 2H), 4.00 (d, J = 13.2 Hz, 6H), 3.79 (s, 6H), 3.70 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 158.2, 156.2, 152.7 (s, 3C), 150.1, 148.6, 135.9, 132.7, 107.2, 104.0, 103.1 (s, 2C), 99.8, 60.2, 57.0, 56.4, 56.1 (s, 2C); HRMS-ESI (m/z): [M+H]+ calcd for C19H21N3O5 - 372.1559; found 372.1542; Tr = 3.26 min, purity > 95%.
4.2 Mass Spectrometry
Samples were analyzed with a hybrid LTQ FT (ICR 7T) (ThermoFisher, Bremen, Germany) mass spectrometer coupled with a Waters Acquity H-class liquid chromatograph system. Samples were introduced via an electrospray source at a flow rate of 0.6 mL/min. Electrospray source conditions were set as: spray voltage 4.7 kV, sheath gas (nitrogen) 45 arb, auxiliary gas (nitrogen) 30 arb, sweep gas (nitrogen) 0 arb, capillary temperature 350°C, capillary voltage 40 V and tube lens voltage 100 V. The mass range was set to 150–2000 m/z. All measurements were recorded at a resolution setting of 100,000.
Separations were conducted on a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μM particle size). LC conditions were set at 100% water with 0.1% formic acid (A) ramped linearly over 9.8 min to 95% acetonitrile with 0.1% formic acid (B) and held until 10.2 min. At 10.21 min the gradient was switched back to 100% A and allowed to re-equilibrate until 11.25 min.
Xcalibur (ThermoFisher, Breman, Germany) was used to analyze the data. Solutions were analyzed at 0.1 mg/mL or less based on responsiveness to the ESI mechanism. Molecular formula assignments were determined with Molecular Formula Calculator (v 1.2.3). Low-resolution mass spectrometry (linear ion trap) provided independent verification of molecular weight distributions. All observed species were singly charged, as verified by unit m/z separation between mass spectral peaks corresponding to the 12C and 13C 12Cc-1 isotope for each elemental composition.
4.3 X-ray Crystallography
Single-crystal x-ray diffraction analyses were performed using a Rigaku FRE+ equipped with either VHF (1, 9, 13, 37) or HF Varimax confocal mirrors (17, 48, 49) and an AFC12 goniometer and HG Saturn 724+ detector equipped with an Oxford Cryosystems low-temperature apparatus operating at T = 100(2) K. CrystalClear-SM Expert 3.1 b27 (CrystalClear, Rigaku Corporation, The Woodlands, Texas, U.S.A., (2008–2014) (1, 9, 13, 37) or CrysAlisPro (CrysAlisPro Software System, Rigaku Oxford Diffraction, Yarnton, Oxford, UK (2016)) (17, 48, 49) was used to record images. CrysAlisPro was used to process all data and apply empirical absorption corrections and unit cell parameters were refined against all data. The structures were solved by intrinsic phasing using SHELXT[35] (1, 9, 13, 17, 37, 48) or charge flipping using SUPERFLIP[36] (49) and refined on Fo2 by full-matrix least-squares refinements using SHELXL-2014[37] as implemented within OLEX2[38]. All non-hydrogen atoms were refined with anisotropic displacement parameters and hydrogen atoms were added at calculated positions except those attached to heteroatoms which were located from the difference map. All hydrogen atoms were refined using a riding model with isotropic displacement parameters based on the equivalent isotropic displacement parameter (Ueq) of the parent atom. Figures were produced using OLEX2. The CIF files for the crystal structures of 1, 9, 13, 17, 37, 48 and 49 have been deposited with the CCDC and have been given the deposition numbers 1534017–1534023 respectively.
4.4 Biological Assays
4.4.1 Differential scanning fluorimetry (DSF) assays
Starting from a 100 μM stock, GAK (14-351) was diluted to 1 μM in buffer 100 mM K2HPO4 pH 7.5 containing 150 mM NaCl, 10% glycerol and 1x dye (Applied Biosystems catalog 4461806). The protein/dye mixture was transferred to a 384-well PCR microplate with 20 μL per well. Compounds at 10 μM in DMSO were added next, in 20 nL volume, using a liquid handling device setup with a pin head to make a final 10 μM compound in the assay plate. The final DMSO concentration in all wells was 0.1%, including the reference well with DMSO only.
Thermal shift data was measured in a qPCR instrument (Applied Biosystems QuantStudio 6) programmed to equilibrate the plate at 25°C for 5 minutes followed by ramping the temperature to 95°C at a rate of 0.05°C per second. Data was processed on Protein Thermal shift software (Applied Biosystems) fitting experimental curves to a Boltzmann function to calculate differential thermal shifts (ΔTm) referenced to protein/dye in 0.1% DMSO only. The compounds were screened against purified GAK (14-351) according to previously reported procedures[20].
4.4.2 Ligand binding displacement assays
Inhibitor binding was determined using a binding-displacement assay, which measures the ability of inhibitors to displace a fluorescent tracer compound from the ATP binding site of the kinase domain. Inhibitors were dissolved in DMSO and dispensed as 16-point, 2× serial dilutions in duplicate into black multi-well plates (Greiner). Each well contained either 0.5 nM or 1 nM biotinylated kinase domain protein ligated to streptavidin-Tb-cryptate (Cisbio), 12.5 nM or 25 nM Kinase Tracer 236 (ThermoFisher Scientific), 10 mM Hepes pH 7.5, 150 mM NaCl, 2 mM DTT, 0.01% BSA, 0.01% Tween-20. Final assay volume for each data point was 5 μL, and final DMSO concentration was 1%. The kinase domain proteins were expressed in E. coli as a fusion with a C-terminal AVI tag (vector pNIC-Bio3, NCBI reference JN792439) which was biotinylated by co-expressed BirA, and purified using the same methods as used previously[34]. After setting up the assay plate it was incubated at room temperature for 1.5 hours and then read using a TR-FRET proto Residue ranges were AAK1: 31-396, BMP2K: 38-345, GAK: 12-347, STK16: 13-305col on a PheraStarFS plate reader (BMG Labtech). The data was normalized to 0% and 100% inhibition control values and fitted to a four parameter dose-response binding curve in GraphPad Software (version 7, La Jolla, CA, USA). The determined IC50 values were converted to Ki values using the Cheng-Prusoff equation and the concentration and Kd values for the tracer (previously determined).
4.4.3 Competition binding assays
Competition binding assays were performed at DiscoverX as described previously [28]. Kinases were produced either as fusions to T7 phage3, or were expressed as fusions to NF-κB in HEK-293 cells and subsequently tagged with DNA for PCR detection. In general, full-length constructs were used for small, single-domain kinases, and catalytic domain constructs including appropriate flanking sequences were used for multidomain kinases. Briefly, for the binding assays, streptavidin-coated magnetic beads were treated with biotinylated affinity ligands to generate affinity resins. The liganded beads were blocked to reduce non-specific binding and washed to remove unbound ligand.
Binding reactions were assembled by combining kinase, liganded affinity beads, and test compounds prepared as 100× stocks in DMSO. DMSO was added to control assays lacking a test compound. Assay plates were incubated at 25°C with shaking for 1 h, and the affinity beads were washed extensively to remove unbound protein. Bound kinase was eluted in the presence of nonbiotinylated affinity ligands for 30 min at 25°C with shaking. The kinase concentration in the eluates was measured by quantitative PCR. The KINOMEscan® panel of approximately 400 wild type human kinase assays was run as a single measurement at 1 μM. Kd values were determined using 11 serial three-fold dilutions of test compound and a DMSO control.
4.4.4 Cell transfection, treatments, and BRET measurements
The N-terminal Nano Luciferase/GAK fusion (NL-GAK) was encoded in pFN31K expression vector, including flexible Gly-Ser-Ser-Gly linkers between NL and GAK (Promega Madison, WI, USA)[39]. For cellular NanoBRET Target Engagement experiments, the NL-GAK fusion construct was diluted with carrier DNA - pGEM-3Zf(-) (Promega, Madison, WI, USA) at a mass ratio of 1:10 (mass/mass), prior to adding FuGENE HD (Promega, Madison, WI, USA). DNA:FuGENE complexes were formed at a ratio of 1:3 (μg DNA: μL FuGENE HD) according to the manufacturer’s protocol (Promega, Madison, WI, USA). The resulting transfection complex (1 part, volume) was then gently mixed with 20 parts (v/v) of HEK-293 cells (ATCC) suspended at a density of 2 × 105 cells/mL in DMEM (Gibco) + 10% FBS (Seradigm/VWR) followed by incubation (37°C / 5% CO2) for 20–24 hours. After 24 hours HEK293 NL-GAK transfected cells were detached from flasks (0.05% Trypsin-EDTA or TrypLE) (Gibco) and re-suspended at a concentration of 2 × 105 cells/mL in Opti-MEM media (Gibco).
Cellular nanoBRET assays were performed in Non-Binding Surface (NBS™)/white, 96-well plates (Corning) by addition of 1.7 × 104 cells per well (85 μL). NanoBRET Tracer 5 (Promega, Madison, WI, USA) was used at a final concentration of 0.25 μM as previously evaluated in a titration experiment. A total of 5 μL per well (20× working stock of nanoBRET Tracer 5 [5 μM]) was added to all wells, except the “no tracer” control wells to which 5 μL per well of tracer dilution buffer alone was added. All test compounds were prepared initially as concentrated stock solutions in 100% DMSO (Sigma). Approximately 1 h prior to addition to assay plates, the concentrated stock solutions were diluted in Opti-Mem media (90%) to prepare 10% DMSO working stock solutions. A total of 10 μL per well of the 10-fold test compound stock solutions (final assay concentration 1% DMSO) were added. For “no compound” and “no tracer” control wells, a total of 10 μL per well of Opti-MEM plus 10% DMSO (9 μL was added, final concentration 1% DMSO). 96-well plates containing cells with nanoBRET Tracer 5 and test compounds (100 μL total volume per well) were equilibrated (37°C / 5% CO2) for 2 h. To measure nanoBRET signal, nanoBRET NanoGlo substrate at a ratio of 1:166 to Opti-MEM media in combination with extracellular NanoLuc Inhibitor diluted 1:500 (10 μL [30 mM stock] per 5 mL Opti-MEM plus substrate) were combined to create a 3-fold stock. A total of 50 μL of the 3× substrate/extracellular NanoLuc inhibitor were added to each well. The plates were read within 15 minutes (GloMax Discover luminometer, Promega, Madison, WI, USA) equipped with 450 nM BP filter (donor) and 600 nM LP filter (acceptor), using 0.3 s integration time instrument utilizing the “nanoBRET 618” protocol.
Test compounds were evaluated at eight concentrations in competition with NanoBRET Tracer 5 in HEK293 cells transiently expressing the GAK fusion protein. Prior to curve fitting, the average BRET ratio for “no tracer” (Opti-MEM + DMSO only) wells was subtracted from all experimental inhibitor well BRET ratio values and converted to mBRET units (x1000). Additional normalization of the NanoBRET assay data was performed by converting experimental values for respective concentrations of experimental inhibitors to relative percent control values (no compound [Opti-MEM + DMSO + Tracer 5 only] wells = 100% Control; no tracer [Opti-MEM + DMSO] wells = 0% Control). The data was normalized to 0% and 100% inhibition control values and fitted to a four parameter dose-response binding curve in Prism Software (version 7, GraphPad, La Jolla, CA, USA).
4.4.5 Multiplexed Inhibitor Bead (MIB) affinity chromatography / MS analysis
SUM159 cells were cultured in a 1:1 mixture of Dulbecco’s Modified Eagle Medium and Nutrient Mixture F-12 medium (GIBCO) supplemented with 5% fetal bovine serum, 1% anti/anti, 5 μg/mL insulin, and 1 μg/mL hydrocortisone. Cells were maintained at 37°C in a humidified 5% CO2 atmosphere. SUM159 cells were grown to 80% confluency, washed twice with PBS, and harvested by scraping cells in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, 10 mM NaF, 2.5 mM Na3VO4, complete protease inhibitor cocktail (Roche), phosphatase inhibitor cocktail 2, and 3 (Sigma). Lysates were sonicated then clarified by centrifugation at 14,000 × g for 15 minutes at 4°C. Lysate was then filtered through a 0.2 μm syringe filter and frozen at −80°C until used. Protein concentration was quantified using a Bradford assay the day of the experiment. DMSO or the indicated concentration of 1 or 49 was added to lysate containing 4 mg of total protein. Lysates were vortexed briefly then incubated for 30 minutes on ice. Kinases were affinity purified as previously described[25]. Briefly, lysates were diluted to 1.33 mg/mL with lysis buffer then NaCl concentration brought to 1M.
Diluted lysates were passed over a mixture of 25 μL settled beads of each of the following inhibitors conjugated to ECH Sepharose beads: Purvalanol B, PP58, VI-16832, UNC21474A, UNC8088A, and 37.5 μL settled beads conjugated to CTx-0294885 and VI-16832. The kinase inhibitor-bead conjugates were previously equilibrated in high salt buffer (50 mM HEPES pH 7.5, 1 M NaCl, 0.5% Triton X-100, 1 mM EDTA, and 1 mM EGTA). MIBs columns were sequentially washed with high salt buffer, low salt buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, and 1 mM EGTA), and SDS buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 0.5% Triton X-100, 1 mM EDTA, 1 mM EGTA, and 0.1% SDS). Proteins were eluted by boiling samples in elution buffer (100 mM Tris-HCl pH 6.8, 0.5% SDS, and 1% β-mercaptoethanol) for 15 minutes twice. Dithiothreitol (DTT) was added to a final concentration of 5 mM and samples were incubated at 60°C for 25 minutes. Samples were then cooled to room temperature on ice and alkylated by adding iodoacetamide to a final concentration of 20 mM for 30 minutes in the dark at room temperature. Samples were then concentrated in 10K Amicon Ultra centrifugal concentrators (Millipore) followed by methanol and chloroform precipitation of proteins. The final protein pellets were re-suspended in 50 mM HEPES pH 8.0 and incubated with trypsin at 37°C overnight. Residual detergent was removed by three sequential ethyl acetate extractions then desalted using Pierce C-18 spin columns (Thermo Scientific) according to the manufacturer’s protocol.
4.4.6 LC/MS/MS Analysis
Each sample was analyzed by LC-MS/MS using an Easy nLC 1000 coupled to a QExactive mass spectrometer equipped with an Easy Spray source (Thermo Scientific). First, samples were reconstituted in loading buffer (5% ACN, 0.1% formic acid), and then loaded onto a PepMap 100 C-18 column (75 μm id × 25 cm, 2 μm particle size) (Thermo Scientific). Peptides were separated over a 165 min gradient consisting of 5–45% mobile phase B at a 250 nL/min flow rate, where mobile phase A was 0.1% formic acid in water and mobile phase B consisted of 0.1% formic acid in ACN. The QExactive was operated in data-dependent mode where the 20 most intense precursors were selected for subsequent fragmentation. For all runs, ESI parameters: 3 × 106 AGC MS1, 80 ms MS1 max inject time, 1 × 105 AGC MS2, 100 ms MS2 max inject time, 20 loop count, 1.8 m/z isolation window, 45 s dynamic exclusion. Spectra were searched against the Uniprot/Swiss-Prot database with the integrated MaxQuant search engine, ANDROMEDA. Kinase abundance was quantitated label-free using the MaxQuant software (Ver. 1.5.1.2). The false discovery rate was set at 5%, and only kinases having two or more unique peptides were considered. The following parameters were used to identify tryptic peptides for protein identification: up to two missed trypsin cleavage sites; carbamidomethylation (C) was set as a fixed modification; and oxidation (M), deamidation (NQ), and phosphorylation (STY) were set as variable modifications. The ratios of label free quantification values of 49 treated samples over DMSO treated samples are reported.
4.5 Molecular Modelling
Molecular modelling was performed using Schrödinger Maestro software package[40]. Structures of small molecules were prepared using and the LigPrep module of Schrodinger suite employing OPLS3 force for all computations. x-ray crystal structure for the GAK (pdb:4Y8D) was pre-processed using the protein preparation wizard of Schrödinger suite in order to optimize the hydrogen bonding network [9a].
Prior to Glide docking, the grid box was centered using corresponding x-ray ligand as template. The ligand docking was performed using default SP settings of Schrodinger Glide with additional hydrogen bond constraints to NH of CYS126 (hinge residue). Graphical illustrations were generated using MOE software[41], Maestro (WaterMap) or PyMOL (The PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC).
4.5.1 Hydration Site Analysis
Hydration site analysis calculated with WaterMap (Schrödinger Release 2016-3: WaterMap, Schrödinger, LLC, New York, NY, 2016.). The 4Y8D structure was prepared with Protein Preparation Wizard (as above). Waters were analyzed within 5 Å of the co-crystallized ligand, and the 2 ns simulation was conducted with OPLS3 force field.
4.5.2 Gas Phase/OPLS3 force field modeling
Torsional barriers of compounds of over N-C linger were estimated using molecular mechanics calculations employing Rapid Torsion Scan functionality of Maestro suite with angle increment of 15 degrees with OPLS3 force field. QM optimized gas phase geometries for compounds shown in table 2 were calculated with Jaguar software. Initial geometries were based on the rapid torsion scan and full geometry optimization was calculated using B3LYP level of theory and 6–31G** basis set with standard geometry optimization setting of Jaguar (Schrödinger Release 2016-3: Jaguar, Schrödinger, LLC, New York, NY, 2016).
Supplementary Material
Acknowledgments
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA), Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and Wellcome Trust. The NIH is acknowledge for support (1U24DK11604-01). David H. Drewry is gratefully acknowledged for stimulating discussions. We also gratefully acknowledge Megan Gray and Matt Robers from Promega and Tammy Havener from UNC for their insight and technical expertise that greatly assisted this research. In addition we thank the EPSRC UK National Crystallography Service for funding and the collection of the crystallographic data. We would also like to thank the Biocenter Finland/DDCB for financial support towards the goals of our work and the CSC-IT Center for Science Ltd. (Finland) for the allocation of computational resources. In addition, we also thank Dr. Brandie M. Ehrmann for LC-MS/HRMS support provided by the Mass Spectrometry Core Laboratory at the University of North Carolina at Chapel Hill.
Footnotes
http://orcid.org/0000-0001-5871-3458 (Christopher Asquith - Orcid)
http://orcid.org/0000-0003-2858-8929 (Jonathan Elkins - Orcid)
http://orcid.org/0000-0003-4799-6792 (Carrow Wells - Orcid)
http://orcid.org/0000-0003-4181-8223 (Timothy Willson - Orcid)
http://orcid.org/0000-0002-9836-0068 (William Zuercher - Orcid)
SUPPLEMENTARY INFORMATION
Supplementary data related to this article can be found at.
References
- 1.a Greener T, Zhao X, Nojima H, Eisenberg E, Greene LE. J Biol Chem. 2000;275(2):1365–1370. doi: 10.1074/jbc.275.2.1365. [DOI] [PubMed] [Google Scholar]; b Kanaoka Y, Kimura SH, Okazaki I, Ikeda M, Nojima H. FEBS Lett. 1997;402(1):73–80. doi: 10.1016/s0014-5793(96)01484-6. [DOI] [PubMed] [Google Scholar]; c Kimura SH, Tsuruga H, Yabuta N, Endo Y, Nojima H. Genomics. 1997;44(2):179–187. doi: 10.1006/geno.1997.4873. [DOI] [PubMed] [Google Scholar]; d Korolchuk VI, Banting G. Traffic. 2002;3(6):428–439. doi: 10.1034/j.1600-0854.2002.30606.x. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu H, Nagamori I, Yabuta N, Nojima H. J Cell Sci. 2009;122(Pt 17):3145–3152. doi: 10.1242/jcs.052795. [DOI] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. Science. 2002;298(5600):1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Barbieri E, Di Fiore PP, Sigismund S. Curr Opin Cell Biol. 2016;39:21–27. doi: 10.1016/j.ceb.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 5.Neveu G, Ziv-Av A, Barouch-Bentov R, Berkerman E, Mulholland J, Einav S. J Virol. 2015;89(8):4387–4404. doi: 10.1128/JVI.02705-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mercer J, Schelhaas M, Helenius A. Annu Rev Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 7.a Aleksandrowicz P, Marzi A, Biedenkopf N, Beimforde N, Becker S, Hoenen T, Feldmann H, Schnittler HJ. J Infect Dis. 2011;204(Suppl 3):S957–967. doi: 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sikka V, Chattu VK, Popli RK, Galwankar SC, Kelkar D, Sawicki SG, Stawicki SP, Papadimos TJ. J Glob Infect Dis. 2016;8(1):3–15. doi: 10.4103/0974-777X.176140. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sloan RD, Kuhl BD, Mesplede T, Munch J, Donahue DA, Wainberg MA. J Virol. 2013;87(14):8110–8123. doi: 10.1128/JVI.00815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. J Cell Biol. 2003;160(3):321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Papatheodorou P, Zamboglou C, Genisyuerek S, Guttenberg G, Aktories K. PLoS One. 2010;5(5):e10673. doi: 10.1371/journal.pone.0010673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Kovackova S, Chang L, Bekerman E, Neveu G, Barouch-Bentov R, Chaikuad A, Heroven C, Sala M, De Jonghe S, Knapp S, Einav S, Herdewijn P. J Med Chem. 2015;58(8):3393–3410. doi: 10.1021/jm501759m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bekerman E, Einav S. Science. 2015;348(6232):282–283. doi: 10.1126/science.aaa3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray MR, Wafa LA, Cheng H, Snoek R, Fazli L, Gleave M, Rennie PS. Int J Cancer. 2006;118(5):1108–1119. doi: 10.1002/ijc.21469. [DOI] [PubMed] [Google Scholar]
- 11.a Sakurai MA, Ozaki Y, Okuzaki D, Naito Y, Sasakura T, Okamoto A, Tabara H, Inoue T, Hagiyama M, Ito A, Yabuta N, Nojima H. PLoS One. 2014;9(6):e100124. doi: 10.1371/journal.pone.0100124. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Susa M, Choy E, Liu X, Schwab J, Hornicek FJ, Mankin H, Duan Z. Mol Cancer Ther. 2010;9(12):3342–3350. doi: 10.1158/1535-7163.MCT-10-0637. [DOI] [PubMed] [Google Scholar]
- 12.a Dzamko N, Zhou J, Huang Y, Halliday GM. Front Mol Neurosci. 2014;7:57. doi: 10.3389/fnmol.2014.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Rhodes SL, Sinsheimer JS, Bordelon Y, Bronstein JM, Ritz B. Ann Hum Genet. 2011;75(2):195–200. doi: 10.1111/j.1469-1809.2010.00616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a Dumitriu A, Latourelle JC, Hadzi TC, Pankratz N, Garza D, Miller JP, Vance JM, Foroud T, Beach TG, Myers RH. PLoS Genet. 2012;8(6):e1002794. doi: 10.1371/journal.pgen.1002794. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dumitriu A, Pacheco CD, Wilk JB, Strathearn KE, Latourelle JC, Goldwurm S, Pezzoli G, Rochet JC, Lindquist S, Myers RH. Hum Mol Genet. 2011;20(8):1478–1487. doi: 10.1093/hmg/ddr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bellei B, Pitisci A, Migliano E, Cardinali G, Picardo M. Cell Signal. 2014;26(4):716–723. doi: 10.1016/j.cellsig.2013.12.023. [DOI] [PubMed] [Google Scholar]
- 15.Fabian MA, Biggs WH, 3rd, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, Galvin M, Gerlach JL, Grotzfeld RM, Herrgard S, Insko DE, Insko MA, Lai AG, Lelias JM, Mehta SA, Milanov ZV, Velasco AM, Wodicka LM, Patel HK, Zarrinkar PP, Lockhart DJ. Nat Biotechnol. 2005;23(3):329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 16.a Tabara H, Naito Y, Ito A, Katsuma A, Sakurai MA, Ohno S, Shimizu H, Yabuta N, Nojima H. PLoS One. 2011;6(10):e26034. doi: 10.1371/journal.pone.0026034. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Takada Y, Matsuo K. Keio J Med. 2012;61(4):120–127. doi: 10.2302/kjm.2011-0009-oa. [DOI] [PubMed] [Google Scholar]
- 17.Drewry DH, Wells CI, Andrews DM, Angell R, Al-Ali H, Axtman AD, Capuzzi SJ, Elkins JM, Ettmayer P, Frederiksen M, Gileadi O, Gray N, Hooper A, Knapp S, Laufer S, Luecking U, Michaelides M, Muller S, Muratov E, Denny RA, Saikatendu KS, Treiber DK, Zuercher WJ, Willson TM. PLoS One. 2017;12(8):e0181585. doi: 10.1371/journal.pone.0181585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nunes M, Shi C, Greenberger LM. Mol Cancer Ther. 2004;3(1):21–27. [PubMed] [Google Scholar]
- 19.Louie J, Hartwig JF. Tetrahedron Lett. 1995;36(21):3609–3612. [Google Scholar]
- 20.Fedorov O, Niesen FH, Knapp S. Methods Mol Biol. 2012;795:109–118. doi: 10.1007/978-1-61779-337-0_7. [DOI] [PubMed] [Google Scholar]
- 21.Robers MB, Dart ML, Woodroofe CC, Zimprich CA, Kirkland TA, Machleidt T, Kupcho KR, Levin S, Hartnett JR, Zimmerman K, Niles AL, Ohana RF, Daniels DL, Slater M, Wood MG, Cong M, Cheng YQ, Wood KV. Nat Commun. 2015;6:10091. doi: 10.1038/ncomms10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machleidt T, Woodroofe CC, Schwinn MK, Mendez J, Robers MB, Zimmerman K, Otto P, Daniels DL, Kirkland TA, Wood KV. ACS Chem Biol. 2015;10(8):1797–1804. doi: 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- 23.Pawar VG, Sos ML, Rode HB, Rabiller M, Heynck S, van Otterlo WA, Thomas RK, Rauh D. J Med Chem. 2010;53(7):2892–2901. doi: 10.1021/jm901877j. [DOI] [PubMed] [Google Scholar]
- 24.a Abel R, Young T, Farid R, Berne BJ, Friesner RA. J Am Chem Soc. 2008;130(9):2817–2831. doi: 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Young T, Abel R, Kim B, Berne BJ, Friesner RA. Proc Natl Acad Sci U S A. 2007;104(3):808–813. doi: 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, Usary J, Kuan PF, Smalley DM, Major B, He X, Hoadley KA, Zhou B, Sharpless NE, Perou CM, Kim WY, Gomez SM, Chen X, Jin J, Frye SV, Earp HS, Graves LM, Johnson GL. Cell. 2012;149(2):307–321. doi: 10.1016/j.cell.2012.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen P, Alessi DR. ACS Chem Biol. 2013;8(1):96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolf AF, Skovgaard T, Knapp S, Jensen LJ, Berthelsen J. PLoS One. 2014;9(6):e98800. doi: 10.1371/journal.pone.0098800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Nat Biotechnol. 2011;29(11):1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 29.Ma H, Deacon S, Horiuchi K. Expert Opin Drug Discov. 2008;3(6):607–621. doi: 10.1517/17460441.3.6.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a Kooistra AJ, Kanev GK, van Linden OP, Leurs R, de Esch IJ, de Graaf C. Nucleic Acids Res. 2016;44(D1):D365–371. doi: 10.1093/nar/gkv1082. [DOI] [PMC free article] [PubMed] [Google Scholar]; b van Linden OP, Kooistra AJ, Leurs R, de Esch IJ, de Graaf C. J Med Chem. 2014;57(2):249–277. doi: 10.1021/jm400378w. [DOI] [PubMed] [Google Scholar]
- 31.a Ravez S, Castillo-Aguilera O, Depreux P, Goossens L. Expert Opin Ther Pat. 2015;25(7):789–804. doi: 10.1517/13543776.2015.1039512. [DOI] [PubMed] [Google Scholar]; b Solomon VR, Lee H. Curr Med Chem. 2011;18(10):1488–1508. doi: 10.2174/092986711795328382. [DOI] [PubMed] [Google Scholar]; c Bridges AJ. Curr Med Chem. 1999;6(9):825–843. [PubMed] [Google Scholar]
- 32.Wu P, Nielsen TE, Clausen MH. Trends Pharmacol Sci. 2015;36(7):422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 33.Haile PA, Votta BJ, Marquis RW, Bury MJ, Mehlmann JF, Singhaus R, Jr, Charnley AK, Lakdawala AS, Convery MA, Lipshutz DB, Desai BM, Swift B, Capriotti CA, Berger SB, Mahajan MK, Reilly MA, Rivera EJ, Sun HH, Nagilla R, Beal AM, Finger JN, Cook MN, King BW, Ouellette MT, Totoritis RD, Pierdomenico M, Negroni A, Stronati L, Cucchiara S, Ziolkowski B, Vossenkamper A, MacDonald TT, Gough PJ, Bertin J, Casillas LN. J Med Chem. 2016;59(10):4867–4880. doi: 10.1021/acs.jmedchem.6b00211. [DOI] [PubMed] [Google Scholar]
- 34.Sorrell FJ, Szklarz M, Abdul Azeez KR, Elkins JM, Knapp S. Structure. 2016;24(3):401–411. doi: 10.1016/j.str.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheldrick GM. Acta Crystallogr A Found Adv. 2015;71:3–8. doi: 10.1107/S2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palatinus L, Chapuis G. J Appl Cryst. 2007;40:786–790. [Google Scholar]
- 37.Sheldrick GM. Acta Crystallogr C Struct Chem. 2015;71:3–8. doi: 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. J Appl Cryst. 2009;42:339–341. doi: 10.1107/S0021889811041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoddart LA, Johnstone EKM, Wheal AJ, Goulding J, Robers MB, Machleidt T, Wood KV, Hill SJ, Pfleger KDG. Nat Methods. 2015;12(7):661–663. doi: 10.1038/nmeth.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Small-Molecule Drug Discovery Suite 2016–3. Schrödinger, LLC; New York, NY: 2016. [Google Scholar]
- 41.Molecular operating environment (MOE), 2016.8. chemical computing group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.