

Abstract
Recent studies point to the prevalence of the evolutionary phenomenon of drastic structural transformation of protein domains while continuing to preserve their basic biochemical function. These transformations span a wide spectrum, including simple domains incorporated into larger structural scaffolds, changes in the structural core, major active site shifts, topological rewiring and extensive structural transmogrifications. Proteins from biological conflict systems, such as toxinantitoxin, restriction-modification, CRISPR/Cas, polymorphic toxin and secondary metabolism systems commonly display such transformations. These include endoDNases, metal-independent RNases, deaminases, ADP ribosyltransferases, immunity proteins, kinases and E1-like enzymes. In eukaryotes such transformations are seen in domains involved in chromatin-related peptide recognition and protein/DNA-modification. Intense selective pressures from “arm-race”-like situations in conflict and macromolecular modification systems could favor drastic structural divergence while preserving function.
Introduction
Since the formulation of the evolutionary theory the question of convergent versus divergent evolution has been widely studied and debated [1]. Analysis of organismal structure revealed that certain structurally similar, archetypal forms have repeatedly evolved from distinct ancestral forms (convergence): for instance, the same “fish-like” body shape has independently evolved among vertebrates on multiple occasions in fishes, ichthyosaurs, and whales [1,2]. In addition to such global convergence of form, organisms also display more limited forms of convergent evolution, such as functionally similar organs constituted from structurally different precursors (e.g. wings of insects, pterosaurs, birds and bats)[1]. With the advent of modern structural studies on proteins the question of convergent versus divergent evolution entered the molecular realm [3].
Protein structure and function: Convergence, divergence, and more complex relationships
From their inception, structural studies furnished clear cases of convergence of biochemical function [3,4], which might be defined as the primary biochemical activity of a given protein, i.e., catalysis or binding of another biomolecule or solute. A classic example are the tRNA synthetases, all of which catalyze the same basic reaction (ATP-dependent ligation of the acyl group of an amino acid of the 2’ or 3’ hydroxyl of the cognate tRNA), but belong to two distinct protein superfamilies with structurally unrelated folds [5]. On the other hand, structural studies also revealed that divergent evolution is rampant among protein domains – enzymatic domains, which were originally considered to be unrelated, such as actin, hexokinase, RNase H, PIWI, and diverse integrases of transposons and retroviruses were shown to contain a common fold (the RNase H fold) with comparable active site residues [6]. Discovery of divergent relationships via structural comparisons has provided a robust framework for understanding the evolutionary “exploration” of substrate space, wherein certain folds have been utilized as platforms for extensive biochemical diversification [7-9]. Development of increasingly sensitive sequence-profile and profile-profile search methods has greatly enabled the detection of such divergent relationships between protein domains [10-12].
The explosion in the number of structures in the past decade is now revealing the prevalence of more complex scenarios beyond straightforward functional convergence and divergent evolution. One of these is local structural convergence in active sites of protein domains with similar functions [13,14], such as glycine rich loops in the nucleotide-binding loops of structurally distinct folds [15,16]. This can be seen as a molecular analog of the localized convergence in functionally comparable body parts of organisms. With the protein structure universe being increasingly populated, more evolutionary intermediates are emerging that help understand better the major structural transmogrifications among domains [7,8]. As a result, an underappreciated phenomenon in the structure-function relationships of proteins has come to light: domains sharing a common ancestry undergoing increasingly drastic divergence of structure while at the same time preserving their ancestral catalytic/binding activity and general substrate specificity. This tendency is distinguishable from conventional divergent evolution, wherein divergence occurs primarily at the level of sequence similarity while generally preserving the core fold [17-19]. Importantly, this situation runs contrary to conventional divergent evolution, wherein sequence divergence goes hand-in-hand with diversification of biochemical function, both in terms of broad substrate specificity and types of reactions catalyzed [13,20-22].
As the above-defined evolutionary phenomenon apparently violates the basic predictive logic used in extrapolating function based on protein sequence/structure divergence it is worth a more detailed documentation. Accordingly, we review the recent literature to illustrate and synthesize information regarding this phenomenon.
The spectrum of structural divergence in the face of biochemical function preservation
The structural divergence of domains sharing a common ancestry but preserving biochemical function is best approximated by a spectrum (Figure 1). This premise stems from the observation that structural change, while objectively measurable in different ways, becomes subjective in terms of the threshold of what might be considered a “drastic” modification. Thus, on one end of this apparent spectrum we have changes that fall with the realm of conventional divergent evolution i.e., sequence divergence and simple elaborations of the core fold, typically in the loops connecting core secondary structure elements. Beyond this “baseline”, for descriptive convenience the spectrum can be broken into the following classes of modifications defining the more drastic changes (Figure 1): 1) A simple, ancestral fold being combined with new secondary structure elements to form structurally distinct scaffolds; 2) Modification of the ancestral fold via deletion/degeneration or insertion or transformation of secondary structure elements in the core; 3) Reorganization of active site residues without change in ancestral catalytic/binding activity; 4) Rewiring of secondary structure elements in the topology while preserving their overall spatial positions; 5) Transmogrification of the ancestral fold with major spatial and/or topological reorganization.
Figure 1. Spectrum of structural divergence which preserves biochemical function.
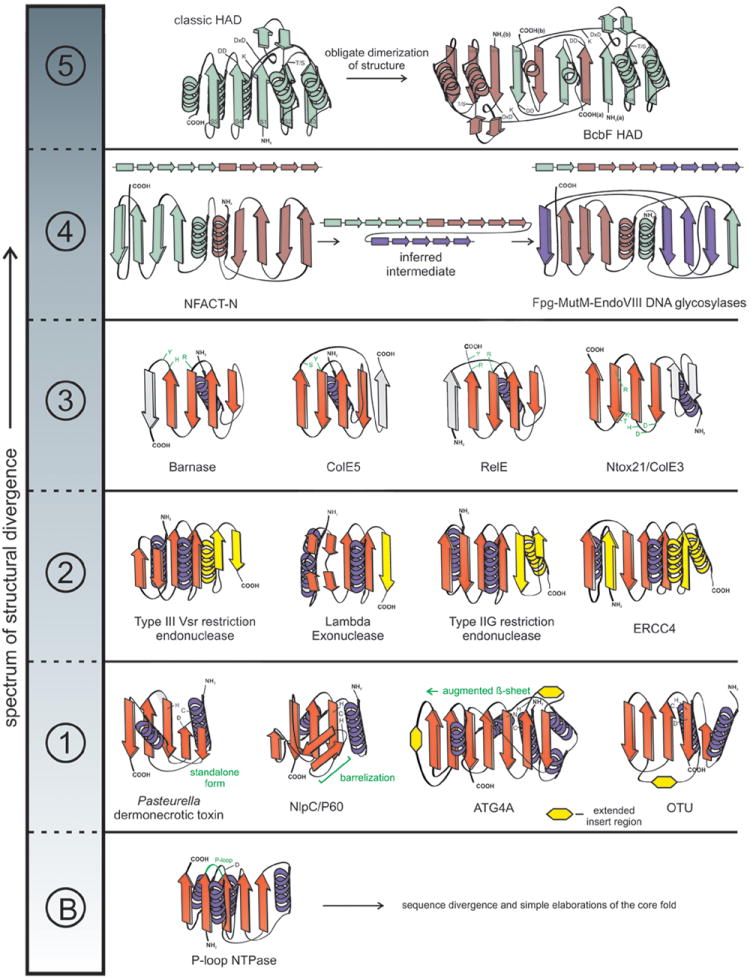
The spectrum is broken up into distinct classes of structural divergence separated by dotted lines. Example structures are depicted as topology diagrams with arrows representing β-strands and coils representing α-helices. ‘B’ represents the ‘baseline’ class of structural divergence. Class 1: representatives of papain-like peptidases; Class 2: restriction endonuclease fold with modified part of the secondary structural elements colored in yellow; Class 3: BECR fold members with divergent active site residues highlighted in green; Class 4: transition between NFACT and Fpg-MutM-EndoVIII DNA glycosylase proteins are accompanied by linear arrays of secondary structural elements to show rewiring, each duplicated basic 4-stranded element is given a distinct color; Class 5: topological transmogrification observed in the obligate dimer-forming BcbF family of HAD domains, strands are labeled, and monomers are given distinct colors.
It must be emphasized that, as these changes span a spectrum of variation, some domains might show more than one of the above classes of modifications. Below we illustrate these classes of modifications using the wealth of examples uncovered by recent structural studies.
Incorporation of simple conserved cores into complex scaffolds
A striking case of this type of structural modification is furnished by the HNH endonucleases that are found in wide variety of DNA-cleaving and, on some occasions, RNA-cleaving enzymes (Figure 2) [23,24]. Simplest versions of this domain display a standalone treble clef fold (Figure 2A), which is stabilized by 4 conserved Zinc-chelating residues and contains an endonuclease active site between the β-hairpin and the C-terminal helix characteristic of this fold (e.g. the restriction endonuclease Hpy99I; PDB 3GOX) [25]. This core is incorporated into increasingly complex structures in various nucleases. In the Cas9 endoDNases of the type-II CRISPR systems it is incorporated into a larger structure formed by α-helices packing around from both the N- and C-termini [26](Figure 2B). In homing endonucleases (e.g. I-PpoI), the original stabilizing Zn2+ of the treble-clef core is lost, but it is incorporated into a larger structure that is stabilized by two newly acquired Zn-chelating sites [27] (Figure 2C). A further variation is seen in the colicin E9 DNase domain, where again the core treble clef has lost its metal and is incorporated into a larger globular domain stabilized by augmentation of the β-hairpin into a sheet by stacking of additional strands (Figure 2D)[28]. Finally, an even more dramatic change is seen in the NucA/non-specific endonuclease family, wherein the treble clef core, which has lost its stabilizing metal, is embedded into an extended sheet formed by two copies of a three-stranded domain with characteristic loop-like C-terminal extensions [24] (Figure 2E). This profusion of structural modifications has often resulted in the relationship between these domains being identified only upon careful sequence structure comparisons [7]. Yet, strikingly, all these domains are rather conservative in terms of their endonuclease activity.
Figure 2. Structural diversity of HNH endonucleases.
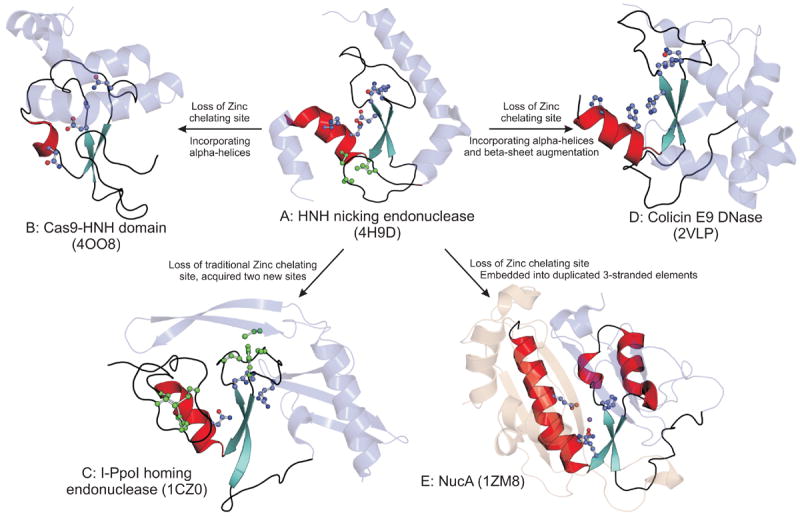
The α-helix and β-sheet of the HNH structural core are shown in red and aquamarine respectively. Metals, active site (blue) and zinc chelating (in green) residues are shown in the ball and stick mode. Other incorporated structural elements are in light blue. The duplicated three-stranded units of NucA are shown in light blue and light brown respectively.
The wider prevalence of this type of modification is indicated by another homologous group of domains, the papain-like peptidases, which catalyze hydrolysis of peptide and related amide linkages (e.g. the isopeptide bond formed by conjugated ubiquitin) (Figure 1) [29]. Here the core fold is formed by a helix bearing the catalytic cysteine, and three strands bearing a histidine and a polar residue, which together constitute the catalytic triad of these enzymes [30]. This core might occur more or less in a simple standalone form (e.g. the Pasteurella Dermonecrotic toxin peptidase [31]) or is incorporated into a wide range of more elaborate scaffolds where the above core is part of an augmented β-sheet (e.g. the ATG8-deconjugating enzyme ATG4B [32]) or β-barrels with varying number of strands (e.g. NlpC/P60 peptidase domains)[30,33]. Indeed, a great diversity of such structural elaborations is observed among papain-like peptidase families, which deconjugate ubiquitin and ubiquitin-like proteins from targets despite the fact they catalyze an essentially equivalent peptidase/isopeptidase reaction [29,30].
Modification of the ancestral fold by degeneration or insertion or transformation of secondary structure elements
Hints for this form of structural transformation were first obtained in the serine/threonine/tyrosine (STY) kinase domains, which are found in enzymes catalyzing a stereotypic reaction of ATP-dependent phosphorylation of hydroxyl or carboxyl groups in peptide side chains, lipid head groups, and various small molecules [34]. This domain displays a complex fold with a 5-stranded unit at the “top”, a smaller central sheet comprised of 3 strands, and a “bottom” formed by a mixed α+β structure related to the equivalent unit of the ATP-grasp fold [7,34]; all three units contribute residues for nucleotide binding or catalysis (Figure 3). Phyletic pattern and structural analysis [35] suggests that the most ancient version of this fold is likely to be the SAICAR synthetase (PurC), which is a kinase catalyzing the formation of a peptide-like bond in purine biosynthesis. Here, the “bottom” element has an extended sheet of 5 strands [36] (Figure 3A). In kinases which phosphorylate aminoglycoside antibiotics, as part of resistance strategies of bacteria, this sheet in the “bottom” element is reduced to 4 strands (Figure 3B)[37,38]. In the Rio1/2 kinases, which are ancient protein kinases shared by archaea and eukaryotes as part of their ribosome maturation apparatus [39], the said sheet is reduced to a mere dyad of strands (Figure 3C) [40]. Finally, in STY kinases, which are the mainstay of signal transduction in eukaryotes and certain bacteria (e.g. myxobacteria, actinobacteria and cyanobacteria), and lipid kinases (e.g. PI3 kinase) the strands are entirely lost in the bottom unit of the domain [34,41,42]. Thus, structures solved to date recapitulate the entire trajectory of structural transformation from the pronounced sheet of the ATP-grasp like precursor to the total loss of the strands (Figure 3D). However, through this major transformation the basic phosphorylation reaction catalyzed by these domains has remained the same in large part because the active site residues lying at the inter-β strand connectors have remained intact, even as the secondary structure of the proximal elements have changed [42]. This change has probably allowed these enzymes to explore a wider substrate space while keeping the basic reaction constant [21].
Figure 3. Cartoon representations of various STY kinase domains illustrating the structural transformations in the superfamily.
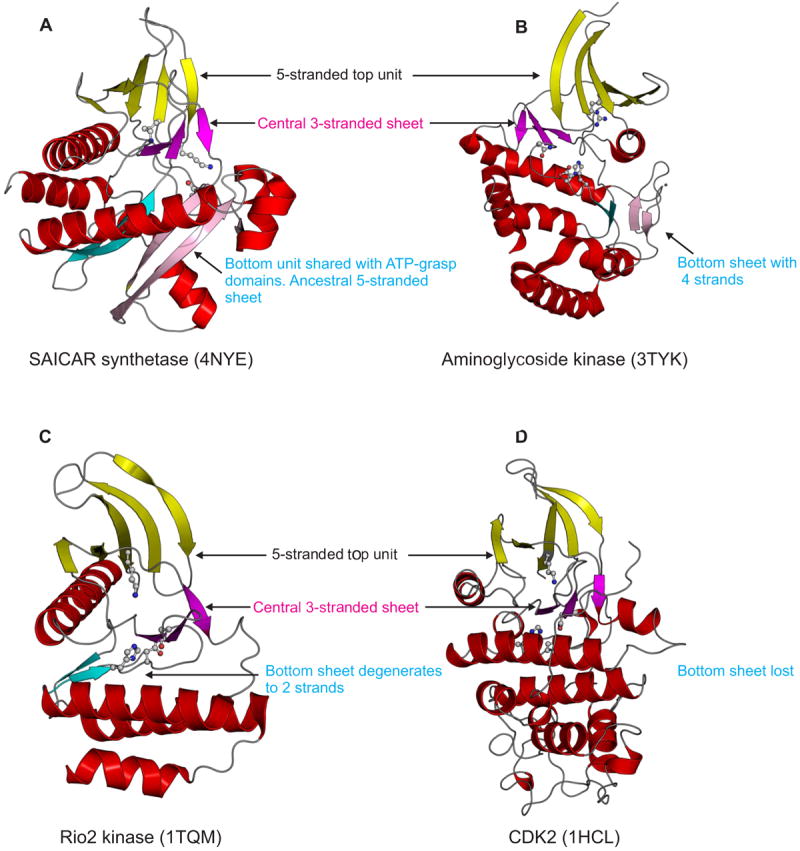
Helices are colored red, whereas strands are colored based on their structural unit. Strands of the top unit are colored yellow and those in the central sheet, magenta. The cyan and pink strands of the bottom unit show the equivalence of these strands between the structures.
New structures and results from sequence analysis suggest that such transformations might be more prevalent, as illustrated by the deaminase-like fold [43]. This fold includes catalytic domains with biochemically distinct activities such as, base deaminase (e.g. the mutagenic and RNA-editing AID/APOBEC deaminases), peptidase, possible nuclease, AICAR transformylase and ADP-ribosyl transferase [43,44]. Despite this diversification the spatial location of the active site is retained throughout the fold along with a core β-sheet with 5 β-strands [43]. However, among these enzymes, recent studies show that deaminases are prone to rampant structural plasticity of the core sheet. First, deaminases might come in two types wherein the 5th strand of the sheet might exist either in parallel or antiparallel configuration [45]. Moreover, the α-helices following this strand might often be lost in several representatives. In other cases not just helices but the entire 5th strand might be lost. Given that other enzymes of this fold closely conform to the ancestral type, these drastic changes appear to be a more recent feature occurring within the deaminases alone [43]. Interestingly, closer examination of their evolution has shown that this rampant structural modification is primarily associated with their diversification as toxins delivered into target cells by diverse bacteria [43]. Comparable structural changes involving strand insertion or deletion are also observed in nucleases of the restriction endonuclease fold that target specific DNA sequences or structures (Figure 1)[46-48]. Other rich sources of such structural modifications are the chromo-like fold and PHD-finger-like domains, which despite their evolutionary plasticity retain a comparable function of binding methylated peptides in chromatin proteins, such as histones [49-52].
Reorganization of active site residues while retaining ancestral catalytic/binding activity
Usually the constellation of residues constituting the active site is the most conserved sequence feature of a superfamily of enzymes catalyzing the same reaction type [17,18,21,22]. Thus, such active sites residues have great predictive value [22] and are typically resilient to a wide range of structural changes in the core fold, as noted in the above-discussed categories. Studies on P-loop NTPases showed that one key active site residue, namely the arginine finger, is not conserved and has independently evolved on multiple occasions, either within the core fold, or in linked domains, or in separate proteins [53]. More drastic alterations of active sites are seen in DNases of the restriction endonuclease fold—the Vsr (very short patch DNA repair)-like endonucleases have undergone a reconfiguration of the ancestral active site via loss of two key residues and acquisition of two new ones from entirely different locations in their structure [47,54-56]. Despite this the spatial position of the active site pocket and their catalytic activity remains comparable to the ancestral versions. Multiple examples of comparable modifications have recently become apparent among RNases of the BECR (Barnase-EndoU-Colicin E5/D-RelE) fold [57]. These RNases include various bacterial and fungal toxins which are deployed against target cells [57,58], toxin components of toxin-antitoxin systems [59,60], and certain RNases involved in splicing such as Endonuclease U [61]. Several families of these RNases possess a histidine that is critical for catalysis [57,62]. While this residue is conserved within a given family (e.g. Ntox21 and colicin E3 families), it is typically not conserved across all members of the BECR fold (Figure 1). In some, such as the RelE family, even the histidine might not be conserved. Yet all families of the BECR fold catalyze a similar metal-independent endoRNase reaction, generating a RNA product with a cyclic 2’-3’ end [62].
Interestingly, at least two other unrelated folds of RNases show a comparable phenomenon. One of these is the recently identified all α-helical HEPN RNase, which is found in type-I and III CRISPR systems and, like the BECR RNases, also in toxin-antitoxin systems [63]. While several of these RNases have a characteristic histidine-containing motif associated with their active site, some families have lost this histidine and acquired distinct catalytic histidines from elsewhere in the sequence [63-65]. Similarly, the RAMP superfamily from the type-I and III CRISPR systems are RNases containing the RRM-like fold and are critical for processing the CRISPR RNAs from their precursor transcript [66,67]. Although several of the individual RAMP families contain conserved histidines, none of these are conserved throughout the superfamily [68]. Structural studies on RAMPs have demonstrated that these independently acquired histidines and other polar residues might constitute distinct but catalytically equivalent active sites for this RNase superfamily [66,68]. Like the BECR RNases, the HEPN and RAMPs are also metal-independent endoRNases that generate products with cyclic 2’-3’ ends [64-66]. Metal-dependent active sites require a precise 3D configuration of metal-chelating residues coming from different parts of the fold probably making them harder to reconfigure. However, the metal-independent RNases are primarily dependent on histidine or other polar residues to facilitate an internal attack on the phosphodiester bond by the 2’OH group in RNA [62]. Hence, it is likely that such residues could independently emerge in a “pre-adapted” RNA-binding scaffold such as those observed in the above-discussed domains.
Rewiring of secondary structure elements while preserving their general spatial positions
Straightforward versions of this type of structural modification are the widely known circular permutations, which are common in domains wherein the N- and C-terminal elements tend to be spatially proximal [13,69]. Circular permutations have occurred on multiple occasions without any change in catalyzed reactions in amide bond-forming ligases of the classical ATP-grasp fold (e.g. glutathione synthetases which ligate glycine to gamma-glutamylcysteine to form glutathione)[70,71]. More dramatic modifications of this type have been reported in the RNA-binding KH domain superfamily [72]. In the ERA GTPases, involved in assembly of the 30S ribosomal subunit, the C-terminal KH domain has undergone a major rewiring of its secondary structure units [73]. However, it still preserves the characteristic sequence motif and overall spatial organization of classic KH domains [72], and binds RNA in a comparable manner [73]. Similar rewiring is also observed in another nucleic acid/nucleotide binding domain, the RAGNYA domain, wherein multiple shifts in inter-secondary structure connectivity are observed [74]. A particularly dramatic case is seen in the nucleic acid ligases [7,75]: whereas in classical amide bond ligases of the ATP-grasp fold a kinase-C terminus-like domain is fused to the C-terminus of a RAGNYA domain, in nucleic acid ligases this domain is inserted into the RAGNYA domain [74]. Despite the consequent rewiring of secondary structure the RAGNYA domain continues to bind ATP just as in the case of unmodified version of the fold.
A recent example of such a modification has come to light with the discovery of the relationship between the Fpg-MutM-EndoVIII family of DNA glycosylases and the NFACT (NEMF-FpbA-caliban- Tae2) proteins that are predicted to be RNA-modifying enzymes [76]. Their shared catalytic domain is a composite fold comprised of an N-terminal 8-stranded β-sandwich with 2 flanking helices and a C-terminal unit formed by two helix-hairpin-helix (HhH) motifs (Figure 1). Here, the respective phyletic patterns of NFACT and DNA glycosylases help in establishing the direction of structural modification – NFACT is found across all three superkingdoms of life, whereas these DNA glycosylases emerged in bacteria and were transferred to eukaryotes but are absent in archaea [76]. This suggests that the latter is likely to have been derived from a precursor like the former. Consistent with this, the β-sandwich in the NFACT domains shows a more ancestral condition of being comprised of a duplication of two 4-stranded elements. Reconstruction of the structural reorganization in the DNA glycosylases indicates that it proceeded via an initial triplication of the basic 4-stranded element, followed by reconstitution of the β-sandwich from secondary structure elements from each of the three copies (Figure 1). The resulting β-sheets are topologically rewired, but the active site is notably conserved across the two versions of the fold, suggesting that RNA modification by NFACT domains is likely to be mechanistically similar to the DNA glycosylases [76]. More generally, while simple circular permutation proceeds via duplicated intermediates, more complex reorganizations might arise from higher order n-plications or domain insertions [69].
Major topological and spatial transmogrifications
The prevalence of these extreme structural modifications is difficult to objectively estimate because evolutionary information necessary for such assessments is typically irretrievably lost [77]. However, in certain cases structural data can be combined with evidence from sequence analysis and contextual information from domain architectures and/or genomic context to make a confident case for such transformations [15,34,72]. Consequently, evidence for several such transformations is emerging from careful case-by-case analysis of structural data. Recently such a transformation was noted in the most common version of the α-helical HEPN RNase domains found in type-IIIA CRISPR systems [78]. Here the HEPN domain is combined with a winged helix-turn-helix domain and has undergone a complex structural reorganization, while retaining the catalytic residues intact [79,80].
Striking examples have emerged from the study of immunity proteins, which neutralize the toxin domains of the recently described prokaryotic polymorphic toxin systems that are deployed by cells to kill intraspecific competitor cells [57]. In these systems there is strong genomic coupling of genes coding for immunity proteins and gene cassettes coding for polymorphic toxin domains. Further, there is evidence for rapid divergence of immunity proteins in response to the polymorphism in the toxin domains that they neutralize [24,57]. Hence, these are fertile grounds for rapid evolutionary changes as immunity proteins diversify to adapt to new toxins. In these systems several distinct types of immunity proteins have been described, of which the SUKH and SuFu domains are the first and second most common immunity proteins [24]. While SUKH and SuFu display superficially different folds [24,81], a closer examination suggests that their core β-sheets are similarly organized (Figure 4): in both cases it is a “split sheet”, i.e. one with a gap between distinct four-stranded and three-stranded elements. This, together with their functional equivalence and cognate genomic organization in the polymorphic toxin loci suggests that they have indeed diverged via drastic structural modification. In the case of the SUKH and SuFu domains this can be reconstructed as involving circular permutation followed by a rotation of the three-stranded element resulting in it being flipped in orientation with respect to the four-stranded sheet (Figure 4). Recently, the structure of a novel immunity protein, CdiI/Imm75, from the Enterobacter cloacae polymorphic toxin system was published [82]. While this was reported as a novel fold, it shows the same structural organization as the SUKH and SuFu domains in being comprised of equivalent four-stranded and three-stranded elements (Figure 4). Thus, CdiI/Imm75 is yet another immunity protein sharing a common ancestry with the SUKH and SuFu domains via major structural modification. In this case it appears to have involved rotation of the three-stranded unit with it now partly stacking with the 4-stranded element to form an open sandwich-like fold (Figure 4).
Figure 4. Topological and spatial transmogrifications in SUKH, SuFu and Imm75 families.
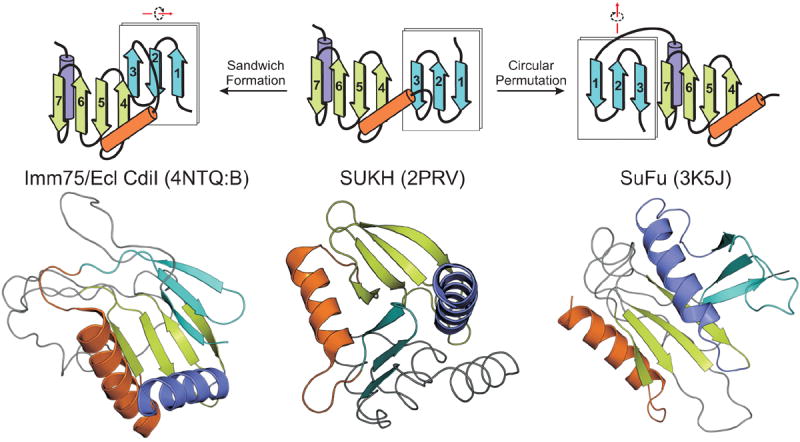
The shared structural elements of these families are equivalently colored in the topological and cartoon representations. The type of structural transition with respect to the SUKH domain is shown above the topology.
Prevalence of function-preserving structural modifications in biological conflict systems and eukaryotic macromolecule modification systems
In the early days of structural studies such drastic modifications were initially regarded as curiosities or rare quirks of evolution [34,70]. However, the sheer wealth of currently-available structure and sequence data are beginning to reveal certain patterns in terms of biological systems where such modifications tend to be overrepresented. While some of the above-mentioned structural modifications, such as that in the nucleic acid ligases and DNA glycosylases, are relatively ancient events in core cellular systems [74,76], the majority of these can be traced to biological conflict systems (Figure 5). These systems are deployed in: 1) intra-genomic conflicts, e.g. toxin-antitoxin and restriction-modification systems [83,84]; 2) inter-genomic conflicts, e.g. the CRISPR/Cas system involved in restricting invasive genomes like bacteriophages [68,78]; 3) intra-specific conflicts, e.g. polymorphic toxin systems [24,57]; 4) inter-specific conflicts, e.g. antibiotics and toxins deployed by bacteria against competitors or hosts, and mechanisms of immunity against them [57,85,86]. Several enzymatic and non-enzymatic domains from these systems, including aminoglycoside kinases, E1-like adenylating enzymes, deaminases, ADP-ribosyltransferases, restriction endonucleases, HNH endonucleases, diverse RNases and immunity proteins from such systems show drastic structural modifications while preserving their biochemical function (Figure 5). Although the majority of instances of such diversification are currently known from prokaryotic conflict systems, traditional immunity systems of animals such as the antibodies and variable lymphocyte receptors also show evidence for such modifications [85,86]. Intense selective pressures found in these conflict systems, which directly impinge on the survival of the organism, have resulted in arms races thereby favoring rapid emergence of innovations [87]. In these situations divergence is often critical for evading counter-adaptations of the rival systems locked in the arms race. Thus, in these systems selection might balance structural divergence (necessary for evasion) with preservation of function (cannot be compromised for organismal survival).
Figure 5. Biological context of proteins showing major structural variations.

Key components of each biological system are illustrated with the proteins and domains showing major structural variations highlighted in light blue.
In eukaryotes such structural diversification is additionally encountered in systems primarily involved in introduction and “reading” of modifications in macromolecules such as DNA, proteins (histones and tubulins) or lipids [49,50,88,89] (Figure 5). In many cases these modifications serve as epigenetic marks that encode information over and beyond the genetic material. Thus, examples of these structural modifications are found in DNA-modifying enzymes, peptide recognition domains and enzymes of the ubiquitin system (Figure 5). The diversification in these systems might be seen as a parallel to the arms race scenario in conflict systems. Here, proliferation of signals based on modified peptides and their utilization as new epigenetic marks probably act as drivers of innovation similar to counter-adaptations of rival systems in biological conflicts.
Concluding remarks
Detection of major structural modifications led to the proposal that the classification of proteins on the basis of folds is not accurate [77,90]. However, as examples of such modifications accumulate it is becoming clear that such an extreme view is unwarranted. Rather, it merely suggests that protein structures can be either plastic while preserving function (Figure 1) or relatively refractory to structural change while diverging in function [17,22]. A closer examination reveals that the former is a strategy that is exploited in specific circumstances, particularly in systems pertaining to biological conflicts or modifications of macromolecules (Figure 5). Thus, plasticity or conservation of a fold is primarily a reflection of the type of selective forces operating on the protein in the context of the biological system in which it functions. In terms of further studies, a better understanding of the interplay between selective forces and such structural transformations would be of particular interest. These transformations could also provide useful information for guiding future protein-engineering efforts. Hence, we hope that this survey of a dramatic but under-appreciated tendency in protein evolution inspires further studies.
Highlights.
Drastic structural transformation of protein domains while retaining their basic biochemical function is a notable phenomenon in protein evolution.
These transformations span a wide spectrum of structural changes including complex modifications of domain structure.
Domains displaying these transformations are most commonly observed in systems involved in biological conflicts, eukaryotic chromatin-related protein/DNA modification and peptide recognition.
These systems show “arm-race”-like situations that could favor drastic structural divergence while preserving function
Acknowledgments
Work by the authors is supported by the funds of the Intramural Research Program of the National Institutes of Health, DHHS, USA. We sincerely apologize to our colleagues whose work could not be cited due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McGhee GR. Convergent evolution : limited forms most beautiful. Cambridge, Mass: MIT Press; 2011. [Google Scholar]
- 2.Benton MJ. Vertebrate Palaeontology. Oxford: Blackwell Publishing; 2009. [Google Scholar]
- 3.Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. doi: 10.1002/pro.5560020104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graumann P, Marahiel MA. A case of convergent evolution of nucleic acid binding modules. Bioessays. 1996;18:309–315. doi: 10.1002/bies.950180409. [DOI] [PubMed] [Google Scholar]
- 5.Perona JJ, Hadd A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry. 2012;51:8705–8729. doi: 10.1021/bi301180x. [DOI] [PubMed] [Google Scholar]
- 6.Majorek KA, Dunin-Horkawicz S, Steczkiewicz K, Muszewska A, Nowotny M, Ginalski K, Bujnicki JM. The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res. 2014 doi: 10.1093/nar/gkt1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Andreeva A, Howorth D, Chothia C, Kulesha E, Murzin AG. SCOP2 prototype: a new approach to protein structure mining. Nucleic Acids Res. 2014;42:D310–314. doi: 10.1093/nar/gkt1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuff AL, Sillitoe I, Lewis T, Clegg AB, Rentzsch R, Furnham N, Pellegrini-Calace M, Jones D, Thornton J, Orengo CA. Extending CATH: increasing coverage of the protein structure universe and linking structure with function. Nucleic Acids Res. 2011;39:D420–426. doi: 10.1093/nar/gkq1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dietmann S, Holm L. Identification of homology in protein structure classification. Nat Struct Biol. 2001;8:953–957. doi: 10.1038/nsb1101-953. [DOI] [PubMed] [Google Scholar]
- 10.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 12.Soding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Research. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Todd AE, Orengo CA, Thornton JM. Plasticity of enzyme active sites. Trends Biochem Sci. 2002;27:419–426. doi: 10.1016/s0968-0004(02)02158-8. [DOI] [PubMed] [Google Scholar]
- 14.Stark A, Sunyaev S, Russell RB. A model for statistical significance of local similarities in structure. J Mol Biol. 2003;326:1307–1316. doi: 10.1016/s0022-2836(03)00045-7. [DOI] [PubMed] [Google Scholar]
- 15.Lupas AN, Ponting CP, Russell RB. On the evolution of protein folds: are similar motifs in different protein folds the result of convergence, insertion, or relics of an ancient peptide world? J Struct Biol. 2001;134:191–203. doi: 10.1006/jsbi.2001.4393. [DOI] [PubMed] [Google Scholar]
- 16.Kozbial PZ, Mushegian AR. Natural history of S-adenosylmethionine-binding proteins. BMC Struct Biol. 2005;5:19. doi: 10.1186/1472-6807-5-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerlt JA, Babbitt PC, Jacobson MP, Almo SC. Divergent evolution in enolase superfamily: strategies for assigning functions. J Biol Chem. 2012;287:29–34. doi: 10.1074/jbc.R111.240945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerlt JA, Babbitt PC. Enzyme (re)design: lessons from natural evolution and computation. Curr Opin Chem Biol. 2009;13:10–18. doi: 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anantharaman V, Aravind L. Diversification of catalytic activities and ligand interactions in the protein fold shared by the sugar isomerases, eIF2B, DeoR transcription factors, acyl-CoA transferases and methenyltetrahydrofolate synthetase. J Mol Biol. 2006;356:823–842. doi: 10.1016/j.jmb.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 20.Murzin AG. How far divergent evolution goes in proteins. Curr Opin Struct Biol. 1998;8:380–387. doi: 10.1016/s0959-440x(98)80073-0. [DOI] [PubMed] [Google Scholar]
- 21.Anantharaman V, Aravind L, Koonin EV. Emergence of diverse biochemical activities in evolutionarily conserved structural scaffolds of proteins. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 22.Torrance JW, Bartlett GJ, Porter CT, Thornton JM. Using a library of structural templates to recognise catalytic sites and explore their evolution in homologous families. J Mol Biol. 2005;347:565–581. doi: 10.1016/j.jmb.2005.01.044. [DOI] [PubMed] [Google Scholar]
- 23.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:49–95. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 24.Zhang D, Iyer LM, Aravind L. A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 2011;39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sokolowska M, Czapinska H, Bochtler M. Crystal structure of the beta beta alpha-Me type II restriction endonuclease Hpy99I with target DNA. Nucleic Acids Res. 2009;37:3799–3810. doi: 10.1093/nar/gkp228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26••.Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343:1247997. doi: 10.1126/science.1247997. The structure of Cas9 is of considerable interest for it illustrates how the core treble clef fold of the HNH endonuclease domain is incorporated into a more complex scaffold and inserted into the RNase H fold DNase domain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eastberg JH, Eklund J, Monnat R, Jr, Stoddard BL. Mutability of an HNH nuclease imidazole general base and exchange of a deprotonation mechanism. Biochemistry. 2007;46:7215–7225. doi: 10.1021/bi700418d. [DOI] [PubMed] [Google Scholar]
- 28.Meenan NA, Sharma A, Fleishman SJ, Macdonald CJ, Morel B, Boetzel R, Moore GR, Baker D, Kleanthous C. The structural and energetic basis for high selectivity in a high-affinity protein-protein interaction. Proc Natl Acad Sci U S A. 2010;107:10080–10085. doi: 10.1073/pnas.0910756107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Love KR, Catic A, Schlieker C, Ploegh HL. Mechanisms, biology and inhibitors of deubiquitinating enzymes. Nat Chem Biol. 2007;3:697–705. doi: 10.1038/nchembio.2007.43. [DOI] [PubMed] [Google Scholar]
- 30.Iyer LM, Koonin EV, Aravind L. Novel predicted peptidases with a potential role in the ubiquitin signaling pathway. Cell Cycle. 2004;3:1440–1450. doi: 10.4161/cc.3.11.1206. [DOI] [PubMed] [Google Scholar]
- 31.Kitadokoro K, Kamitani S, Miyazawa M, Hanajima-Ozawa M, Fukui A, Miyake M, Horiguchi Y. Crystal structures reveal a thiol protease-like catalytic triad in the C-terminal region of Pasteurella multocida toxin. Proc Natl Acad Sci U S A. 2007;104:5139–5144. doi: 10.1073/pnas.0608197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28:1341–1350. doi: 10.1038/emboj.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Xu Q, Chiu HJ, Farr CL, Jaroszewski L, Knuth MW, Miller MD, Lesley SA, Godzik A, Elsliger MA, Deacon AM, et al. Structures of a bifunctional cell wall hydrolase CwlT containing a novel bacterial lysozyme and an NlpC/P60 DL-endopeptidase. J Mol Biol. 2014;426:169–184. doi: 10.1016/j.jmb.2013.09.011. This new structure of the NlpC/P60 endopeptidase domain illustrates how the ancestral core of the papain-like fold is incorporated into a barrel-like structure. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grishin NV. Phosphatidylinositol phosphate kinase: a link between protein kinase and glutathione synthase folds. J Mol Biol. 1999;291:239–247. doi: 10.1006/jmbi.1999.2973. [DOI] [PubMed] [Google Scholar]
- 35.Brown AM, Hoopes SL, White RH, Sarisky CA. Purine biosynthesis in archaea: variations on a theme. Biol Direct. 2011;6:63. doi: 10.1186/1745-6150-6-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levdikov VM, Barynin VV, Grebenko AI, Melik-Adamyan WR, Lamzin VS, Wilson KS. The structure of SAICAR synthase: an enzyme in the de novo pathway of purine nucleotide biosynthesis. Structure. 1998;6:363–376. doi: 10.1016/s0969-2126(98)00038-0. [DOI] [PubMed] [Google Scholar]
- 37.Shi K, Houston DR, Berghuis AM. Crystal structures of antibiotic-bound complexes of aminoglycoside 2″-phosphotransferase IVa highlight the diversity in substrate binding modes among aminoglycoside kinases. Biochemistry. 2011;50:6237–6244. doi: 10.1021/bi200747f. [DOI] [PubMed] [Google Scholar]
- 38.Stogios PJ, Shakya T, Evdokimova E, Savchenko A, Wright GD. Structure and function of APH(4)-Ia, a hygromycin B resistance enzyme. J Biol Chem. 2011;286:1966–1975. doi: 10.1074/jbc.M110.194266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39•.Widmann B, Wandrey F, Badertscher L, Wyler E, Pfannstiel J, Zemp I, Kutay U. The kinase activity of human Rio1 is required for final steps of cytoplasmic maturation of 40S subunits. Mol Biol Cell. 2012;23:22–35. doi: 10.1091/mbc.E11-07-0639. The Rio family kinases which have two strands in the bottom unit (Figure 3) were shown to possess protein kinase activity similar to the kinases lacking strands in this unit. This established that the basic catalytic activity of these enzymes remained the same despite the structural modification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.LaRonde-LeBlanc N, Wlodawer A. Crystal structure of A. fulgidus Rio2 defines a new family of serine protein kinases. Structure. 2004;12:1585–1594. doi: 10.1016/j.str.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 41.Williams R, Berndt A, Miller S, Hon WC, Zhang X. Form and flexibility in phosphoinositide 3-kinases. Biochem Soc Trans. 2009;37:615–626. doi: 10.1042/BST0370615. [DOI] [PubMed] [Google Scholar]
- 42.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Iyer LM, Zhang D, Rogozin IB, Aravind L. Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 2011;39:9473–9497. doi: 10.1093/nar/gkr691. Several new deaminase domains were discovered for the first time and the major structural modifications of the deaminase fold involving loss of C-terminal secondary structure elements were demonstrated. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Conticello SG. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008;9:229. doi: 10.1186/gb-2008-9-6-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prochnow C, Bransteitter R, Klein MG, Goodman MF, Chen XS. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature. 2007;445:447–451. doi: 10.1038/nature05492. [DOI] [PubMed] [Google Scholar]
- 46.Newman M, Murray-Rust J, Lally J, Rudolf J, Fadden A, Knowles PP, White MF, McDonald NQ. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 2005;24:895–905. doi: 10.1038/sj.emboj.7600581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aravind L, Makarova KS, Koonin EV. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen BW, Xu D, Chan SH, Zheng Y, Zhu Z, Xu SY, Stoddard BL. Characterization and crystal structure of the type IIG restriction endonuclease RM.BpuSI. Nucleic Acids Res. 2011;39:8223–8236. doi: 10.1093/nar/gkr543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Khorasanizadeh S. Recognition of methylated histones: new twists and variations. Curr Opin Struct Biol. 2011;21:744–749. doi: 10.1016/j.sbi.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 50.Sanchez R, Zhou MM. The PHD finger: a versatile epigenome reader. Trends Biochem Sci. 2011;36:364–372. doi: 10.1016/j.tibs.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.He F, Umehara T, Saito K, Harada T, Watanabe S, Yabuki T, Kigawa T, Takahashi M, Kuwasako K, Tsuda K, et al. Structural insight into the zinc finger CW domain as a histone modification reader. Structure. 2010;18:1127–1139. doi: 10.1016/j.str.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 52.Kim D, Blus BJ, Chandra V, Huang P, Rastinejad F, Khorasanizadeh S. Corecognition of DNA and a methylated histone tail by the MSL3 chromodomain. Nat Struct Mol Biol. 2010;17:1027–1029. doi: 10.1038/nsmb.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iyer LM, Makarova KS, Koonin EV, Aravind L. Comparative genomics of the FtsK-HerA superfamily of pumping ATPases: implications for the origins of chromosome segregation, cell division and viral capsid packaging. Nucleic Acids Res. 2004;32:5260–5279. doi: 10.1093/nar/gkh828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor GK, Heiter DF, Pietrokovski S, Stoddard BL. Activity, specificity and structure of IBth0305I: a representative of a new homing endonuclease family. Nucleic Acids Res. 2011;39:9705–9719. doi: 10.1093/nar/gkr669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dynan W, Fox K, Stoddard B. Editorial: NAR surveys the past, present and future of restriction endonucleases. Nucleic Acids Res. 2014;42:1–2. doi: 10.1093/nar/gkt1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kinch LN, Ginalski K, Rychlewski L, Grishin NV. Identification of novel restriction endonuclease-like fold families among hypothetical proteins. Nucleic Acids Res. 2005;33:3598–3605. doi: 10.1093/nar/gki676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57••.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Polymorphic toxin systems: Comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. This comprehensive survey of polymorphic toxin systems uncovered over 150 toxin domains and 90 immunity proteins illustrating their extraordinary biochemical diversification. The transformations in the HNH nucleases and BECR fold RNases are also described. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ng CL, Lang K, Meenan NA, Sharma A, Kelley AC, Kleanthous C, Ramakrishnan V. Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nat Struct Mol Biol. 2010;17:1241–1246. doi: 10.1038/nsmb.1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miallau L, Jain P, Arbing MA, Cascio D, Phan T, Ahn CJ, Chan S, Chernishof I, Maxson M, Chiang J, et al. Comparative proteomics identifies the cell-associated lethality of M. tuberculosis RelBE-like toxin-antitoxin complexes. Structure. 2013;21:627–637. doi: 10.1016/j.str.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60•.Boggild A, Sofos N, Andersen KR, Feddersen A, Easter AD, Passmore LA, Brodersen DE. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure. 2012;20:1641–1648. doi: 10.1016/j.str.2012.08.017. This new structure of the RelE helped establish that they possess an active site distinct from other structurally distinct BECR fold RNases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Renzi F, Caffarelli E, Laneve P, Bozzoni I, Brunori M, Vallone B. The structure of the endoribonuclease XendoU: From small nucleolar RNA processing to severe acute respiratory syndrome coronavirus replication. Proc Natl Acad Sci U S A. 2006;103:12365–12370. doi: 10.1073/pnas.0602426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang W. Nucleases: diversity of structure, function and mechanism. Q Rev Biophys. 2011;44:1–93. doi: 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63•.Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, Aravind L. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct. 2013;8:15. doi: 10.1186/1745-6150-8-15. The HEPN domain was established as being an RNase domain and its active site was identified using sensitive sequence analysis. The structural transformations and active site remodelling of the HEPN domains are also described. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Klaiman D, Steinfels-Kohn E, Krutkina E, Davidov E, Kaufmann G. The wobble nucleotideexcising anticodon nuclease RloC is governed by the zinc-hook and DNA-dependent ATPase of its Rad50-like region. Nucleic Acids Res. 2012;40:8568–8578. doi: 10.1093/nar/gks593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meineke B, Shuman S. Determinants of the cytotoxicity of PrrC anticodon nuclease and its amelioration by tRNA repair. RNA. 2012;18:145–154. doi: 10.1261/rna.030171.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66••.Niewoehner O, Jinek M, Doudna JA. Evolution of CRISPR RNA recognition and processing by Cas6 endonucleases. Nucleic Acids Res. 2014;42:1341–1353. doi: 10.1093/nar/gkt922. Multiple structures of RAMP RNases of the Cas6 family reported here illustrate the phenomenon of active site plasiticity among these nucleases. Despite catalyzing a very similar reaction, the active site residues differ even among related members of the family. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reeks J, Naismith JH, White MF. CRISPR interference: a structural perspective. Biochem J. 2013;453:155–166. doi: 10.1042/BJ20130316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Makarova KS, Aravind L, Wolf YI, Koonin EV. Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Peisajovich SG, Rockah L, Tawfik DS. Evolution of new protein topologies through multistep gene rearrangements. Nat Genet. 2006;38:168–174. doi: 10.1038/ng1717. [DOI] [PubMed] [Google Scholar]
- 70.Polekhina G, Board PG, Gali RR, Rossjohn J, Parker MW. Molecular basis of glutathione synthetase deficiency and a rare gene permutation event. EMBO J. 1999;18:3204–3213. doi: 10.1093/emboj/18.12.3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L. Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol Biosyst. 2009;5:1636–1660. doi: 10.1039/b917682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grishin NV. KH domain: one motif, two folds. Nucleic Acids Res. 2001;29:638–643. doi: 10.1093/nar/29.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tu C, Zhou X, Tropea JE, Austin BP, Waugh DS, Court DL, Ji X. Structure of ERA in complex with the 3′ end of 16S rRNA: implications for ribosome biogenesis. Proc Natl Acad Sci U S A. 2009;106:14843–14848. doi: 10.1073/pnas.0904032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balaji S, Aravind L. The RAGNYA fold: a novel fold with multiple topological variants found in functionally diverse nucleic acid, nucleotide and peptide-binding proteins. Nucleic Acids Res. 2007;35:5658–5671. doi: 10.1093/nar/gkm558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nandakumar J, Shuman S, Lima CD. RNA ligase structures reveal the basis for RNA specificity and conformational changes that drive ligation forward. Cell. 2006;127:71–84. doi: 10.1016/j.cell.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 76.Burroughs AM, Aravind L. A highly conserved family of domains related to the DNA-glycosylase fold helps predict multiple novel pathways for RNA modifications. RNA Biol. 2014;11 doi: 10.4161/rna.28302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrey D, Fischer M, Honig B. Structural relationships among proteins with different global topologies and their implications for function annotation strategies. Proc Natl Acad Sci U S A. 2009;106:17377–17382. doi: 10.1073/pnas.0907971106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Makarova KS, Anantharaman V, Grishin NV, Koonin EV, Aravind L. CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front Genet. 2014 doi: 10.3389/fgene.2014.00102. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79•.Kim YK, Kim YG, Oh BH. Crystal structure and nucleic acid-binding activity of the CRISPRassociated protein Csx1 of Pyrococcus furiosus. Proteins. 2013;81:261–270. doi: 10.1002/prot.24183. This structure shows a remarkable structural modification of the HEPN domain while preserving the ancestral active site residues. [DOI] [PubMed] [Google Scholar]
- 80.Lintner NG, Frankel KA, Tsutakawa SE, Alsbury DL, Copie V, Young MJ, Tainer JA, Lawrence CM. The structure of the CRISPR-associated protein Csa3 provides insight into the regulation of the CRISPR/Cas system. J Mol Biol. 2011;405:939–955. doi: 10.1016/j.jmb.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Das D, Finn RD, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Cai X, Carlton D, Chen C, Chiu HJ, et al. The crystal structure of a bacterial Sufu-like protein defines a novel group of bacterial proteins that are similar to the N-terminal domain of human Sufu. Protein Sci. 2010;19:2131–2140. doi: 10.1002/pro.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beck CM, Morse RP, Cunningham DA, Iniguez A, Low DA, Goulding CW, Hayes CS. CdiA from Enterobacter cloacae Delivers a Toxic Ribosomal RNase into Target Bacteria. Structure. 2014 doi: 10.1016/j.str.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hayes F, Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit Rev Biochem Mol Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 84.Leplae R, Geeraerts D, Hallez R, Guglielmini J, Dreze P, Van Melderen L. Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011;39:5513–5525. doi: 10.1093/nar/gkr131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCurley N, Hirano M, Das S, Cooper MD. Immune related genes underpin the evolution of adaptive immunity in jawless vertebrates. Curr Genomics. 2012;13:86–94. doi: 10.2174/138920212799860670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86••.Wang F, Ekiert DC, Ahmad I, Yu W, Zhang Y, Bazirgan O, Torkamani A, Raudsepp T, Mwangi W, Criscitiello MF, et al. Reshaping antibody diversity. Cell. 2013;153:1379–1393. doi: 10.1016/j.cell.2013.04.049. This study of bovine antibodies illustrates how new globular modules with extraordinary structural diversity are generated within the heavy chain immunoglobulin domain. In this process modules with several distinct structures are generated from a single germline precursor in the mature B-cell via hypermutation of the original glycine- and tyrosine-rich sequence, probably via action of an error-prone polymerase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Aravind L, Anantharaman V, Zhang D, de Souza RF, Iyer LM. Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front Cell Infect Microbiol. 2012;2:89. doi: 10.3389/fcimb.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hashimoto H, Pais JE, Zhang X, Saleh L, Fu ZQ, Dai N, Correa IR, Jr, Zheng Y, Cheng X. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature. 2014;506:391–395. doi: 10.1038/nature12905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89••.Hu L, Li Z, Cheng J, Rao Q, Gong W, Liu M, Shi YG, Zhu J, Wang P, Xu Y. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell. 2013;155:1545–1555. doi: 10.1016/j.cell.2013.11.020. The TET enzymes generate oxidized derivatives of methylcytosine in DNA which act as epigenetic marks or intermediates for demethylation. This was the first structure of a TET enzyme and illustrated how the core double-stranded β-helix fold is heavily structurally modified in animals by the de novo acquitsition of multiple metal-chelating elements. However, its activity is identical to the stucturally simpler enzyme described in reference 88. [DOI] [PubMed] [Google Scholar]
- 90.Honig B. Protein structure space is much more than the sum of its folds. Nat Struct Mol Biol. 2007;14:458. doi: 10.1038/nsmb0607-458. [DOI] [PubMed] [Google Scholar]