

Abstract
In the adult brain, the neural stem cell (NSC) pool comprises quiescent and activated populations with distinct roles. Transcriptomic analysis revealed that quiescent and activated NSCs exhibited differences in their protein homeostasis network. Whereas activated NSCs had active proteasomes, quiescent NSCs contained large lysosomes. Quiescent NSCs from young mice accumulated protein aggregates, and many of these aggregates were stored in large lysosomes. Perturbation of lysosomal activity in quiescent NSCs affected protein-aggregate accumulation and the ability of quiescent NSCs to activate. During aging, quiescent NSCs displayed defects in their lysosomes, increased accumulation of protein aggregates, and reduced ability to activate. Enhancement of the lysosome pathway in old quiescent NSCs cleared protein aggregates and ameliorated the ability of quiescent NSCs to activate, allowing them to regain a more youthful state.
Preservation of a pristine proteome is critical for maintaining cell function over long periods of time, and decline in proteome health is associated with aging and neurodegenerative disease (1, 2). Three primary mechanisms are used to maintain protein homeostasis (proteostasis): molecular chaperones, the proteasome proteolytic system, and the lysosome-autophagy proteolytic system (2, 3). Proteostasis is particularly important for stem cell proliferation and differentiation (4). However, somatic stem cell pools contain not only proliferating (activated) stem cells but also quiescent stem cells. How these different types of adult stem cells maintain protein homeostasis and deal with protein aggregation is largely unknown.
Protein aggregates are particularly relevant in the brain, where their accumulation is associated with neurodegenerative disease (1). The adult brain contains pools of regenerative neural stem cells (NSCs) that can generate new neurons (neurogenesis) (5, 6). In one of the neurogenic niches—the subventricular zone (SVZ)—a subset of quiescent astrocytes are the source of quiescent NSCs (qNSCs), which in turn give rise to activated NSCs (aNSCs) and neural progenitor cells (NPCs) (5, 7). These NPCs generate new neurons, oligodendrocytes, and astrocytes, contributing to the maintenance of olfactory function, as well as repair after brain injury (5, 8). The ability of qNSCs to activate declines with age (9–11). Consistently, aging is associated with a decline in neurogenesis and a reduction in olfactory discrimination and memory (6, 9). Understanding the age-dependent changes in proteostasis in the NSC pool could help identify ways to enhance the function of old NSCs and maintain brain health.
To gain an unbiased overview of the proteostasis network in different cell types in a regenerative brain region during aging, we used transcriptomic profiling. We aged cohorts of transgenic mice expressing green fluorescent protein (GFP) under the control of the human glial fibrillary acidic protein (GFAP) promoter, which drives expression in astrocytes and NSCs (7). Using fluorescence-activated cell sorting (FACS) with a combination of GFP and cell surface markers, we simultaneously purified five cell types from the SVZ of paired young (3- to 4-month-old) and old (19- to 22-month-old) mice: endothelial cells (which provide key environmental signals to NSCs), SVZ astrocytes (hereafter, astrocytes), qNSCs, aNSCs, and NPCs (Fig. 1A and fig. S1A) (7). Each population expressed well-established markers (fig. S1B) and showed expected cell-cycle characteristics (fig. S1C). We used RNA-sequencing (RNA-seq) to globally profile the transcriptomes of four biological replicates per cell type and per age (tables S1 and S2 and fig. S2, A to C). The transcriptomes of endothelial cells, quiescent cells (astrocytes and qNSCs), and activated cells (aNSCs and NPCs) could be distinguished by principal component analysis (PCA) (Fig. 1B) and hierarchical clustering (fig. S2D). By contrast, young and old samples from the same cell type were not easily separated when all cell types were considered together (Fig. 1B and fig. S2D), though age-dependent differences were revealed when cell types were analyzed individually (see below, Fig. 4 and fig. S8).
Fig. 1. qNSCs exhibit a protein quality-control signature distinct from their activated progeny, with enlarged lysosomes.
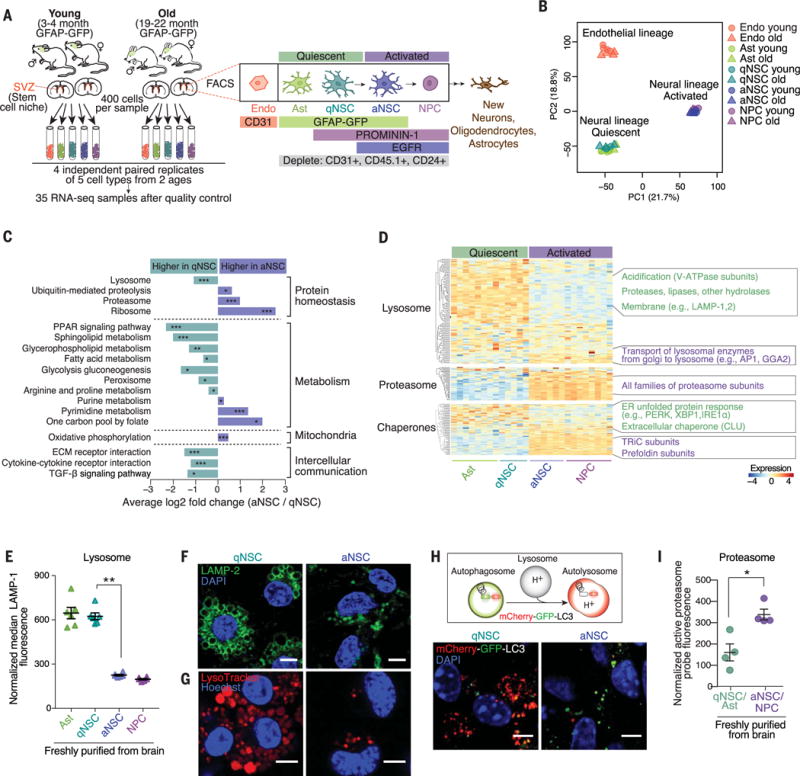
(A) Experimental design. Endo, endothelial cell; Ast, astrocyte; EGFR, epidermal growth factor receptor. (B) PCA of five cell types and two ages, performed on variance stabilizing transformation (VST)–normalized read counts of all detected genes. PC, principal component. (C) Selected KEGG pathways significantly upregulated in qNSCs or aNSCs. P values determined by bootstrap sampling, with Benjamini-Hochberg correction. *False discovery rate (FDR)–adjusted P ≤ 0.05; **FDR-adjusted P ≤ 0.001; ***FDR-adjusted P ≤ 0.0001. See also fig. S3 and table S5. (D) Heatmap showing expression levels of all genes in pathways from the three main branches of proteostasis: proteasome, lysosome, and chaperones. VST-normalized read counts, scaled row-wise. V-ATPase, vacuolar H+–adenosine triphosphatase. See also table S6. (E) Median fluorescence of LAMP-1 normalized to cell size, as measured by flow cytometry. Mean ± SEM of values from six different mice (3 to 4 months old) analyzed on a single day. Each point represents FACS-sorted cells from a single mouse. P value determined by one-sided Wilcoxon rank sum test. **P ≤ 0.01. (F and G) Representative immunofluorescence images of primary cultures of qNSCs and aNSCs from 3-month-old mice stained with (F) LAMP-2 (green, lysosomal membranes) and 4′,6-diamidino-2-phenylindole (DAPI) (blue, nuclei) or (G) LysoTracker (red, acidic organelles) and Hoechst (blue, nuclei). Scale bars, 5 μm. See also figs. S5B and S9B. (H) Top: In cells expressing mCherry-GFP-LC3, autophagosomes display both GFP and mCherry fluorescence (yellow-green), whereas autolysosomes display only mCherry fluorescence (red) because GFP is denatured by the acidity of the lysosome. Bottom: Representative images of GFP and mCherry fluorescence in primary cultures of qNSCs and aNSCs from 3-month-old mice. Scale bars, 5 μm. See also fig. S5C. (I) Fluorescence level of Me4BodipyFL-Ahx3Leu3VS, a probe that covalently binds active proteasomes, in populations directly isolated from the brain. Mean ± SEM of values from four different mice (3 to 4 months old) sorted and analyzed on two different days. Each point represents cells from a single mouse. P value determined by one-sided Wilcoxon rank sum test. *P ≤ 0.05.
Fig. 4. Old qNSCs show lysosome defects, increased protein aggregates, and decreased activation, but these can be counteracted by lysosomal activation.
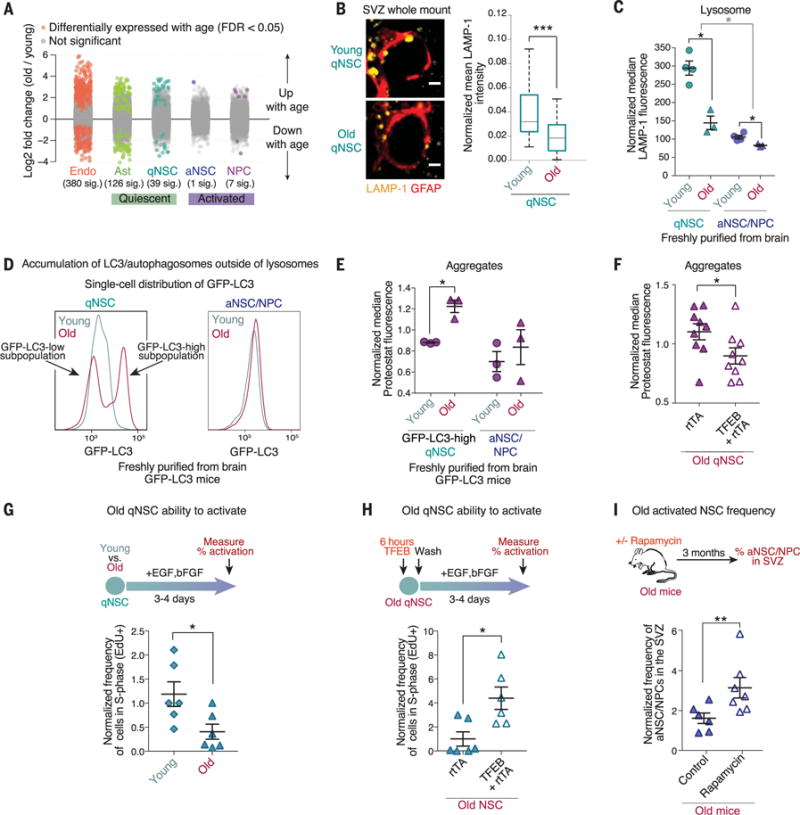
(A) Log2 fold changes between young (3- to 4-month-old) and old (19- to 22-month-old) samples of each cell type for all detected genes using DESeq2 (36). Each point represents one gene. Genes in color are significantly (“sig.”) up- or down-regulated with age with FDR = 0.05. Genes in gray are not significantly up- or down-regulated with age. (B) SVZ whole-mount staining of young (3-month-old) and old (21-month-old) qNSCs. Left: LAMP-1 (lysosomes, yellow) staining in qNSCs identified by the combination of a KI67-negative cell soma and a GFAP+ (red) projection through ependymal cell pinwheels at the ventricular surface (KI67, proliferation marker). Scale bar, 2 μm. Right: Quantification of LAMP-1 fluorescence intensity in qNSCs normalized for cell size from three young and three old mice (n = 65 qNSCs). (C) Median fluorescence of LAMP-1 normalized for cell size. Mean ± SEM of values from three to four young and old mice, analyzed by flow cytometry on a single day. Each point represents FACS-sorted cells from a single mouse. (D) Representative histograms of the GFP-LC3 fluorescence per cell in qNSC and aNSC populations from a 5-month-old versus 25-month-old transgenic GFP-LC3 mouse, as measured by flow cytometry. See also fig. S9, C and D. (E) Median fluorescence of Proteostat in GFP-LC3-high qNSCs and aNSCs and NPCs from young (2- to 5-month-old) and old (25-month-old) GFP-LC3 mice normalized to the median value per replicate to combine samples prepared at different times. Mean ± SEM of values from three to four different mice analyzed by flow cytometry on a single day. Each point represents cells FACS-sorted from a single mouse. (F) Median fluorescence of Proteostat normalized to the median value per replicate. Mean ± SEM of values from nine biological replicates analyzed on three different days. Each point represents one independent primary culture derived from a 17- to 22-month-old mouse. (G) Percentage of cells in an individual culture that incorporated EdU (cells in S phase) during a 3-hour pulse, as measured by intracellular flow cytometry. Mean ± SEM of values from six different biological replicates analyzed on two different days. Each point represents one independent primary culture derived from two 3-month-old or 20-month-old mice. (H) Percentage of cells that incorporated EdU measured as in (G). Mean ± SEM of values from six different biological replicates analyzed on two different days. Each point represents one independent primary culture derived from a 17- to 22-month-old mouse. (I) Old mice (22 months old) were treated for 3 months with rapamycin in mouse chow. SVZ composition was then analyzed by FACS. Each point represents the abundance of aNSCs and aNPCs (EGFR+CD24−CD31−CD45− cells) in a single mouse, expressed as the percentage of total live cells in the SVZ. Mean ± SEM of values from six to seven different mice. P values [except panel (B)] determined by one-sided Wilcoxon rank sum test; Panel (B) P values determined by two-sided Wilcoxon rank sum test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.01.
Analysis of Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways that are up-regulated or down-regulated between qNSCs and aNSCs showed differences in several homeostatic and metabolic pathways (e.g., lipid metabolism, DNA repair, cell cycle) (7, 12–15) as well as strong differences in the protein homeostasis network (Fig. 1C; figs. S3, A to D, and S4A; and tables S3 to S6). aNSCs exhibited increased expression of proteasome-associated genes (Fig. 1D, fig. S4B, and tables S5 and S6) (7, 15). By contrast, qNSCs had increased expression of lysosome-associated genes (Fig. 1D, fig. S4B, and tables S5 and S6). A motif for the transcription factor EB (TFEB), a master regulator of lysosomes and autophagy (16, 17), was enriched in lysosomal genes of qNSCs (fig. S4C). More generally, TFEB target genes were more highly expressed in qNSCs than in aNSCs (fig. S4C) (17), and TFEB was among the top five most significant upstream regulators of qNSC gene expression (fig. S4D). qNSCs and aNSCs also expressed different types of genes that encode protein chaperones (Fig. 1D, fig. S4E, and table S6): endoplasmic reticulum (ER) unfolded-protein response genes were more highly expressed in qNSCs, whereas the TCP-1 ring complex (TRiC) and prefoldin complex genes were more highly expressed in aNSCs (fig. S4E). Similar differences in branches of the proteostasis network were observed between the transcriptomes of quiescent and activated stem cells of other tissues (muscle and hematopoietic) (fig. S4F). Thus, quiescent and activated stem cells mobilize different branches of the protein quality-control network.
We experimentally assessed the status of the lysosomes and proteasomes in qNSCs and aNSCs. Staining for the lysosomal membrane protein LAMP-1 revealed that qNSCs freshly isolated from the adult brain had more LAMP-1 staining than did their activated counterparts (Fig. 1E). This was confirmed by staining for LAMP-1 in whole mounts of the SVZ (fig. S5A). Staining for the lysosomal membrane protein LAMP-2 in a primary culture system for NSC quiescence and activation (18) showed that qNSCs not only had more lysosomes but also larger lysosomes than aNSCs (Fig. 1F and fig. S5B). The large qNSC lysosomes were labeled with LysoTracker, a fluorescent probe specific to acidic organelles (Fig. 1G), suggesting that these lysosomes had the acidity required for proteolytic activity. Lysosomes can fuse with autophagosomes, thereby forming autolysosomes that, in turn, degrade autophagosome contents (3). Using a system in which LC3 (a marker of autophagosomes) is fused to both GFP and mCherry (Fig. 1H) (19), we found that qNSCs contained numerous large lysosomes that had fused with autophagosomes to form autolysosomes but had not yet degraded their contents (Fig. 1H and fig. S5C). Consistently, qNSCs freshly isolated from the SVZ showed less accumulation of LC3 than did aNSCs in response to inhibitors of lysosomal acidification (bafilomycin A and chloroquine) (fig. S5D), indicative of slower degradation of autophagosomes by qNSC lysosomes. Thus, although the lysosomes of qNSCs are acidified, they degrade their contents relatively slowly at steady state.
We then tested whether proteasome activity was different between qNSCs and aNSCs. As predicted by our RNA-seq data, qNSCs directly isolated from the brain had reduced staining for catalytically active proteasomes (Fig. 1I) (20) and conjugated ubiquitin (fig. S5E) compared to that of aNSCs. Furthermore, lysates from primary cultured qNSCs had less proteasome activity than did lysates of aNSCs (fig. S5F). Thus, qNSCs and aNSCs use different branches of the proteostasis network, with qNSCs exhibiting large lysosomes and aNSCs displaying active proteasomes.
The differences between protein quality-control pathways in qNSCs and aNSCs raise the question of each cell type’s ability to regulate proteome health (fig. S6A). We stained young adult qNSCs and aNSCs freshly purified from the brain and in primary culture with Proteostat dye, which becomes highly fluorescent upon binding to the amyloid-type β-sheet tertiary structure of protein aggregates (21). qNSCs, even from young adults, exhibited increased Proteostat staining compared to that of aNSCs (Fig. 2, A and B). The accumulation of protein aggregates in young qNSCs was surprising, given that cellular quiescence is accompanied by reduced protein synthesis (14, 15), and indeed, freshly isolated qNSCs had less protein synthesis than did aNSCs (fig. S6B). A large proportion of protein aggregates in qNSCs were contained within lysosomes (Fig. 2A), consistent with the observation that qNSC lysosomes degrade the contents of autophagosomes more slowly.
Fig. 2. qNSCs have insoluble protein aggregates localized in large lysosomes.
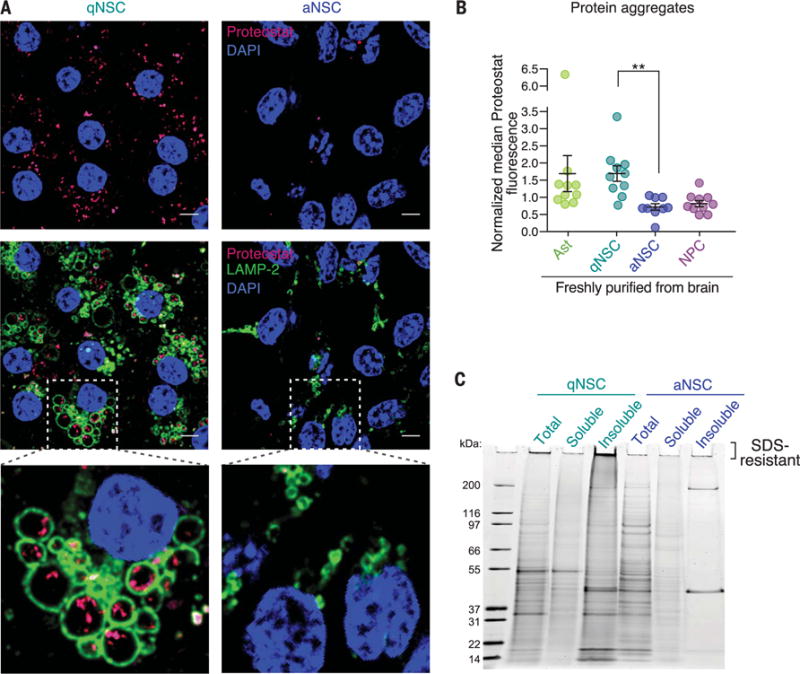
(A) Representative immunofluorescence images of qNSC and aNSC primary cultures from 3-month-old mice stained with Proteostat (magenta, protein aggregates), LAMP-2 (green, lysosomal membranes), and DAPI (blue, nuclei). Scale bars, 5 μm. (B) Median fluorescence of Proteostat normalized to cell size and to the median value on the day of measurement (to combine independent experiments). Mean ± SEM of values from 10 different mice (3 to 4 months old) analyzed by flow cytometry on four different days. Each point represents FACS-sorted cells from a single mouse. P value determined by two-sided Wilcoxon rank sum test. **P ≤ 0.01. (C) Representative SDS-PAGE gel stained with SYPRO Ruby for detection of protein in the total, soluble, and insoluble fractions of qNSCs and aNSCs. See also fig. S6, C to E.
Biochemical fractionation of lysates from qNSCs and aNSCs to enrich for protein aggregates confirmed that qNSCs contained significantly more insoluble protein aggregates than did aNSCs (Fig. 2C and fig. S6, C to E). The presence of proteins at a variety of molecular weights in the qNSC insoluble fraction suggests that qNSC aggregates contain a wide array of proteins (Fig. 2C). Additionally, accumulation of qNSC insoluble proteins at the top of the SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel is consistent with the presence of highly ordered amyloid-like aggregates (22) (Fig. 2C and fig. S6E). Thus, qNSCs contain more protein aggregates than their activated counterparts, and this aggregation appears to affect many proteins. The accumulation of protein aggregates in healthy young adult qNSCs raises the possibility that these aggregates play a physiological role.
The primary function of qNSCs is to generate aNSCs in response to growth factor signals. We tested whether manipulations that regulate lysosomal activity could affect protein aggregates in qNSCs and, in turn, influence activation of qNSCs in response to activation signals [epidermal growth factor (EGF) and basic fibroblast growth factor (bFGF)]. Treatment with bafilomycin A, which blocks lysosomal acidification, led to increased accumulation of protein aggregates (Fig. 3, A and B, and fig. S7A) and to decreased activation in response to growth factors (Fig. 3C). To induce lysosomal degradation, we deprived cells of nutrients by incubating them in Hank’s balanced salt solution (HBSS) (23). Nutrient deprivation of qNSCs led to a significant decrease in lysosomal staining (fig. S7B) and in the amount of protein aggregates (Fig. 3, A and D, and fig. S7, C and D). A brief pulse of nutrient deprivation in qNSCs enhanced their subsequent activation by growth factors, and this was abolished by bafilomycin A (Fig. 3E). Enhanced qNSC activation did not result from direct effects of nutrient deprivation on proliferation: Nutrient deprivation did not affect proliferation of qNSCs in the absence of growth factors, and indeed, it decreased proliferation of aNSCs (fig. S7E). To activate lysosome function more selectively, we used expression of TFEB—a master regulator of lysosome biogenesis and autophagy (16, 17). Brief expression of an activated form of TFEB in qNSCs (24) (fig. S7F) led to decreased abundance of protein aggregates in qNSCs (Fig. 3F) and to enhanced qNSC activation in response to growth factors (Fig. 3G). Thus, activating the lysosome-autophagy pathway in qNSCs cleared protein aggregates and improved qNSC activation in response to growth factors.
Fig. 3. Modulation of lysosomal activity in qNSCs affects protein aggregates and the transition from quiescence to activation.
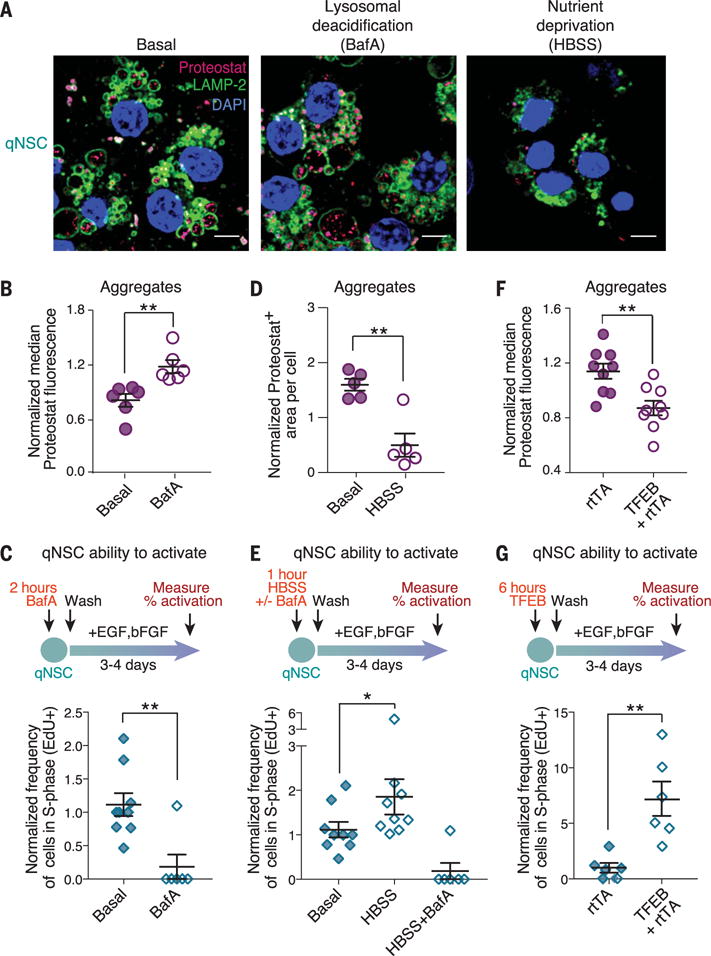
(A) Representative immunofluorescence images of primary cultures of qNSCs from 3-month-old mice after 18 hours of treatment with 50 nM bafilomycin A (BafA, a lysosomal V-ATPase inhibitor) or 3 hours of nutrient deprivation in HBSS. Cells are stained with Proteostat (magenta, protein aggregates), LAMP-2 (green, lysosomal membranes), and DAPI (blue, nuclei). Scale bars, 5 μm. Basal, quiescence media. (B) Median Proteostat fluorescence normalized to the median value per replicate to combine samples prepared at different times. Mean ± SEM of values from six biological replicates analyzed by flow cytometry on two different days. Each point represents an independent primary culture derived from two 3- to 4-month-old mice. (C) Percentage of cells that incorporated 5-ethynyl-2′-deoxyuridine (EdU) (cells in S phase of the cell cycle) during a 3-hour pulse, as measured by intracellular flow cytometry. Mean ± SEM of values from nine different biological replicates analyzed on four different days. Each point represents one independent primary culture derived from two 3- to 4-month-old mice. (D) Differences in Proteostat+ area quantified from images. Mean ± SEM of values from five independent biological replicates from two independent experiments. (E) Percentage of cells that incorporated EdU measured as in (C). Mean ± SEM of values from nine different biological replicates analyzed on four different days. (F) Proteostat fluorescence measured as in (B). Mean ± SEM of values from nine biological replicates analyzed on three different days. rtTA, reverse tetracycline-controlled transactivator. (G) Percentage of cells that incorporated EdU measured as in (C). Mean ± SEM of values from six different biological replicates analyzed on two different days. For all panels, P values determined by one-sided Wilcoxon rank sum test. *P ≤ 0.05; **P ≤ 0.01.
The collapse of protein homeostasis is a key feature of aging in differentiated cells (1). To understand how aging affects protein homeostasis in stem cells, we analyzed our RNA-seq data sets for changes in global transcriptional profiles as well as specific transcriptional signatures in qNSCs and aNSCs freshly purified from old (19- to 22-month-old) versus young (3- to 4-month-old) mice (Fig. 1A). Quiescent cell types (qNSCs and astrocytes) displayed more and larger-magnitude transcriptional changes with age than did their activated counterparts (aNSCs and NPCs) (Fig. 4A, fig. S8A, and tables S7 and S8). This was not caused by technical variability between biological replicates (fig. S8B) and was also observed with a different transcript quantification and analysis method (fig. S8, C and D). Consistently, the transcriptome of young and old qNSCs and astrocytes could be distinguished by PCA, whereas that of young and old aNSCs and NPCs could not (fig. S8, E and F). The age-associated differences in qNSCs could reflect changes that arise from a heterogeneous population of qNSCs (5, 11, 15). The expression of genes associated with lysosome function, but not proteasome function, was significantly changed with age in qNSCs but not in aNSCs (fig. S8G). Thus, qNSCs undergo more transcriptional changes with age than do aNSCs and NPCs, and the expression of lysosome genes changes with age.
We experimentally examined the status of lysosomes and proteasomes in old qNSCs and aNSCs. Old qNSCs in their in vivo niches or freshly purified from the brain exhibited less lysosomal staining (LAMP-1) than young qNSCs (Fig. 4, B and C), and this decrease was significantly greater than that observed with age in aNSCs (Fig. 4C). This reduction in lysosomal staining was also observed in primary qNSC cultures from old mice (fig. S9, A and B). The age-dependent decrease in lysosome levels could result in fewer lysosomes available to fuse with autophagosomes (and/or endosomes). Indeed, using old GFP-LC3 reporter mice, we identified a distinct subpopulation of old qNSCs that had increased amounts of GFP-LC3 fluorescence (Fig. 4D and fig. S9, C and D), indicative of reduced autophagic flux into acidified lysosomes (where GFP is normally denatured) (25). There was heterogeneity within the qNSC population with age (Fig. 4D and fig. S9, C and D), indicating that a subset of old qNSCs have defects in their lysosomes. By contrast, proteasome activity was not significantly altered with age in qNSCs or aNSCs (fig. S9E).
We tested how protein aggregates are affected by aging in qNSCs. The subpopulation of old qNSCs with high amounts of GFP-LC3 exhibited an increased amount of protein aggregates in vivo (Fig. 4E). Consistently, primary cultures of qNSCs from old mice also exhibited an increase in abundance of protein aggregates (fig. S9F). The age-dependent accumulation of GFP-LC3 and protein aggregates in vivo was reduced by subjecting mice to fasting for 48 hours (fig. S9G), indicating that these aggregates can be modulated by nutrient deprivation. The increased amount of protein aggregates in old qNSCs could also be reduced by expression of constitutively active TFEB (Fig. 4F). Thus, old qNSCs exhibit increased protein aggregation, which may result from age-dependent reduction in lysosome number or function, and this can be counteracted by TFEB expression.
The ability of qNSCs to activate declines with age (9–11). Consistently, old qNSCs in primary culture activated less efficiently than young qNSCs in response to growth factors (Fig. 4G). Interestingly, a pulse of expression of the active form of TFEB in old qNSCs restored activation in response to growth factors (Fig. 4H), suggesting that increased lysosomal function helps promote activation of old NSCs. Furthermore, systemic treatment of old mice with rapamycin, a mechanistic target of rapamycin (mTOR) inhibitor that enhances the lysosome-autophagy pathway (26, 27), resulted in increased abundance of aNSCs in the SVZ (Fig. 4I). Although rapamycin could also exert its effects indirectly by acting in cells other than NSCs and/or by regulating other pathways (26), these data are consistent with the possibility that increasing lysosomal function can clear protein aggregates and counteract the decline in activation of old qNSCs (fig. S9H).
Our study identifies a previously uncharacterized organelle state in qNSCs: an enlarged lysosome that contains a large amount of insoluble protein aggregates. The clearance of qNSC aggregates by lysosomal activation may provide a burst of energy for activation or prevent the propagation of potentially toxic aggregates to NSC progeny. Although changes in autophagy during aging have been observed in hematopoietic and muscle stem cells (27–29), these studies did not examine the lysosomes and their protein aggregate content in these stem cells during aging. As lysosomes are central hubs for key cellular trafficking, signaling, and metabolic pathways, targeting of lysosome biogenesis and/or activation to clear aggregates in qNSCs may provide broad strategies to allow activation of old mammalian cells in vivo. This possibility is intriguing in light of the benefits of dietary restriction or starvation followed by refeeding to regenerative stem cell pools (30, 31) and the effects of the lysosome-autophagy pathway on protein aggregates (32) and life span (33) in Caenorhabditis elegans. Identifying the conserved mechanisms by which protein aggregates are regulated in aging stem cells should help identify specific ways to restore function to old stem cells and tissues.
Supplementary Material
Acknowledgments
We thank P. Codega, V. Silva-Vargas, and F. Doetsch for help with the NSC FACS protocol; C. Di Malta and A. Ballabio for the constitutively active TFEB vector; M. Hansen for the mCherry-GFP-LC3 vector and for helpful discussions; A. Roux and C. Kenyon for help with the protein aggregate protocol; R. Zoncu for suggestions regarding the lysosome; T. Palmer, S. Artandi, M. Winslow, C. Jan, A. Freund, C. Sidrauski, K. Rainbolt, and M. Pech for helpful discussions and input on the manuscript; D. Gaviño and J. Brett for sharing rapamycin-treated mice; the Stanford Shared FACS Facility and, in particular, C. Carswell-Crumpton for technical support; B. Benayoun, L. Booth, I. Harel, and other members of the Brunet lab for helpful discussions and feedback on the manuscript; C.-K. Hu and E. Noblin for advice on confocal microscopy and image analysis; and J. Butterfield for help with mouse husbandry and genotyping. This work is supported by NIH grant P01 AG036695 (A.B. and T.A.R.), Stanford Bio-X seed grant (A.B. and J.F.), the NSF (D.S.L. and E.A.P.), a National Defense Science and Engineering Graduate Fellowship (D.S.L.), NIH grant F31 AG043232 (E.A.P.), the Stanford Cancer Biology training grant T32 CA09302 (D.S.L. and E.A.P.), NIH training grant T32 GM7365 (B.W.D.), and the Glenn/American Federation for Aging Research (AFAR) Postdoctoral Fellowship Program for Translational Research on Aging (X.Z.).
Footnotes
Author contributions: D.S.L. and A.B. planned the study. D.S.L. performed the experiments, analyzed the data, and wrote the manuscript with help from A.B. K.H. analyzed all RNA-seq data and performed all statistical analyses. T.R. designed, performed, and analyzed the TFEB experiments. A.E.W. and E.A.P. helped optimize RNA-seq, and A.E.W. generated the RNA-seq libraries. A.E.W. provided input into the analysis of proteostasis differences and subcloned mCherry-GFP-LC3 in a lentiviral vector. A.M. optimized and performed SVZ whole-mount staining and image analysis. E.A.P. helped with FACS protocol development and the generation of RNA-seq data. E.A.P. and B.W.D. helped with quantitative PCR validation. E.A.P. and B.W.D. optimized the culture system for NSC quiescence and activation. X.Z. and R.W.Y. helped optimize FACS. X.Z. helped with aggregate gels. R.W.Y. performed TFEB motif analysis. T.T.H. and E.P. provided young and old transgenic GFP-LC3 mice and input on GFP-LC3 sort. S.M. performed the upstream regulator analysis on RNA-seq data. K.D. helped with proteasome activity assay, LC3 antibody, and staining for FACS. T.A.R. provided intellectual contribution on stem cells and provided rapamycin-treated mice. J.F. provided intellectual contribution on proteostasis. All authors discussed the results and commented on the manuscript.
Competing interests: None declared.
Data and materials availability: Sequencing data have been deposited to ArrayExpress under accession number E-MTAB-5172 and to the Sequence Read Archive (SRA) under accession number SRP075993. The normalized RNA-seq read counts are also available in table S2. All code used for the RNA-seq analysis is available on Github (https://github.com/brunetlab/Leeman_et_al_2017.git).
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Balch WE, Morimoto RI, Dillin A, Kelly JW. Science. 2008;319:916–919. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- 2.Hipp MS, Park SH, Hartl FU. Trends Cell Biol. 2014;24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Wong E, Cuervo AM. Cold Spring Harb Perspect Biol. 2010;2:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vilchez D, Simic MS, Dillin A. Trends Cell Biol. 2014;24:161–170. doi: 10.1016/j.tcb.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Lim DA, Alvarez-Buylla A. Cold Spring Harb Perspect Biol. 2016;8:a018820. doi: 10.1101/cshperspect.a018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonçalves JT, Schafer ST, Gage FH. Cell. 2016;167:897–914. doi: 10.1016/j.cell.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 7.Codega P, et al. Neuron. 2014;82:545–559. doi: 10.1016/j.neuron.2014.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kernie SG, Parent JM. Neurobiol Dis. 2010;37:267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enwere E, et al. J Neurosci. 2004;24:8354–8365. doi: 10.1523/JNEUROSCI.2751-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capilla-Gonzalez V, Cebrian-Silla A, Guerrero-Cazares H, Garcia-Verdugo JM, Quiñones-Hinojosa A. Glia. 2014;62:790–803. doi: 10.1002/glia.22642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Giachino C, et al. Stem Cells. 2014;32:70–84. doi: 10.1002/stem.1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Knobloch M, Jessberger S. Curr Opin Neurobiol. 2017;42:45–52. doi: 10.1016/j.conb.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Obermair FJ, et al. Stem Cell Res. 2010;5:131–143. doi: 10.1016/j.scr.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Valcourt JR, et al. Cell Cycle. 2012;11:1680–1696. doi: 10.4161/cc.19879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Llorens-Bobadilla E, et al. Cell Stem Cell. 2015;17:329–340. doi: 10.1016/j.stem.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Sardiello M, et al. Science. 2009;325:473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 17.Palmieri M, et al. Hum Mol Genet. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 18.Martynoga B, et al. Genes Dev. 2013;27:1769–1786. doi: 10.1101/gad.216804.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pankiv S, et al. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 20.Berkers CR, et al. Mol Pharm. 2007;4:739–748. doi: 10.1021/mp0700256. [DOI] [PubMed] [Google Scholar]
- 21.Shen D, et al. Cell Biochem Biophys. 2011;60:173–185. doi: 10.1007/s12013-010-9138-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, et al. Cell Res. 2013;23:508–523. doi: 10.1038/cr.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Di Malta C, et al. Science. 2017;356:1188–1192. doi: 10.1126/science.aag2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lamming DW, Ye L, Sabatini DM, Baur JA. J Clin Invest. 2013;123:980–989. doi: 10.1172/JCI64099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.García-Prat L, et al. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 28.Ho TT, et al. Nature. 2017;543:205–210. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang AH, Rando TA. EMBO J. 2014;33:2782–2797. doi: 10.15252/embj.201488278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brandhorst S, et al. Cell Metab. 2015;22:86–99. doi: 10.1016/j.cmet.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valdez G, et al. Proc Natl Acad Sci USA. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohnert KA, Kenyon C. Nature. 2017;551:629–633. doi: 10.1038/nature24620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lapierre LR, et al. Nat Commun. 2013;4:2267. doi: 10.1038/ncomms3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.