

Abstract
Novel pyrazole-based EZH2 inhibitors have been prepared through a molecular pruning approach from known inhibitors bearing a bicyclic moiety as a central scaffold. The hit compound 1o (N-((4,6-dimethyl-2-oxo-1,2-dihydropyridin-3-yl)methyl)-5-methyl-1-phenyl-1H-pyrazole-4-carboxamide) showed low micromolar EZH2/PRC2 inhibition and high selectivity towards a panel of other methyltransferases. Moreover, 1o displayed cell growth arrest in breast MDA-MB231, leukaemia K562, and neuroblastoma SK-N-BE cancer cells joined to reduction of H3K27me3 levels and induction of apoptosis and autophagy.
This article is part of a discussion meeting issue ‘Frontiers in epigenetic chemical biology’.
Keywords: epigenetics, enhancer of zeste homologue 2 inhibitors, apoptosis, autophagy
1. Introduction
Enhancer of zeste homologues 1 and 2 (EZH1 and EZH2) represent the two catalytic subunits of the polycomb repressive complex 2 (PRC2), a pivotal chromatin-modifying complex conserved from Drosophila to humans that includes, in addition to EZH1 or EZH2, also suppressor of zeste 12 (SUZ12), embryonic ectoderm development (EED), histone binding proteins RbAp46/48, and additional protein subunits such as AEBP2, PCLs and JARID2 [1–6]. EZH2, the most efficient catalytic subunit, does not have its own lysine methyltransferase (MT) activity but needs at least two other protein subunits (EED and SUZ12) to catalyse lysine methylation [7]. EZH2 is able to specifically catalyse methylation of histone H3 on lysine 27 up to trimethylation (H3K27me3) by using S-adenosyl-l-methionine (SAM) as the co-substrate. Since H3K27me3 is a repressive chromatin mark [2], the polycomb complex mediates gene silencing of target genes involved in fundamental cellular processes, such as cell cycle regulation, cell fate and differentiation, senescence and cancer [1,8]. Several molecular mechanisms leading to increased levels of H3K27me3 have been observed in human cancers. Overexpression of EZH2 was first linked to cancer by microarray studies of prostate [9] and breast [10] cancer, and then reported in other numerous and diverse cancer types, from endometrial to melanoma, from glioblastoma to ovarian cancers [11]. Somatic EZH2 mutations and deletions have been reported, some of them being gain-of-function mutations (such as Y641 to N, F, C, S or H, and A677G) and associated with many kinds of lymphomas, others being loss-of-function and typically shown in myelodysplasia and myeloproliferative disorders [12].
The first described EZH2 inhibitor was the carbocyclic adenosine analogue 3-deazaneplanocin (DZNep, figure 1), a derivative of the natural antibiotic neplanocin-A [13]. Owing to its inhibition of S-adenosyl-l-homocysteine (SAH) hydrolase, the effect of DZNep is due to SAH accumulation and its EZH2 inhibition is indirect and not very specific [14]. However, inhibition of EZH2 by either DZNep or RNA interference (RNAi) leads to reduced proliferation and invasiveness in breast and prostate cancer cell lines [15,16]. Since 2012, a number of potent, selective and SAM-competitive small molecules have been described, active at nanomolar levels against mutated and/or wild-type EZH2 (figure 1) [1,11,12]. Treatment of cells with EPZ005687 (figure 1) resulted in concentration-dependent reduction of H3K27 methylation without major decreases in other histone methyl marks [17]. When EPZ005687 was studied in lymphoma cells bearing EZH2 Y641 or A677 mutation, concentration-dependent cell killing was observed, with only minimal effects in lymphoma cell lines containing wild-type EZH2 [17]. Differently, GSK126 (figure 1), actually in Phase I clinical trial for the treatment of lymphomas, solid tumours and multiple myeloma, selectively and potently inhibited both wild-type and mutant EZH2 activity, and effectively inhibited the growth of mutant EZH2 diffuse large B-cell lymphoma xenografts in mice [18]. Recently, two novel EZH2 inhibitors, tazemetostat [19] and CPI-1205 [20] (figure 1), entered Phase I/II clinical trials for treatment of B-cell lymphoma, malignant mesothelioma, synovial sarcoma and other solid tumours. Thus, EZH2 inhibitors represent a great clinical promise for cancer treatment. Nevertheless, additional efforts are needed for the development of novel chemotypes, since many of these drugs suffer from poor bio-distribution and limited brain penetration [21,22], and for some of them potential acquired resistance has been described [23,24].
Figure 1.

Known EZH2 inhibitors. GSK126, tazemetostat and CPI-1205 are in clinical trials for treatment of blood and/or solid tumours.
At the beginning of our work, only EZH2 inhibitors bearing a bicyclic heterocyclic moiety (indazole, indole) as a central core were reported. As part of our ongoing research to develop inhibitors of histone/protein MTs [25–34], we planned to perform structural simplification and modification of the indazole scaffold featured by EPZ005687 and related compounds (such as GSK343 [35] and UNC1999 [36]; figure 2). Our approach was based on: (i) simplification of the indazole ring using the monocyclic pyrazole one; (ii) shifting of the 4,6-dimethyl-3-methylamino-2-pyridinone group, crucial for the binding of the molecule to the enzyme [37], from the C4-indazole to the C4-pyrazole position; (iii) N1-pyrazole substitutions with alkyl (iso-propyl, cyclopentyl), aryl (phenyl) and arylalkyl (benzyl) groups; (iv) addition of alkyl (methyl, n-propyl) and of different aromatic (phenyl, 1-pyrrolyl, styryl) substituents at the C5-pyrazole position. All the newly obtained derivatives, 1a–r, have been screened against the EZH2 MT activity by using the PRC2 multicomponent protein in order to determine their inhibition activity (IC50 values or per cent of inhibition at fixed dose, table 1). Afterwards, we tested the capability of the hit compound 1o to arrest proliferation in four different cancer cell lines (human breast cancer MDA-MB231, myelogenous leukaemia K562, prostate cancer PC3 and neuroblastoma SK-N-BE), as well as to decrease the histone H3K27me3 levels in the most responsive K562 and SK-N-BE cells. Moreover, the cell death mechanism (apoptosis, autophagy) of 1o was investigated in the same cell lines.
Figure 2.
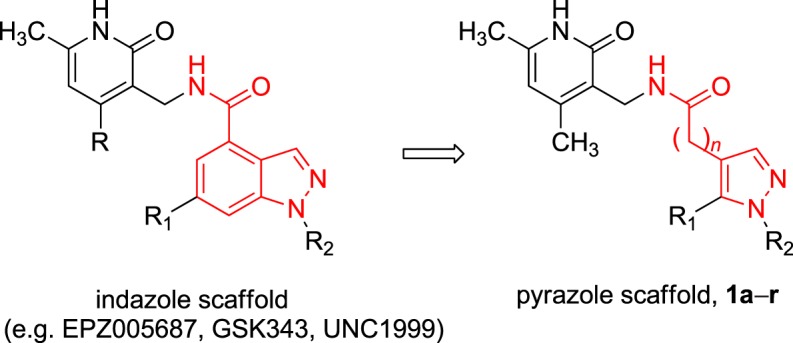
Design of the novel pyrazole-based EZH2 inhibitors 1a–r. (Online version in colour.)
Table 1.
Inhibiting activity of compounds 1a–r against EZH2/PRC2. 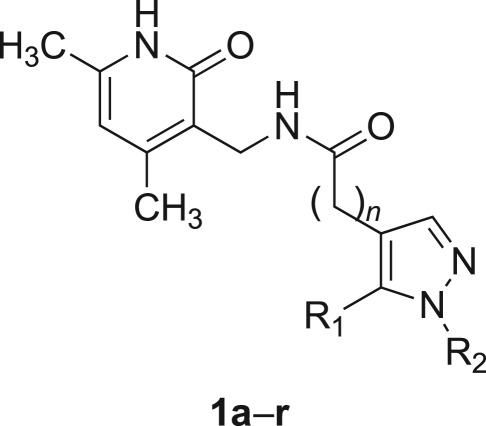
| compound | n | R1 | R2 | % inhibition at 50 µM | IC50 (μM) |
|---|---|---|---|---|---|
| 1a | 0 | 1H-pyrrol-1-yl | isopropyl | 0 | |
| 1b | 0 | 1H-pyrrol-1-yl | cyclopentyl | 4.7 | |
| 1c | 0 | 1H-pyrrol-1-yl | phenyl | 0 | |
| 1d | 0 | 1H-pyrrol-1-yl | benzyl | 5.8 | |
| 1e | 0 | phenyl | isopropyl | 0 | |
| 1f | 0 | phenyl | cyclopentyl | 2.7 | |
| 1g | 0 | phenyl | phenyl | 6.9 | |
| 1h | 0 | phenyl | benzyl | 20 | |
| 1i | 0 | styryl | isopropyl | 0 | |
| 1j | 0 | styryl | cyclopentyl | 2.9 | |
| 1k | 0 | styryl | phenyl | 0 | |
| 1l | 0 | styryl | benzyl | 11.8 | |
| 1m | 0 | H | phenyl | 25.3 | >400 |
| 1n | 0 | methyl | isopropyl | 16.7 | 342.7 |
| 1o | 0 | methyl | phenyl | 71.5 | 15.4 |
| 1p | 0 | methyl | benzyl | 68.3 | 19.6 |
| 1q | 0 | n-propyl | phenyl | 29.0 | 151 |
| 1r | 1 | methyl | phenyl | 4.5 | >400 |
| SAH | 34.7 | ||||
| GSK126 | 0.009 |
2. Material and methods
(a). EZH2/PRC2 assays
The EZH2 substrate (0.05 mg ml−1 core histone, or 5 µM H3/H4 octamer, or 5 µM H3/H4 tetramer, or 5 µM histone H3) was added in the freshly prepared reaction buffer (50 mM Tris-HCl (pH 8.0), 50 mM NaCl, 1 mM EDTA, 1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1% dimethyl sulfoxide (DMSO)). The PRC2 complex (complex of human EZH2, human EED, human SUZ12, human AEBP2 and human RbAp48) was delivered into the substrate solution and the mixture was mixed gently. Afterwards, the tested compounds dissolved in DMSO were delivered into the enzyme/substrate reaction mixture by using Acoustic Technology (Echo 550, LabCyte Inc., Sunnyvale, CA, USA) in the nanolitre range, and 3H-SAM was added into the reaction mixture to initiate the reaction. The reaction mixture was incubated for 1 h at 30°C and then it was delivered to filter-paper for detection. The data were analysed using Excel and GraphPad Prism software for IC50 curve fits. For SAM competition experiments, the same procedure was used with different SAM concentration (1, 2.5, 5 and 10 µM) and H3/H4 octamer as the substrate.
(b). EZH1 complex, DOT1 L, G9a, MLL1 complex, SET7/9, PRMT1 and DNMT1 assays
The appropriate methyltransferase substrate (0.05 mg ml−1 core histone for EZH1 complex, 0.05 mg ml−1 oligonucleosomes for DOT1 L, 5 µM histone H3 (aa 1–21) peptide for G9a, 0.05 mg ml−1 nucleosomes for MLL1 complex, 0.05 mg ml−1 core histone for SET7/9, 5 µM histone H4 for PRMT1, and 0.001 mg ml−1 poly(dI-dC) for DNMT1) was added in freshly prepared reaction buffer (50 mM Tris-HCl (pH 8.5), 5 mM MgCl2, 50 mM NaCl, 0.01% Brij35, 1 mM DTT, 1% DMSO). The MT enzyme was delivered into the substrate solution and the mixture was mixed gently. Afterwards, the tested compounds dissolved in DMSO were delivered into the enzyme/substrate reaction mixture by using Acoustic Technology (Echo 550, LabCyte Inc., Sunnyvale, CA, USA) in the nanolitre range, and 1 µM 3H-SAM was also added into the reaction mixture to initiate the reaction. The reaction mixture was incubated for 1 h at 30°C and then it was delivered to filter-paper for detection. The data were analysed using Excel and GraphPad Prism software for IC50 curve fits.
(c). Cell cultures
The MDA-MB-231 human breast carcinoma cell line, the human neuroblastoma cell line SK-N-BE, and the human chronic myelogenous leukaemia cell line K562 were maintained in RPMI 1640 medium (Sigma-Aldrich, St Louis, MO, USA). The human prostate adenocarcinoma cell line PC-3 was maintained in DMEM medium (Sigma-Aldrich). Both media were supplemented with 100 units ml−1 penicillin, 0.1 mg ml−1 streptomycin, and 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich). All the cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
(d). Cell viability
Cells were seeded in a 96 well plate. The next day cells were treated as described and cell viability was measured by using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) (Promega, Milan, Italy), following manufacturer's instructions. Cell viability was also assessed by trypan blue exclusion. Following treatment, cells were stained by addition of trypan blue. Cells were then visualized and counted with a phase contrast microscope (NIKON Eclipse TE2000U). Finally, cell viability and morphology were also documented by taking pictures of the cells after 2, 4 and 6 days of treatment by using a digital camera mounted on an EclipseNet 2000 microscope (Nikon Instruments, Florence, Italy).
(e). Protein extraction and western blot assays
GSK126 was dissolved in DMSO and added to a final concentration of 1 µM. Compound 1o was dissolved in DMSO and added to PMSF final concentrations of 0.5, 1 and 5 µM for the indicated times. Cells were harvested, washed twice in phosphate-buffered saline and resuspended in lysis buffer (50 mM Tris pH 7.4, 5 mM EDTA, 250 mM NaCl, 50 mM NaF, 0.1% Triton X-100, 10 µg ml−1 leupeptin and 1 mM PMSF). After 30 min on ice, the lysates were clarified by centrifugation (10 min at 4°C) and the supernatant fraction collected. Protein concentration was measured by the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA). Proteins were applied to sodium dodecyl sulfate (SDS)-polyacrylamide gels. The gels were blotted (1.5 h at 230 mA) onto a Hybond-ECL nitrocellulose filter (Amersham Life Science, Inc.). A Dual Color prestained protein solution (Bio-Rad Laboratories) was used as a molecular weight standard. The filter was washed twice with Tris-buffered saline (TBS)–0.1% Tween-20 buffer (TBS-T), before blocking non-specific binding sites with 5% milk/TBS-T for 1 h. The filter was then incubated with the specific primary antibody (mouse anti-human H3K27me3, anti-human H3K4me3, anti-human H3, anti-caspase 3 antibody (Abcam, Cambridge, UK), rabbit anti-human LC3-I/II (Novus Biologicals, Abingdon, UK), mouse anti-β-actin (Sigma), or rabbit anti-PARP (Santa Cruz Biotechnology, Santa Cruz, CA, USA)) diluted in 5% milk/TBS-T overnight at 4°C. The nitrocellulose filter was washed twice and incubated with horseradish peroxidase-conjugated appropriate secondary antibody (mouse anti-rabbit HRP, goat anti-mouse HRP (Amersham Biosciences, Piscataway, NJ, USA)). Detection was performed at room temperature using the enhanced chemiluminescent detection system (ECL kit) purchased from Euroclone (Milan, Italy). Experiments were repeated three times. Densitometric analysis of the bands relative to β-actin was performed using ImageJ software (NIH).
3. Results and discussion
The synthetic procedures used to obtain the pyrazole compounds 1a–r are reported in the electronic supplementary material.
(a). Enzyme assays
All new synthesized compounds have been tested against the human histone five-component PRC2, containing EZH2, EED, SUZ12, RbAp48 and AEBP2. The assay was performed using core histone and SAM as substrate and co-substrate, respectively. SAH and GSK126 were used as reference compounds. All compounds were initially screened at the fixed 50 µM concentration, then selected compounds were profiled in 10-dose IC50 mode with twofold serial dilution starting from 200 µM solutions (table 1 and electronic supplementary material, figure S1).
As a part of our molecular pruning approach, we first decided to synthesize compounds in which the indazole nucleus was replaced by a 5-arylpyrazole ring. First we inserted at the pyrazole-C5 position a 1H-pyrrol-1-yl (1a–d), phenyl (1e–h) or styryl (1i–l) group, and an isopropyl, cyclopentyl, phenyl or benzyl substituent at N1, obtaining compounds displaying very low EZH2/PRC2 inhibition (table 1). The shift of the phenyl ring from pyrazole-C5 to -N1 position (1m) led to the first pyrazole analogue with greater than 25% inhibition at 50 µM (table 1). However, its negligible IC50 value (greater than 400 µM) forced us to perform additional structural modifications. The introduction of a methyl group at the pyrazole-C5, with compound 1o, turned in a nice improvement of the inhibitory potency against EZH2 (IC50 = 15.4 µM). Replacing the N1-phenyl of 1o with the N1-benzyl group gave the equipotent analogue 1p (IC50 = 19.6 µM). Conversely, changing the N1-phenyl for an isopropyl group (1n) as well as lengthening the C5-methyl of 1o with C5-n-propyl (1q) or homologation of its carboxamide to acetamide linkage (1r) caused a severe drop of potency or a return to null EZH2 inhibition (table 1).
Further experiments on 1o using different substrates (H3/H4 octamer, H3/H4 tetramer and histone H3) confirmed its inhibitory ability against EZH2/PRC2 (IC50 values = 6.3, 5.3 and 5.7 µM, respectively), as well as its SAM-dependent mechanism of action (electronic supplementary material, figures S1 and S2). Moreover, 1o proved to be a selective inhibitor of EZH2 among different MTs, it being 22-fold less potent against the closely related EZH1 complex, and practically inactive against a panel of MTs including the arginine MT PRMT1 and the DNA MT DNMT1 (table 2).
Table 2.
IC50 or percentage of inhibition of 1o against a panel of methyltransferases.
| MT | IC50 (µM) | % inhibition at 100 µM |
|---|---|---|
| EZH1 complex | 138.5 | 43 |
| DOT1L | 6 | |
| G9a | 7 | |
| MLL1 complex | 7 | |
| SET7/9 | 0.8 | |
| PRMT1 | 8 | |
| DNMT1 | 0 |
(b). Antiproliferative effect of 1o on a panel of cancer cells
Compounds 1o and GSK126, used as the reference drug, were tested in MDA-MB231, K562, PC3 and SK-N-BE cells at three different concentrations (1, 10 and 25 µM) for up to 4 or 5 days of treatment (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay). In this assay, 1o showed a dose- and time-dependent reduction of cell growth, detectable from 1 µM in SK-N-BE and from 10 µM in MDA-MB231 and in K562 cells. Differently, GSK126 was very potent against the leukaemia K562 cells, but displayed significant effects in the tested solid tumour cells only at 25 µM (figure 3).
Figure 3.

Antiproliferative effects of 1o (top row) and of GSK126 (bottom row) on a panel of cancer cell lines, treated with 1, 10 and 25 µM of the compound for 4 or 5 days (MTS Cell Proliferation Assay). Error bars represent standard deviation. (Online version in colour.)
When tested in non-cancer human embryonic kidney cells HEK293, 1o proved to be less toxic than GSK126, showing only moderate toxicity after 5 days at 10 µM (electronic supplementary material, figure S3).
The effects of 1o on cell growth and morphology were assessed in SK-N-BE and K562 cells, chosen as representative of non-adherent and adherent cell lines, respectively, using GSK126 as the reference drug. With respect to the control, 1o increased cell detachment in SK-N-BE and reduced cell growth in K562 cells. Alternatively, cell viability was also measured by trypan blue exclusion assay and results obtained were similar to those of MTS (not shown). Finally, similar results demonstrating increased number of dead cells were obtained with GSK126 at 1 µM (electronic supplementary material, figure S4).
(c). Effect of 1o on H3K27me3 levels in cancer cells
As a functional read-out of EZH2 inhibition in cells, we determined the levels of H3K27me3 in SK-N-BE and in K562 cells treated with 1o (0.5 and 1 µM) for 6 days, in comparison with GSK126 (1 µM) as the reference compound. β-Actin was used for equal loading. Western blots in figure 4a show a clear concentration-dependent reduction of the H3K27me3 levels after treatment with 1o, particularly in SK-N-BE cells. As expected, GSK126 displayed the strongest effect in K562 cells (see figure 4b for quantitation by densitometry analysis). When tested in the same cell lines in order to check H3K4me3 and histone H3 levels after treatment, neither 1o nor GSK126 altered such levels (electronic supplementary material, figure S5).
Figure 4.

(a) Western blot analysis showing decrease of H3K27me3 levels by 1o in SK-N-BE and K562 cells. (b) Relative densitometric analysis (arbitrary units). β-Actin was used for normalization. Error bars represent standard deviation.
(d). Effect of 1o on autophagy induction in myelogenous leukaemia K562 and neuroblastoma SK-N-BE cell lines
Autophagy represents a physiological mechanism used by cells to recycle intracellular components or remove damaged organelles. However, excessive activation of autophagy can result in cell death. EZH2 has been recently reported to epigenetically repress negative regulators of the mTOR pathway such as TSC2 leading to inhibition of autophagy [38]. Accordingly, the EZH2 inhibitors GSK343 [35] and UNC1999 [36] have been shown to induce autophagic cell death in MDA-MB-231 cells [39], and co-treatment of colon cancer HT-29 and HCT-15 cells with UNC1999 and gefitinib, an EGFR inhibitor, gave not only a significant decrease in cell number and increased apoptosis as compared with single pathway inhibition, but also induction of autophagy, suggesting that autophagy may play a role in the observed synergy [40]. From a molecular point of view, induction of autophagy can be evaluated by studying the expression of proteins that represent markers of such a process. The most studied and used marker of autophagy is a protein called MAP1LC3A or microtubule associated protein 1 light chain 3 alpha or LC3. In fact, the full length form of LC3 is cleaved to a smaller LC3-II form that is then lapidated and associates with the forming autophagosome. Therefore, accumulation of LC3-II is used as an indicator of autophagy induction.
In this context, we investigated the capability of 1o to display autophagic effect in K562 and SK-N-BE cells, evaluated through LC3-II protein accumulation. After treating K562 (3 days) and SK-N-BE (4 days) cells with 1o (0.5 and 1 µM), western blot was performed showing a clear dose-dependent induction of LC3-II protein (figure 5a), even stronger than with the reference compound GSK126 (used at 1 µM) in both cell lines. Quantitation of these data by densitometric analysis is shown in figure 5b.
Figure 5.

Effect of 1o on autophagy induction. (a) Western blot analysis for LC3-II protein accumulation in K562 and SK-N-BE cells after 3 and 4 days' treatment, respectively, with 0.5 and 1 µM 1o. GSK126 (1 µM) was added as the reference drug. (b) Relative densitometric analysis (arbitrary units). β-Actin was used for normalization. Error bars represent standard deviation.
(e). Apoptosis induction by 1o in neuroblastoma SK-N-BE cell lines
To verify the induction of apoptosis events by 1o in SK-N-BE cells, the levels of poly(ADP-ribose) polymerase (PARP) cleavage, a mark of late event in apoptosis, and caspase 3 induction were determined by western blot followed by densitometric analysis. Tested at doses from 0.5 to 5 µM for 4 days, despite it being less potent than GSK126 (tested at 1 µM as the reference drug), 1o induced dose-dependent PARP cleavage (determined by anti-PARP antibody) as well as caspase 3 induction (figure 6).
Figure 6.

Western blot (a) and related densitometric analysis (b) for PARP cleavage and caspase 3 induction by 1o at 0.5, 1 and 5 µM for 4 days in SK-N-BE cells. GSK126 (1 µM) was added as the reference compound. β-Actin was used as loading control. Error bars represent standard deviation.
4. Conclusion
Numerous studies highlighted the role of the PRC2 catalytic component EZH2 in cancer development and progression, and mutation and/or overexpression of EZH2 have been shown in a growing number of neoplasia. In this study, we report the design, synthesis and biological validation of the novel pyrazole-based EZH2 inhibitors 1a–r. In some known EZH2 inhibitors (figure 1), we replaced the indole/indazole bicyclic ring with the simpler pyrazole group, and we found the insertion of a phenyl or benzyl portion at the N1 and a methyl group at the C5 position of the pyrazole ring to afford the highest inhibition. The most potent compound, 1o (IC50 = 5.3–15.4 µM), displayed arrest of cell proliferation in SK-N-BE neuroblastoma, MDA-MB231 breast cancer, and K562 leukaemia cells during 4/5 days of treatment. GSK126, used as the reference drug because it bears a bicyclic (indole) central core and is actually in clinical trials for blood cancers, was more potent against K562 but less efficient against solid tumour cell lines. Western blot analysis performed in K562 and SK-N-BE cells treated with 1o revealed a reduction of the levels of H3K27me3, the epigenetic mark for EZH2 activity, and induction of autophagy, detected by an increase of the LC3-II levels higher than that obtained with GSK126, used as the reference drug. When tested in SK-N-BE cells for induction of apoptosis, 1o caused PARP cleavage and induction of caspase 3, in this case lower than those obtained with GSK126, suggesting that in this cell line autophagy rather than apoptosis induction may be involved in the arrest of proliferation. The anti-cancer activities of 1o will be investigated in other contexts and, at the same time, medicinal chemistry studies will be performed to improve its potency in in vitro and in vivo assays.
Supplementary Material
Data accessibility
The datasets supporting this article (chemistry, physical and chemical data for intermediate compounds 2–5 and final compound 1a–r, dose–response curves for 1n, 1o, 1p and 1q against EZH2, SAM-dependent EZH2 inhibition by 1o, cytotoxicity of 1o and GSK126 in HEK293 cells, morphology studies of SK-N-BE and K562 cells treated with 1o or GSK126, western blot analysis for detection of H3K4me3 levels in SK-N-BE and K562 cells treated with 1o or GSK126) have been uploaded as part of the electronic supplementary material.
Authors' contributions
M.T., S.V. and A.M. designed the study. P.M., B.M., D.B., G.S., C.Z. and P.T. did the synthetic work. B.M., G.S. and C.Z. analysed the enzyme data. L.P., I.C., S.S. and E.F. performed the cellular experiments and western blot analyses. S.V. and A.M. drafted the manuscript, with contributions by P.M., C.Z., P.T. and M.T.
Competing interests
We have no competing interests.
Funding
This work was supported by EC 7th Framework Program project REGPOT-CT-2013-316149-InnovaBalt (P.T. and D.B.), by COST Action CM1406 (P.T., A.M. and S.V.), by the IIT-Sapienza Project (E.F. and A.M.), by RF-2010-2318330 (A.M.), by PRIN 2016 (prot. 20152TE5PK) (A.M.), by AIRC 2016 (no. 19162) (A.M.), and by NIH (no. R01GM114306) (A.M.) funds.
References
- 1.Comet I, Riising EM, Leblanc B, Helin K. 2016. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer 16, 803–810. ( 10.1038/nrc.2016.83) [DOI] [PubMed] [Google Scholar]
- 2.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. 2002. Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043. ( 10.1126/science.1076997) [DOI] [PubMed] [Google Scholar]
- 3.Kim H, Kang K, Kim J. 2009. AEBP2 as a potential targeting protein for polycomb repression complex PRC2. Nucleic Acids Res. 37, 2940–2950. ( 10.1093/nar/gkp149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nekrasov M, et al. 2007. Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J. 26, 4078–4088. ( 10.1038/sj.emboj.7601837) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker E, Chang WY, Hunkapiller J, Cagney G, Garcha K, Torchia J, Krogan NJ, Reiter JF, Stanford WL. 2010. polycomb-like 2 associates with PRC2 and regulates transcriptional networks during mouse embryonic stem cell self-renewal and differentiation. Cell Stem Cell 6, 153–166. ( 10.1016/j.stem.2009.12.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Margueron R, Ku M, Chambon P, Bernstein BE, Reinberg D. 2010. Jarid2 and PRC2, partners in regulating gene expression. Genes Dev. 24, 368–380. ( 10.1101/gad.1886410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao R, Zhang Y. 2004. Suz12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol. Cell 15, 57–67. ( 10.1016/j.molcel.2004.06.020) [DOI] [PubMed] [Google Scholar]
- 8.Sauvageau M, Sauvageau G. 2010. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7, 299–313. ( 10.1016/j.stem.2010.08.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varambally S, et al. 2002. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629. ( 10.1038/nature01075) [DOI] [PubMed] [Google Scholar]
- 10.Kleer CG, et al. 2003. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl Acad. Sci. USA 100, 11 606–11 611. ( 10.1073/pnas.1933744100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim KH, Roberts CW. 2016. Targeting EZH2 in cancer. Nat. Med. 22, 128–134. ( 10.1038/nm.4036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu B, Konze KD, Jin J, Wang GG. 2015. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 43, 698–712. ( 10.1016/j.exphem.2015.05.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glazer RI, Hartman KD, Knode MC, Richard MM, Chiang PK, Tseng CK, Marquez VE. 1986. 3-Deazaneplanocin: a new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun. 135, 688–694. ( 10.1016/0006-291X(86)90048-3) [DOI] [PubMed] [Google Scholar]
- 14.Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE, Jones PA. 2009. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 8, 1579–1588. ( 10.1158/1535-7163.MCT-09-0013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tan J, et al. 2007. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 21, 1050–1063. ( 10.1101/gad.1524107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bryant RJ, Cross NA, Eaton CL, Hamdy FC, Cunliffe VT. 2007. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate 67, 547–556. ( 10.1002/pros.20550) [DOI] [PubMed] [Google Scholar]
- 17.Knutson SK, et al. 2012. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896. ( 10.1038/nchembio.1084) [DOI] [PubMed] [Google Scholar]
- 18.McCabe MT, et al. 2012. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112. ( 10.1038/nature11606) [DOI] [PubMed] [Google Scholar]
- 19.Kuntz KW, et al. 2016. The importance of being Me: magic methyls, methyltransferase inhibitors, and the discovery of tazemetostat. J. Med. Chem. 59, 1556–1564. ( 10.1021/acs.jmedchem.5b01501) [DOI] [PubMed] [Google Scholar]
- 20.Vaswani RG, et al. 2016. Identification of (R)-N-((4-methoxy-6-methyl-2-oxo-1,2-dihydro-pyridin-3-yl)methyl)-2-methyl-1-(1-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)ethyl)-1H-indole-3-carboxamide (CPI-1205), a potent and selective inhibitor of histone methyltransferase EZH2, suitable for Phase I clinical trials for B-cell lymphomas. J. Med. Chem. 59, 9928–9941. ( 10.1021/acs.jmedchem.6b01315) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar A, et al. 2015. EZH2 inhibitor GSK126: metabolism, drug transporter and rat pharmacokinetic studies. Med. Res. Archiv. 1, 1–28. [S.l.], n. 3, june 2015. See http://www.journals.ke-i.org/index.php/mra/article/view/273 (accessed 14 March 2018). [Google Scholar]
- 22.Zhang P, de Gooijer MC, Buil LC, Beijnen JH, Li G, van Tellingen O. 2015. ABCB1 and ABCG2 restrict the brain penetration of a panel of novel EZH2 inhibitors. Int. J. Cancer 137, 2007–2018. ( 10.1002/ijc.29566) [DOI] [PubMed] [Google Scholar]
- 23.Gibaja V, et al. 2016. Development of secondary mutations in wild-type and mutant EZH2 alleles cooperates to confer resistance to EZH2 inhibitors. Oncogene 35, 558–566. ( 10.1038/onc.2015.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoumacher M, Le Corre S, Houy A, Mulugeta E, Stern MH, Roman-Roman S, Margueron R. 2016. Uveal melanoma cells are resistant to EZH2 inhibition regardless of BAP1 status. Nat. Med. 22, 577–578. ( 10.1038/nm.4098) [DOI] [PubMed] [Google Scholar]
- 25.Mai A, et al. 2007. Synthesis and biological validation of novel synthetic histone/protein methyltransferase inhibitors. ChemMedChem 2, 987–991. ( 10.1002/cmdc.200700023) [DOI] [PubMed] [Google Scholar]
- 26.Ragno R, et al. 2007. Small molecule inhibitors of histone arginine methyltransferases: homology modeling, molecular docking, binding mode analysis, and biological evaluation. J. Med. Chem. 50, 1241–1253. ( 10.1021/jm061213n) [DOI] [PubMed] [Google Scholar]
- 27.Mai A, et al. 2008. Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors . J. Med. Chem. 51, 2279–2290. ( 10.1021/jm701595q) [DOI] [PubMed] [Google Scholar]
- 28.Palacios D, et al. 2010. TNF/p38α/polycomb signaling to Pax7 locus in satellite cells links inflammation to the epigenetic control of muscle regeneration. Cell Stem Cell 7, 455–469. ( 10.1016/j.stem.2010.08.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castellano S, et al. 2010. Design, synthesis and biological evaluation of carboxy analogues of arginine methyltransferase inhibitor 1 (AMI-1). ChemMedChem 5, 398–414. ( 10.1002/cmdc.200900459) [DOI] [PubMed] [Google Scholar]
- 30.Cheng D, Valente S, Castellano S, Sbardella G, Di Santo R, Costi R, Bedford MT, Mai A. 2011. Novel 3,5-bis(bromohydroxybenzylidene)piperidin-4-ones as coactivator-associated arginine methyltransferase 1 inhibitors: enzyme selectivity and cellular activity. J. Med. Chem. 54, 4928–4932. ( 10.1021/jm200453n) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valente S, et al. 2012. Identification of PR-SET7 and EZH2 selective inhibitors inducing cell death in human leukemia U937 cells. Biochimie 94, 2308–2313. ( 10.1016/j.biochi.2012.06.003) [DOI] [PubMed] [Google Scholar]
- 32.Castellano S, et al. 2012. Identification of small-molecule enhancers of arginine methylation catalyzed by coactivator-associated arginine methyltransferase 1. J. Med. Chem. 55, 9875–9890. ( 10.1021/jm301097p) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ciarapica R, et al. 2014. Pharmacological inhibition of EZH2 as a promising differentiation therapy in embryonal RMS. BMC Cancer 14, 139 ( 10.1186/1471-2407-14-139) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ciarapica R, et al. 2014. The polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx). Oncogene 33, 4173–4184. ( 10.1038/onc.2013.471) [DOI] [PubMed] [Google Scholar]
- 35.Verma SK, et al. 2012. Identification of potent, selective, cell-active inhibitors of the histone lysine methyltransferase EZH2. ACS Med. Chem. Lett. 3, 1091–1096. ( 10.1021/ml3003346) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konze KD, et al. 2013. An orally bioavailable chemical probe of the lysine methyltransferases EZH2 and EZH1. ACS Chem. Biol. 8, 1324–1334. ( 10.1021/cb400133j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooun A, et al. 2016. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat. Commun. 7, 11384 ( 10.1038/ncomms11384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei FZ, et al. 2015. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 11, 2309–2322. ( 10.1080/15548627.2015.1117734) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu TP, Lo HL, Wei LS, Hsiao HH, Yang PM. 2015. S-Adenosyl-L-methionine-competitive inhibitors of the histone methyltransferase EZH2 induce autophagy and enhance drug sensitivity in cancer cells. Anticancer Drugs 26, 139–147. ( 10.1097/CAD.0000000000000166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katona BW, Liu Y, Ma A, Jin J, Hua X. 2014. EZH2 inhibition enhances the efficacy of an EGFR inhibitor in suppressing colon cancer cells. Cancer Biol. Ther. 15, 1677–1687. ( 10.4161/15384047.2014.972776) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting this article (chemistry, physical and chemical data for intermediate compounds 2–5 and final compound 1a–r, dose–response curves for 1n, 1o, 1p and 1q against EZH2, SAM-dependent EZH2 inhibition by 1o, cytotoxicity of 1o and GSK126 in HEK293 cells, morphology studies of SK-N-BE and K562 cells treated with 1o or GSK126, western blot analysis for detection of H3K4me3 levels in SK-N-BE and K562 cells treated with 1o or GSK126) have been uploaded as part of the electronic supplementary material.