

ABSTRACT
Macroautophagy/autophagy is a highly conserved process in which subcellular components destined for degradation are sequestered within autophagosomes. The selectivity of autophagy is determined by autophagy receptors, such as Pichia pastoris Atg30 (autophagy-related 30), which controls the selective degradation of peroxisomes (pexophagy) through the assembly of a receptor-protein complex (RPC). Previously, we proved that the peroxisomal acyl-CoA-binding protein, Atg37, and the highly conserved peroxin, Pex3, are required for RPC formation and efficient pexophagy. Here, we describe how Atg37 and Pex3 regulate the assembly and activation of the pexophagic RPC. We demonstrate that Atg30 requires both Atg37 and Pex3 to recruit Atg8 and Atg11 to the pexophagic RPC, because Atg37 depends on Pex3 for its localization at the peroxisomal membrane. We establish that due to close proximity of Atg37- and Pex3-binding sites in the middle domain of Atg30, the binding of these proteins to Atg30 is mutually exclusive within this region. We also show that direct binding of Pex3 and Atg37 to Atg30 regulates its phosphorylation by the Hrr25 kinase, negatively and positively, respectively. Based on these results we present a model that clarifies the assembly and activation of the pexophagic RPC through the phosphoregulation of Atg30.
KEYWORDS: Atg30, Atg37, autophagic receptor, peroxin, peroxisome, Pex3, pexophagy, receptor-protein complex, selective autophagy
Abbreviations
- 5-FOA
5-fluoroorotic acid
- Atg
autophagy related
- PAS
phagophore assembly site
- Pex3m
Pex3L320P,N325D
- PMP
peroxisomal membrane protein
- RPC
receptor protein complex
- Y2H
yeast 2-hybrid
- Y3H
yeast 3-hybrid
Introduction
Autophagy is an evolutionarily conserved and regulated “self-eating” process prevalent in all eukaryotes from yeasts to plants and mammals [1]. In this process, cargo destined for degradation is targeted to the lytic compartment (lysosome/vacuole) via double-membrane vesicles, named autophagosomes, which originate from the phagophore membranes that elongate from a phagophore assembly site (PAS) [2]. Depending on the nature of degraded cargos and morphological properties of the process, several distinct types of autophagy can be distinguished [3]. For instance, pexophagy is a type of autophagy where peroxisomes are selectively recognized by appropriate autophagic receptor(s), which bridge peroxisomes to the autophagic machinery, causing their sequestration within autophagosomes and subsequent lysosomal degradation [4]. Many insights regarding the autophagic pathways, autophagic receptors, signaling components and their regulation have come from studies of yeasts [5], which are excellent models to decipher the mechanisms underlying autophagy. However, the pexophagy process is still not clearly understood even in these model organisms and the molecular basis for regulation and specificity, as well as the players required at each step of pexophagy, still require further studies.
To date there are 2 known pexophagy-specific receptors in yeast: Atg30 from Pichia pastoris and Atg36 from Saccharomyces cerevisiae [6–8]. These 2 receptors are not structural homologs, but they share high functional similarity. These proteins interact with some peroxisomal membrane protein(s) (PMPs) that also function as peroxisome biogenesis factors or peroxins (Atg36 binds to the PMP Pex3, and Atg30 binds to Pex3 and other PMPs, Pex14 and Atg37) and localize on the peroxisome membrane. Additionally, these receptors are phosphorylated to recruit the autophagic scaffold protein, Atg11, which recruits other autophagy-related (Atg) proteins to the PAS, and the phagophore and autophagosome protein, Atg8, necessary for phagophore membrane elongation [9]. Whereas the kinase(s) involved in phosphoregulation of Atg30 is still unknown, the CSNK1D/casein kinase 1δ homolog, Hrr25 kinase, regulates Atg11 binding to Atg36 [10].
Recently, our laboratory demonstrated that Atg30 interacts with other Atg proteins, besides Atg8 and Atg11. Atg30 interacts with another autophagic scaffold protein, Atg17 [6], and with Atg37 [11,12], an acyl-CoA-binding protein that is conserved from yeast to human, but is not present in S. cerevisiae. Atg37 is an integral PMP that positively regulates the receptor protein complex (RPC) during pexophagy [11,12]. Our previous studies on the formation of the pexophagic RPC in P. pastoris revealed that Atg37 contributes a new functional unit required for regulation of pexophagy [11]. We have shown previously that recruitment of Atg11 to Atg30 is Atg37 dependent, because the Atg30 protein binds to Atg11 only in the presence of Atg37 [12]. Interestingly, another study from our group demonstrated that Pex3, like Atg37, is required for pexophagy in P. pastoris and regulates the phosphostatus of Atg30, as well as the recruitment of Atg11 to the pexophagic RPC [13].
Because both Atg37 and Pex3 appeared to regulate Atg30 in the same manner by modulating the formation of the phagophore around a peroxisome(s), we wondered whether these proteins function in the same linear pathway or participate in 2 independent and parallel mechanisms. In this study, we sought to understand the roles of Atg37 and Pex3 in assembling the pexophagic RPC. We demonstrate that Atg30 requires Atg37, as well as functional Pex3, for the recruitment of both Atg11 and Atg8 to the pexophagic RPC. We establish that Atg37 and Pex3 depend on each other for their proper localization at the peroxisome membrane. Atg37 determines the proper Pex3 localization on the peroxisomal surface, and, conversely, Atg37 is recruited to peroxisomes in a Pex3-dependent manner. We also show that a Pex3 mutant, Pex3m, which is defective in binding Atg30 and impairs the phosphorylation of Atg30, is unable to bind Atg37, thereby causing the mislocalization of Atg37 and impairing the formation of the pexophagic RPC [13]. We characterize Atg37- and Pex3-binding sites in Atg30 and establish whether their close proximity causes a competition between Atg37 and Pex3 for binding to middle domain of Atg30. We define a new role for Pex3 directly bound to Atg30 in pexophagic RPC formation. We prove also that Atg30, like other selective receptors, Atg19 and Atg36 [10,14-16], is phosphorylated by the Hrr25 kinase. However, Pex3 directly bound to Atg30 negatively regulates its interaction with Hrr25, whereas Atg37 facilitates it. Finally, based on our new results, we offer an expanded model that clarifies the sequence of steps in the formation and regulation of the pexophagic RPC in P. pastoris.
Results
Atg30 requires Atg37, as well as Pex3, for recruitment of Atg8 and Atg11 to the pexophagic RPC
Triggering pexophagy in P. pastoris requires an active pexophagic RPC, where the pexophagy receptor, Atg30, recruits autophagic machinery crucial for engulfment of peroxisomes by the phagophore. During pexophagy conditions, Atg30 interacts with Atg8, as well as with 2 autophagy scaffold proteins, Atg11 and Atg17 [6,9]. Our previous work demonstrated that binding of Atg8 and Atg11, but not of Atg17, to Atg30 depends on proper phosphorylation of Atg30 [9,12]. Phosphorylation at S71 and S112 in Atg30 modulates its interactions with Atg8 and Atg11, respectively [9]. Because Atg30 is not efficiently phosphorylated in the absence of Atg37, or in pex3m cells (where Pex3 wild-type [WT] was replaced by the Pex3L320P,N325D mutant—called Pex3m for simplicity), thereby impairing Atg11 recruitment to Atg30 [12,13], we asked if Atg37 and/or Pex3 is also required for the recruitment of Atg8 to the pexophagic RPC.
We conducted co-immunoprecipitation experiments under pexophagy conditions using atg37Δ, pex3m and WT cells defective in autophagosome-vacuole fusion (ypt7Δ) to prevent rapid degradation of peroxisomes during pexophagy. As an additional control, an atg8Δ strain was used for the identification in WT cells of the Atg8 protein using immunoblots. We observed Pex3, Atg11 and Atg8 co-immunoprecipitation in immunoprecipitates of Atg30, and corresponding stronger phosphorylation of Atg30 in WT cells. However, whereas deletion of the ATG37 gene did not affect Pex3 binding in atg37Δ cells, it severely decreased both Atg8 and Atg11 binding to Atg30 (Figure 1A). Similarly, weaker phosphorylation of Atg30 and severely impaired affinity isolation of Atg8 and Atg11 with Atg30 were observed in co-immunoprecipitation studies using cells expressing Pex3m (Figure 1B), with which Atg30 did not interact. Based on these data, we conclude that Atg37 and Pex3 regulate the recruitment of both Atg8 and Atg11 to the pexophagic RPC.
Figure 1.
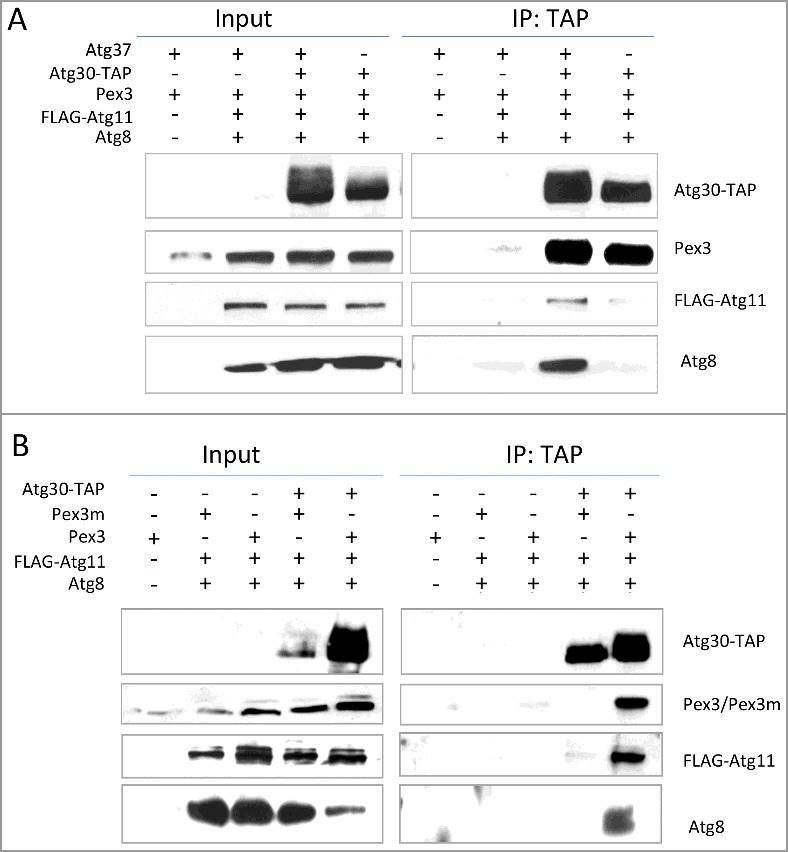
Atg30 requires both Atg37 and Pex3 for Atg8 recruitment to the pexophagic RPC. (A) Atg8 (and Atg11) does not co-immunoprecipitate with Atg30 in the absence of Atg37. Methanol-grown atg8Δ cells (sJCF925; see Table S3) and ypt7Δ cells expressing FLAG-Atg11 and GFP-Atg17 (not shown) with or without Atg30-TAP and deletion of endogenous ATG3712 (sTN632, sTN634, sTN641, sTN642; Table S3), were adapted to SD-N medium for 0.5 h to induce pexophagy before “IP: TAP” experiments were conducted. (B) Atg8 (and Atg11) does not co-immunoprecipitate with Atg30 in pex3m cells. Methanol-grown atg8Δ cells (sJCF925; Table S3) and ypt7Δ cells expressing FLAG-Atg11 with or without Atg30-TAP and replacement of the endogenous PEX3 gene by PEX3m (sSFB431, sSFB432, sSFB435, sSFB436; Table S3) were adapted to SD-N medium for 0.5 h to induce pexophagy before “IP: TAP” experiments were performed. In atg37Δ and pex3m cells, impaired recruitment of Atg8 (and Atg11) to Atg30 correlates with weaker phosphorylation of Atg30. Immunoprecipitation was performed using IgG-agarose beads. Immunoprecipitated proteins were visualized with anti-calmodulin-binding peptide, anti-P. pastoris Pex3, anti-FLAG and anti-P. pastoris Atg8 antibodies. IP, immunoprecipitate.
Proper localization of Pex3 on peroxisomes depends on Atg37, which competes with Atg30 for binding to Pex3
To distinguish between the roles of Atg37 and Pex3 in triggering pexophagy, we studied the interdependence between Atg30, Atg37 and Pex3 proteins and their mutual interactions to clarify the Atg37 and Pex3 roles in the assembly of the pexophagic RPC.
We showed earlier that Pex3, besides binding Atg30, also directly interacts with Atg37 [12]. This interaction occurs under both peroxisome proliferation and pexophagy conditions and is Atg30 independent [12]. We also showed previously that Atg37 localization on the peroxisome periphery depends on Atg30 [12]. Because both Atg30 and Atg37 interact with Pex3 and with each other, we examined if Atg30 or Atg37 are necessary for the localization of Pex3 on peroxisome clusters. In WT and in atg8Δ cells, under peroxisome proliferation conditions, Pex3 was generally at the periphery of peroxisome clusters, where phagophore formation is initiated. However, in both atg30Δ and atg37Δ mutants, Pex3 was often mislocalized to the middle of the peroxisome cluster (Figure 2A). Interestingly, mislocalization was not more severe in the atg30Δ atg37Δ double mutant (Figure 2B). Because, as we have previously reported, the localization of Atg37 to the peroxisome cluster periphery depends on Atg30, we conclude that the proper localization of Pex3 on the peroxisome cluster, and consequently its role in triggering pexophagy, depends on Atg37 (which in turn requires Atg30 for its proper localization).
Figure 2.
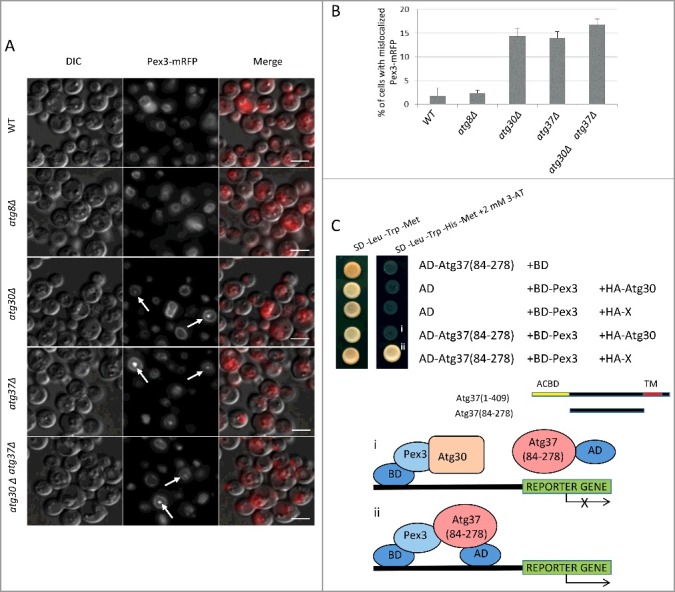
Proper localization of Pex3 on peroxisomes depends on Atg37, which competes with Atg30 for binding to Pex3. (A) Atg37 (directly) and Atg30 (indirectly) are required for localization of Pex3-mRFP at the peroxisome cluster periphery. Cells from strains sTN406, sTN408, sTN410, sTN412 and sTN414 (Table S3) were grown in methanol medium for 15 h. Arrows point to the peroxisome cluster with the mislocalization of Pex3-mRFP in the middle. Scale bars: 5 µm. (B) Quantification of data from panel (A). Data are averages from 3 independent experiments. (C) Atg37(84-278) competes with Atg30 for binding to Pex3 in a Y3H assay. Atg37(84-278) was fused to the Gal4-activation domain (AD) and used with previously described BD-Pex3.12 Lack of methionine in the medium induces expression of HA-Atg30 or irrelevant protein (X) from the MET25 promoter, and lack of histidine in the medium, with or without 3-AT, was used to assess protein-protein interaction. Empty plasmids, pGBKT7 with Gal4 DNA-binding domain (BD) and pGAD-GH with Gal4-activation domain (AD), were used to monitor the levels of self-activation of each analyzed construct. 3-AT, 3-amino-1,2,4-triazole; ACBD, acyl-CoA-binding domain; TM, transmembrane domain. i. Schematic depicting competition between AD-Atg37(84-278) and HA-Atg30 for Pex3 in the Y2H assay; ii. schematic depicting interaction between BD-Pex3 and AD-Atg37(84-274) in the Y3H assay. Interaction between BD-Pex3 and HA-Atg30 affects reporter gene expression.
Next, we tested if Atg37 and Atg30 jointly bind Pex3 or if these interactions are mutually exclusive by using the yeast 3-hybrid (Y3H) system, where a third protein is expressed from the MET25 promoter, which responds to methionine levels in the medium. We examined Atg30-Pex3 complex formation in the presence of Atg37 amino acids 1-278 (Atg37[1-278]), or a protein that is irrelevant for complex formation (not recognized by any of the examined proteins), and noticed a severe reduction of Atg30 binding to Pex3 (Figure S1A). This was true when Atg37(1-278) was expressed moderately in low-level methionine medium (20 mM Met) (Figure S1A and B). The effect was magnified to a complete inhibition of the Atg30-Pex3 interaction after strong induction of expression of Atg37(1-278) in the absence of methionine (Figure S1B). Because Atg37(1-278) binds also to Atg30, we could not exclude the possibility that Atg37 competes not only with Atg30 for Pex3, but also with Pex3 for Atg30. Therefore, we used the truncated Atg37(84-278) lacking the acyl-CoA-binding domain crucial for Atg30 binding and tested its ability to interact with Pex3, both in the absence and presence of Atg30. The interaction between Atg37(84-278) and Pex3 was severely reduced when Atg30 was co-expressed (Figure 2C), relative to that in the absence of Atg30, but not in the presence of an irrelevant protein (Figure 2Cii). This observation led us to conclude that Atg37 competes with Atg30 for binding to Pex3. Consistent with this conclusion, slightly more Pex3 was pulled down with Atg37 in atg30Δ cells, than in the WT cells [12].
Pex3 mutant protein defective in binding Atg30 is also unable to bind Atg37
The observed competition between Atg30 and Atg37 prompted us to examine if Pex3m, which is defective in binding Atg30, also fails to interact with Atg37. We conducted 3 independent analyses of Pex3m interactions with Atg37.
First, using the yeast 2-hybrid (Y2H) assay we found that while WT Pex3 maintained its interaction with Atg37(84-278), Pex3m did not (Figure 3A). To verify this result, we performed an in vitro protein binding assay on glutathione Sepharose beads with recombinant, purified, cytosolic domains of the truncated proteins, HIS-Atg37(1-278), GST-Pex3, GST-Pex3m and free GST [12,13]. We noticed that Atg37(1-278) was pulled down with Pex3, but not with Pex3m or with free GST (Figure 3B).
Figure 3.
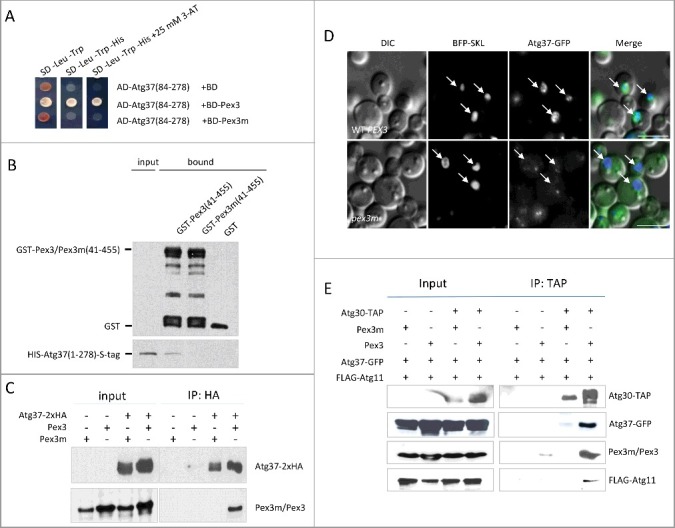
Impaired Pex3m binding to Atg37 affects the localization of Atg37 to peroxisomes and the formation of the pexophagic RPC. (A) Atg37 interacts with Pex3, but not with Pex3m in Y2H; the AD-Atg37(84-278) fusion was used with the previously described BD-Pex3 and BD-Pex3m fusions.12,13 Abbreviations as in Figure 1C. (B) Atg37 does not bind Pex3m in vitro. There was equivalent loading in the input and bound lanes. (C) Atg37 does not co-immunoprecipitate with Pex3m. Cells from strains sKZR007 and sKZR008 (Table S3) were induced in methanol medium for 15 h. Immunoprecipitation was performed using HA-agarose beads. Immunoprecipitated proteins were visualized with anti-HA and anti-P. pastoris Pex3 antibodies. (D) Atg37-Pex3 binding is required for peroxisomal localization of Atg37-GFP. Cells from strains sKZR001 and sKZR002 (Table S3) were grown in methanol medium for 15 h; Arrows indicate peroxisomes marked by BFP-SKL. Scale bars: 5 µm. (E) Atg37-GFP does not co-immunoprecipitate with Atg30 in pex3m cells. This lack of binding correlates with weak phosphorylation of Atg30 and impaired recruitment of Atg11 to Atg30. Methanol-grown cells from strains sKZR003, sKZR004, sKZR005 and sKZR006 (Table S3) were adapted to glucose medium without nitrogen for 0.5 h. Immunoprecipitation was performed using IgG-agarose beads. Immunoprecipitated proteins were visualized with anti-calmodulin-binding peptide, anti-GFP, anti-P. pastoris Pex3, and anti-FLAG antibodies.
Finally, we confirmed using a co-immunoprecipitation assay, the absence of the Atg37-Pex3m complex in P. pastoris cells under peroxisome proliferation conditions in ypt7Δ cells. We used ypt7Δ cells defective in autophagosome-vacuole fusion to prevent any basal degradation of peroxisomes during peroxisome proliferation conditions. As in previous experiments, Atg37 co-immunoprecipitated with Pex3, but not with Pex3m (Figure 3C). These multiple lines of evidence strongly indicate that mutations in Pex3m disrupt not only its interaction with Atg30, but also Atg37 binding to this mutant form of Pex3.
Peroxisomal localization of Atg37 and its binding to Atg30 is affected in pex3m cells
Our previous studies showed that Atg37 binds Pex3, and the 2 proteins colocalize on the peroxisomal membranes in WT cells, as well as at the ER in pex19Δ cells [12]. Because mutations in Pex3m eliminate Atg37-Pex3 complex formation, we examined the localization of Atg37 in pex3m cells. Under peroxisome proliferation conditions, Atg37 was detected on the peroxisome cluster surface in WT cells. However, in pex3m cells, peroxisomal localization of Atg37 was strongly reduced (Figure 3D).
To form the pexophagic RPC, Atg30 must recruit the autophagic machinery, represented by Atg8, Atg11 and Atg17. Because Atg37 regulates the recruitment of Atg8 and Atg11 to Atg30, the absence of Atg37 on peroxisomal membranes should affect the formation of a functional pexophagic RPC. In accordance with our previous results (Figure 1) [12], under pexophagy conditions, we co-immunoprecipitated Atg37, Pex3 and Atg11 with strongly phosphorylated Atg30 from lysates expressing WT Pex3. However, in pex3m cells, none of these proteins were pulled down with Atg30, which was noticeably weakly phosphorylated (Figure 3E). In sum, our data demonstrate that the pexophagic RPC was not formed in pex3m cells due to impaired Atg37 recruitment to peroxisomes and lack of Atg37-Atg30 complex formation required for proper phosphoregulation of Atg30 and subsequent recruitment of the autophagic machinery.
Identification of Atg37- and Pex3-binding sites in Atg30
Atg30 has 1 binding site for Atg37 and 2 for Pex3
Because our results suggest that in pex3m cells the pexophagy phenotype can be explained by Atg37 mislocalization, rather than by lack of direct interaction between Pex3m and Atg30, the precise role of Pex3 bound to Atg30 needed clarification. Thus, to understand mechanistically how the pexophagic RPC is formed, we mapped Atg37- and Pex3-binding sites on Atg30. We constructed 6 different Atg30 fragments (Atg30-1 through Atg30-6) (Figure 4A) and compared their Atg37-binding efficiencies to that of the full-length Atg30 using Y2H. Atg37 bound only to the Atg30 constructs that retained the fragment (amino acids 194-271) represented in Atg30-5 (Figure 4B). In parallel, we generated Atg30 truncations to map the Pex3-binding motif. Using Y2H analysis, we identified 2 fragments: Atg30-5(194-271) and Atg30-6(267-384), containing the C terminus of Atg30 that interacted with Pex3 as strongly as did the full-length Atg30 (Figure 4C,D).
Figure 4.

Identification of Atg37- and Pex3-binding regions in Atg30. (A) Schematic of truncated forms of Atg30 used to map Atg37- and Pex3- binding sites in Atg30. (B) Atg37 interacts with Atg30 constructs containing the Atg30-5 fragment of Atg30 in a Y2H assay; AD fusions of truncated forms of Atg30 (Atg30-1-6) were used with previously described BD-Atg37(1-278).12 (C) Pex3 interacts with the Atg30-5 fragment of Atg30 in a Y2H assay. The listed AD-Atg30 fusions were used with the previously described BD-Pex3 and BD-Pex3m fusions.12,13 (D) AD-Atg30-5 and AD-Atg30-6 interact with BD-Pex3 as strongly as does full-length AD-Atg30 in a Y2H assay.
One of the Pex3-binding sites in Atg30 is close to the Atg37-binding motif
Precise mapping of Atg37 and Pex3-binding sites was necessary because both Atg37 and Pex3 recognized one fragment of Atg30. As we showed previously, Atg30 competes with palmitoyl-CoA for its binding to Atg37 [12], which means that Atg37 probably recognizes a motif in Atg30 similar to the palmitoyl-CoA-binding site. Therefore, we modeled the acyl-CoA binding pocket in Atg37, analyzed what features of palmitoyl-CoA might be crucial for its binding to Atg37 and extrapolated these findings to identify similar features in the Atg30 amino acid sequence. Consistent with literature data, our in silico analysis revealed that palmitoyl-CoA occupies the acyl-CoA binding pocket using its hydrophobic adenosine and surrounding phosphates (data not shown). In Atg30-5, which still interacts with Atg37, only one sequence (amino acids 202-215) with such features (a hydrophobic amino acid[s] surrounded by negatively charged residues) was present and, in addition, was evolutionarily conserved in Atg30 homologs (Figure S2). We substituted several different amino acids within this motif to alanine (A) in full-length Atg30, and tested the resulting Atg30 mutants for loss of Atg37 interaction by using the Y2H assay. The strongest interaction defect was observed after substitution in Atg30 of W210 or I212, or both residues simultaneously, whereas substitutions of D202, D205, L206 and I207 had no effect on Atg37 recognition (Figure 5A, 5E). In contrast, none of these substitutions had any effect on Pex3 binding to Atg30 (Figure 5A, 5E). This finding demonstrates that these residues are not involved in Pex3 binding and that the Atg37-binding site in Atg30 is distinct from that of Pex3.
Figure 5.

Identification of Atg37- and Pex3-binding sites in Atg30. (A) Substitutions at W210 or I212 in Atg30 affect interaction of Atg30 with Atg37, but not with Pex3. Y2H assays between AD-Atg30 WT or its mutants (Atg30D202A, Atg30D205A, Atg30L206A, Atg30I207A, Atg30W210A and Atg30I212A) and BD-Atg37(1-278) or BD-Pex3. (B) Substitutions at Y236, L241 and I252 in Atg30-5 affect the interaction of Atg30-5 with Pex3, but not with Atg37. Y2H assays between AD-Atg30-5 WT or its mutants (Atg30-5Y236A, Atg30-5L241A, Atg30-5Y244A and Atg30-5I252A) and BD-Atg37(1-278) or BD-Pex3. (C) The Atg30-1W210,I212A mutant does not bind Atg37 in vitro. Binding studies revealed that only the Atg30-1W210,I212A mutant was deficient in Atg37 binding, whereas the Atg30-1 WT and other mutants (Atg30-1W210A, Atg30-1I212A, Atg30-1Y236A, Atg30-1L241A, Atg30-1I252A and Atg30-1Δ236-244) were still pulled down with GST-Atg37(1-278). Free GST protein was used as a control. There was equivalent loading in the input and bound lanes. (D) Atg30-1Y236A, Atg30-1I252A and Atg30-1Δ236-244 mutants do not bind Pex3 in vitro. Binding studies revealed that only Atg30-1Y236A, Atg30-1I252A and Atg30-1Δ236-244 mutants were deficient in Pex3 binding, whereas Atg30-1 WT and other mutants (Atg30-1W210,212A, Atg30-1I212A, Atg30-1L241A) were still pulled down with GST-Pex3(41-455). The GST-Pex3m(41-455) protein was used as a control. There was equivalent loading in the input and bound lanes. (E) Simultaneous substitution of W210 and I212 in Atg30 strongly affects its interaction with Atg37, but not with Pex3. AD-Atg30W210,212A was used with the previously described BD-Atg37(1-278) and BD-Pex3.12 (F) Deletion of amino acids 236-244 in AD-Atg30-5 disrupts its interaction with BD-Pex3 in a Y2H assay; AD-Atg30-5 and AD-Atg30-5Δ236-244 were used with previously described BD-Pex3.12 (G) Schematic depicting the identified Atg37- (green) and Pex3- (light and dark orange) binding sites in Atg30. Asterisks indicate amino acids in the Atg30-5 region crucial for Atg37 or Pex3 binding.
Because Atg30 residues crucial for Atg37 interaction were not required for binding Pex3, we performed a “reverse” Y2H library screening assay on a mutagenized library of Atg30-5(194-271) to map the Pex3-binding site in the truncated form of Atg30. The reverse Y2H system relies upon usage of a reporter gene that is toxic to growing yeast cells, so the selective pressure is against the formation of a 2-hybrid complex. Thus the “reverse” Y2H, contrary to the classical Y2H approach, specifically facilitates identification of events that dissociate the protein-protein interaction. We used yeast strain MaV203 with URA3 as a reporter gene because the Ura3 protein converts 5-fluoroorotic acid (5-FOA) into a toxic compound, 5-fluorouracil [17]. Thus, the growth of clones with interacting AD- and BD-fusion proteins is inhibited in SD -Leu -Trp medium containing 5-FOA.
Following random mutagenesis using a PCR-based approach, the Atg30-5(194-271) sequences were inserted into an appropriate Y2H vector to generate a library of mutant AD-Atg30-5 chimeras. The library was screened for clones with impaired binding to Pex3, which appeared as 5-FOA-resistant colonies. After screening 5 × 106 clones, we obtained ∼200 clones with impaired binding to Pex3. We further analyzed those with the strongest effect. Binding of Pex3 to the Atg30-5 fragment was inhibited with mutations of residues from amino acids 229-259, suggesting the importance of this region for Pex3 interaction (Figure S3A). Four residues were often mutated in analyzed clones. We substituted them to A and showed that residues: Y236, L241 and I252, but not Y244, were critical for interaction of Atg30 with Pex3 (Figure 5B). We also proved that deletion of amino acids 236-244 of Atg30 had a strong effect on Pex3 binding (Figure 5F). However, due to another Pex3-binding site at the C terminus of Atg30, none of these mutations abolished Pex3 binding by the full-length Atg30 (Figure S3C). We also tested these mutants for Atg37 interaction. All designed Atg30 mutants that lost the ability to bind Pex3 still retained binding to Atg37 (Figure 5B).
Similar results were obtained using in vitro protein binding assays on glutathione Sepharose beads for the most promising mutants from the Y2H analyses. We tested recombinant, HIS-tagged, Atg30-1 truncated forms containing the mutations mentioned above for their interactions with the cytosolic domains of GST-Atg37 and GST-Pex3, and used as controls GST-Pex3m and free GST. We noticed that in the in vitro protein-binding assay, the Atg30-1W210,I212A mutant, which in Y2H studies was clearly recognized by Pex3 but not by Atg37, was also pulled down with GST-Pex3, but not by GST-Atg37. In contrast, mutants Atg30-1Y236A, Atg30-1I252A and Atg30-1Δ236-244, behaved as they did in Y2H studies, gave opposite results, and were bound by Atg37, but not by Pex3 (Figure 5C, 5D).
These results, and the fact that one of the random mutants (mut7) with a stop codon at amino acid 230 was able to bind Atg37, but not Pex3 (Figure S3A), let us conclude that Atg37- and Pex3-binding sites within this middle domain of Atg30 are close, but distinct and separable (Figure 5G).
Close proximity of Atg37- and Pex3-binding sites creates competition between Atg37 and Pex3 for binding to Atg30
As Atg37 and Pex3 have closely-positioned binding sites in the Atg30-5 region, we determined if these separable binding sites are mutually exclusive or not. We performed a Y3H assay and analyzed the interactions between Pex3 and a full-length Atg30 (with both Pex3-binding sites) or an Atg30-2 fragment (with only one Pex3-bindng site that is located near the Atg37–binding site) in the presence or absence of Atg37. Because we showed previously that Atg37 binds both Atg30 and Pex3 (Figure 3, 4B, [12]) and competes with Atg30 for Pex3 (Figure 2C), we eliminated this competition by expressing from the MET25 promoter Atg37(1-125), a truncated Atg37 containing mainly the acyl-CoA domain crucial for Atg30 binding (Figure 6A) [12]. Whereas Pex3 interaction with full-length Atg30 was almost unaffected by the presence of Atg37(1-125), Pex3 interaction with the Atg30-2 truncated form was completely inhibited (Figure 6B). These data suggest hindrance between Atg37 and Pex3 in their binding to Atg30 in the middle region where their binding sites are close to each other. However, Atg37 did not affect Pex3 binding to the C-terminal, Pex3-binding site in Atg30.
Figure 6.
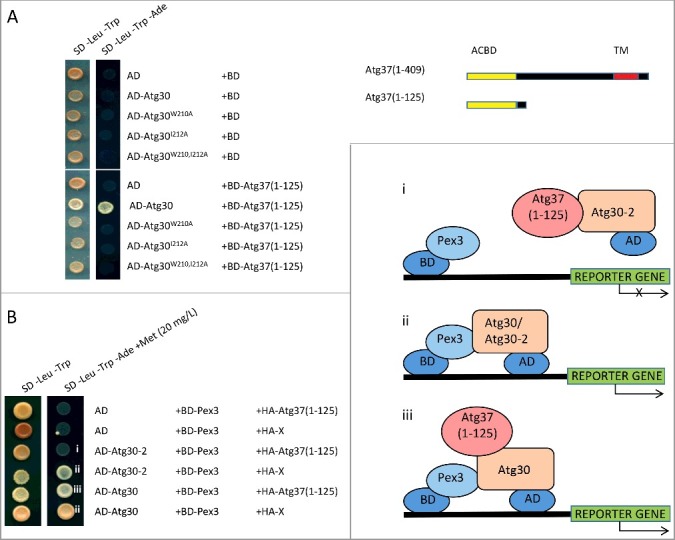
Atg37 competes with Pex3 for binding to Atg30. (A) The acyl-Co domain of Atg37 interacts with the Atg30-5 fragment, but not mutated forms of Atg30 (Atg30-5W210A, AD-Atg30-5I212A or Atg30-5W210,212A) in a Y2H assay. (B) Atg37 displaces Pex3 from the central binding site in Atg30, but does not affect its binding to the C-terminal, Pex3-binding site in Atg30 in a Y3H assay; AD fusions of full-length Atg30 and its truncated form Atg30-2(64-271) were used with the previously described BD-Pex3.12 Expression of HA-Atg37(1-125) and irrelevant protein (X) for protein complex formation was controlled by the MET25 promoter. i. competition between AD-Atg37(1-125) and BD-Pex3 for binding to AD-Atg30-2 in the Y3H assay. There is reduced interaction between AD-Atg30-2 and BD-Pex3 due to competitive binding of HA-Atg37(1-125) to Atg30-2; ii. interaction between AD-Atg30 or AD-Atg30-2 and BD-Pex3 in the Y2H assay; iii. Schematic depicting interactions between AD-Atg30, HA-Atg37(1-125) and BD-Pex3 in the Y2H assay.
Competition between Atg37 and Pex3 was additionally supported by Y3H experiments with Atg30 mutants defective in binding Pex3 or Atg37 (Figure S4). We proved that Atg30-Pex3, or Atg30-Atg37 complex formation, was restored even in the presence of the third protein (Atg37 or Pex3, respectively) by using proper Atg30 mutants that are not recognized by Atg37 or Pex3, respectively. For instance, whereas the interaction between WT Atg30-5 or Atg30-5Y244A, harboring a mutation that had no effect on Pex3 binding, and Atg37(1-278) was strongly inhibited by overexpressed Pex3 (Figure S4A), the Atg30-5Y236A and Atg30-5Δ236-244 mutants affected in Pex3 binding still formed strong complexes with Atg37(1-278) (Figure S4A). Corresponding results were obtained in studies with WT and mutants of full-length Atg30 (Figure S4A). Similarly, whereas the interaction between WT Atg30 and Pex3 was completely inhibited by Atg37(1-278) (Figure S4B), the Atg30W210A and Atg30I212A mutants, defective in binding Atg37, still formed complexes with Pex3 (Figure S4B). However, due to competition between Atg37 and Atg30 for binding to Pex3, the efficiencies of Atg30W210A and Atg30I212A binding to Pex3 were affected by the Atg37(1-278)-Pex3 interaction (Figure S4B).
Functional Atg37- and Pex3-binding sites in the Atg30 middle domain are required for pexophagy
To gain insight into the regulation of Atg30 by its mutually exclusive interaction with Atg37 and Pex3 via its middle domain, we tested the Atg30 mutants affecting each of these binding sites in Atg30 in vivo for their effects on Atg30 phosphorylation and pexophagy.
Consistent with previous data demonstrating that Atg37 is required for proper Atg30 phosphorylation, pexophagy and Atg11 localization on peroxisomes [12], Atg30W210A I212A, which cannot bind Atg37 in vivo (Figure 7D), was weakly phosphorylated (Figure 7A), severely affected in its rate of pexophagy (Figure 7B), and noticeably impaired in the localization of GFP-Atg11 (Figure 7E). This confirms our previous conclusion [12,13] that binding Atg37 to Atg30 activates Atg30 by allowing its phosphorylation and the recruitment of Atg11 to the pexophagic RPC.
Figure 7.
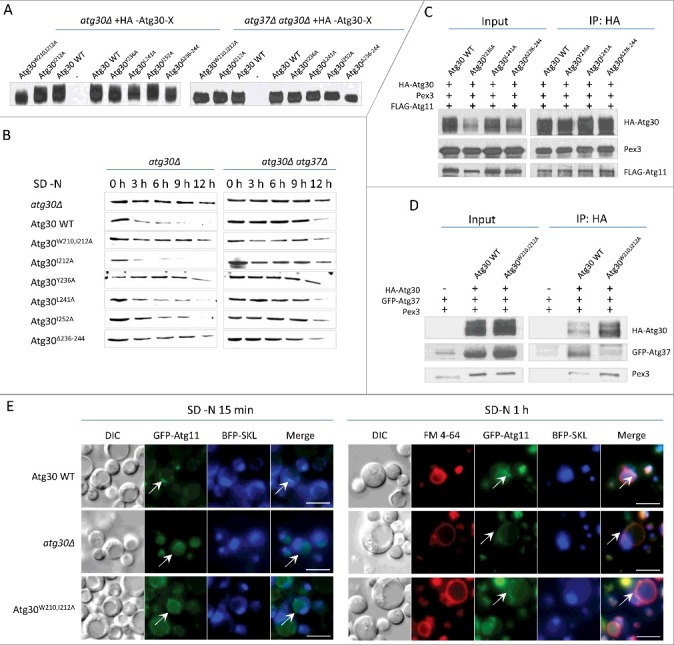
Mutations in Atg30 affect pexophagy, Atg30 phosphorylation and Atg11 recruitment. (A) Phosphorylation status of Atg30 WT and different Atg30 mutants expressed in atg30Δ or atg30Δ atg37Δ cells (sKZR009-20 and sKZR030-31; Table S3) was analyzed by western blot after 15 h of methanol induction. (B) Pexophagy assay for Atg30 mutants. Methanol-grown cells expressing Atg30 WT or different Atg30 mutants expressed in atg30Δ and atg30Δ atg37Δ cells (sKZR009-20 and sKZR030-31; Table S3) were adapted to SD-N medium. Cell lysates were prepared as described in Materials and Methods and resolved by SDS-PAGE. Western blotting was performed with antibodies against P. pastoris Aox1. (C) Atg30Y236A, Atg30L241A and Atg30Δ236-244 mutants co-immunoprecipitate with Atg11. Methanol-grown ypt7Δ cells expressing FLAG-Atg11 and Atg30WT, Atg30Y236A, Atg30L241A or Atg30Δ236-244 mutants (sKZR026-29 - see Table S3), were adapted to SD-N medium for 0.5 h to induce pexophagy before the co-IP:HA experiment was conducted. Immunoprecipitated proteins were visualized with anti-HA, anti-FLAG and anti-P. pastoris Pex3 antibodies. (D) The Atg30W210,212A mutant has severely reduced ability to co-immunoprecipitate with Atg37, but not with Pex3. Methanol-grown ypt7Δ atg30Δ cells expressing GFP-Atg37 and cells expressing GFP-Atg37 and Atg30 WT or the Atg30W210,212A mutant (sTN566, sKZR021and sKZR022; Table S3), were adapted to SD-N medium for 0.5 h to induce pexophagy before the co-IP:HA experiment was conducted. Immunoprecipitated proteins were visualized with anti-HA, anti-GFP and anti-P. pastoris Pex3 antibodies. (E) Interaction between Atg37 and Atg30 is required for localization of Atg11 in close proximity to peroxisomes. Methanol-grown Atg30 WT, atg30Δ and Atg30W210,212A cells expressing BFP-SKL and GFP-Atg11 (sTN640, sKZR023and sKZR024; Table S3) were adapted to SD-N for 15 min or 1 h and stained with FM 4-64 for vacuole visualization. White arrowheads indicate GFP-Atg11 localization. Scale bars: 5 µm.
In addition, strains expressing the Atg30Y236A and Atg30Δ236-244 mutants, which showed a greatly reduced ability to bind Pex3 in the middle domain in Y2H and in vitro binding studies, had impaired pexophagy (Figure 7B). However, these mutants did not exhibit as strong and significant differences in phosphorylation of Atg30 as did the strain expressing Atg30W210,I212A (Figure 7A). Interestingly, Atg11 recruitment to the RPC was not affected in these mutants (Figure 7C). Therefore, we conclude that Pex3 binding to the middle domain of Atg30 is not required for the initiation of pexophagy, at least until Atg11 binds to Atg30, but is essential downstream of this step.
Finally, because in the absence of Atg37, Pex3-binding mutants did not show improved phosphorylation of Atg30 (Figure 7A) or enhanced rates of pexophagy (Figure 7B), Atg37 must play an additional role in pexophagy than just the displacement of Pex3 from Atg30.
Atg30 binds to, and is phosphorylated by, Hrr25 kinase
We reported previously that Atg30 is phosphorylated by an unknown kinase(s) that triggers pexophagy. We proposed consensus sequences for Atg11- and Atg8-binding sites in P. pastoris Atg30 and Atg32, as well as in S. cerevisiae Atg36 and Atg32, which undergo phosphoregulation [9]. Although the kinase(s) involved in the Atg8-binding site phosphorylation remains undiscovered, the Hrr25-mediated phosphorylation of serine residues located within the Atg11-binding motif of ScAtg36 promotes its interaction with Atg11 [10]. Because the amino acid phosphorylated by Hrr25 within the Atg11-binding motif of Atg36 is conserved in Atg30, we asked whether Atg30 function is also controlled by Hrr25-dependent phosphorylation. First, we asked if PpHrr25 selectively recognizes bona fide substrates in vivo in the Y2H system. Whereas ScAtg36 interacted with Hrr25 from P. pastoris, ScAtg32 did not, suggesting conservation of the mechanism between the 2 yeast species (Figure 8A). We also found that Hrr25 interacted with Atg30 (Figure 8B).
Figure 8.
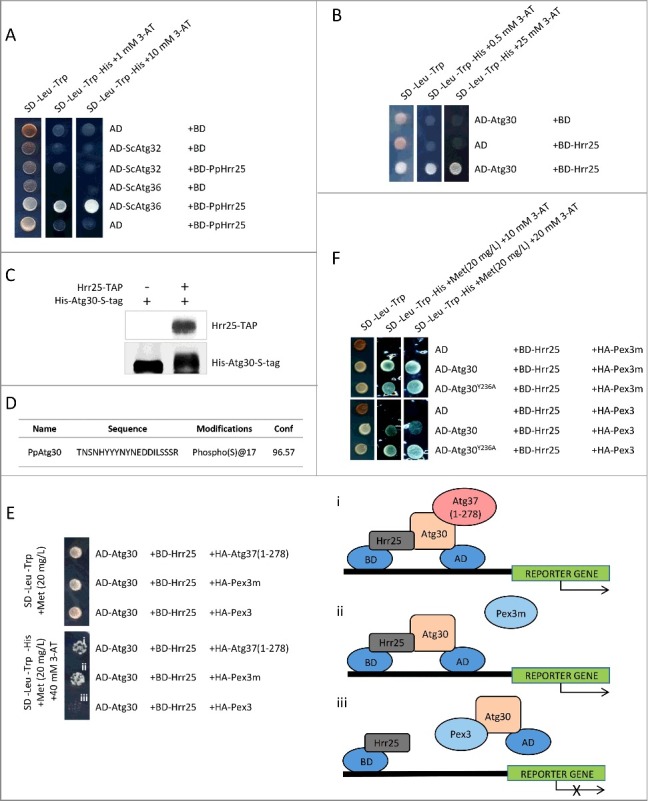
Hrr25 phosphorylates Atg30 and Pex3 inhibits the interaction of Atg30 with Hrr25. (A) Hrr25 from P. pastoris interacts with the pexophagy receptor, Atg36, but not with the mitophagy receptor, Atg32, from S. cerevisiae in a Y2H assay; BD-PpHrr25 was used with the previously described AD-ScAtg36 and AD-ScAtg32.9 (B) Atg30 interacts with Hrr25 in a Y2H assay; AD-Atg30 was used with BD-Hrr25. (C) Atg30 mobility shift on an SDS-PAGE gel after in vitro phosphorylation with Hrr25 kinase. Proteins were visualized with anti-calmodulin-binding peptide and anti-HIS tag antibodies; (D) Atg30 is phosphorylated at S112 by Hrr25 in vitro; MS2 data revealing the S112 phosphopeptide. (E) Pex3, but not Pex3m or Atg37, affects Atg30 interaction with Hrr25 in a Y3H assay. Expression of HA-Atg37(1-278), HA-Pex3 or HA-Pex3m was controlled by the MET25 promoter and a low level of methionine in the medium (20 mg/L) was used to express moderate levels of these proteins. i. Schematic depicting interaction between BD-Hrr25 and AD-Atg30 in the presence of HA-Atg37(1-278) in a Y3H assay. Binding of Atg37(1-278) to AD-Atg30 does not affect Atg30-Hrr25 complex formation and, therefore, reporter gene expression. ii. Schematic depicting interaction between BD-Hrr25 and AD-Atg30, which is unaffected by the presence of HA-Pex3m in the Y3H assay. iii. Schematic depicting depletion of interaction between BD-Hrr25 and AD-Atg30 in the presence of HA-Pex3 in the Y3H assay. (F) Disruption of the Pex3-binding site located in close proximity to the Atg37-binding site in Atg30 restores recruitment of Hrr25 to Atg30 even in the presence of Pex3 in a Y3H assay; AD-fusions of WT Atg30 or its mutants (Atg30Y236A, Atg30L241A and Atg30Y244A) were used with BD-Hrr25. Expression of HA-Pex3 or HA-Pex3m was controlled by the MET25 promoter.
Next we addressed whether Hrr25 phosphorylates Atg30 directly and at which sites. We subjected Atg30, purified from E. coli, to in vitro phosphorylation by TAP-Hrr25 kinase precipitated from P. pastoris cells grown in pexophagy conditions. Atg30 was intensively phosphorylated by TAP-Hrr25 in vitro as visualized by the slower mobility of Atg30 during SDS-PAGE, in contrast to Atg30 from a control sample lacking TAP-Hrr25 (Figure 8C). Moreover, mass spectrometry analysis detected phosphorylation at S112, previously suggested as a crucial site in Atg30 for Atg11 binding (Figure 8D) [9]. This result demonstrates that in P. pastoris, similar to S. cerevisiae, the Hrr25 kinase phosphorylates the pexophagy receptor at a site that is not only important for its interaction with Atg11 but is also a prerequisite for triggering pexophagy.
Pex3 binding to the middle domain of Atg30 inhibits the interaction of Atg30 with Hrr25
We showed previously that phosphorylation of Atg30 is important for its interaction with Atg11, and Atg37 binding to Atg30 facilitates this phosphorylation. To understand why the replacement of Pex3 by Atg37 on Atg30 is crucial for the pexophagic RPC, we conducted a Y3H assay. We tested Atg30 binding to Hrr25 in the presence of Atg37, Pex3 or Pex3m, which was used here as a reference control because Pex3m is defective in binding Atg30 (Figure 8E). We noticed that co-expression of Atg37, as well as Pex3m, did not affect Atg30 binding to Hrr25. However, expression of Pex3 significantly decreased the interaction of Atg30 with Hrr25 (Figure 8E).
Next, we asked if interaction between Atg30 and Hrr25 kinase in the presence of Pex3 could be restored by mutation of the Pex3-binding site located in close proximity to the Atg37-binding site. To address this question, we replaced Y236 in Atg30 and tested the latter's interaction with Hrr25 co-expressed with either Pex3 or Pex3m. Previously, we showed that single substitution at Y236 disrupts the Pex3-binding site, which is next to the Atg37-binding site, without affecting the C-terminal Pex3-binding site in Atg30 (Figure 5 and Figure S3). As expected, WT Atg30 interacted with Hrr25 in the presence of Pex3m, but failed to interact with Hrr25 in the presence of Pex3 (Figure 8F). However, the Atg30Y236A mutant regained its interaction with Hrr25, even in the presence of Pex3, to the level similar to that observed in the presence of Pex3m (Figure 8F).
In sum, these observations shed light regarding how Atg37 and Pex3 regulate Atg30 during the formation of the active pexophagic RPC (Figure 9). These results indicate that Pex3 binding to Atg30 negatively regulates the Atg30 interaction with Hrr25. This interaction might prevent premature initiation of pexophagy, but might also be required to terminate pexophagy. When Atg37 binds to Atg30, displacing Pex3 in the process from the vicinity of this Atg37-binding region in Atg30, then the negative regulation of Atg30 phosphorylation by Pex3 is bypassed, thereby allowing Hrr25 recruitment to Atg30, followed by Atg30 phosphorylation at S112, culminating in Atg11 recruitment. However, downstream of this step, displacing Atg37 by Pex3 seems to be crucial for completion and termination of pexophagy.
Figure 9.
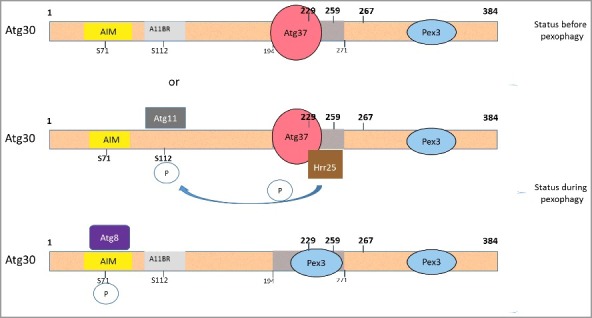
Pex3 competes with Atg37 for Atg30 to oppose the interaction between Atg30 and Hrr25 kinase. Schematic diagrams of Atg30 with its interacting proteins and steps during pexophagy. When Atg37 is recruited to the peroxisomal membrane in a Pex3-dependent manner, Atg30 engages Atg37 at the pexophagic RPC and Pex3 is displaced or excluded from the Atg30 middle domain (amino acids 194-271). It is currently unknown whether the 2 Pex3-binding sites on Atg30 are occupied by 2 distinct, or a single, Pex3 molecule. The displacement of Pex3 by Atg37 at their adjacent binding sites in the middle domain on Atg30 enables binding of Hrr25 to Atg30, allowing thereby the phosphorylation of Atg30 at the Atg11-binding site, followed by the recruitment of Atg11. When Atg30 interacts with Pex3, via its middle domain, it creates a state that is unfavorable for Hrr25 binding, allowing a second phosphorylation at S71, by an unknown kinase to permit Atg8 recruitment to Atg30. In our model, the uppermost scheme represents the situation before pexophagy; and the next 2 schemes are during pexophagy. AIM, Atg8-interaction motif; A11BR, Atg11 binding region.
Discussion
Atg37 and Atg37-bound Pex3 cooperate in the same linear pathway to activate Atg30
We studied here the role of Atg37 and Pex3 in the assembly of the pexophagic RPC. We have shown previously that in atg37Δ and pex3m cells pexophagy is disrupted due to weak phosphorylation of Atg30, which, in turn, impairs recruitment of Atg11 to the pexophagic RPC. Based on the atg37Δ mutant phenotype, we speculated previously that Atg37 might be also involved in facilitating Atg30-Atg8 binding for efficient phagophore formation. Here, we found that both Atg37 and Pex3 are required for the recruitment of both autophagic proteins, Atg11 and Atg8, to Atg30.
In the absence of Atg37, as well as in pex3m cells, Atg30 was only weakly phosphorylated and unable to recruit these proteins. Although our previous studies linked the pexophagy defect in pex3m cells with direct inhibition of Atg30-Pex3 complex formation [13], our analysis here shows that the pexophagy phenotype in pex3m cells could be explained by impairment of subcomplex formation between Atg37 and Pex3m, rather than by lack of direct binding between Atg30 and Pex3m. This conclusion explains why we observed identical phenotypes—weak phosphorylation of Atg30, inability to recruit Atg8 and Atg11 to Atg30, lack of phagophore elongation and pexophagy defects—in cells either lacking Atg37 or in those expressing Pex3m. In other words, these phenotypes simply reflect the failure of each side of a pairwise interaction required for pexophagy.
What is the role of this Atg37-Pex3 interaction in pexophagy? Atg37-Pex3 complex formation is crucial for the delivery of Atg37 to the peroxisome membrane. We observed that in pex3m cells the level of Atg37 protein was lower, its transport to the peroxisomal membrane was strongly diminished and, in consequence, peroxisomal localization of Atg37 was noticeably reduced. Because Atg37 is required at the peroxisome to be involved in phagophore formation during pexophagy, mislocalization of Atg37 impairs both pexophagic RPC assembly and pexophagy in pex3m cells. We also noticed that proper localization of some, but not the all, Pex3 on the peroxisome cluster periphery depended on Atg37 (Figure 2), which itself requires Atg30 for such localization [12]. These findings shed light on pexophagic RPC assembly in P. pastoris and demonstrate that Atg37 and Atg37-bound Pex3 cooperate in the same linear pathway to activate Atg30.
Another important outcome from these studies was that Atg30, Atg37 and Pex3 have pairwise interactions that compete with each other. This explains our previous results, which showed that, similar to Atg30, Atg37 directly binds Pex3 and the co-immunoprecipitation of Atg37 with Pex3 is even stronger in the absence of Atg30 [12]. We proved that Atg37 competes with Atg30 for Pex3. Finally, the binding of Pex3 or Atg37 to Atg30 has distinct consequences in regulating the recruitment of Hrr25 kinase to Atg30.
These conclusions emphasize a positive role for Pex3 in the initiation of pexophagy; namely, the recruitment of Atg37 to peroxisome membranes and, subsequently, the proper assembly of the RPC, which is crucial for pexophagy.
Atg30-bound Pex3 downregulates the binding of Atg30 to Hrr25
Previous work from our group revealed the existence of a consensus sequence for Atg11 binding in several autophagic receptors, including Atg30 from P. pastoris. Recent studies showed that Hrr25-mediated phosphorylation of autophagic receptors in the Atg11-binding motif increased the interactions of these receptors with Atg11. This interaction of the cargo-receptor complex with Atg11 is necessary for the activation of the Atg1 kinase, which is necessary for autophagy [18,19]. It was shown that S97 of ScAtg36, which is conserved in Atg30 (S112), is phosphorylated by Hrr25 kinase and mutation at this position causes severe defects in the Atg11 recruitment to the pexophagic RPC, and thus affects pexophagy [10]. We show here that Hrr25 is the kinase that phosphorylates Atg30 at S112 and some other sites, but not at the Atg8-binding site in vitro (Figure 8C, 8D), but our inability to generate a Hrr25-deficient strain prevents the extension of this conclusion definitively in vivo. We demonstrated that Hrr25 intensely phosphorylated Atg30 in vitro, also at S112, which was necessary for Atg11 binding. This shows that pexophagy in P. pastoris may be regulated by a conserved and conceptually identical mechanism as in S. cerevisiae.
Our data also demonstrate why recruitment of Atg37 is required for pexophagy, uncovering its mechanistic role and shedding light on the role for Pex3 in counteracting the influence of Atg37. We mapped the Atg37-binding site and identified 2 amino acids (W210 and I212) crucial for interaction of Atg30 with Atg37 (Figure 5). We also proved the existence of 2 Pex3-binding regions (amino acids 229-259 and 267-384) in Atg30 (Figure 4). The Atg37-binding motif is located in close proximity to a centrally located binding site for Pex3 (amino acids 229-259) and recruitment of both Atg37 and Pex3 to this region of Atg30 is mutually exclusive (Figure 6, S4). Because Atg37 binding to Atg30 is instrumental for positive Atg30 phosphoregulation and pexophagic RPC assembly, we studied how Pex3 regulates the Hrr25 interaction with Atg30 and found that Pex3 bound to Atg30 severely inhibited the recognition of Atg30 by Hrr25 (Figure 8E, 8F). We suggest that recruitment of Atg37 to the pexophagic RPC displaces Pex3 and relieves this inhibition of Hrr25 recruitment to Atg30 by Pex3, thereby allowing phosphorylation of S112 by Hrr25 and further recruitment of Atg11. However, at a later step of pexophagy, Pex3 rebinding to the middle domain of Atg30 may be a prerequisite for Hrr25 dissociation from the pexophagic RPC. This point is suggested by the fact that Atg30 mutants that could bind Atg37, but not Pex3 in the middle domain, were still impaired in pexophagy (Figure 7B), despite the fact that Atg11 recruitment, and by implication Hrr25 phosphorylation at Atg30(S112), were normal (Figure 7C). These data strongly suggest the requirement of an additional Pex3-binding step to the middle domain of Atg30, perhaps to displace Hrr25, and allow phosphorylation at Atg30 (S71) and/or Atg8 recruitment.
In conclusion, our results allow us to expand our previous model to explain how pexophagic RPC formation is regulated. We propose a new model (Figure 9). When Atg37 is recruited to the peroxisomal membrane in a Pex3-dependent manner, Atg30 engages Atg37 at the RPC. The displacement of Pex3 by Atg37 in the middle domain of the Atg30 protein enables binding of Hrr25, phosphorylation of Atg30 at S112 and recruitment of Atg11 to the pexophagic RPC. Therefore, we suggest that Atg37 and, indirectly, the Atg37-bound Pex3 regulate the phosphostatus of Atg30 by allowing recognition of Atg30 by at least one kinase, Hrr25. Further, since we know that Atg8 and Atg11 cannot interact simultaneously with the autophagic receptors [9], Pex3 rebinding to the middle domain of Atg30 might cause Atg37 and Hrr25 dissociation, allowing further Atg30 modulations, such as phosphorylation/dephosphorylation events culminating in Atg8 recruitment.
Our study on the assembly of the pexophagic RPC in P. pastoris raises several new questions. For instance, the pexophagic RPC architecture might be of major importance to understand the biological role of its components. In the future, more attention should be dedicated to the stoichiometry of this protein complex to clarify how many Pex3 molecules bind to each Atg30. It is plausible that 2 distinct Pex3 molecules occupy 2 Pex3-binding sites, or one Pex3 molecule interacts simultaneously with both sites in Atg30. It is still unknown what kinase phosphorylates the Atg8-binding site in Atg30 (and other selective autophagic receptors) and how recruitment of this kinase is regulated precisely. Further investigation of phosphorylation/dephosphorylation events on Atg30 is crucial for proper understanding of the consecutive steps of pexophagy.
Materials and methods
Plasmid construction
Plasmids used in this work are listed in Tables S1 and S2. Details on their construction are available upon request. For pDEST plasmid construction, cDNA cloning into pENTR™/D-TOPO vector and Gateway LR recombination reactions were done as described in the Gateway® Technology—a universal technology to clone DNA sequence for functional analysis and expression in multiple systems (Invitrogen, 12535-019 and 12535-027). For site-directed mutagenesis the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, 200523) was used. All plasmids were checked by restriction digestion and/or by DNA sequencing. Conventional techniques were used for Escherichia coli transformation [20].
Yeast cells and transformation
The P. pastoris strains used in this work are in Table S3. Media used to grow strains include: YPD (1% wt:vol yeast extract, 2% wt:vol peptone, and 2% wt:vol glucose); nitrogen starvation medium or SD-N (1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 2% wt:vol glucose); and methanol medium (1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate, 0.05% wt:vol yeast extract, 0.5% wt:vol ammonium sulfate, 0.5% vol:vol methanol). Histidine (50 mg/L) and/or arginine (50 mg/L) were added when needed. All cultures were grown at 30°C. YNB solution (1.7 g/L yeast nitrogen base without amino acids and ammonium sulfate) was used to wash cells. Cells were transformed by electroporation, as described previously [21].
Protein in vitro binding assay
Protein in vitro binding assay was done as described previously [12,13]. In brief, HIS-tagged proteins (Table S2) were purified from E. coli BL21 (DE3) cells or Rosetta (DE3) cells by Ni-NTA affinity chromatography. After cells reached OD600 = 0.4-0.6 IPTG (Sigma-Aldrich, I6758) at a concentration of 0.4 mM was used for 3 h to induce protein expression. Next, cells were collected, resuspended in ice-cold lysis buffer (50 mM Tris-HCl, pH 7.5, 300 mM NaCl) with protease inhibitors (Sigma-Aldrich, P-8465), disrupted by sonication and centrifuged at 10,000 g for 10 min at 4°C. The supernatant was incubated 1 h at 4°C with Ni-NTA agarose (Qiagen, 30210). After that the beads were washed 5 times with lysis buffer containing 50 mM imidazole and bound protein was collected using elution buffer (300 mM imidazole in 50 mM Tris-Cl, pH 7.5). To purify GST-tagged proteins (Table S2) the E. coli Rosetta (DE3) cells were used and glutathione affinity chromatography. Cells were induced with 0.4 mM IPTG for 2 h. Next cells were collected, resuspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% NP-40 [Abcam, ab142227]) with protease inhibitors (Sigma-Aldrich, P-8465), disrupted by sonication and centrifuged at 10,000 g for 10 min at 4°C. The supernatant was incubated 1 h at 4°C with the glutathione Sepharose 4B medium (GE Healthcare, 17075601). Then, the beads were washed 5 times with lysis buffer, and bound protein was collected using elution buffer (10 mM reduced L-glutathione (Sigma-Aldrich, G4251) in 50 mM Tris-HCl, pH 8.0).
Purified recombinant proteins HIS-Atg37(1-278), GST-Pex3(41-455) and GST-Pex3m(41-455) were quantified by Coomassie Brilliant Blue staining. For the Atg37-Pex3 in vitro binding assay, we mixed 56 pmol of HIS-Atg37ΔC (0.28 µM) and 14 pmol of GST-Pex3(41-455) or GST-Pex3m(41-455) (0.07 µM) in 200-µl binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% vol:vol NP-40) for 1 h at 4°C. For the in vitro binding assay between Atg30-1 mutants and Atg37 or Pex3, equal volumes (200 µl) of each whole protein lysate containing HIS-Atg30-1WT or different HIS-Atg30-1 mutant proteins were mixed with 21 pmol of GST-Atg37(1-278), free GST, GST-Pex3(41-455) or GST-Pex3m(41-455) in 200-µl binding buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% vol:vol NP-40) for 1 h at 4°C.
For each binding assay, proteins were incubated 1 h at 4°C with 50 μl of prewashed glutathione-Sepharose 4B (GE Healthcare, 17075601) to bind the GST-tagged proteins. Then, the beads were washed 5 times with ice-cold binding buffer. Proteins were eluted with 50 µl of 2 × SDS loading buffer and boiled for 5 min. Input samples were mixed separately with 2 × SDS loading buffer and boiled for 5 min. Samples were loaded on an SDS-PAGE gel and analyzed by immunoblotting with anti-HIS·tag (Roche Life Science, 11922416001) or anti-GST antibodies (Novagen, 71097).
Immunoprecipitation of proteins
Immunoprecipitation assays were performed as described previously [6,12]. In brief, for immunoprecipitation of Atg30-TAP or HA-Atg30, cells were grown in methanol medium for 17 h or 15 h, respectively, and 100 OD or 50 OD, respectively, of cells were transferred to glucose medium without nitrogen for 0.5 h. For immunoprecipitation of Atg37-2xHA 100 OD of cells were induced in methanol medium for 4 h. Then cells were washed in Dulbecco's PBS (Gibco, 14190), lysed as above with 250 µl of glass beads in 1 ml of IP buffer (1% wt:vol CHAPS [Sigma-Aldrich, C3023], 50 mM HEPES-KOH, pH 7.2, 150 mM NaCl, 1 mM EDTA, 10% vol:vol glycerol, 50 mM NaF, 1 mM protease inhibitors [Sigma-Aldrich, P8215]), and centrifuged at 500 g for 10 min. Protease inhibitors used were: PMSF (Roche, 10837091001), leupeptin (Sigma-Aldrich, L2884), aprotinin (Sigma-Aldrich, A-3428), and a protease inhibitor cocktail (Sigma-Aldrich, P8215). To break the cells, samples were thawed on ice followed by 10 cycles of vortexing (2 min at maximum speed followed by 2 min on ice). The supernatant was incubated at 4°C for 30 min with rotation to solubilize membrane proteins and centrifuged at 21,000 g for 10 min. Cleared cell lysate was incubated 1 h at 4°C with 100 µl of washed human IgG-agarose (Sigma-Aldrich, A6284) (IP:TAP) or with 50 µl of washed EZview Red Anti-HA Affinity Gel (Sigma-Aldrich, E6779) (IP:HA). Then, beads were washed 5 times with IP buffer, mixed with 100 µl (IP-TAP) or 50 µl (IP-HA), respectively, of 2 × SDS-loading buffer, and boiled for 5 min. Samples were analyzed by immunoblotting with input and IP corresponding to 0.2 and 7.5 OD of cells per lane, respectively. Protein gel blots were performed with appropriate antibodies: Anti-HA High Affinity Antibody (Roche Life Science, 11867423001), anti-FLAG, M2 antibody (Sigma-Aldrich, F3165), anti-calmodulin binding protein epitope tag antibody (Upstate, 05-173), and anti-GFP (Roche Life Science, 11814460001).
Phosphorylation and pexophagy studies
For pexophagy assays, yeast cells were pre-grown in YPD, washed twice with YNB solution and transferred to 25 ml of methanol medium to induce peroxisome biogenesis with a starting OD600 = 0.3 for 15 h. Then, cells were washed twice with YNB solution and transferred to 25 ml of SD-N at an OD600 = 2 to induce pexophagy. At each time-point, 1 ml culture samples were taken for TCA precipitation (described previously [22]). Western blotting was performed with antibodies against P. pastoris AOX1. For phosphorylation studies, samples from the 0-h time-point were analyzed by western blotting with anti-HA antibody (Roche Life Science, 11867423001).
In vitro phosphorylation assay and sequencing
Recombinant HIS-Atg30 was induced and purified as described above. P. pastoris cells expressing or lacking Hrr25-TAP were grown in methanol medium for 15 h and 100 OD of cells were transferred to SD-N medium for 0.5 h, then 100 OD of cells were washed in Dulbecco's PBS (Gibco, 14190). These cells were lysed with 250 µl of acid-washed glass beads in 1 ml of IP buffer described above and centrifuged at 500 g for 10 min. The supernatant was incubated at 4°C for 30 min with rotation, centrifuged at 21,000 g for 10 min and incubated 1 h at 4°C with 100 µl of washed human IgG-agarose (Sigma-Aldrich, A6284). Next, beads were washed 5 times with IP buffer followed by 5 times with kinase buffer (20 mM HEPES-KOH, pH 7.2, 150 mM NaCl, 1 mM MgCl2, 0.2 mM DTT, 1 mM ATP [Sigma-Aldrich, A1852]). Then, purified HIS-Atg30 protein (0.28 µM) was added to the TAP-bead complex and incubated at 30°C for 1 h. Finally, samples were eluted with 100 µl of 2× loading buffer, boiled and subjected to SDS-PAGE gel and immunoblotting. Protein gel blots were performed with appropriate antibodies: Anti-calmodulin binding protein epitope tag antibody (Upstate, 05-173), anti-HIS tag antibody (Roche Life Science, 11922416001). Bands corresponding to HIS-Atg30 were excised from the gel and subjected to mass spectrometry.
Protein interactions in Y2H, Y3H and “reverse” Y2H systems
For Y2H and Y3H analysis the GAL4-based Matchmaker yeast 2-hybrid system (Clontech Laboratories Inc.) was used. Full-length open reading frames or truncated forms were inserted in pGAD-GH (Clontech Laboratories, 638853), pGADT7-AD (Clontech Laboratories, 630442) or pDEST-GADT7 (Arabidopsis Biological Resource Center, CD3-763; Shaw Laboratory) (AD) and pGBT9 (Clontech Laboratories, K1605-A), pGBKT7 (Clontech Laboratories, 630443), pDEST-GBKT7 (Arabidopsis Biological Resource Center, CD3-764; Shaw Laboratory) (BD) plasmids. The pBridge plasmid (Clontech Laboratories, 630404) for co-expression of a BD fusion protein and a third protein with a nuclear localization sequence and HA-tag was used for the 3-hybrid assay. All plasmids are listed in Table S1. The S. cerevisiae strains AH109 and Y2H gold were used. For transformation of yeast the LiAc-single-stranded carrier DNA-PEG method [23] was used following the “Quick and Easy TRAFO Protocol” (https://home.cc.umanitoba.ca/≫gietz/Quick.html). After transformation yeast were spread on synthetic dropout (SD) medium (-Leu, -Trp) (Sunrise Science, 1719) to select for transformants containing the introduced plasmids. Plates were incubated at 30°C for up to 5 d. Next 3 representative transformants from each strain were plated on proper selective media: SD medium (-Leu, -Trp) (Sunrise Science, 1719), SD medium (-His, -Leu, -Trp) (Sunrise Science, 1725) w/ or w/o 3-amino-1,2,4-triazole (Sigma-Aldrich, A8056) at the concentration indicated in the figure and/or SD medium (-Leu, -Trp, -Ade) (Sunrise Science, 1754) to test protein interactions. Additionally, in Y3H assays expression of the third protein was achieved by using media with low (20 mg/L) (Sunrise Science, 1719) or lack of methionine (Met) (Sunrise Science, 1781, 1776-supplemented with Ade (20 mg/L)). Results were analyzed after 3-7 d.
For the “reverse” Y2H analysis Atg30-5 (amino acids 194-271) was randomly mutagenized by a PCR-based random mutagenesis approach described previously [24], and inserted into the pDEST-GADT7 vector (Arabidopsis Biological Resource Center, CD3-763; Shaw Laboratory) to generate a library of mutant AD-Atg30-5 chimeras. The S. cerevisiae strain MaV203 was first transformed by the LiAc-single-stranded carrier DNA-PEG method [23] following the “Quick and Easy TRAFO Protocol” with pKSN65 (Table S1). After selection and verification MaV203 yeast cells containing BD-Pex3 were transformed with the library using the lithium acetate transformation method described in the Yeast Protocols Handbook (BD Biosciences Clontech, PT3024-1). After transformation yeast were spread on SD medium (-Leu -Trp) (Sunrise Science, 1719) supplemented with 0.2% 5-fluoroorotic acid (5-FOA; Sigma-Aldrich, F5013) to select transformants with disrupted interaction between BD-Pex3 and AD-Atg30-5. Plates were incubated at 30°C for up to 7 d.
Fluorescence microscopy
P. pastoris cells were grown to late exponential phase in YPD medium, diluted to OD600 of 0.2 with fresh YPD, and grown to the early/mid-exponential phase. Next, cells were washed twice with YNB solution and inoculated into peroxisome proliferation (methanol) medium for 15 h and, if necessary, washed as above and switched to SD-N for 15 min or 1 h with FM 4-64 (Life Technologies, T3166) for vacuole visualization and immediately taken for microscopy observation. Images were captured at room temerature using a motorized fluorescence microscope (Axioskop 2 MOT) with a Plan-Apochromat 100 × /1.40 NA oil differential interference contrast (DIC) objective lens and monochrome digital camera (AxioCam MRm; all from Carl Zeiss). Optimal exposition times were automatically applied to capture images. All images were acquired and processed using the AxioVision software (Carl Zeiss), version 4.8.2.
In silico analysis
A sequence similarity search for the Atg30 protein was performed as a Standard Protein BLAST analysis on a dataset of the nonredundant (nr) protein sequences, using the default parameter and the algorithm blastp (protein-protein BLAST) at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/BLAST/). The organism names, abbreviations and GenBank accession numbers of Atg30 homologs are the following: Ogataea polymorpha (Op, XP_018208520.1), Candida arabinofermentans (Ca, ODV85315.1), Kuraishia capsulata (Kc, CDK26512.1), Pachysolen tannophilus (Pt, ODV98102.1) and Cyberlindnera fabianii (Cf, CDR46266.1). Sequences were aligned using Multalin (http://multalin.toulouse.inra.fr/multalin/) [25]. The primary structure of the peptide was drawn using PepDraw (http://pepdraw.com/).
Supplementary Material
Funding Statement
This work was supported by the HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) [grant number DK41737]; UC San Diego Frontiers of Innovation Scholars Program (FISP) [grant number P0034]; European Molecular Biology Organization (EMBO) [grant number EMBO ALTF 1238-2014]; HHS | NIH | National Institute of General Medical Sciences (NIGMS) [grant number GM119571].
Acknowledgements
This work was supported by NIH grants DK41737 (to SS) and GM119571 (to TYN). KZR was supported by an EMBO fellowship and a UCSD FISP grant. The yeast strain overexpressing Hrr25-TAP was provided by Dr. Wei Wang. We thank Dr. Jean-Claude Farré for strains, advice and critical reading of the manuscript.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Yorimitsu T, Klionsky DJ. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol Biol Cell. 2005;16:1593–1605. doi: 10.1091/mbc.E04-11-1035. PMID:15659643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24:24–41. doi: 10.1038/cr.2013.168. PMID:24366339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kraft C, Reggiori F, Peter M. Selective types of autophagy in yeast. Biochim Biophys Acta. 2009;1793:1404–1412. doi: 10.1016/j.bbamcr.2009.02.006. PMID:19264099 [DOI] [PubMed] [Google Scholar]
- [4].Zientara-Rytter K, Subramani S. Autophagic degradation of peroxisomes in mammals. Biochem Soc Trans. 2016;44:431–440. doi: 10.1042/BST20150268. PMID:27068951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17:537–552. doi: 10.1038/nrm.2016.74. PMID:27381245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Farre JC, Manjithaya R, Mathewson RD, et al. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev Cell. 2008;14:365–376. doi: 10.1016/j.devcel.2007.12.011. PMID:18331717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Motley AM, Nuttall JM, Hettema EH. Atg36: the Saccharomyces cerevisiae receptor for pexophagy. Autophagy. 2012;8:1680–1681. doi: 10.4161/auto.21485. PMID:22874561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Motley AM, Nuttall JM, Hettema EH. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 2012;31:2852–2868. doi: 10.1038/emboj.2012.151. PMID:22643220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Farre JC, Burkenroad A, Burnett SF, et al. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 2013;14:441–449. doi: 10.1038/embor.2013.40. PMID:23559066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Tanaka C, Tan LJ, Mochida K, et al. Hrr25 triggers selective autophagy-related pathways by phosphorylating receptor proteins. J Cell Biol. 2014;207:91–105. doi: 10.1083/jcb.201402128. PMID:25287303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nazarko TY. Atg37 regulates the assembly of the pexophagic receptor protein complex. Autophagy. 2014;10:1348–1349. doi: 10.4161/auto.29073. PMID:24905344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Nazarko TY, Ozeki K, Till A, et al. Peroxisomal Atg37 binds Atg30 or palmitoyl-CoA to regulate phagophore formation during pexophagy. J Cell Biol. 2014;204:541–557. doi: 10.1083/jcb.201307050. PMID:24535825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Burnett SF, Farre JC, Nazarko TY, et al. Peroxisomal Pex3 activates selective autophagy of peroxisomes via interaction with the pexophagy receptor Atg30. J Biol Chem. 2015;290:8623–8631. doi: 10.1074/jbc.M114.619338. PMID:25694426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pfaffenwimmer T, Reiter W, Brach T, et al. Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO Rep. 2014;15:862–870. doi: 10.15252/embr.201438932. PMID:24968893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mochida K, Ohsumi Y, Nakatogawa H. Hrr25 phosphorylates the autophagic receptor Atg34 to promote vacuolar transport of alpha-mannosidase under nitrogen starvation conditions. FEBS Lett. 2014;588:3862–3869. doi: 10.1016/j.febslet.2014.09.032. PMID:25281559 [DOI] [PubMed] [Google Scholar]
- [16].Nakatogawa H. Hrr25: an emerging major player in selective autophagy regulation in Saccharomyces cerevisiae. Autophagy. 2015;11:432–433. doi: 10.1080/15548627.2015.1017195. PMID:25700828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vidal M, Brachmann RK, Fattaey A, et al. Reverse two-hybrid and one-hybrid systems to detect dissociation of protein-protein and DNA-protein interactions. Proc Natl Acad Sci U S A. 1996;93:10315–10320. doi: 10.1073/pnas.93.19.10315. PMID:8816797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kamber RA, Shoemaker CJ, Denic V. Receptor-bound targets of selective autophagy use a scaffold protein to activate the Atg1 kinase. Mol Cell. 2015;59:372–381. doi: 10.1016/j.molcel.2015.06.009. PMID:26166702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Torggler R, Papinski D, Brach T, et al. Two independent pathways within selective autophagy converge to activate Atg1 kinase at the vacuole. Mol Cell. 2016;64:221–235. doi: 10.1016/j.molcel.2016.09.008. PMID:27768871 [DOI] [PubMed] [Google Scholar]
- [20].Sambrook J, Frisch EF, Maniattis T. Molecular cloning: a laboratory manual. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- [21].Cregg JM, Russel KA. Transformation. Methods Mol Biol. 1998;103:27–39. PMID:9680631 [DOI] [PubMed] [Google Scholar]
- [22].Baerends RJ, Faber KN, Kram AM, et al. A stretch of positively charged amino acids at the N terminus of Hansenula polymorpha Pex3p is involved in incorporation of the protein into the peroxisomal membrane. J Biol Chem. 2000;275:9986–9995. doi: 10.1074/jbc.275.14.9986. PMID:10744674 [DOI] [PubMed] [Google Scholar]
- [23].Gietz RD, Woods RA. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/S0076-6879(02)50957-5. PMID:12073338 [DOI] [PubMed] [Google Scholar]
- [24].Lin-Goerke JL, Robbins DJ, Burczak JD. PCR-based random mutagenesis using manganese and reduced dNTP concentration. Biotechniques. 1997;23:409–412. PMID:9298207 [DOI] [PubMed] [Google Scholar]
- [25].Corpet F. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1988;16:10881–10890. doi: 10.1093/nar/16.22.10881. PMID:2849754 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.