

Abstract
MicroRNAs are important epigenetic regulators of protein expression by triggering degradation of target mRNAs and/or inhibiting their translation. Dysregulation of microRNA expression has been reported in several cancers, including prostate cancer (PC). We comprehensively characterized the proteomic footprint of a panel of 12 microRNAs that are potently Suppressed in Metastatic PC (SiM-miRNAs: miR-1, miR-133a, miR-133b, miR-135a, miR-143-3p, miR-145-3p, miR-205, miR-221-3p, miR-221-5p, miR-222-3p, miR-24-1-5p and miR-31) using Reverse Phase Proteomic Arrays (RPPA). Re-expression of these SiM-miRNAs in PC cells suppressed cell proliferation and targeted key oncogenic pathways, including cell cycle, apoptosis, Akt/mTOR signaling, metastasis and the AR axis. However, only 12%, at most, of these observed protein expression changes could be explained by predicted direct binding of miRNAs to corresponding mRNAs, suggesting that the majority of these proteomic effects result indirectly. AR and its Steroid Receptor Coactivators (SRC-1, -2, and -3) were recurrently affected by these SiM-miRNAs. In agreement, we identified inverse correlations between expression of these SiM-miRNAs and early clinical recurrence, as well as with AR transcriptional activity in human PC tissues. We also identified robust induction of miR-135a by androgen and strong direct binding of AR to the miR-135a locus. As miR-135a potently suppresses AR expression, this results in a negative feedback loop that suppresses AR protein expression in an androgen-dependent manner, while de-repressing AR expression upon androgen deprivation. Our results demonstrate that epigenetic silencing of these SiM-miRNAs can result in increased AR axis activity and cell proliferation, thus contributing to disease progression. We further demonstrate that a negative feedback loop involving miR-135a can restore AR expression under androgen deprivation conditions, thus contributing to the upregulation of AR protein expression in CRPC. Finally, our unbiased proteomic profiling demonstrates that the majority of actual protein expression changes induced by SiM-miRNAs cannot be explained based on predicted direct interactions.
Keywords: MicroRNAs, prostate cancer, androgen receptor, SRC-1, SRC-2, SRC-3
INTRODUCTION
Despite significant advances in prostate cancer (PC) treatment, progression eventually occurs, highlighting an unmet need for a better understanding of the driving mechanisms of castration-resistant PC (CRPC). CRPC frequently still expresses AR and AR-target genes, such as PSA1, suggesting the AR axis is reactivated in situ and drives CRPC, despite castrate levels of peripheral testosterone2, 3. Several mechanisms contribute to persistent AR signaling2–4, including the frequent overexpression of AR protein5–8 and its coactivators, Steroid Receptor Coactivator (SRC)-1, -2 and -39–14. Interestingly, AR and SRC gene amplification occurs in only a subset of these tumors15–17, suggesting possible epigenetic mechanisms being responsible as well. MicroRNAs (miRNAs) represent a key epigenetic regulatory mechanism, which we examined in detail in this study.
MicroRNAs bind preferentially to specific sequences in the 3′-untranslated region (3′UTR) of mRNAs, and regulate protein output through translational repression and/or mRNA destabilization18, 19. Most protein coding genes and virtually every biological process are subject to miRNA-mediated regulation18–20. Several studies have examined the dysregulated expression of miRNAs in PC (mainly in primary tumors), and have reported suppression of several miRNAs including miR-31, miR-1, miR-133a/b, miR-143, and miR-145 with variable overlap in their findings21–31. Conversely, several miRs have been found to be overexpressed in PC22, including miR-32, miR-148a24, miR-2132 and the miR-106b-25 cluster33.
While the expression patterns of miRNAs in PC (in particular primary tumors) have been reported extensively, their targets are not well defined, and usually limited to the study of a handful of selected proteins and/or guided by bioinformatic predictions of miRNA/mRNA interactions using algorithms that frequently give incomplete or divergent results and can miss important effects34–36. Consequently, a comprehensive map of the actual (not predicted) proteomic footprint of miRNAs in PC, in particular metastatic PC (metPC), is still needed. Utilizing Reverse Phase Proteomic Arrays (RPPA), we characterized the proteomic footprint of miRNAs that are suppressed in metPC (SiM-miRNAs). We determined that the majority of the proteomic effects observed result from indirect mechanism(s), whereas only 12% of the protein alterations could be explained by direct binding of miRNAs to the corresponding mRNA. AR itself and its Steroid Receptor Coactivators (SRC-1, -2, and -3) were observed to be recurrently affected by these SiM-miRNAs. We also identified a negative feedback loop involving miR-135a-5p that regulates AR protein expression by androgen and de-represses AR expression upon androgen deprivation, thus contributing to the upregulation of AR protein expression in CRPC.
RESULTS
SiM-miRNAs are consistently downregulated in multiple PC datasets
To characterize the microRNA landscape of metPC, we examined a publicly available dataset of 28 normal prostate, 99 primary PC and 14 metastatic PC samples [GSE2103617]. We selected the top 14 miRNAs suppressed in metPC (SiM-miRNAs) for further evaluation (Figure 1A). Thirteen of the 14 were also downregulated in primary PC in the same dataset, compared to normal prostate (t-test, P<0.01, Figure 1A). We also examined three additional, independent datasets and found that many of the fourteen SiM-miRNAs were significantly downregulated in primary PC (compared to normal prostate) in the GSE36803 dataset21 (t-test, p<0.01; Fig. S1A); in the GSE45604 dataset37 (t-test, p<0.01, Fig. S1B); and in the GSE6636 dataset (t-test, p<0.01, Fig. S1C). Next, we examined DNA methylation differences between normal prostate tissue and metPC, and found that the genomic loci corresponding to twelve out of these fourteen SiM-miRNAs are hypermethylated in metPC samples (Figure 1B, t-test, p<0.05).
Figure 1. Analysis of microRNAs suppressed in metPC (SiM-miRNAs).
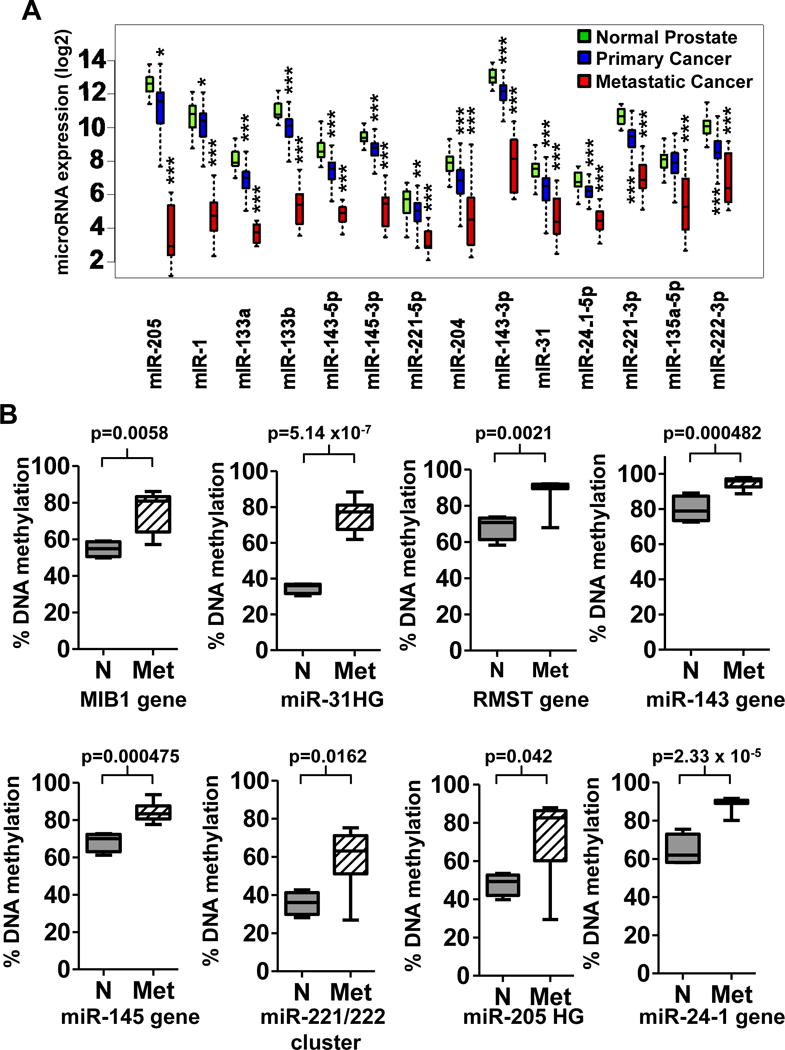
A. Bioinformatic analysis of the miRNA expression levels in 14 metPC samples compared to 28 normal prostate samples from the GSE21036 dataset selected fourteen miRNAs significantly downregulated in metPC (SiM-miRNAs, P<0.05 for all by t-test) for further analysis. Of these 14 microRNAs, thirteen were also downregulated in the 99 primary PC samples of the same dataset, compared to normal prostate. * indicates p values < 0.01; ** indicates p values < 0.001; *** indicates p values < 0.0001 as determined by two-tailed t-test. B. Analysis of DNA methylation within the promoter of each SiM-miRNA gene locus in metPC samples compared to normal prostate samples in the GSE38240 dataset, that contains 4 normal (N) and 8 metPC (Met) patient samples assayed with the Illumina Infinium Human Methylation 450K BeadChip Kit. Our results include 12 SiM-miRNAs in 8 boxplots, as some of the SiM-miRNAs occur in the same host gene or in clusters. DNA methylation levels are significantly increased (p < 0.05) in metPC at twelve out of fourteen SiM-miRNA loci: MIB1 (host gene of miR-1 and miR-133a), the miR-31HG (host gene of miR-31), RMST (host gene of miR-135a-5p), MIR143HG (host gene of miR-143-5p, miR-143-3p and miR-145-3p), the miR-221-5p/miR-221-3p/miR-222 cluster, the miR-205 HG (host gene of miR-205), and the host gene for miR-24-1-5p. There were no significant CpG methylation differences observed within the loci analyzed for miR-133b or miR-204 in this dataset (p >0.05).
Copy number alterations in SiM-miRNAs
To assess the potential role of copy number alterations (CNAs), particularly deletions, in dysregulation of SiM-miRNA expression, we integrated CNA data from patient cohorts containing primary and metastatic PC. Frequency of deletion of the corresponding loci in 157 primary and 37 metastatic specimens from the Taylor et al cohort17, and in 11 primary and 50 metastatic specimens from the Grasso et al cohort38 are presented in Fig. S1D and S1E, respectively. In 14 metastatic specimens from the Taylor et al cohort17 where both CNA and SiM-miRNA expression datasets were available, we found that SiM-miRNA expression was suppressed in the majority of specimens, even in the absence of locus deletion, suggesting epigenetic silencing (Fig. S1F).
Prognostic significance of the SiM-miRNAs in primary PC specimens
We next examined for possible association between SiM-miRNA expression and clinical outcomes. Using the 99 primary PC patient samples of the Taylor et al. cohort17, we found that, for nine SiM-miRNAs, low expression (bottom quartile) was associated with worse biochemical recurrence (BCR)-free survival (p<0.05, Figure 2A, vs Fig. S2 for SiM-miRNAs without prognostic significance). For miR-1 and miR-31, our findings confirm previously published individual reports from other groups21, 25. To examine whether this prognostic significance is related to the Gleason score (or if the SiM-miRNAs can provide additional prognostic insight), we examined whether expression of the SiM-miRNAs is correlated with the Gleason score (GS). As shown in Fig. S1G, only two (miR-205 and miR-31) of the 14 SiM-miRNAs exhibited any (specifically, negative) correlation with GS (PC samples with high GS exhibited lower miR-205 and miR-31 expression, one way ANOVA, p<0.05). This suggests that the prognostic information provided by most SiM-miRNAs is not linked to their relationship to GS.
Figure 2. A. Expression of SiM-miRNAs is associated with clinical outcome in primary PC.
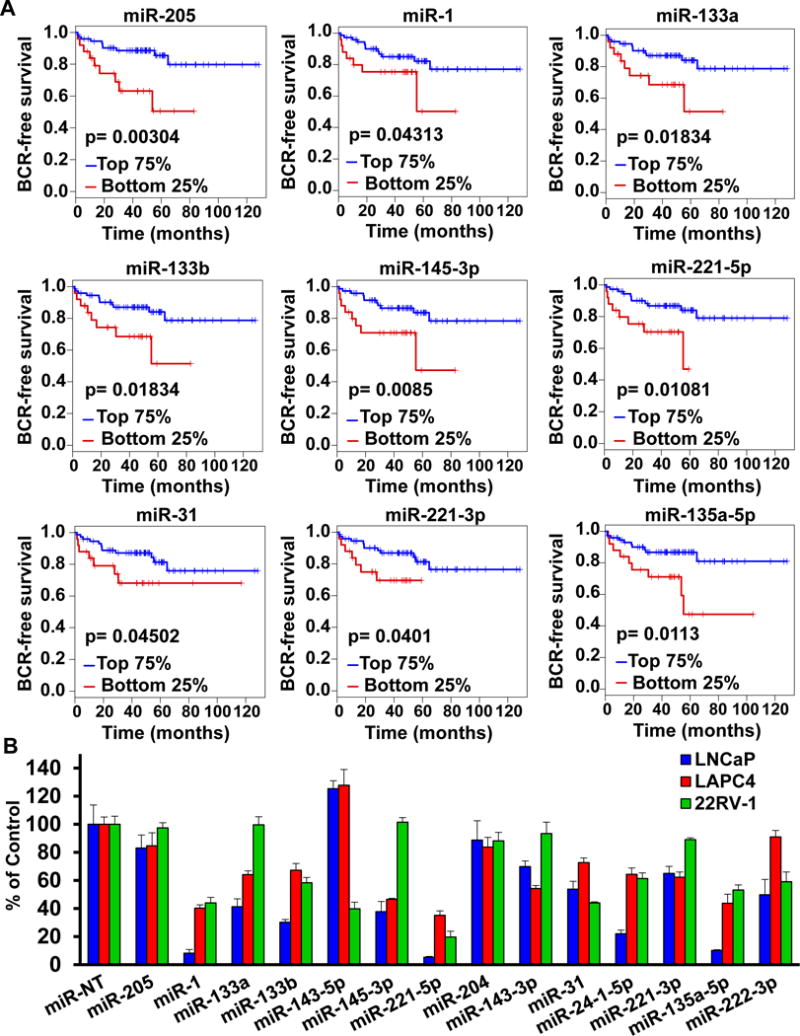
We ranked the 99 primary PC patient samples of the Taylor et al cohort (GSE21036,17) according to their individual miRNA z-score (compared to normal prostate tissue) for each SiM-miRNA. We then compared the biochemical recurrence (BCR)-free survival between the bottom quartile and the top 3 quartiles of the ranked specimens using the log-rank test. We observed that for nine SiM-miRNA (miR-205, miR-1, miR-133a, miR-133b, miR-145-3p, miR-221-5p, miR-31, miR-221-3p, and miR-135a-5p), low expression of the microRNA (bottom quartile) was associated with worse BCR-free survival (p<0.05). B. Transfection of SiM-miRNA mimetics into PC cell lines suppresses cell number. LNCaP, LAPC4 and 22Rv1 cells were transfected with SiM-miRNA mimetics or negative control non-target miR (miR-NT) to a final concentration of 30 nM and incubated for 96 hours. MTT assays were performed and are reported as mean cell viability ± S.D.
Anticancer effect of SiM-miRNAs in PC cell lines
To determine the functional impact of the fourteen SiM-miRNAs on PC cells, we transfected the corresponding miRNA mimetics into the androgen-dependent LNCaP and LAPC4 and the AR-dependent, androgen-independent 22Rv1 PC cells for 96 hrs, followed by MTT assay. We found that the SiM-miRNAs significantly reduced PC cell number, with particularly robust effects in the cases of miR-1, miR-221-5p, miR-135a-5p, miR-31, mir-133b, miR-24-1-5p, while the others induced variable reductions across the cell lines used (Figure 2B).
Proteomic footprint of the SiM-miRNAs in PC cells
We next sought to define a comprehensive proteomic profile of the molecular footprint of the SiM-miRNAs in PC cells. We transfected LNCaP cells with mimetics corresponding to 12 SiM-miRNAs (miR-143-5p and miR-204 were excluded due to a lack of prognostic significance in the patient cohort and minimal anti-cancer activity in LNCaP cells) and performed RPPA analysis. Overall, combining the proteomic footprint of the 12 SiM-miRNAs, we determined 466 SiM-miRNA-induced protein expression changes. Because miRNAs can lead to mRNA degradation and/or translation inhibition via direct binding to the 3′UTR of the corresponding genes, we estimated what percentage of the observed proteomic changes could be possibly explained by direct SiM-miRNA/mRNA interactions. Using the union of five leading prediction algorithms (for most comprehensive and inclusive predictions), we determined which down-regulated proteins were also predicted to be direct targets of each of the 12 SiM-miRNAs (Table S2). Only 12.01% of the protein expression changes measured using RPPA could be potentially attributed to predicted direct binding of SiM-miRNAs to mRNAs, even when the most inclusive predictions were used. This result led us to focus on the pathways and processes that were recurrently affected by re-expression of the SiM-miRNAs, rather than on particular miRNA/mRNA binding.
Pathways and processes affected by the 12 SiM-miRNAs
We determined pathways and processes enriched in the proteomic signatures of the 12 SiM-miRNAs using the ConsensusPathDB resource. Of note, one of the top terms was “Prostate Cancer”. Other common processes and pathways concordantly affected by the SiM-miRNAs in metPC include cell cycle, apoptosis, Akt/mTOR signaling, migration, and the AR axis (Fig. 3A: pathways containing member proteins affected by at least 4 SiM-miRNAs; Fig. S3A: 10 pathways significantly enriched (p<0.05, hypergeometric distribution) in the proteomic footprint of at least 4 miRNAs). Setting a threshold of concordant change by at least 4 SiM-miRNAs for each protein/target, followed by pathway enrichment analysis using ConsensusPathDB, revealed effects on mTOR signaling, protein translation, PKB/Akt signaling, cell cycle/growth arrest (CDKN1A [p21] and CDKN1B [p27]) and apoptosis signaling through BIM (Figure 4A-C and Fig. S4A-M).
Figure 3. Restoration of expression of SiM-miRNAs markedly depletes AR expression and inhibits androgen-mediated induction of AR target genes KLK3 and TMPRSS2.
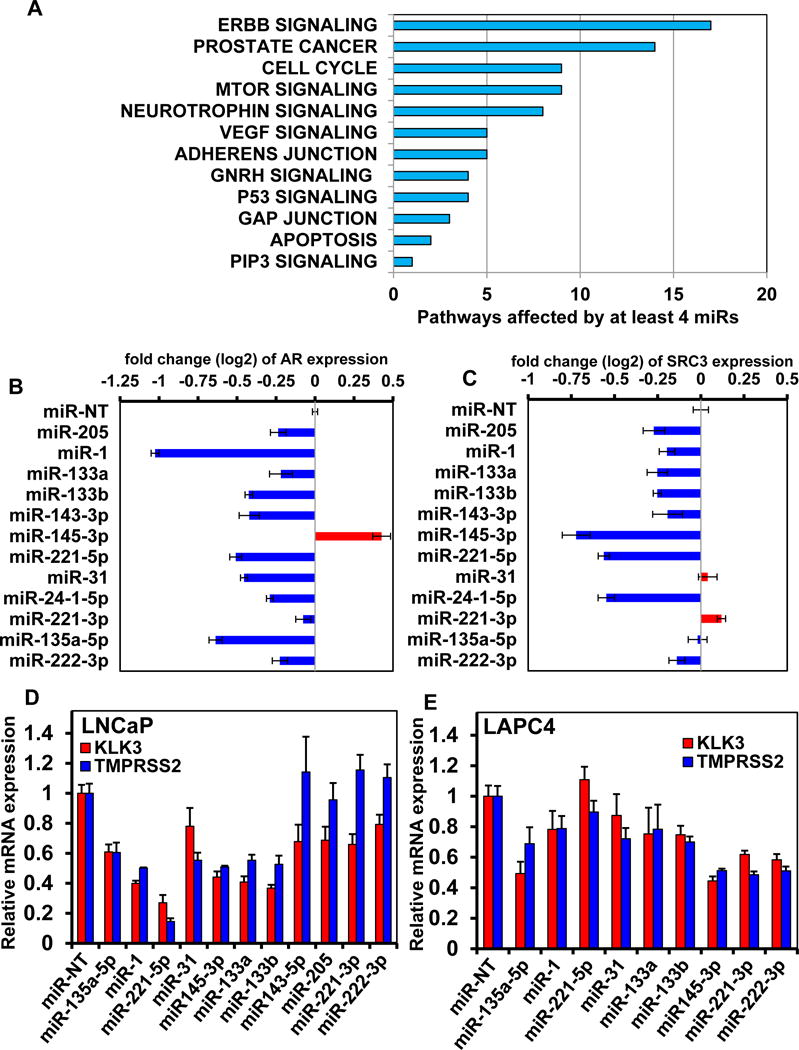
A. LNCaP cells were transfected with SiM-miRNA mimetics or non-target miR (miR-NT) for 48 hours, followed by RPPA. Pathway analysis was run on the identified target protein changes. The graph displays the cancer-related KEGG pathways for proteins altered in the same direction by at least four of the twelve SiM-miRNAs. B-C. LNCaP cells were transfected with SiM-miRNA mimetics for 48 hours, followed by RPPA. The log2 fold change in AR and SRC3 expression following transfection with each SiM-miRNA is shown. Restoration of expression of miR-1, miR-135a-5p, miR-221-5p and miR-31 results in the greatest repression of AR protein expression. Columns represent the average of 3 independent transfections; Bars, S.E. D-E. LNCaP (D) and LAPC4 (E) cells were transfected with the indicated SiM-miRNA mimetics in charcoal stripped serum for 24 hours. Following this, the cells were treated with 1 nM of R1881 for 24 more hours. At the end of treatment, total RNA was isolated and reverse transcribed. The resulting cDNA was utilized for qRT-PCR to determine the expression levels of KLK3 and TMPRSS2 in the cells. Relative mRNA expression is normalized to the expression of β-Actin mRNA; Bars, S.E.
Figure 4. Proteomic analysis of PC cells following restoration of expression of SiM microRNAs.
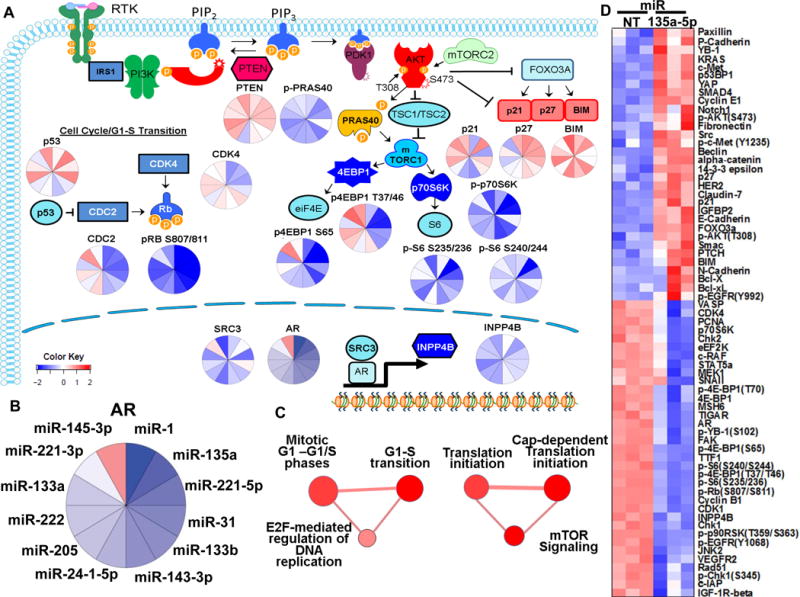
A. Diagrammatic and pie chart representation of cell cycle, G1/S transition, AR axis and PI3K/PTEN/AKT/mTOR signaling pathways and proteins altered by expression of the SiM-miRNA mimetics in LNCaP cells. The order of the SiM-miRNAs in each pie chart follows that depicted in (B) for AR. C. Pathway analysis of the RPPA results indicates that many of the targets altered by the SiM-miRNA mimetics (at least six in the same direction) are involved in G1/S transition, DNA replication, eukaryotic translation initiation, activation of post-cap dependent binding to ribosomes, AKT and mTOR signaling. The AKT/mTOR pathway was significantly targeted, with phospho-AKT(pSer473 and Thr308), phospho-GSK3(Ser9), phospho-S6(Ser235/236 and Ser240/244), phospho-p90RSK(pThr359/Ser363) and phospho-4EBP1(pSer65, Thr37/46/70) being suppressed by SiM-miRNA mimetic transfection. Interestingly, mTOR (phospho and total) tended to be increased. D. Heatmap of RPPA results from LNCaP cells transfected with miR135a-5p mimetics in biologic triplicates for 48 hours. Each column represents a separate transfection for miR-NT (non-target miR) or miR-135a-5p.
SiM-miRNAs suppress AR signaling
Considering the known importance of AR for PC, it was particularly interesting that 10 of the 12 SiM-miRNAs suppressed AR protein expression (Figure 3B [log2 fold change] and confirmation by immunoblot is shown in Figure 5). Moreover, 6 of the 12 SiM-miRNAs suppressed the protein expression of the AR coactivator SRC-3 (Figure 3C [log2 fold change], and confirmation by immunoblot is shown in Figure 5). The AR-inducible protein INPP4B was also suppressed by 6 of the 12 SiM-miRNAs (Table S3). This concordant effect on AR signaling prompted us to examine the impact of SiM-miRNAs on the other AR coactivators SRC-1 and SRC-2 (not included in the RPPA panel). We observed variable but robust suppression of both SRC1 and SRC2 by SiM-miRNAs (Figure 5). Moreover, re-expression of SiM-miRNAs in LNCaP cells significantly depleted the expression levels of c-Myc and SKP2, and induced PARP cleavage, a marker for caspase-mediated apoptosis.
Figure 5. Restoration of expression of SiM-miRNAs depletes AR and steroid receptor coactivators in PC cells.
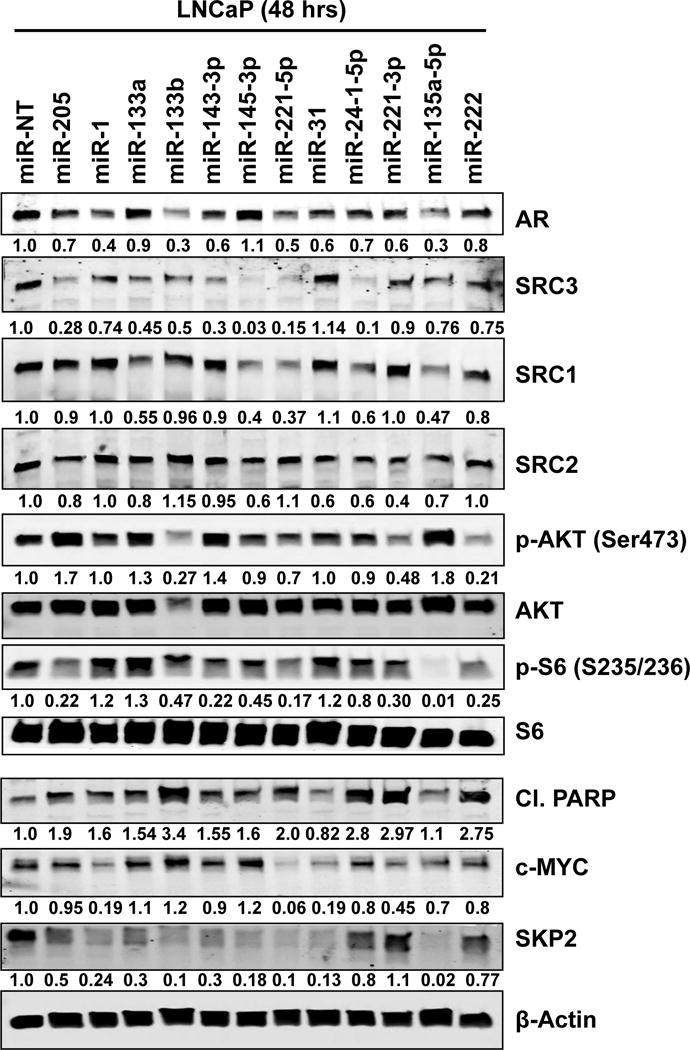
LNCaP cells were transfected with the indicated SiM-miRNA mimetics for 48 hours. Then, cells were harvested and total cell lysates were prepared. Immunoblot analyses were conducted for the expression levels of AR, SRC-1, SRC-2, SRC-3, p-AKT(S473), total AKT, p-S6(S235/236), total S6, cleaved PARP, c-Myc, SKP2 and β-Actin in the cell lysates. The numbers beneath the bands represent densitometry analysis of band intensity relative to the expression of β-Actin.
To understand the potential mechanism behind these effects, we utilized multiple prediction algorithms to examine if any of the SiM-miRs could directly target AR and the p160 co-activators. Interestingly, miR-135a-5p, miR-1-5p, and miR-143-5p were predicted to target SRC1 3′UTR via multiple miRNA/mRNA prediction algorithms (Table S4). Following this, we interrogated over 50 Argonaute-HITS-CLIP and PAR-CLIP experiments from the Starbase compendium for evidence of SiM-miRNA binding to AR and p160 SRC mRNAs (Table S5)39–44. Focusing on miR-135a-5p, we identified evidence for a direct interaction with SRC1 (and ROCK1, which has been previously reported to be a direct target45), but not for AR, SRC2 or SRC3. In agreement, we next observed significant depletion (p <0.05) of SRC1 but not of AR, SRC2 or SRC3 mRNA in LNCaP and LAPC4 cells transfected with miR-135a-5p mimic (Fig. S5A and S5B, respectively).
To further examine the impact of SiM-miRNAs on AR signaling, we re-expressed SiM-miRNAs in LNCaP and LAPC4 cells and confirmed that SiM-miRNAs, in particular miR-135a-5p, miR-1 and miR-221-5p, could suppress known AR-dependent mRNAs (KLK3/PSA and TMPRSS2, Figure 3D and 3E).
We next examined the relationship between SiM-miRNA expression and AR transcriptional activity utilizing mRNA and miRNA expression data from primary PC samples17. We applied a transcriptomic signature that we previously derived from LNCaP cells treated with two different AR siRNAs46 and calculated the gene signature score for each PC specimen. We then examined its correlation with the expression levels of each of the 12 SiM-miRNAs (Pearson correlation coefficient was computed as previously47, p<0.05). For 11 of 12 SiM-miRNAs, miRNA expression was positively correlated with presence of a gene signature generated by silencing AR (i.e. inversely correlated with AR transcriptional activity), further supporting the role of SiM-miRNAs in the regulation of the AR axis in PC (Fig. S6A-C). The one notable exception was miR-135a-5p, despite having potently suppressed AR expression and transcriptional activity in our experiments (Figure 3, Figure 4, and Figure 5). To further examine the regulatory relationship between miR-135a-5p and AR signaling, we compared (by GSEA) a publicly available gene expression signature of LNCaP cells after re-expression of miR-135a-5p (GSE45620)45 with our previously derived signatures from LNCaP cells treated with two different AR siRNAs46. We found that AR-induced genes (genes down-regulated by AR siRNA A or B) are significantly enriched among the genes suppressed by miR-135a-5p (Fig. S6D), confirming that acute treatment with miR-135a-5p suppresses AR transcriptional activity. This led us to the hypothesis that the regulatory relationship between miR-135a-5p and AR signaling is complex and distinct from the relationship of the other 11 SiM-miRNAs. Notably, miR-135a-5p was also an outlier in regards to its lack of downregulation in three of four primary PC datasets (Figure 1A, Fig. S1A-C), further suggesting that its epigenetic silencing in metPC may be related to a mechanism distinct from those of other SiM-miRNAs.
Re-expression of miR-135a-5p suppresses genes related to metastasis in PC cells
We next examined whether miR-135a-5p would affect the expression of metastasis-related genes in PC cells by comparing publicly available datasets of metastatic PC specimens with the transcriptional impact of re-expression of miR-135a-5p on LNCaP cells. GSEA demonstrated that genes upregulated in metastatic PC are suppressed by re-expression of miR-135a-5p in LNCaP cells (NES score of −1.88, q<0.09, Fig. S7). These data are in line with a recently reported elegant study in which re-expression of miR-135a-5p reduced metastasis-promoting genes in cells in vitro. Furthermore, the re-expression of miR-135a-5p significantly inhibited tumor growth and dramatically reduced spontaneous metastases of the breast cancer cells to the bone of mice implanted with the MDA-MB-231 cells48.
Androgen induces expression of miR-135a-5p in PC cells
We next determined the impact of androgen (R1881) on miR-135a-5p expression in LNCaP, VCaP, and LAPC4 cells. Treatment of androgen-starved cells with 1 nM of R1881 robustly induced miR-135a-5p expression in LNCaP and VCaP cells (Figure 6A) and to a lesser degree in LAPC4 cells (Fig. S8). In line with these effects, treatment of androgen-exposed cells with the androgen receptor antagonist MDV3100 suppressed miR-135a-5p expression in LNCaP and VCaP cells (Figure 6B). These results demonstrate that miR-135a-5p has a distinct regulatory interplay with the AR signaling axis. Examination of our previously published ChIP-Seq datasets46 demonstrated strong binding of AR, its pioneer factors FOXA1 and GATA2, the coactivator SRC-2, CBP, p300 and RNA Pol II at a downstream enhancer of RMST, the host gene of miR-135a-5p (Figure 6C). The enhancer role of this complex was supported by strong co-localization of H3K4me1, H3K4me2, and H3K27ac, but not H3K4me3 (Figure 6C); this chromatin mark profile has been established to correspond to an enhancer role49, 50.
Figure 6. Androgen induces miR-135a-5p expression in PC cells.
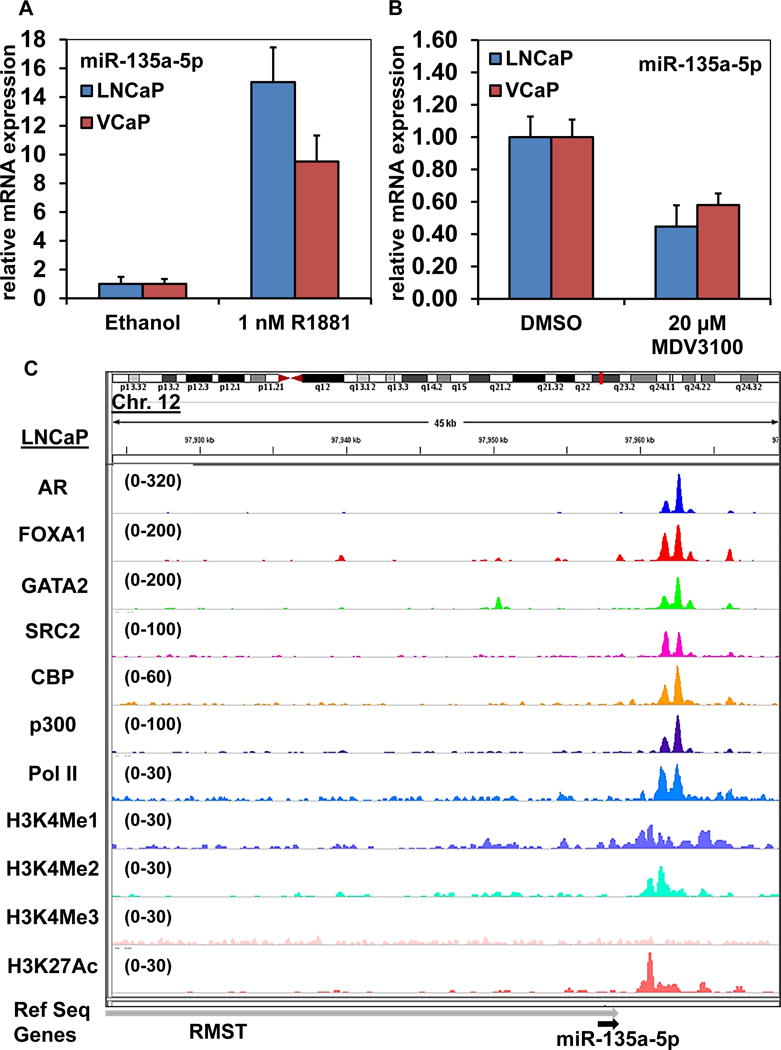
A. LNCaP and VCaP cells were plated in media supplemented with 10% charcoal stripped FBS for 48 hours. Then, R1881 (1 nM) was added and the cells were incubated for an additional 24 hours. At the end of treatment, total RNA was isolated by the Trizol method and reverse transcribed. Stem loop-mediated reverse transcription of mRNA was performed with 250 ng of total RNA for miR-135a-5p and RNU6B. Relative expression of miR-135a-5p in each cell line was determined by quantitative PCR and normalized to RNU6B; Bars, S.E. B. LNCaP and VCaP cells were treated with MDV3100, as indicated, for 48 hours. Total RNA was isolated and reverse transcribed utilizing stem loop-mediated reverse transcription for miR-135a-5p and RNU6B. Relative expression of miR-135a-5p in each cell line was determined by quantitative PCR and normalized to RNU6B; Bars, S.E. C. We examined the ChIP-Seq profiles for AR, FOXA1, GATA2, SRC-2, CBP, p300, RNA Pol II, H3K4me1, H3K4me2, H3K4me3 and H3K27Ac at the RMST gene locus in LNCaP cells utilizing the IGV browser. We observed significant binding/localization of these proteins to a region approximately 5 kb downstream of the miR-135a-5p coding sequence within the RMST gene (gray arrow), which is the host gene for miR-135a-5p (black arrow).
SiM-miRNAs are epigenetically silenced in PC
We next examined the mechanism(s) behind the downregulation of SiM-miRNAs in PC, utilizing ChIP-Seq data (GSE38685) for the active transcription mark H3K4me3 and the repressive mark H3K27me3 in prostate epithelial cells (PrEC) and LNCaP cells. Compared to PrEC cells, the sequence tag density of H3K4me3 was markedly reduced in LNCaP cells at the RMST gene locus (host gene of miR-135a, p < 0.05). Conversely, the H3K27me3 signal tag density was significantly increased at the same locus in LNCaP compared to PrEC cells (p < 0.05, Figure 7A and Table S6). In addition, at the miR-221/miR-222 locus, the H3K4me3 sequence tag density was decreased in LNCaP compared to PrEC cells (p<0.05, Fig. S9A and Suppl Table 6), with a trend for increased H3K27me3 in LNCaP that did not reach statistical significance. Upstream of the MIB1 gene (host gene for miR-1 and miR-133a) the H3K27me3 sequence tag density was significantly greater in LNCaP than PrEC cells (p<0.05, Fig. S9B and Table S6), whereas the H3K4me3 sequence tag density was not significantly different between the two cell types. As previously observed in PC cells21, the miR-31HG locus exhibits both lower H3K4me3 and higher H3K27me3 sequence tag density in LNCaP compared to PrEC cells (p<0.05, Fig. S9C and Table S6). These results point to epigenetic regulation of the SiM-miRNAs miR-135a-5p, miR-221-5p and miR-1 in PC and are in agreement with prior findings for miR-3121.
Figure 7. Epigenetic features and chromatin structure determines expression and accessibility of miR-135a-5p in PC versus normal prostate epithelial cells.
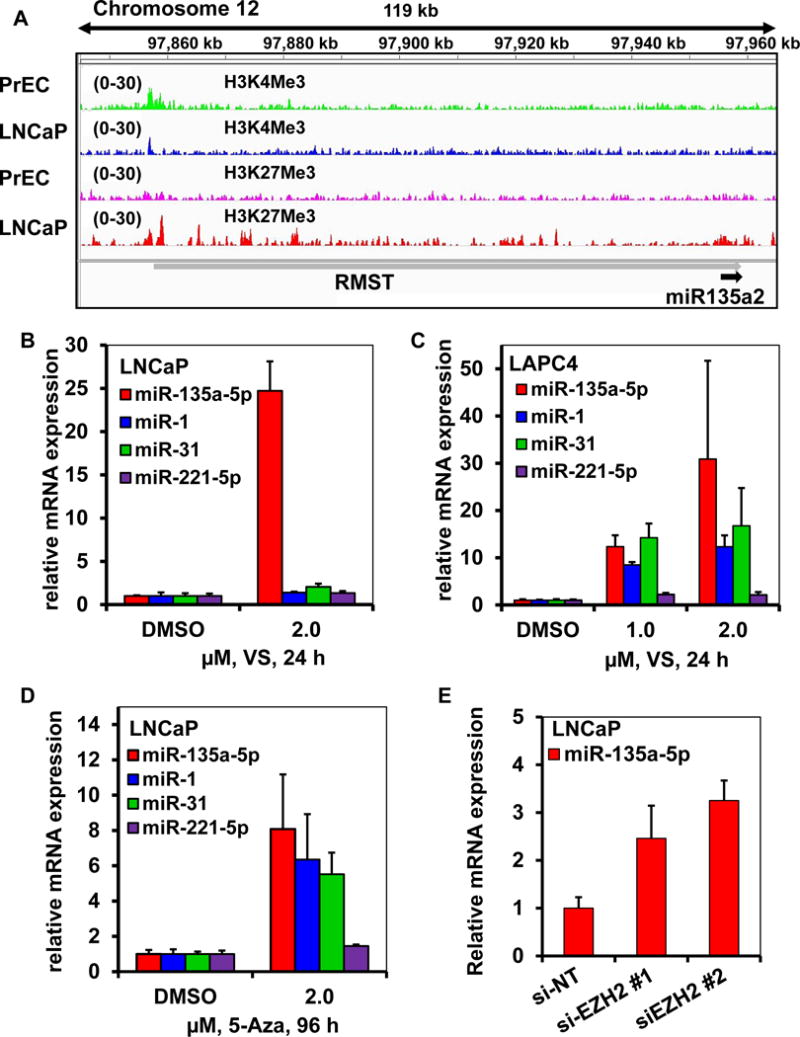
A. ChIP-Seq datasets for chromatin marks (H3K4me3 and H3K27me3) from PrEC and LNCaP cells were visualized utilizing the IGV browser. A. In LNCaP cells, the RMST gene (the host gene for miR-135a-5p) locus exhibits stronger H3K27me3 signal and weaker H3K4me3 signal compared to PrEC cells. B-D. Treatment with pan-histone deacetylase inhibitor and DNA demethylating agents causes reactivation of miR-135a-5p, miR-1, miR-31 and miR-221-5p expression in PC cells. B-D. LNCaP and LAPC4 cells were treated as indicated for 24 hours with vorinostat (VS) or 96 hours with the DNA methyltransferase inhibitor 5-azacitidine (5-Aza). Total RNA was isolated and reverse transcribed utilizing a Stem loop-mediated reverse transcription kit for miR-135a-5p, miR-1, miR-31 and miR-221-5p. Relative expression of each miR was determined by quantitative PCR and normalized to RNU6B; Bars, S.E. E Silencing of EZH2 activates the expression of miR-135a-5p in PC cells. LNCaP cells were transfected with si-NT or si-EZH2 for 48 hours. At the end of treatment, total RNA was isolated by the Trizol method. Stem loop-mediated reverse transcription of miR-135a-5p was performed with 250 ng of total RNA. Relative expression of miR-135a-5p was determined by quantitative PCR and normalized to RNU6B; Bars, S.E.
Epigenetic modifying agents modulate SiM-miRNA expression in PC cells
Having established that several SiM-miRNAs are epigenetically silenced in PC, we next determined the effects of treatment with the pan-HDAC inhibitor vorinostat and the DNA methyltransferase inhibitor 5-azacytidine on miR-135a-5p, miR-1 and miR-221-5p expression in PC cells. Vorinostat markedly and significantly increased miR-135a-5p expression in LNCaP, LAPC4 and VCaP cells (Figure 7B-C and Fig. S10A). Vorinostat also variably increased miR-1, miR-221-5p and miR-31 levels (Figure 7B-C and Fig. S10A). The DNA methyltransferase inhibitor 5-azacytidine also markedly induced miR-135a-5p, miR-1, and miR-31 expression in PC cells (Figure 7D and Fig S10B). The induction of miR-31 by vorinostat and by 5-azacytidine served as a positive control, as has been previously reported21. Having previously determined that the promoter of the host gene for miR-135a is highly marked by H3K27 trimethylation, which is catalyzed by the histone lysine methyltransferase EZH2 (a component of the Polycomb Repressive Complex 2), we also determined the role of EZH2 on the expression of miR-135a in PC cells. Silencing EZH2 resulted in the de-repression of miR-135a-5p expression in LNCaP and LAPC4 cells (Figure 7E and Figure S10C).
DISCUSSION
Dysregulation of miRNA expression has been reported in PC21–33, in particular primary PC, but their actual protein targets are less well defined. We comprehensively profiled the proteomic footprint of miRNAs suppressed in metPC and delineated their role in a series of processes, including cell cycle, apoptosis, AKT/mTOR signaling and the AR axis. We mined microRNA data from a large PC patient cohort17 and focused our attention on those most suppressed in metPC (SiM-miRNAs). The majority of the SiM-miRNAs were also suppressed in primary PCs in the same dataset17 and in additional, independent datasets (Refs 21, 37 and GSE6636). For nine SiM-miRNAs, their expression levels were associated with clinical outcome (higher levels were inversely associated with biochemical recurrence after prostatectomy). In agreement with a significant regulatory role in PC biology, we found that most of these SiM-miRNAs exerted potent growth-inhibitory effects upon their re-introduction into PC cell lines.
Our comprehensive analysis of these SiM-miRNAs agrees with and consolidates individual prior reports on miR-3121 and miR-125. Interestingly, we found miR-221 and miR-222 to be downregulated in PC in this present study and the same was reported in other studies24, 51, yet their expression is increased in the androgen-independent cell line LNCaP-Abl, compared to the androgen-dependent parental LNCaP cells, and they have been reported to promote the CRPC phenotype52–54.
As the proteomic footprint of microRNAs in PC had not previously been defined comprehensively, we performed RPPA analysis in LNCaP cells transfected with SiM-miRNA mimetics. Importantly, only 12%, at most, of the SiM-miRNA effects measured via RPPA could be explained by direct miRNA binding onto the corresponding mRNAs. This suggests that most of the effects of these SiM-mRNAs in cells are indirect. We focused our analysis on common pathways concordantly regulated by the SiM-miRNAs in metPC, including cell cycle, apoptosis, Akt/mTOR signaling, migration, and the AR axis. In agreement with the anti-proliferative effect of the SiM-miRNAs seen in our MTT experiments, positive regulators of cell cycle, including phospho-Rb(pSer807/811), cyclin B1, PCNA, CDK1 and c-Myc, were suppressed, whereas the negative regulators Cyclin E1 and p27 were increased. The increase in p27 is consistent with the significant decrease in Skp2 (Figure 5). Receptor tyrosine kinase (RTK) cell cycle activators, such as HER3, pEGFR(pTyr1068), IGF1R and VEGFR2, as well as the AKT/mTOR pathway were frequently affected. Positive regulators of apoptosis, such as BIM and SMAC, were increased, in agreement with our finding of increased cleaved PARP levels. Moreover, cell junction and cell adhesion proteins that generally suppress cell migration and metastasis, such as claudin7 and E-cadherin, were concordantly upregulated upon re-expression of the SiM-miRNAs mimetics. MicroRNA-205 has been previously reported to upregulate E-cadherin and induce a mesenchymal to epithelial transition (MET)55. Collectively, our findings indicate that the SiM-miRNAs induce a combinatorial proteomic profile of suppressed cell cycle, AKT/mTOR signaling and metastatic potential, with increased apoptosis, cell adhesion and MET signaling. Thus, the silencing of these miRNAs in metPC would have a broad impact across cellular functions, including increased mitosis, AKT/mTOR signaling, and metastatic potential, while suppressing apoptosis and cell adhesion.
Due to the critical role of the AR axis in PC, it is intriguing that the SiM-miRNAs concordantly suppressed AR signaling. Ten out of 12 SiM-miRNAs suppressed AR at the protein level. Moreover, the AR p160 coactivators SRC-1, SRC-2, and SRC-3 were recurrent targets of the SiM-miRNAs. Overexpression of AR and SRC-1, -2 and -3 proteins is very common in CRPCs and can contribute to persistent AR signaling5–14. Elevated expression of all three SRCs is associated with shorter time to recurrence, resistance to androgen deprivation therapy and overall more aggressive PC9–13, 17. AR and SRC-2 (NCOA2) gene amplifications occur in small subsets of PCs, mainly CRPCs15–17, while SRC-1 (NCOA1) and SRC-3 (NCOA3) gene amplifications are not commonly encountered17, 38, 56. Consequently, a large part of the overexpression of AR and SRC-2 proteins and almost the entire overexpression of SRC-1 and SRC-3 proteins has to be attributed to epigenetic and post-transcriptional mechanisms. Our study now identifies an additional epigenetic mechanism regulating expression of AR and its SRC coactivators, namely miRNAs that are downregulated in metPC, leading to de-repression of the AR signaling axis.
Next, we examined the relationship between SiM-miRNAs and AR transcriptional activity in PC tissue. We determined that for 11 of 12 SiM-miRNAs, the miRNA levels were positively correlated with the presence of a gene signature generated by silencing AR (i.e. inversely correlated with AR transcriptional activity). These data further supported the role of SiM-miRNAs in the regulation of the AR axis in PC. The single outlier was miR-135a-5p, a SiM-miRNA that potently suppresses AR expression and activity, suggesting a more complex relationship to AR. Moreover, miR-135a-5p expression is generally not suppressed in primary PC (only one out of four primary PC datasets indicated miR-135a-5p downregulation). This further suggested that silencing of miR135a-5p in metPC may be related to mechanism(s) distinct from those of the other SiM-miRNAs. Indeed, we documented that androgen potently induced the expression of miR-135a-5p, consistent with a previous report by Kroiss et al45 who identified an androgen response element (ARE) in the miR-135a promoter region. While we also found AR recruitment to the same ARE under androgen stimulation of LNCaP cells with 1 nM R1881 (data not shown), we additionally identified stronger co-recruitment of AR, its pioneer factors FOXA1 and GATA2, the coactivator SRC-2, CBP, p300 and RNA Pol II to a region immediately downstream of the miR-135a-5p gene in LNCaP cells cultured without the addition of synthetic androgen. The robust presence of H3K4me1, H3K4me2, and H3K27ac, but not H3K4me3, at the latter site further suggests that it functions as an enhancer, even under normal growth conditions (medium supplemented with 10% FBS). Combined with the inhibitory effect of miR-135a-5p on expression of AR, SRC-1, SRC-2 and SRC-3, this suggests a negative feedback loop that regulates AR axis transcriptional output and can de-repress it upon androgen deprivation. These findings also explain why miR-135a-5p was the sole outlier that lacked an inverse correlation of its levels with AR transcriptional activity in PC tissues (because the negative feedback nature of this dual interaction stabilizes this network and blunts any correlation under steady-state conditions) and why miR-135a-5p expression is not significantly downregulated in three of the four primary PC datasets that we analyzed (because they had not yet been exposed to androgen deprivation).
Our findings add to previously reported negative feedback loops through which AR can auto-regulate its expression: a) AR directly binds its own gene to repress its own expression through recruitment of lysine specific demethylase 1 (LSD5/KDM1A) and H3K4me2 and H3K4me1 demethylation57; and b) androgen starvation upregulates GATA2 expression, which then binds to the AR gene promoter to induce AR expression46. Of note, Lin et al. have reported a different epigenetic loop that involves regulation of AR by a miRNA: miR-31 and AR can mutually repress each other21. Thus, in the context of androgen deprivation, both of them will be increased, resulting in a positive feedback loop, in contrast to the negative feedback loop of miR-135a-5p with AR. It is obviously possible that additional miRNAs can also regulate AR signaling: Östling et al. identified 71 miRNAs that influenced AR levels in human PC cells (52 decreasing and 19 increasing AR protein)58, although with very little overlap with the findings of our study.
We observed significant increases in CpG methylation in metPC patient samples compared to normal prostate samples at or within SiM-miRNA promoters and host genes, suggesting an additional layer of epigenetic regulation. In agreement, this epigenetic silencing was relieved by treatment with the DNMT1 inhibitor 5-azacytidine. Similar to prior observations for miR-3121, we also observed that SiM-miRNAs miR-1, miR-135a-5p, and miR-221-5p are also epigenetically (dys)regulated at the level of histone methylation marks in PC. Specifically, PC LNCaP cells exhibit decreased levels of H3K4me3 (active transcription mark) and/or increased H3K27me3 (transcriptional repression mark) at the gene loci of these SiM-miRNAs, compared to PrEC cells. This epigenetic silencing was relieved by depleting the histone lysine methyltransferase EZH2 (that catalyzes H3K27 trimethylation), as well as by treatment with the pan-HDAC inhibitor vorinostat.
Therefore, epigenetic silencing appears to be a widespread phenomenon that suppresses the expression of SiM-miRNAs in metPC, which, in turn, results in diverse impact across cellular functions, including increased AR activity, mitosis, AKT/mTOR signaling, EMT and metastatic potential, while suppressing apoptosis and cell adhesion. As previously proposed for miR-3121, these findings further support that epigenetic therapies could complement existing hormonal agents in order to inhibit the AR axis in CRPC.
MATERIALS AND METHODS
Human PC datasets
To characterize the microRNA landscape of metPC, we examined a publicly available dataset of 28 normal prostate samples, 99 primary PC samples and 14 metastatic PC samples profiled by Agilent-019118 Human miRNA Microarray 2.0 G4470B (GSE2103617). We first selected miRNAs suppressed at least 4-fold in metPC compared to the normal prostate samples. Next, we sorted the miRNAs by their final expression in the metastatic data in increasing order, and chose the top fifteen microRNAs for further investigation. One of them, hsa-miR-886-3p, was determined, according to miRBase, to be a fragment of Vault RNA and not a microRNA, so it was excluded from further analysis, leaving fourteen microRNAs. We also studied the primary PC datasets GSE3680321, GSE4560437 and GSE6636. Statistical differences between normal samples and metastatic tumor samples, or normal samples and primary tumor samples were determined utilizing a two-tailed t-test. P-values <0.01 were considered significant.
Prognostic significance of SiM-miRNA expression in PC patients
We evaluated the prognostic significance of SiM-miRNA expression in the Taylor et al17 patient cohort. For each SiM-miRNA and for each PC specimen, we computed the z-score for its expression within each cohort, as described previously17. Specimens were then ranked according to their individual miRNA z-score, and association with biochemical recurrence (BCR)-free survival was evaluated by comparing the bottom quarter of the ranked specimens with the rest of the specimens (upper three quarters) using the log-rank test. Survival significance was assessed by employing the package survival59 in the R statistical system. P-values < 0.05 were considered significant. Moreover, we divided the patients in three groups according to the Gleason score: low (<7), intermediate (=7), and high (>7). Significant association with the Gleason score was assessed independently for each of the SiM-miRNAs using the one-way ANOVA test (p<0.05) implemented in the Graphpad Prism statistical analysis software (GraphPad Software, Inc., La Jolla, CA).
Reagents
All reagents used in this study were obtained from commercial vendors. Detailed descriptions are provided in the Supplement.
Cell Culture
Human cell lines were obtained and cultured as previously described60.
Analysis of DNA methylation in human PC specimens
We analyzed DNA methylation data obtained with the Illumina Infinium Human Methylation 450K BeadChip Kit (GSE3824061). This dataset contains profiles of four normal prostate samples and eight metPC samples. We analyzed methylation differences at the level of individual CpG probes. Statistical significance of DNA methylation changes was assessed using the t-test (p<0.05) implemented in the R statistical system.
Statistical Analysis
In cell viability assays, each experimental point was set up in at least triplicate wells and each assay was repeated identically and independently at least twice. Data were expressed as percentage of the value obtained from control wells. Values are expressed as mean ± SD. Statistical significance was assessed using the t-test. P-values < 0.05 were considered significant.
Supplementary Material
Acknowledgments
The authors acknowledge the joint participation by Adrienne Helis Malvin Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with Baylor College of Medicine.
Funding/Support: This work was also supported by the American Cancer Society RSG-14-218-01-TBG (to N.M.), the Prostate Cancer Foundation (to B.W.O. and N.M.); the Conquer Cancer Foundation of the American Society of Clinical Oncology Young Investigator and Career Development Awards (both to N.M.); NICHD 8818 and Department of Defense Breast Cancer Research Program Innovator Award (B.W.O.); and the Pilot/Feasibility Program of the Diabetes & Endocrinology Research Center (P30-DK079638) at Baylor College of Medicine (N.M.), and an Alkek Foundation for Molecular Discovery Pilot grant (C.C.). N.M. is a Dan L. Duncan Scholar, a Caroline Wiess Law Scholar and a member of the Dan L. Duncan Cancer Center (supported by the NCI Cancer Center Support Grant P30CA125123) and the Center for Drug Discovery at Baylor College of Medicine. The authors also would like to acknowledge the assistance of the Shared Resources of the Dan L. Duncan Cancer Center (supported by the NCI Cancer Center Support Grant P30CA125123) and the Functional Proteomics RPPA Core Facility (The University of Texas M.D. Anderson Cancer Center, Houston, Texas).
Footnotes
Conflict of Interest: All authors declare that they have no financial conflicts of interest to disclose.
Supplementary Information: Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2012;30:644–646. doi: 10.1200/JCO.2011.39.1300. [DOI] [PubMed] [Google Scholar]
- 2.Mitsiades N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer research. 2013;73:4599–4605. doi: 10.1158/0008-5472.CAN-12-4414. [DOI] [PubMed] [Google Scholar]
- 3.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 4.Debes JD, Tindall DJ. Mechanisms of androgen-refractory prostate cancer. N Engl J Med. 2004;351:1488–1490. doi: 10.1056/NEJMp048178. [DOI] [PubMed] [Google Scholar]
- 5.Gregory CW, He B, Johnson RT, Ford OH, Mohler JL, French FS, et al. A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 2001;61:4315–4319. [PubMed] [Google Scholar]
- 6.Linja MJ, Savinainen KJ, Saramaki OR, Tammela TL, Vessella RL, Visakorpi T. Amplification and overexpression of androgen receptor gene in hormone-refractory prostate cancer. Cancer Res. 2001;61:3550–3555. [PubMed] [Google Scholar]
- 7.Waltering KK, Helenius MA, Sahu B, Manni V, Linja MJ, Janne OA, et al. Increased expression of androgen receptor sensitizes prostate cancer cells to low levels of androgens. Cancer Res. 2009;69:8141–8149. doi: 10.1158/0008-5472.CAN-09-0919. [DOI] [PubMed] [Google Scholar]
- 8.Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 9.Agoulnik IU, Vaid A, Bingman WE, 3rd, Erdeme H, Frolov A, Smith CL, et al. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005;65:7959–7967. doi: 10.1158/0008-5472.CAN-04-3541. [DOI] [PubMed] [Google Scholar]
- 10.Agoulnik IU, Vaid A, Nakka M, Alvarado M, Bingman WE, 3rd, Erdem H, et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006;66:10594–10602. doi: 10.1158/0008-5472.CAN-06-1023. [DOI] [PubMed] [Google Scholar]
- 11.Yan J, Yu CT, Ozen M, Ittmann M, Tsai SY, Tsai MJ. Steroid receptor coactivator-3 and activator protein-1 coordinately regulate the transcription of components of the insulin-like growth factor/AKT signaling pathway. Cancer Res. 2006;66:11039–11046. doi: 10.1158/0008-5472.CAN-06-2442. [DOI] [PubMed] [Google Scholar]
- 12.Yan J, Erdem H, Li R, Cai Y, Ayala G, Ittmann M, et al. Steroid receptor coactivator-3/AIB1 promotes cell migration and invasiveness through focal adhesion turnover and matrix metalloproteinase expression. Cancer Res. 2008;68:5460–5468. doi: 10.1158/0008-5472.CAN-08-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou HJ, Yan J, Luo W, Ayala G, Lin SH, Erdem H, et al. SRC-3 is required for prostate cancer cell proliferation and survival. Cancer Res. 2005;65:7976–7983. doi: 10.1158/0008-5472.CAN-04-4076. [DOI] [PubMed] [Google Scholar]
- 14.Tien JC, Liu Z, Liao L, Wang F, Xu Y, Wu YL, et al. The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer. Cancer Res. 2013;73:3997–4008. doi: 10.1158/0008-5472.CAN-12-3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer research. 2012;72:6142–6152. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koivisto P, Kononen J, Palmberg C, Tammela T, Hyytinen E, Isola J, et al. Androgen receptor gene amplification: a possible molecular mechanism for androgen deprivation therapy failure in prostate cancer. Cancer Res. 1997;57:314–319. [PubMed] [Google Scholar]
- 17.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidigal JA, Ventura A. The biological functions of miRNAs: lessons from in vivo studies. Trends in cell biology. 2014 doi: 10.1016/j.tcb.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer research. 2011;71:5950–5954. doi: 10.1158/0008-5472.CAN-11-1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Kelnar K, Vlassov AV, Brown D, Wang J, Tang DG. Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer research. 2012;72:3393–3404. doi: 10.1158/0008-5472.CAN-11-3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin PC, Chiu YL, Banerjee S, Park K, Mosquera JM, Giannopoulou E, et al. Epigenetic repression of miR-31 disrupts androgen receptor homeostasis and contributes to prostate cancer progression. Cancer research. 2013;73:1232–1244. doi: 10.1158/0008-5472.CAN-12-2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porkka KP, Pfeiffer MJ, Waltering KK, Vessella RL, Tammela TL, Visakorpi T. MicroRNA expression profiling in prostate cancer. Cancer research. 2007;67:6130–6135. doi: 10.1158/0008-5472.CAN-07-0533. [DOI] [PubMed] [Google Scholar]
- 23.Ambs S, Prueitt RL, Yi M, Hudson RS, Howe TM, Petrocca F, et al. Genomic profiling of microRNA and messenger RNA reveals deregulated microRNA expression in prostate cancer. Cancer research. 2008;68:6162–6170. doi: 10.1158/0008-5472.CAN-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fang YX, Gao WQ. Roles of microRNAs during prostatic tumorigenesis and tumor progression. Oncogene. 2014;33:135–147. doi: 10.1038/onc.2013.54. [DOI] [PubMed] [Google Scholar]
- 25.Hudson RS, Yi M, Esposito D, Watkins SK, Hurwitz AA, Yfantis HG, et al. MicroRNA-1 is a candidate tumor suppressor and prognostic marker in human prostate cancer. Nucleic acids research. 2012;40:3689–3703. doi: 10.1093/nar/gkr1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart M, Nolte E, Wach S, Szczyrba J, Taubert H, Rau TT, et al. Comparative microRNA profiling of prostate carcinomas with increasing tumor stage by deep sequencing. Molecular cancer research: MCR. 2014;12:250–263. doi: 10.1158/1541-7786.MCR-13-0230. [DOI] [PubMed] [Google Scholar]
- 27.Saini S, Majid S, Shahryari V, Arora S, Yamamura S, Chang I, et al. miRNA-708 control of CD44(+) prostate cancer-initiating cells. Cancer research. 2012;72:3618–3630. doi: 10.1158/0008-5472.CAN-12-0540. [DOI] [PubMed] [Google Scholar]
- 28.Formosa A, Lena AM, Markert EK, Cortelli S, Miano R, Mauriello A, et al. DNA methylation silences miR-132 in prostate cancer. Oncogene. 2013;32:127–134. doi: 10.1038/onc.2012.14. [DOI] [PubMed] [Google Scholar]
- 29.Formosa A, Markert EK, Lena AM, Italiano D, Finazzi-Agro E, Levine AJ, et al. MicroRNAs, miR-154, miR-299-5p, miR-376a, miR-376c, miR-377, miR-381, miR-487b, miR-485-3p, miR-495 and miR-654-3p, mapped to the 14q32.31 locus, regulate proliferation, apoptosis, migration and invasion in metastatic prostate cancer cells. Oncogene. 2014;33:5173–5182. doi: 10.1038/onc.2013.451. [DOI] [PubMed] [Google Scholar]
- 30.Hulf T, Sibbritt T, Wiklund ED, Patterson K, Song JZ, Stirzaker C, et al. Epigenetic-induced repression of microRNA-205 is associated with MED1 activation and a poorer prognosis in localized prostate cancer. Oncogene. 2013;32:2891–2899. doi: 10.1038/onc.2012.300. [DOI] [PubMed] [Google Scholar]
- 31.Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27:1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- 32.Ribas J, Ni X, Haffner M, Wentzel EA, Salmasi AH, Chowdhury WH, et al. miR-21: an androgen receptor-regulated microRNA that promotes hormone-dependent and hormone-independent prostate cancer growth. Cancer research. 2009;69:7165–7169. doi: 10.1158/0008-5472.CAN-09-1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hudson RS, Yi M, Esposito D, Glynn SA, Starks AM, Yang Y, et al. MicroRNA-106b-25 cluster expression is associated with early disease recurrence and targets caspase-7 and focal adhesion in human prostate cancer. Oncogene. 2013;32:4139–4147. doi: 10.1038/onc.2012.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yue D, Liu H, Huang Y. Survey of Computational Algorithms for MicroRNA Target Prediction. Curr Genomics. 2009;10:478–492. doi: 10.2174/138920209789208219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Witkos TM, Koscianska E, Krzyzosiak WJ. Practical Aspects of microRNA Target Prediction. Curr Mol Med. 2011;11:93–109. doi: 10.2174/156652411794859250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reyes-Herrera PH, Ficarra E. One decade of development and evolution of microRNA target prediction algorithms. Genomics Proteomics Bioinformatics. 2012;10:254–263. doi: 10.1016/j.gpb.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casanova-Salas I, Rubio-Briones J, Calatrava A, Mancarella C, Masia E, Casanova J, et al. Identification of miR-187 and miR-182 as Biomarkers of Early Diagnosis and Prognosis in Patients with Prostate Cancer Treated with Radical Prostatectomy. The Journal of urology. 2014 doi: 10.1016/j.juro.2014.01.107. [DOI] [PubMed] [Google Scholar]
- 38.Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erhard F, Haas J, Lieber D, Malterer G, Jaskiewicz L, Zavolan M, et al. Widespread context dependency of microRNA-mediated regulation. Genome Res. 2014;24:906–919. doi: 10.1101/gr.166702.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, et al. Ago HITS-CLIP expands understanding of Kaposi’s sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog. 2012;8:e1002884. doi: 10.1371/journal.ppat.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kishore S, Jaskiewicz L, Burger L, Hausser J, Khorshid M, Zavolan M. A quantitative analysis of CLIP methods for identifying binding sites of RNA-binding proteins. Nat Methods. 2011;8:559–564. doi: 10.1038/nmeth.1608. [DOI] [PubMed] [Google Scholar]
- 42.Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–338. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- 43.Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe. 2011;10:515–526. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012;8:e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kroiss A, Vincent S, Decaussin-Petrucci M, Meugnier E, Viallet J, Ruffion A, et al. Androgen-regulated microRNA-135a decreases prostate cancer cell migration and invasion through downregulating ROCK1 and ROCK2. Oncogene. 2014;0 doi: 10.1038/onc.2014.222. [DOI] [PubMed] [Google Scholar]
- 46.He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proceedings of the National Academy of Sciences of the United States of America. 2014 doi: 10.1073/pnas.1421415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer research. 2014;74:5631–5643. doi: 10.1158/0008-5472.CAN-14-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taipaleenmaki H, Browne G, Akech J, Zustin J, van Wijnen AJ, Stein JL, et al. Targeting of Runx2 by miR-135 and miR-203 Impairs Progression of Breast Cancer and Metastatic Bone Disease. Cancer research. 2015;75:1433–1444. doi: 10.1158/0008-5472.CAN-14-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–394. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tong AW, Fulgham P, Jay C, Chen P, Khalil I, Liu S, et al. MicroRNA profile analysis of human prostate cancers. Cancer gene therapy. 2009;16:206–216. doi: 10.1038/cgt.2008.77. [DOI] [PubMed] [Google Scholar]
- 52.Sun T, Wang Q, Balk S, Brown M, Lee GS, Kantoff P. The role of microRNA-221 and microRNA-222 in androgen-independent prostate cancer cell lines. Cancer research. 2009;69:3356–3363. doi: 10.1158/0008-5472.CAN-08-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun T, Wang X, He HH, Sweeney CJ, Liu SX, Brown M, et al. MiR-221 promotes the development of androgen independence in prostate cancer cells via downregulation of HECTD2 and RAB1A. Oncogene. 2014;33:2790–2800. doi: 10.1038/onc.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun T, Yang M, Chen S, Balk S, Pomerantz M, Hsieh CL, et al. The altered expression of MiR-221/-222 and MiR-23b/-27b is associated with the development of human castration resistant prostate cancer. The Prostate. 2012;72:1093–1103. doi: 10.1002/pros.22456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gandellini P, Folini M, Longoni N, Pennati M, Binda M, Colecchia M, et al. miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer research. 2009;69:2287–2295. doi: 10.1158/0008-5472.CAN-08-2894. [DOI] [PubMed] [Google Scholar]
- 56.Demichelis F, Setlur SR, Beroukhim R, Perner S, Korbel JO, Lafargue CJ, et al. Distinct genomic aberrations associated with ERG rearranged prostate cancer. Genes Chromosomes Cancer. 2009;48:366–380. doi: 10.1002/gcc.20647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20:457–471. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ostling P, Leivonen SK, Aakula A, Kohonen P, Makela R, Hagman Z, et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer research. 2011;71:1956–1967. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- 59.Therneau TM, Grambsch PM. Modeling survival data: extending the Cox model. Springer; New York: 2000. [Google Scholar]
- 60.He B, Lanz RB, Fiskus W, Geng C, Yi P, Hartig SM, et al. GATA2 facilitates steroid receptor coactivator recruitment to the androgen receptor complex. Proc Natl Acad Sci U S A. 2014;111:18261–18266. doi: 10.1073/pnas.1421415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Science translational medicine. 2013;5:169ra110. doi: 10.1126/scitranslmed.3005211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.