

ABSTRACT
Pseudomonas aeruginosa is an opportunistic Gram-negative pathogen that requires iron for growth and virulence. Under low-iron conditions, P. aeruginosa transcribes two highly identical (95%) small regulatory RNAs (sRNAs), PrrF1 and PrrF2, which are required for virulence in acute murine lung infection models. The PrrF sRNAs promote the production of 2-akyl-4(1H)-quinolone metabolites (AQs) that mediate a range of biological activities, including quorum sensing and polymicrobial interactions. Here, we show that the PrrF1 and PrrF2 sRNAs promote AQ production by redundantly inhibiting translation of antR, which encodes a transcriptional activator of the anthranilate degradation genes. A combination of genetic and biophysical analyses was used to define the sequence requirements for PrrF regulation of antR, demonstrating that the PrrF sRNAs interact with the antR 5′ untranslated region (UTR) at sequences overlapping the translational start site of this mRNA. The P. aeruginosa Hfq protein interacted with UA-rich sequences in both PrrF sRNAs (Kd [dissociation constant] = 50 nM and 70 nM). Hfq bound with lower affinity to the antR mRNA (0.3 μM), and PrrF was able to bind to antR mRNA in the absence of Hfq. Nevertheless, Hfq increased the rate of PrrF annealing to the antR UTR by 10-fold. These studies provide a mechanistic description of how the PrrF1 and PrrF2 sRNAs mediate virulence traits, such as AQ production, in P. aeruginosa.
IMPORTANCE The iron-responsive PrrF sRNAs play a central role in regulating P. aeruginosa iron homeostasis and pathogenesis, yet the molecular mechanisms by which PrrF regulates gene expression are largely unknown. In this study, we used genetic and biophysical analyses to define the interactions of the PrrF sRNAs with Hfq, an RNA annealer, and the antR mRNA, which has downstream effects on quorum sensing and virulence factor production. These studies provide a comprehensive mechanistic analysis of how the PrrF sRNAs regulate virulence trait production through a key mRNA target in P. aeruginosa.
KEYWORDS: Pseudomonas aeruginosa, PrrF, PQS, Hfq, iron, antR
INTRODUCTION
Pseudomonas aeruginosa is a Gram-negative opportunistic pathogen that causes life-threatening infections in diverse patient populations. P. aeruginosa virulence depends on a multitude of virulence factors that include exotoxins (1–3), cell-to-cell communication via quorum-sensing factors (4–6), and nutrient acquisition systems (7). Nutrients, such as iron, are sequestered from invading pathogens by the innate immune system through a process referred to as nutritional immunity (8). Iron is critical for maintaining P. aeruginosa growth and virulence, and P. aeruginosa uses multiple pathways to acquire the metal from its host, including siderophore-dependent systems, heme acquisition pathways, and ferrous iron transporters (7, 9–11). In order to limit uptake of this redox-active metal, P. aeruginosa also possesses an intricate regulatory cascade that regulates the expression of iron uptake systems in response to available iron sources (12). This system is largely presided over by the ferric uptake regulator (Fur) protein, an iron-binding transcriptional repressor that blocks the expression of P. aeruginosa's iron uptake regulon under iron-replete conditions. The Fur protein also regulates the expression of numerous virulence factors, including secreted proteases and exotoxin A (13–17). This response allows P. aeruginosa to deploy its arsenal of virulence factors upon host-employed iron starvation, highlighting iron as a major environmental regulator of pathogenesis.
P. aeruginosa virulence and iron homeostasis also depend on the Fur-regulated PrrF1 and PrrF2 sibling small regulatory RNAs (sRNAs) (18, 19). The PrrF1 and PrrF2 sRNAs are 95% identical to one another, and they are transcribed from genes that are located in tandem on the genome in all sequenced strains of P. aeruginosa (20). The PrrF sRNAs are transcribed in response to iron depletion and contribute to iron homeostasis by blocking the production of nonessential iron-containing proteins (21). The PrrF sRNAs have also been shown to promote the production of several 2-alkyl-4(1H)-quinolone metabolites (AQs) (14, 18), which mediate a variety of biological activities that are important for virulence. These AQs include the Pseudomonas quinolone signal (PQS) and 2-heptyl-4-hydroxyquinolone (HHQ) quorum-sensing molecules, which induce the expression of multiple virulence genes (22–24). The AQ-biosynthetic pathway also produces an N-oxide-substituted AQ, 2-heptyl-4-hydroxyquinolone N-oxide (HQNO). HQNO promotes virulence by exerting toxic effects on mammalian cells and may also limit the growth of competing microbial species during polymicrobial infections (25–27). The PrrF sRNAs increase the production of each of these AQs, indicating their effects occur at the initiation of AQ synthesis (18). PrrF is thought to promote AQ synthesis by blocking the expression of the antABC and catBCA genes, which encode enzyme complexes that mediate degradation of the AQ precursor, anthranilate (Fig. 1A). Supplementation of ΔprrF1,2 mutant cultures with anthranilate restores production of PQS (14), supporting the model in which PrrF regulation of anthranilate metabolism underlies the requirement for PrrF in AQ biosynthesis (28).
FIG 1.

Model of PrrF-regulated production of 2-alkyl-4-quinolones. (A) PrrF sRNAs are predicted to promote alkylquinolone production by regulating the metabolism of anthranilate. In this model, the PrrF sRNAs repress the expression of antR, encoding a LysR-type transcriptional activator that induces expression of antABC, and catBCA genes for anthranilate catabolism. This prevents the degradation of anthranilate, allowing the metabolite to instead be used for the synthesis of numerous 2-akyl-4(1H)-quinolone metabolites via the pqsABCDE, pqsH, and pqsL gene products, as previously described. R indicates the presence of an alkyl chain, which varies in length and saturation. (B) Predicted complementarity between the PrrF1 and PrrF2 sRNAs and the antR leader sequence. The numbers indicate the orientation in relation to the transcriptional start site (PrrF1 and PrrF2) or the translational start site (antR leader). The predicted start codon of the antR mRNA is underlined. The asterisks indicate the region of antR that was protected by PrrF1 in RNase protection analysis performed in the present study. Vertical lines indicate canonical base pairing. Colons indicate noncanonical (G-U) base pairing. Dashes indicate gaps in the base pairing.
Bacterial sRNAs typically function by complementary base pairing with mRNA molecules, which can lead to either positive or negative effects on mRNA stability and expression (29–32). Base pairing of the sRNA near the ribosomal binding site (RBS) of the mRNA most often results in decreased mRNA stability and reduced translation efficiency. In contrast, base pairing of the sRNA upstream of the translational start site has been shown to alter the secondary structure of some mRNAs, resulting in increased translation efficiency. Both the PrrF1 and PrrF2 sRNAs share significant complementarity with the RBS and surrounding region of the antR mRNA (Fig. 1B), encoding a LysR-type transcriptional activator of the antABC and catBCA operons (14). A recent study demonstrated that overexpression of the PrrF2 sRNA resulted in decreased translation of the antR mRNA in P. aeruginosa (33). The authors of the study further showed that this regulation depended on the P. aeruginosa Hfq RNA binding protein, which we previously found interacts with the PrrF sRNAs (34). This finding is in line with what has been shown for the Hfq proteins of many other bacterial species, including Escherichia coli, where the Hfq protein accelerates annealing of sRNAs with their target mRNA molecules (33, 35–39). However, neither the biophysical basis of this regulation nor its impact on PrrF-regulated AQ production has been determined.
In the current study, we sought to determine the mechanisms underlying PrrF regulation of AQ production. We show that the PrrF1 and PrrF2 sRNAs function redundantly to repress antR expression and promote AQ production. We further show that this regulation occurs through posttranscriptional regulation by the PrrF sRNAs via the 5′ untranslated region (UTR) of the antR mRNA. We mapped the nucleotides required for interaction of the PrrF sRNAs with the antR mRNA through a combination of in vivo and in vitro analyses. We further showed that the P. aeruginosa Hfq protein binds tightly to the PrrF sRNAs and increases their rate of annealing to the antR mRNA leader sequence. These data provide a comprehensive model based on genetic, biophysical, and metabolite analyses for how the iron-responsive PrrF sRNAs promote the production of AQs by P. aeruginosa.
RESULTS
Anthranilate supplementation restores production of most alkylquinolones to the ΔprrF1,2 mutant.
Previous studies using thin-layer chromatography (TLC) showed that anthranilate supplementation restored PQS production to the ΔprrF1,2 mutant (14). This led to the model (Fig. 1A) in which PrrF repression of the antABC and catBCA genes spared anthranilate from degradation so that it could be used to synthesize PQS. If this is the case, then anthranilate supplementation should also restore production of other AQs, as anthranilate is a precursor for all of these metabolites. However, TLC does not detect the production of other AQ metabolites. We therefore determined the effect of anthranilate supplementation on AQ production, using a liquid chromatography-tandem mass spectrometry (LC–MS-MS) protocol previously developed in our laboratory (40, 41). The wild-type (WT) and ΔprrF1,2 mutant strains were grown for 18 h in dialyzed tryptic soy broth (DTSB), an iron-depleted medium used extensively by our laboratory to analyze P. aeruginosa AQ production (14, 18, 40–42). This assay quantifies the levels of both the C7 and C9 congeners of the PQS, HHQ, and HQNO metabolites, which represent the most abundant congeners under a variety of culture conditions (43). As previously observed, the ΔprrF1,2 mutant showed a significant reduction in the production of almost all the AQs tested by LC–MS-MS (Table 1). The one exception was the C7 congener of PQS (C7-PQS), which showed only a small and insignificant reduction in the ΔprrF1,2 mutant (Table 1). With the exception of the C9-PQS congener, supplementation of the ΔprrF1,2 mutant cultures with 500 μM anthranilate restored production of AQs to levels at or above those of the wild-type strain (Table 1). Interestingly, anthranilate supplementation of ΔprrF1,2 mutant cultures did not restore C7-PQS and C9-PQS production as robustly as previously observed by TLC (14). There was no significant difference in the concentration of individual AQ species produced by the ΔprrF1 and ΔprrF2 strains, indicating redundant effects of PrrF1 and PrrF2 sRNAs in regulating AQ production. This may be due to the production of multiple PQS congeners, with varying alkyl chain lengths and levels of saturation, that cannot be differentiated by TLC. Regardless, these data indicate that the ΔprrF1,2 mutant defect in AQ production is due, at least in part, to deregulated anthranilate metabolism.
TABLE 1.
Anthranilate supplementation restores AQ production to the ΔprrF1,2 mutant
| Strain | Normalized concn [(μg/μl)/OD600]a |
|||||
|---|---|---|---|---|---|---|
| C7-PQS | C9-PQS | HHQ | NHQ | HQNO | NQNO | |
| Wild type | 0.37 ± 0.03 | 0.52 ± 0.03 | 0.10 ± 0.04 | 0.25 ± 0.09 | 1.56 ± 0.05 | 6.59 ± 0.34 |
| ΔprrF1,2 mutant | 0.33 ± 0.03 | 0.27 ± 0.05d | 0.04 ± 0.01b | 0.11 ± 0.02b | 0.83 ± 0.21d | 3.73 ± 0.84d |
| ΔprrF1,2 mutant + anthranilate | 0.53 ± 0.03d,f | 0.17 ± 0.02d,e | 0.40 ± 0.09c,f | 0.37 ± 0.05b,f | 2.93 ± 0.31d,f | 6.07 ± 0.76e |
The concentration of each AQ in the supernatants of the indicated strains grown in DTSB, with or without 500 μM anthranilate, as indicated, was determined by LC–MS-MS and normalized by culture density, as described in Materials and Methods. Significant differences were determined by two-tailed Student's t test. OD600, optical density at 600 nm.
P < 0.05 for the ΔprrF1,2 mutant, with or without anthranilate, compared to the wild-type strain.
P < 0.005 for the ΔprrF1,2 mutant, with or without anthranilate, compared to the wild-type strain.
P < 0.0005 for the ΔprrF1,2 mutant, with or without anthranilate, compared to the wild-type strain.
P < 0.005 for the ΔprrF1,2 mutant with versus without anthranilate.
P < 0.0005 for the ΔprrF1,2 mutant with versus without anthranilate.
The PrrF sRNAs posttranscriptionally regulate expression of antR.
We next investigated the genetic mechanism by which the PrrF sRNAs affect AQ production by analyzing expression of antR, which encodes a LysR-type transcriptional activator of the antABC and catBCA genes for anthranilate degradation. We first identified the antR transcriptional start site (TSS) by 5′ rapid amplification of cDNA ends (RACE) using RNA isolated from the wild-type PAO1 strain grown in DTSB with 100 μM FeCl3 supplementation. This growth condition was previously shown to allow high levels of antR mRNA in PAO1 (14). The TSS was located within a string of G residues adjacent to sequence 95 to 98 nucleotides (nt) upstream of the translational start site (see Fig. S1 in the supplemental material). This TSS was distinct from that previously identified for antR, potentially due to the high-iron growth conditions used for the analysis, which produce a much higher level of antR transcription than was seen in the earlier study (44).
We next cloned the sequence corresponding to the predicted iron-dependent antR promoter, the antR untranslated region (UTR), and the portion of the antR coding sequence that is predicted to pair with the PrrF sRNAs and fused this fragment to a promoterless lacZY operon that lacked its native Shine-Dalgarno sequence (Fig. 2A). The resulting fusions were integrated onto the PAO1 and ΔprrF1,2 chromosomes at the att site. The antR reporter strains were grown in DTSB, with or without supplementation with 100 μM FeCl3, for 18 h. As expected, β-galactosidase activity was strongly repressed when the wild-type strain was grown under low-iron conditions. Also as expected, we observed significant induction of β-galactosidase activity in the ΔprrF1,2 mutant reporter strain compared to the wild-type reporter strain when both were grown in low iron (Fig. 2A). Interestingly, iron supplementation still caused a small but statistically significant induction of β-galactosidase activity from the antR reporter in the ΔprrF1,2 mutant (Fig. 2A), indicating that iron activates antR expression through both PrrF-dependent and PrrF-independent pathways.
FIG 2.

The PrrF sRNAs mediate iron-regulated posttranscriptional repression of antR. The indicated strains carrying the antR transcriptional and translational fusions (A), the antR translational fusion (B), or the antR transcriptional fusion (C) were grown for 18 h at 37°C in DTSB without (white bars) or with (gray bars) 100 μM FeCl3 supplementation. Diagrams of the corresponding reporter fusions are shown above the graphs. In each diagram, +1 denotes the transcriptional start site, SD denotes the Shine-Dalgarno site, and AUG indicates the translational start site. In panels A and B, the region of PrrF complementarity (PrrF C′) with the antR UTR is indicated above the diagram by a horizontal bar. The error bars indicate the standard deviations of β-galactosidase activity from three (A) or six (B and C) independent experiments. The asterisks indicate a significant difference when comparing each of the mutants to PAO1 grown under low-iron conditions or as indicated by the horizontal bars. *, P < 0.05; **, P < 0.001.
We next determined whether iron activation of antR expression occurs by transcriptional or posttranscriptional mechanisms. An antR translational fusion consisting of the native lac promoter followed by the antR 5′UTR fused to the lacZY operon, as shown in Fig. 2B, was integrated at the att site of PAO1 and the ΔprrF1,2 mutant. The β-galactosidase activity from this translational reporter showed 7.3-fold induction by iron, and this induction was eliminated in the ΔprrF1,2 mutant (Fig. 2B). We also constructed an antR transcriptional fusion consisting of the antR promoter fused to lacZY preceded by its native Shine-Dalgarno site (Fig. 2C). The antR transcriptional reporter was also activated by iron in wild-type PAO1, but only by 2.3-fold (Fig. 2C). Moreover, while a slight reduction in antR promoter activity was observed in the ΔprrF1,2 mutant compared to the wild type, deletion of the prrF1-prrF2 locus did not reduce iron activation of this reporter (Fig. 2C). Combined, these data demonstrate that PrrF-dependent iron activation of antR expression occurs via the antR 5′ UTR, while PrrF-independent iron activation of antR occurs at the antR promoter.
PrrF1 and PrrF2 promote AQ production through redundant repression of antR expression.
Previous studies showed that the PrrF1 and PrrF2 sRNAs are redundant for regulation of many targets, including antR and antA (14, 21). However, the impact of the individual PrrF sRNAs on AQ production was not known. We therefore determined the impacts of the individual PrrF1 and PrrF2 sRNAs on AQ production by LC–MS-MS. The PAO1, ΔprrF1, and ΔprrF2 single mutants and the ΔprrF1,2 double mutant were grown in DTSB for 18 h and analyzed for AQ production by LC–MS-MS. Individual deletion of either the prrF1 or prrF2 gene exerted no effect on the production of C7-PQS, HHQ, HQNO, or NQNO (2-nonyl-4-hydroxyquinoline n-oxide) (Table 2) and caused only small (less than 50%) reductions in C9-PQS and NHQ production (Table 2). There was also no significant difference in the concentrations of individual AQ species produced by the ΔprrF1 and ΔprrF2 strains, indicating redundant effects of PrrF1 and PrrF2 sRNAs in regulating AQ production. The antR reporter fusion was also integrated onto the chromosomes of the ΔprrF1 and ΔprrF2 single mutants, and the resulting strains were grown for 18 h in DTSB and analyzed for β-galactosidase activity. Similar to what was observed for the production of most AQs, deletion of either prrF1 or prrF2 resulted in no significant change in β-galactosidase activity from the antR reporter (Fig. 2B). These data demonstrate that the PrrF1 and PrrF2 sRNAs redundantly regulate antR expression to promote AQ production.
TABLE 2.
PrrF1 and PrrF2 are redundant in their regulation of AQ production
| Strain | Normalized concn [(μg/μl)/OD600]a |
|||||
|---|---|---|---|---|---|---|
| C7-PQS | C9-PQS | HHQ | NHQ | HQNO | NQNO | |
| Wild type | 0.64 ± 0.06 | 1.01 ± 0.10 | 0.08 ± 0.02 | 0.27 ± 0.07 | 2.39 ± 0.28 | 12.21 ± 1.03 |
| ΔprrF1 mutant | 0.54 ± 0.08 | 0.65 ± 0.05d | 0.05 ± 0.01 | 0.14 ± 0.03b | 2.06 ± 0.62 | 9.82 ± 0.01 |
| ΔprrF2 mutant | 0.55 ± 0.07 | 0.80 ± 0.06c | 0.06 ± 0.01 | 0.17 ± 0.05 | 2.50 ± 0.61 | 11.10 ± 1.99 |
| ΔprrF1,2 mutant | 0.50 ± 0.07b | 0.35 ± 0.06d | 0.04 ± 0.01b | 0.06 ± 0.01d | 0.95 ± 0.27d | 3.71 ± 0.00d |
The concentration of each AQ in the supernatants of the indicated strains grown in DTSB was determined by LC–MS-MS and normalized by culture density, as described in Materials and Methods.
P < 0.05, comparing the ΔprrF1, ΔprrF2, or ΔprrF1,2 mutant to the wild type.
P < 0.005, comparing the ΔprrF1, ΔprrF2, or ΔprrF1,2 mutant to the wild type.
P < 0.0005, comparing the ΔprrF1, ΔprrF2, or ΔprrF1,2 mutant to the wild type.
PrrF represses antR via sequences in the antR UTR.
We next sought to identify the specific nucleotides of the antR leader sequence that are required for regulation by the PrrF1 and PrrF2 sRNAs. A series of mutations were designed and introduced into the antR reporter (Fig. 3A), and the altered antR reporters were integrated at the att site of wild-type strain PAO1. While mutation of the sequence adjacent to the RBS ablated β-galactosidase activity from the antR reporter under high-iron conditions (alt-antRA and alt-antRC mutants [Fig. 3B]), mutation of the sequence further upstream of the RBS had no significant effect on the β-galactosidase activity of the antR reporter under high-iron conditions (alt-antRB mutation [Fig. 3B]). We further explored the effects of the alt-antRB mutant, which altered 3 nucleotides approximately 20 nt upstream of the antR RBS, on β-galactosidase activity from the antR reporter under low-iron conditions. These data showed a small but statistically significant derepression of β-galactosidase activity from the alt-antRB reporter compared to the wild-type antR reporter when grown in iron-depleted medium (Fig. 3B). Despite showing reduced translation efficiency under high-iron conditions, the alt-antRC reporter also caused a small but statistically significant increase in β-galactosidase activity under low-iron conditions (Fig. 3B). Combined, these data suggest that PrrF repression of antR depends upon a significant stretch of complementarity between the PrrF sRNAs and the antR 5′ UTR. We attempted to restore PrrF regulation of the altered alt-antRB reporter by transforming the alt-antRB reporter strain with a plasmid carrying a compensatory prrF1-prrF2 allele designed to restore PrrF1-antR and PrrF2-antR complementarity. However, this construct had no effect on β-galactosidase activity from the alt-antRB reporter, potentially due to altered structures of the PrrF or antR UTR (data not shown). Further analysis of the PrrF-antR interaction, including the role of the P. aeruginosa Hfq protein in their association, was therefore conducted in vitro.
FIG 3.
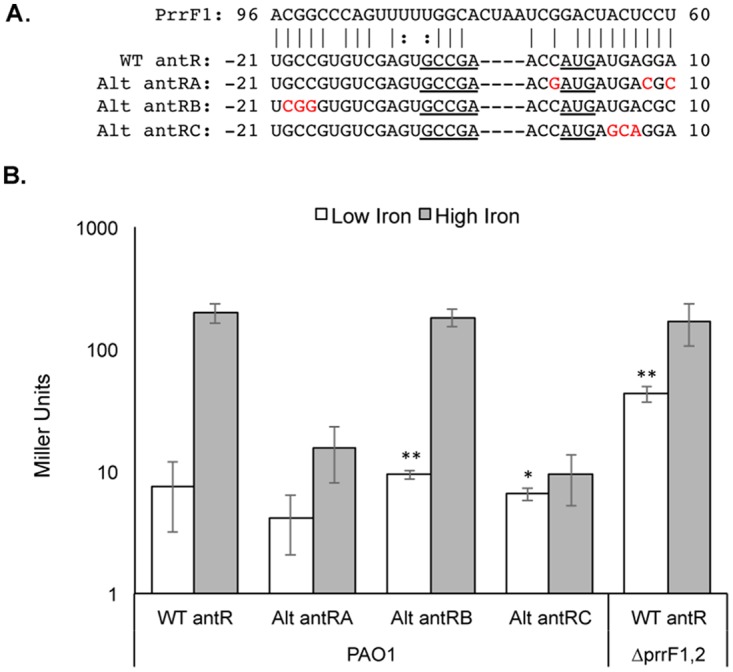
Mutations in the antR 5′ UTR reduce PrrF repression. (A) Mutations were generated in the antR translational fusion to disrupt the predicted interaction between the PrrF sRNAs and the antR 5′ UTR. (B) The indicated strains carrying either the WT or altered antR translational reporters shown in panel A were grown for 18 h in DTSB without (white bars) or with (gray bars) 100 μM FeCl3 supplementation, and the cultures were assayed for β-galactosidase activity. The error bars represent the standard deviations from three independent experiments. The asterisks indicate a significant difference in Miller units when comparing the low-iron values to those of the WT antR translational reporter in strain PAO1 grown under low-iron conditions. *, P < 0.01; **, P < 0.005.
PrrF sRNAs bind to the arginine patch of Hfq.
The PrrF sRNAs were previously shown to interact with the P. aeruginosa Hfq protein (34), indicating a role for Hfq in PrrF regulatory function. Studies of E. coli and Salmonella sRNAs showed that the 3′-terminal U's associated with the Rho-independent transcription terminator bind the proximal pore of Hfq (45, 46). An internal AU-rich motif additionally interacts with the arginine patch on the rim of Hfq (47, 48), which is necessary for efficient annealing with its mRNA targets (36). To probe the secondary structure of the PrrF sRNAs and locate the P. aeruginosa Hfq binding site, we partially digested 32P-labeled PrrF sRNAs with RNase If. The RNase If digestion pattern of PrrF1 in the absence of Hfq [Fig. 4A, lane (−) Hfq] agrees with the predicted secondary structure of PrrF1 (Fig. 4E), except that the regions between nt 60 and 65 and nt 80 and 90 were more protected than expected, suggesting that the region between stem-loop 2 and stem-loop 3 forms one or more alternative secondary structures.
FIG 4.

RNase If footprinting of PrrF1 and antR. (A) RNase If digestion pattern of 5′-32P-PrrF1 sRNA at 30°C with no Hfq [(−) Hfq], 0.17 to 0.67 μM P. aeruginosa (Pa) Hfq, 0.17 to 0.33 μM E. coli (Ec) Hfq, and 0.33 μM E. coli R16A Hfq, as shown. Lanes T1, Alk lys, and FL represent control reactions in urea with RNase T1 (T1), alkaline hydrolysis (Alk lys), and no treatment (FL). E. coli Hfq binds sRNA more strongly than P. aeruginosa Hfq, and binding of a second E. coli Hfq hexamer may account for some differences between the protection pattern in 0.17 and 0.33 μM E. coli Hfq. (B) RNase If digestion patterns of 5′-32P-labeled antR mRNA at 30°C with no Hfq (lane A), 0.5 μM P. aeruginosa Hfq (lane A H), 0.5 μM PrrF1 (lane A P1), and 0.5 μM P. aeruginosa Hfq plus 0.5 μM PrrF1 (lane A P1 H). Control reactions were as for panel A. Numbers to the left of the images in panels A and B indicate the nucleotide numbers of the RNA molecule where cleavage occurs to produce the observed bands. (C) Relative digestion of PrrF1 sRNA by RNase If in the absence and presence of P. aeruginosa Hfq (see Materials and Methods). The inset shows the region between nt 90 and 95. (D) Relative digestion of antR mRNA by RNase If in the absence and presence of P. aeruginosa Hfq and PrrF1 sRNA. (E and F) Summary of nuclease digestion patterns on the predicted secondary structures of PrrF1 (E) and antR 5′ UTR (F), as indicated in the key. The schematics summarize the results of several footprinting experiments for each RNA. Weak (<2×) and strong (∼2×) protection were determined by quantitation of the footprinting gels with SAFA (71), as illustrated in panels C and D. A plus sign indicates increased nuclease cleavage upon Hfq (red) or PrrF1 (blue) binding, suggesting structural rearrangements of the RNA molecules. The secondary structures of the PrrF1 sRNA and antR mRNA shown in panels E and F, respectively, were generated using a combination of computational prediction (MFold) (73) and experimental results (A and B).
The presence of P. aeruginosa Hfq weakly protected AU-rich segments of PrrF1, A58 to U59 and A69 to A72, and more strongly protected the U-rich motifs, U76 to A81 and U85 to U89, from digestion by RNase If (Fig. 4A, lanes Pa Hfq, and E, red symbols). The extent of protection increased with increasing Hfq concentrations, suggesting that these internal A/U- and U-rich motifs bind Hfq, as observed in other sRNAs. For comparison, we also probed the structure of PrrF1 RNA in the presence of E. coli Hfq, which also protected nt 76 to 89 of PrrF1, as we found with P. aeruginosa Hfq (Fig. 4A, Ec Hfq, lane 0.17 μM WT). Interestingly, when R16 on the rim of E. coli Hfq was replaced with alanine, this internal A/U-rich region was no longer protected from RNase digestion (Fig. 4A, Ec Hfq, lane 0.33 μM R16A), suggesting that this region interacts with the arginine patch. PrrF2 sRNA, which differs by only a few nucleotides from PrrF1, exhibited a similar RNase If digestion pattern in the absence and presence of P. aeruginosa, WT E. coli, and R16A Hfq (see Fig. S2 and S3 in the supplemental material). These results indicate that P. aeruginosa Hfq recognizes the PrrF1 and PrrF2 sRNAs in a manner similar to that of Hfq proteins from E. coli and Salmonella.
Hfq and PrrF1 bind different domains of the antR mRNA leader.
To map the binding sites for P. aeruginosa Hfq and PrrF1 on the antR mRNA leader, a 32P-labeled fragment of the antR mRNA from the transcription start site through the 14th codon was also partially digested with RNase If in the presence of P. aeruginosa Hfq and PrrF1. GC-rich sequences at the 5′ end of antR mRNA were completely protected from RNase digestion, consistent with the formation of stable secondary structure at the 5′ end of the mRNA (Fig. 4B). In the presence of Hfq, the A-rich loop between nt 42 and 50 of the antR mRNA was moderately protected from RNase digestion (Fig. 4B, lane A H and lane A H P1), suggesting this loop provides a weak binding site for Hfq in the antR mRNA (Fig. 4F, red circles). P. aeruginosa Hfq also protected residues downstream of the PrrF binding site (U112), but not when PrrF1 was present (Fig. 4F, red squares). When PrrF1 sRNA was allowed to bind antR mRNA, a downstream region spanning nt 91 to 110 became strongly protected from RNase If (Fig. 4C, lane A P and lane A H P1, and F, blue symbols). These residues comprise part of the predicted region of complementarity between PrrF1 and antR mRNA (Fig. 3A) and coincide with the sequence required for full PrrF repression of PantR translational activity in vivo (Fig. 3B, alt-antRC), indicating that this region is a likely binding site for PrrF1. The region was also protected when PrrF1 and P. aeruginosa Hfq were added to antR mRNA simultaneously, suggesting that PrrF1 can bind to antR mRNA in the absence or presence of P. aeruginosa Hfq.
P. aeruginosa Hfq binds to PrrF sRNA with higher affinity than to antR mRNA.
To determine whether P. aeruginosa Hfq can facilitate base pairing between sRNAs and mRNAs by simultaneously binding both RNAs in a ternary complex, we measured the affinity of P. aeruginosa Hfq for PrrF sRNAs and antR mRNA by native gel electrophoretic mobility shift assays (EMSA). When 32P-labeled PrrF1 or PrrF2 was titrated with P. aeruginosa Hfq, we observed sequential gel shifts corresponding to the binding of one Hfq hexamer [P-H(I)] or two or more hexamers [P-H(II)] (Fig. 5A; see Fig. S4 in the supplemental material). The dissociation constant for each PrrF-Hfq complex was determined by fitting the fraction of bound PrrF sRNA versus the Hfq concentration to a partition function for two independent sites on PrrF (Fig. 5B). The resulting dissociation constants for P-H(I) ranged from 51 to 65 nM Hfq hexamer for PrrF1 (Table 3) and 70 to 74 nM Hfq6 for PrrF2 (Table 3; see Fig. S4 in the supplemental material). Thus, PrrF1 and PrrF2 sRNAs not only have similar sequences, they interact similarly with P. aeruginosa Hfq.
FIG 5.

Equilibrium binding of Hfq with PrrF1 and antR. (A) Incubation of 32P-labeled PrrF1 with 0 to 667 nM Hfq6 resulted in a specific mobility shift [P1-H(I)], followed by an additional diffuse mobility shift [P1-H(II)] arising from two or more Hfq hexamers binding to PrrF1. (B) Fractions of P1-H(I) and P1-H(II) complexes were fit to equations 1 and 2 to obtain the dissociation constants (Table 3). (C) Titration of 32P-antR mRNA with 0 to 1,667 nM Hfq6 resulted in three distinct RNPs, A1-H(I), A1-H(II,) and A1-H(III), with one to three Hfq hexamers, respectively. (D) Fractions of A1-H(I) and the total of A1-H(II) plus A1-H(III) complexes, as in panel B.
TABLE 3.
Equilibrium binding constantsa for PrrF1, PrrF2, Hfq, and antR
| Complex | Kd1 (nM) | Kd2 (nM) |
|---|---|---|
| PrrF1 + Hfq | 65 ± 7, 51 ± 2 | 135 ± 12, 183 ± 407 |
| PrrF2 + Hfq | 70 ± 2, 74 ± 3 | 207 ± 7, 480 ± 50 |
| antR + Hfq | 295 ± 9, 302 ± 24 | 440 ± 500, 407 ± 943 |
| PrrF1 + antR | 2.5 ± 1, 7 ± 1 | NDb |
P. aeruginosa Hfq bound to the 32P-labeled antR mRNA leader 4 to 6 times less tightly than to PrrF1 or PrrF2, with dissociation constants of 295 nM and 440 nM Hfq6 for the first and second Hfq complexes, respectively (Fig. 5C and D and Table 3). This low affinity of P. aeruginosa Hfq for antR mRNA may be due to the lack of repeated (AAN)4 motifs, which are necessary for efficient interaction of the distal face of Hfq with mRNAs in E. coli (29). In support of this idea, the short A-rich binding sequence (AAAACAGA) at the loop of SLII was only weakly protected by P. aeruginosa Hfq in our footprinting experiments (Fig. 4D). In contrast, many E. coli mRNAs contain three or four AAN triplets, which bind E. coli Hfq tightly (29). The weak affinity of P. aeruginosa Hfq for antR mRNA was not due to the inability of P. aeruginosa Hfq to recognize AAN motifs, because the Kd (dissociation constant) for P. aeruginosa Hfq binding to A18 RNA was 8 nM Hfq6 (see Fig. S5 in the supplemental material), very similar to the Kd of 7.5 nM for E. coli Hfq (49). Moreover, P. aeruginosa Hfq has been reported to bind AAN repeats in amiE mRNA and CrcZ sRNA in P. aeruginosa (50, 51).
P. aeruginosa Hfq forms a ternary complex with PrrF1 and antR mRNA.
To determine whether Hfq is needed to stabilize base pairing between PrrF sRNA and antR mRNA, we measured the binding affinity of our antR mRNA fragment for PrrF1 sRNA by EMSA and found that even at 0.1 nM PrrF1, 30% of antR mRNA formed a complex with PrrF1 sRNA. The fraction of antR mRNA-PrrF1 complex as a function of the PrrF1 concentration was fit to a two-state quadratic equation, yielding a dissociation constant of 2.5 nM (Fig. 6B). Thus, PrrF1 sRNA binds tightly to antR mRNA in the absence of Hfq and may not require Hfq for binding to antR mRNA in P. aeruginosa.
FIG 6.

Effect of Hfq on sRNA-mRNA duplex stability. (A) Addition of 0 to 100 nM PrrF1 sRNA to 5 nM 32P-labeled antR mRNA produces PrrF1-antR mRNA duplex. (B) A fraction of PrrF1-antR mRNA duplex (Fig. 5A) with increasing PrrF1 concentrations was fit to a 2-state quadratic equation (equation 3). (C) A preformed complex of 5 nM 32P-antR mRNA and 10 nM PrrF1 sRNA was titrated with 0 to 200 nM Hfq6 to compare the stabilities of the RNA duplex at different Hfq concentrations. An increase in the Hfq concentrations resulted in an increase of antR-PrrF1 (A-P1) band intensity up to 50 nM. A stable ternary complex (antR-PrrF1-Hfq) was visible above 50 nM Hfq6. High Hfq concentrations destabilized the antR-PrrF1 duplex, resulting in free antR RNA. (D) The fraction of antR mRNA-PrrF1 complexes as a function of the Hfq concentration was fit to equation 4.
To determine the effect of P. aeruginosa Hfq on the stability of the sRNA-mRNA duplex, we titrated a preformed 32P-antR mRNA-PrrF1 complex with Hfq. Low concentrations of Hfq slightly increased retention of the antR mRNA-PrrF1 complex in the native gel. Around 50 nM Hfq, which is comparable to the dissociation constant for PrrF1 (51 nm) (Table 3), a stable ternary complex between Hfq, PrrF1, and antR mRNA appeared (Fig. 6D). Above 100 nM Hfq, the antR mRNA-PrrF1 duplexes fell apart, releasing free antR mRNA (Fig. 6D). This shift in the stability of the sRNA-mRNA duplex suggests that excess Hfq may sequester PrrF1 sRNA in an inactive complex that cannot pair with antR mRNA. A similar phenomenon was observed for E. coli Hfq binding to rpoS mRNA (52).
Hfq increases the rate of sRNA-mRNA annealing.
We previously observed that P. aeruginosa Hfq accelerates the annealing of RNA oligomers about 10-fold (53), which is about 10-fold less than E. coli Hfq, a very active RNA annealer. We attribute this to replacement of an arginine (R17) with a lysine (K17) on the rim of P. aeruginosa Hfq (53). We next asked whether P. aeruginosa Hfq can also accelerate the annealing of PrrF1 sRNA with our 32P-labeled antR mRNA fragment. Although PrrF1 bound to antR mRNA in the absence of Hfq, the rate of duplex formation was low (0.1 min−1) (Fig. 7). In the presence of 100 nM Hfq, antR, PrrF1, and Hfq rapidly formed a ternary complex (k1 = ∼3 min−1), which was followed by a slower phase of annealing (k2 = 0.2 min−1) and accumulation of antR mRNA-PrrF1 binary complex. In 200 nM Hfq, the magnitude of the fast phase was smaller, but more antR mRNA-PrrF1 complex accumulated within the first few minutes of the reaction than in the absence of Hfq (Fig. 7B). These data suggested that P. aeruginosa Hfq can increase sRNA-mRNA duplex formation by up to ∼30-fold, despite weak binding of Hfq to antR mRNA.
FIG 7.
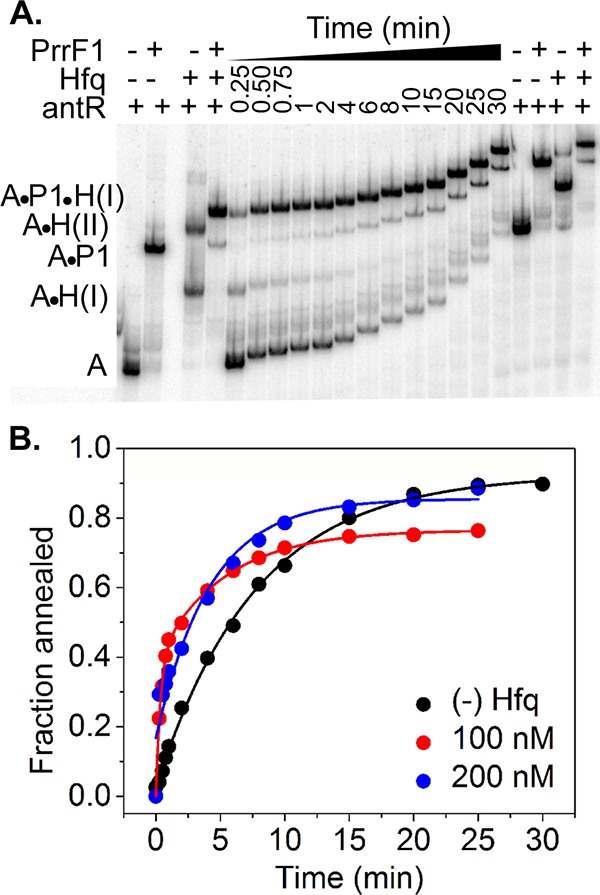
Effect of P. aeruginosa Hfq on RNA annealing kinetics. (A) 5 nM 32P-antR RNA, 30 nM PrrF1 sRNA, and 100 nM Hfq were mixed rapidly in TNK buffer (10 mM Tris-HCl, pH 7.5, 50 mM NaCl, 50 mM KCl) and loaded on 6% native PAGE while running continuously. (B) Formation of antR RNA-PrrF1 binary [(−) Hfq] or the sum of antR RNA-PrrF1 and antR RNA-PrrF1-Hfq complexes (100 and 200 nM Hfq) over time were fit to a biexponential rate equation (equation 5). The controls were incubated for an hour to allow the reactions to reach equilibrium. The standard deviation among four technical replicates of each reaction was <10%.
DISCUSSION
The PrrF sRNAs play a central role in P. aeruginosa iron homeostasis and pathogenesis (14, 18, 19, 21), yet the molecular basis for ΔprrF1,2 mutant virulence attenuation remains unclear. In this study, we aimed to determine the genetic and biochemical bases on which the PrrF sRNAs promote the production of AQs, which play multiple roles in P. aeruginosa virulence (24, 41, 54–56). We demonstrated that the PrrF1 and PrrF2 sRNAs redundantly affect antR translational activity to promote AQ production (Fig. 2 and Table 2). We also identified two distinct regions of the antR leader that are required for full repression of the PantR-′lacZ reporter in vivo (Fig. 3). One of these regions (mutated in the alt-antRB fusion) was not protected from RNase digestion in the presence of the PrrF1 sRNA (Fig. 4), suggesting these nucleotides do not directly pair with the PrrF sRNAs. Mutation of this region may instead cause structural changes to the antR mRNA that are important for interaction with the PrrF sRNAs, particularly since these mutations are expected to alter the stability of the stem-loop adjacent to the PrrF1 binding site. The second site identified in our in vivo analysis (mutated in the alt-antRC fusion) overlaps the antR translational start site (Fig. 3) and was protected from RNase digestion in the presence of PrrF1 (Fig. 4), indicating that the region pairs with the PrrF sRNAs in vivo. The latter region is predicted to anneal to a sequence that is completely conserved between PrrF1 and PrrF2 (Fig. 1B), providing a mechanistic rationale for the redundancy of the PrrF sRNAs in regulating antR expression. Notably, both the antRA and antRC mutations resulted in loss of translation under high-iron conditions, presumably due to changes in the antR UTR structure. In line with this idea, MFold analysis of these mutant UTRs resulted in stable hairpin structures that sequestered the Shine-Dalgarno site and translational start site of antR (data not shown).
Our studies also build on the current model of how P. aeruginosa Hfq contributes to PrrF regulation of antR. We showed that P. aeruginosa Hfq binds with high affinity to the PrrF sRNAs through the arginine patch on the Hfq rim (Fig. 4A), similar to what has been reported for several E. coli sRNAs (47, 48). While P. aeruginosa Hfq was not required for formation of the antR-PrrF1 complex, it increased the rate of formation of the complex and stabilized the preformed antR-PrrF1 complex. This biophysical evidence supports previous in vivo work by Sonnleitner et al., which demonstrated that a Δhfq mutant was defective for PrrF-mediated regulation of antR (33) and that the binding potential of P. aeruginosa Hfq largely accounts for catabolite repression control (Crc) regulation of antR. Interestingly, P. aeruginosa Hfq bound weakly to the antR mRNA (295 nM) in our studies (Fig. 5D), presumably owing to a shorter A-rich motif (AAN) in contrast to the multiple AAN motifs that mediate Hfq recognition and binding to mRNAs in E. coli (57). This suggests either that Hfq can act via transient interactions with mRNA targets or that other RNA binding proteins contribute to mRNA recognition by P. aeruginosa Hfq. Nevertheless, these studies clearly demonstrate that Hfq plays an important role in PrrF-mediated regulation of antR expression in P. aeruginosa.
This study further demonstrated that iron regulates antR through both PrrF-dependent and PrrF-independent mechanisms (Fig. 2). This finding is consistent with previous work showing that iron activates antA expression in the ΔprrF1,2 mutant (14), demonstrating that iron regulates anthranilate metabolism via AntR through at least two distinct mechanisms. In addition to serving as a precursor for AQ synthesis, anthranilate is a central metabolite in P. aeruginosa, and thus, its catabolism likely requires extensive control. Anthranilate is synthesized by anthranilate synthase from chorismate, which also serves as a precursor for siderophore and amino acid biosynthesis (58). Anthranilate is also formed as an intermediate in the kynurenine pathway, which converts tryptophan to NAD (NAD+) (28, 59). Anthranilate degradation via the antABC-encoded anthranilate dioxygenase, which is dependent upon iron (60), feeds into the catechol ortho-cleavage pathway to form the tricarboxylic acid (TCA) cycle intermediate succinyl-coenzyme A (CoA) (61). Expression of the antABC operon is dependent upon the presence of anthranilate, which serves as a coinducer of the LysR-type AntR regulator (14). Thus, while PrrF regulation of antR expression is clearly important for AQ production in low-iron environments (Table 1) (18), many additional factors likely converge to regulate anthranilate metabolism and AQ production.
Our studies additionally identified a new TSS for the antR mRNA that is upstream of that observed for P. aeruginosa strain PA14 grown in L broth (44). Since the TSS of the longer transcript was identified under our high-iron growth conditions and not in previous studies, we hypothesize that the transcript is dependent upon iron supplementation of the growth medium. In line with this hypothesis, our data show that iron increases the activity of this upstream promoter (Fig. 2C), as well as translation of the resulting mRNA transcript (Fig. 2B). At this time, it is unclear how iron regulates the antR promoter, as we did not identify an obvious Fur-binding site in the antR-antA intergenic region. Notably, the shorter antR transcript in the PA14 transcriptome lacks the Hfq binding site described here, indicating that Hfq cannot facilitate PrrF-mediated regulation of this shorter transcript. Kim and colleagues previously identified an AntR binding site immediately upstream of the TSS for the shorter antR transcript (62), and a potential RpoN-dependent promoter is immediately upstream of this shorter TSS (63) (shown in Fig. S1 in the supplemental material). Thus, it is possible that the shorter antR transcript is responsive to nitrogen and amino acid metabolism via anthranilate while the longer transcript is specifically responsive to iron. The iron-dependent transcript is additionally subject to catabolite repression control due to the ability of the CrcZ sRNA to sequester Hfq when preferred carbon sources have been depleted (33). These combined studies further demonstrate the complexity of anthranilate catabolism regulation by P. aeruginosa in diverse environments.
Surprisingly, our study showed disparate effects of anthranilate supplementation on C9-PQS and C7-PQS production by the ΔprrF1,2 mutant (Table 1), further underlining the complexity of AQ production regulation. The alkyl chains of HHQ and C7-PQS are derived from octanoic acid (64), and it is assumed that longer and shorter chains of different AQ congeners are derived from correspondingly longer or shorter fatty acids. We previously found that iron regulation of AQ production was variable between different congeners of each AQ metabolite (40), indicating that iron regulation of fatty acid metabolism or availability may play an important role in modulating AQ production. Combined with our current data, these studies suggest that numerous metabolic and iron-regulatory pathways modulate AQ production, highlighting the interplay of P. aeruginosa's metabolic diversity, iron dependency, and pathogenic potential.
The redundancy of PrrF1 and PrrF2 observed in this work further begs the question of why the prrF1 and prrF2 genes are duplicated in tandem in all sequenced strains of P. aeruginosa. It is possible that slight differences between the PrrF1 and PrrF2 sequences allow differential regulation of some mRNA targets. Alternatively, differential expression of the PrrF1 and PrrF2 sRNAs may allow diverse environmental signals, in addition to iron depletion, to affect PrrF regulation of metabolism and virulence. Indeed, recent work by Little and colleagues demonstrated that the P. aeruginosa AlgZR two-component regulatory system activates PrrF expression through specific interaction with the prrF2, but not the prrF1, promoter (65). The tandem duplication of the prrF genes also results in the production of a longer heme response sRNA named PrrH (66). PrrH interacts with the P. aeruginosa Hfq protein (34), but the significance and function of this longer sRNA remain unknown. Future biophysical and genetic studies of the PrrH sRNA should yield novel insights into the physiological significance of the tandem arrangement of the prrF genes in P. aeruginosa.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The strains used in these studies are summarized in Table S1 in the supplemental material. E. coli strains were maintained in LB medium prepared as described by Miller (67). P. aeruginosa strains were maintained in brain heart infusion (BHI) medium. For β-galactosidase reporter and AQ quantification studies, BHI overnight liquid cultures were inoculated from five colonies isolated on BHI agar plates. The overnight BHI cultures were diluted to an absorbance (A600) of 0.05 in Chelex-treated DTSB prepared as previously described (14), with or without supplementation with 100 μM FeCl3. The DTSB cultures were grown for 18 h at 37°C. Antibiotics were added at the following concentrations: for E. coli, ampicillin, 100 μg/ml; tetracycline, 10 or 15 μg/ml; and chloramphenicol, 12.5 μg/ml; for P. aeruginosa, carbenicillin, 250 μg/ml; tetracycline, 150 μg/ml.
Quantitation of AQs.
AQs were quantified by LC–MS-MS as previously described (40, 41) with some modifications. Specifically, AQs were extracted from 300 μl of culture supernatants and spiked with 10 μl of 25 μM nalidixic acid as an internal standard, as previously described (6). Organic extracts were transferred to new microcentrifuge tubes and dried using a SpeedVac concentrator (Thermo-Fisher). The AQ extracts were resuspended in a mobile phase of 1:1 acetonitrile-water with 0.1% formic acid and 200 μM EDTA and separated using a dual-pump Dionex Ultimate 3000 Rapid Separation ultra-high performance liquid chromatography (UHPLC) device (Thermo-Fisher). Separation was attained using an Ascentis Express C8 column (100 by 2.1 mm; 2.7-μm particle size; Sigma-Aldrich) maintained at 30°C. Mobile phases A and B were water with 0.1% formic acid and 200 μM EDTA, and acetonitrile with 0.1% formic acid, respectively. The mobile phase gradient was maintained at a constant flow rate of 0.4 ml/min as follows: 20% acetonitrile for 0.5 min, followed by a linear gradient from 20% to 95% acetonitrile over 3.5 min and then 95% acetonitrile for 1.5 min. The liquid chromatography (LC) column was reequilibrated to 20% acetonitrile for 2 min between samples. LC-separated metabolites were quantified on a TSQ Quantum Ultra tandem-quadrupole mass spectrometer (Thermo-Fisher) equipped with an electrospray ionization (ESI) source in positive ion electrospray mode at a spray voltage of 3 kV maintained at 450°C. Parameters for multiple-reaction monitoring (MRM) (see Table S2 in the supplemental material) were optimized using Thermo TSQ EZ-tune software automation. Ten-point calibration curves were prepared with known concentrations of all six representative AQ species (C7-PQS, C9-PQS, HHQ, NHQ, HQNO, and NQNO), including a nalidixic acid standard. Unknown AQ concentrations in bacterial culture extracts were calculated by comparing the peak area ratio of a spiked internal standard to those of individual AQ species using Xcalibur version 3.0 software in Quan Browser mode (Thermo).
Determination of the antR transcriptional start site.
The TSS of antR was determined using the 5′ RACE system (Invitrogen) according to the manufacturer's instructions. Briefly, total RNA was isolated from P. aeruginosa strain PAO1 grown in DTSB supplemented with 100 μM FeCl3 for 18 h, and cDNA was synthesized by reverse transcription using a reverse primer specific to the antR mRNA (antR.rev) (see Table S3 in the supplemental material). After dC tailing and amplification with a nested antR reverse primer (antR.nested) (see Table S3 in the supplemental material), the PCR product was sequenced. The TSS of the gene was determined by looking for a series of C residues at the 5′ end of the reverse sequence. As the cDNA ends adjacent to a GGGG in the PAO1 genomic sequence, the exact TSS within this G tract is ambiguous.
Construction of antR reporters.
The Mini-CTX1-lacZY-SD vector, which lacks the lacZ Shine-Dalgarno site, was constructed by PCR amplification of the lacZY operon, starting at the +1 translational site, from the Mini-CTX1-lacZY vector using the LacZ-SD-HindIII-for and LacZ-SD-AatI-rev primers (see Table S3 in the supplemental material). The fragment was cloned into PCR2.1 (Invitrogen), confirmed by sequencing, and then ligated into Mini-CTX1 using HindIII and AatI restriction fragments. The antR reporter (transcriptional and translational) (Fig. 2A) was constructed by PCR amplification of the leader and promoter sequence, spanning 67 nt upstream of the transcription start site to 15 nt downstream of the translational start site, using oligonucleotides antR(67)-EcoRI-for and antR-HindIII-rev (see Table S3 in the supplemental material) and PAO1 chromosomal DNA as the template. The PCR product was subsequently cloned into PCR2.1 and confirmed by sequencing. EcoRI-HindIII-digested fragments of the antR promoter and 5′ UTR were ligated into Mini-CTX1-lacZY-SD digested with the same enzymes to generate Mini-CTX1-PantR-′lacZY-SD. Altered antR reporters were generated using the QuikChange II XL site-directed mutagenesis kit (Agilent) following the manufacturer's instructions, with PCR2.1-antR as the template and primers alt-antR-EcoRI-for and antR-HindIII-rev (see Table S3 in the supplemental material). The antR translational fusion (Fig. 2B) was generated by PCR amplification of the antR 5′ UTR, from the +1 transcriptional site to 15 nt downstream of the translational start site, using oligonucleotides antR-UTR.for and antR-UTR.rev (see Table S3 in the supplemental material). The PCR product was subsequently cloned into PCR2.1 and confirmed by sequencing. The EcoRI-HindIII-digested fragment of the antR 5′ UTR was ligated into Mini-CTX-Plac-lacZY-SD digested with the same enzymes to generate Mini-CTX-Plac-UTRantR-′lacZY-SD. The antR transcriptional fusion was generated by PCR amplification of the antR promoter (from 67 nt upstream of the transcriptional start site to the transcriptional start site) using oligonucleotides antR-promoter-EcoRI-for and antR-promoter-HindIII-rev (see Table S3 in the supplemental material) and PAO1 chromosomal DNA as the template. The PCR product was subsequently cloned into PCR2.1 and confirmed by sequencing. The fragment was digested with EcoRI-HindIII and ligated into Mini-CTX1-lacZY digested with the same enzymes to generate the Mini-CTX1-PantR′-lacYZ plasmid. All the reporter constructs were integrated at the att site of the PAO1, ΔprrF1, ΔprrF2, and ΔprrF1,2 chromosomes as previously described (68).
β-Galactosidase activity.
β-Galactosidase activity was measured as described previously (69). Briefly, the absorbances (A600) of cultures were determined, and cells were harvested by centrifugation and resuspended in potassium phosphate buffer, followed by 1:10 dilution in Z buffer. Potassium phosphate and Z buffer were prepared as previously described (69). The cells were lysed using chloroform and 0.1% sodium dodecyl sulfate (SDS). ONPG (o-nitrophenyl-β-d-galactopyranoside) substrate was added to the solution, and the reaction was stopped with sodium carbonate after 10 to 40 min, when a clear color change was observed. The reaction mixtures were briefly centrifuged, and the absorbance (A420) of the supernatant was determined. The β-galactosidase activity was calculated in Miller units: (1,000 × A420)/(time [minutes] × volume [milliliters] × A600).
Hfq purification.
The P. aeruginosa Hfq protein was purified using the Impact protein purification system (NEB, Ipswitch, MA) with an N-terminal intein tag from plasmid pTYB21. Overnight cultures of Rosetta 2(DE3) cells (NEB) carrying the pTYB21 vector with the hfq allele, previously cloned into the multiple-cloning site (MCS) (34), were diluted 1:100 in 1 liter of LB medium containing ampicillin and chloramphenicol and grown to mid-logarithmic phase (optical density [OD], ∼0.5) at 37°C with shaking. P. aeruginosa Hfq protein expression was then induced by addition of 1 mM IPTG (isopropyl β-d-1-thiogalactopyranoside), and cultures were grown overnight at 18°C. Cells were harvested by centrifugation at 5,000 rpm for 10 min at 4°C. The cell pellets were resuspended in 40 ml 50 mM Tris-HCl, pH 8.5, 1 M NaCl, 1 mM EDTA. The homogeneous solution was treated with 1 ml protease inhibitor cocktail (Sigma) and lysed by sonication. DNase I and RNase A were added to the sonicated solution, which was then placed on ice for 1 h. The lysates were cleared by centrifugation at 17,600 × g for 30 min and run through a column (10-ml bed volume) containing the chitin-binding domain (NEB; S6651). To cleave the intein tag, the column was quickly flushed with three bed volumes of cleavage buffer (50 mM Tris-HCl, pH 8.5, 1 M NaCl, 1 mM EDTA, 50 mM dithiothreitol [DTT]), column flow was stopped, and the column was incubated overnight at 4°C. The P. aeruginosa Hfq protein was eluted from the column by adding an additional three bed volumes of cleavage buffer to the column. The P. aeruginosa Hfq protein was further cleared of nucleic acid contaminants using a cation exchange column (UNO-S6; Bio-Rad) as described previously (35). After extensive dialysis in storage buffer (50 mM Tris-HCl, pH 7.5, 250 mM NH4Cl, 1 mM EDTA, 10% glycerol), small aliquots of P. aeruginosa Hfq were flash frozen and stored at −80°C. The purity of the protein was determined based on SDS-PAGE and the ratio of absorbance at 260 nm to 280 nm being less than 0.7.
RNase If footprinting.
5′-32P-PrrF1 (0.1 μM) and PrrF1-Hfq complexes (8 μl) containing 0.5 μM PrrF1 and 0.17 μM to 0.67 μM P. aeruginosa Hfq, E. coli Hfq, or E. coli Hfq-R16A were prepared as described previously (53) and incubated for 30 min at 30°C. Samples were treated with 2 μl 1-U/μl RNase If for 1 min at 37°C, and 10 μl buffered phenol was added to stop the reaction. After extraction with phenol and chloroform and precipitation with ethanol, RNA was dissolved in 8 μl formamide loading dye (90% [vol/vol] formamide, 1× Tris-borate-EDTA [TBE], 0.1% [wt/vol] bromophenol blue, 0.1% [wt/vol] xylene cyanol) and subsequently loaded on an 8% polyacrylamide sequencing gel. Sequence ladders were obtained by nuclease digestion under denaturing conditions (70). Band intensities were integrated with SAFA (71) and normalized to bands with constant intensity in different experiments. PrrF2 samples were prepared and analyzed using the same protocol. Complexes with an antR mRNA fragment (nt −98 to +42 from the translational start site) were prepared as described above and treated with 2 μl 0.5-U/μl RNase If (0.75 U/μl for samples containing PrrF1) for 1 min at 37°C. Samples were processed and analyzed as described above.
Native gel mobility shift assays for PrrF sRNAs and antR mRNA.
PrrF1 and PrrF2 sRNAs and antR mRNAs were transcribed in vitro with T7 RNA polymerase (RNAP) from a PCR template. The RNA sequences are shown in Fig. 2 and Fig. S1 in the supplemental material. Binding affinities of P. aeruginosa Hfq variants with 32P-labeled RNAs at 30°C were measured by native gel mobility shift assays as previously described (52). The fraction of RNA in complex with one or two Hfq hexamers, R-H I or R-H II, were fit to a partition function for two nonidentical independent sites:
| (1) |
| (2) |
in which Kd1 and Kd2 are the dissociation constants for single and multiple Hfq hexamer binding to the RNA and n is the Hill coefficient.
The equilibrium association between 5 nM 32P-antR mRNA and unlabeled PrrF sRNAs in the absence or presence of P. aeruginosa Hfq was measured at 30°C as described previously (72). The fraction of antR mRNA-PrrF complexes as a function of the PrrF concentration was fit to a quadratic equation:
| (3) |
where KDR is the dissociation constant for the antR mRNA-PrrF complex. To study the effects of Hfq on the stability of antR mRNA-PrrF complexes, different concentrations of P. aeruginosa Hfq were added to a preformed complex of 5 nM 32P-antR mRNA and 10 nM PrrF1 and incubated at 30°C for 1 h before loading the samples on a native 6% polyacrylamide gel. The fraction of antR mRNA-PrrF complexes as a function of the Hfq concentration was empirically fit to a modified binding equation:
| (4) |
in which A0 is the fraction of antR mRNA-PrrF complex without Hfq; A1, A2, and A3 are the changes in the fraction of antR mRNA-PrrF complex; and n1, n2, and n3 are the Hill coefficients at different Hfq concentrations.
The association kinetics of 5 nM 32P-labeled antR mRNA with 100 nM PrrF sRNAs in the absence or presence of Hfq was measured at 30°C by native gel mobility shift assay as described previously (52, 53, 72). The appearances of antR mRNA-PrrF duplex and antR mRNA-Hfq-sRNA ternary complex over time were fit to a pseudo-first-order biphasic rate equation:
| (5) |
Supplementary Material
ACKNOWLEDGMENTS
We thank Cassandra Nelson for careful reading of the manuscript and Jace Jones for technical assistance with the mass spectrometry analysis.
This work was supported by NIH grants R01 AI123320 (to A.G.O.-S., S.A.W., and M.A.K.) and R01 GM120425 (to S.A.W.) and a University of Maryland School of Pharmacy Merit Award Fellowship (to L.D.).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00704-17.
REFERENCES
- 1.Pollack M. 1983. The role of exotoxin A in Pseudomonas disease and immunity. Rev Infect Dis 5(Suppl 5):S979–S984. doi: 10.1093/clinids/5.Supplement_5.S979. [DOI] [PubMed] [Google Scholar]
- 2.Vasil ML, Graham LM, Ostroff RM, Shortridge VD, Vasil AI. 1991. Phospholipase C: molecular biology and contribution to the pathogenesis of Pseudomonas aeruginosa. Antibiot Chemother 44:34–47. doi: 10.1159/000420295. [DOI] [PubMed] [Google Scholar]
- 3.Ohman DE, Burns RP, Iglewski BH. 1980. Corneal infections in mice with toxin A and elastase mutants of Pseudomonas aeruginosa. J Infect Dis 142:547–555. doi: 10.1093/infdis/142.4.547. [DOI] [PubMed] [Google Scholar]
- 4.Brint JM, Ohman DE. 1995. Synthesis of multiple exoproducts in Pseudomonas aeruginosa is under the control of RhlR-RhlI, another set of regulators in strain PAO1 with homology to the autoinducer-responsive LuxR-LuxI family. J Bacteriol 177:7155–7163. doi: 10.1128/jb.177.24.7155-7163.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latifi A, Winson MK, Foglino M, Bycroft BW, Stewart GS, Lazdunski A, Williams P. 1995. Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol Microbiol 17:333–343. doi: 10.1111/j.1365-2958.1995.mmi_17020333.x. [DOI] [PubMed] [Google Scholar]
- 6.Collier DN, Anderson L, McKnight SL, Noah TL, Knowles M, Boucher R, Schwab U, Gilligan P, Pesci EC. 2002. A bacterial cell to cell signal in the lungs of cystic fibrosis patients. FEMS Microbiol Lett 215:41–46. doi: 10.1111/j.1574-6968.2002.tb11367.x. [DOI] [PubMed] [Google Scholar]
- 7.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, Facchini M, Pasquali P, Bragonzi A, Visca P. 2016. Role of iron uptake systems in Pseudomonas aeruginosa virulence and airway infection. Infect Immun 84:2324–2335. doi: 10.1128/IAI.00098-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hood MI, Skaar EP. 2012. Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10:525–537. doi: 10.1038/nrmicro2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meyer JM, Neely A, Stintzi A, Georges C, Holder IA. 1996. Pyoverdine is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takase H, Nitanai H, Hoshino K, Otani T. 2000. Requirement of the Pseudomonas aeruginosa tonB gene for high-affinity iron acquisition and infection. Infect Immun 68:4498–4504. doi: 10.1128/IAI.68.8.4498-4504.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takase H, Nitanai H, Hoshino K, Otani T. 2000. Impact of siderophore production on Pseudomonas aeruginosa infections in immunosuppressed mice. Infect Immun 68:1834–1839. doi: 10.1128/IAI.68.4.1834-1839.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinhart AA, Oglesby-Sherrouse AG. 2016. Regulation of Pseudomonas aeruginosa virulence by distinct iron sources. Genes (Basel) 7:E126. doi: 10.3390/genes7120126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiens JR, Vasil AI, Schurr MJ, Vasil ML. 2014. Iron-regulated expression of alginate production, mucoid phenotype, and biofilm formation by Pseudomonas aeruginosa. mBio 5:e01010-. doi: 10.1128/mBio.01010-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oglesby AG, Farrow JM III, Lee JH, Tomaras AP, Greenberg EP, Pesci EC, Vasil ML. 2008. The influence of iron on Pseudomonas aeruginosa physiology: a regulatory link between iron and quorum sensing. J Biol Chem 283:15558–15567. doi: 10.1074/jbc.M707840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banin E, Vasil ML, Greenberg EP. 2005. Iron and Pseudomonas aeruginosa biofilm formation. Proc Natl Acad Sci U S A 102:11076–11081. doi: 10.1073/pnas.0504266102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. 2002. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 99:7072–7077. doi: 10.1073/pnas.092016999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilderman PJ, Vasil AI, Johnson Z, Wilson MJ, Cunliffe HE, Lamont IL, Vasil ML. 2001. Characterization of an endoprotease (PrpL) encoded by a PvdS-regulated gene in Pseudomonas aeruginosa. Infect Immun 69:5385–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhart AA, Nguyen AT, Brewer LK, Bevere J, Jones JW, Kane MA, Damron FH, Barbier M, Oglesby-Sherrouse AG. 2017. The Pseudomonas aeruginosa PrrF small RNAs regulate iron homeostasis during acute murine lung infection. Infect Immun 85:e00764-. doi: 10.1128/IAI.00764-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinhart AA, Powell DA, Nguyen AT, O'Neill M, Djapgne L, Wilks A, Ernst RK, Oglesby-Sherrouse AG. 2015. The prrF-encoded small regulatory RNAs are required for iron homeostasis and virulence of Pseudomonas aeruginosa. Infect Immun 83:863–875. doi: 10.1128/IAI.02707-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FS. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilderman PJ, Sowa NA, FitzGerald DJ, FitzGerald PC, Gottesman S, Ochsner UA, Vasil ML. 2004. Identification of tandem duplicate regulatory small RNAs in Pseudomonas aeruginosa involved in iron homeostasis. Proc Natl Acad Sci U S A 101:9792–9797. doi: 10.1073/pnas.0403423101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim K, Kim YU, Koh BH, Hwang SS, Kim SH, Lepine F, Cho YH, Lee GR. 2010. HHQ and PQS, two Pseudomonas aeruginosa quorum-sensing molecules, down-regulate the innate immune responses through the nuclear factor-kappaB pathway. Immunology 129:578–588. doi: 10.1111/j.1365-2567.2009.03160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deziel E, Lepine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. 2004. Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A 101:1339–1344. doi: 10.1073/pnas.0307694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Calfee MW, Coleman JP, Pesci EC. 2001. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lemma E, Simon J, Schagger H, Kroger A. 1995. Properties of the menaquinol oxidase (Qox) and of qox deletion mutants of Bacillus subtilis. Arch Microbiol 163:432–438. doi: 10.1007/BF00272132. [DOI] [PubMed] [Google Scholar]
- 26.Tynecka Z, Malm A. 1985. The effect of DCCD, HQNO or Cd2+ on glutamate oxidation in Staphylococcus aureus. Ann Univ Mariae Curie Sklodowska Med 40:149–155. [PubMed] [Google Scholar]
- 27.Van Ark G, Berden JA. 1977. Binding of HQNO to beef-heart sub-mitochondrial particles. Biochim Biophys Acta 459:119–127. doi: 10.1016/0005-2728(77)90014-7. [DOI] [PubMed] [Google Scholar]
- 28.Farrow JM III, Pesci EC. 2007. Two distinct pathways supply anthranilate as a precursor of the Pseudomonas quinolone signal. J Bacteriol 189:3425–3433. doi: 10.1128/JB.00209-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soper T, Mandin P, Majdalani N, Gottesman S, Woodson SA. 2010. Positive regulation by small RNAs and the role of Hfq. Proc Natl Acad Sci U S A 107:9602–9607. doi: 10.1073/pnas.1004435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dimastrogiovanni D, Frohlich KS, Bandyra KJ, Bruce HA, Hohensee S, Vogel J, Luisi BF. 2014. Recognition of the small regulatory RNA RydC by the bacterial Hfq protein. Elife 2014:3. doi: 10.7554/eLife.05375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottesman S, McCullen CA, Guillier M, Vanderpool CK, Majdalani N, Benhammou J, Thompson KM, FitzGerald PC, Sowa NA, FitzGerald DJ. 2006. Small RNA regulators and the bacterial response to stress. Cold Spring Harbor Symp Quant Biol 71:1–11. doi: 10.1101/sqb.2006.71.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Masse E, Escorcia FE, Gottesman S. 2003. Coupled degradation of a small regulatory RNA and its mRNA targets in Escherichia coli. Genes Dev 17:2374–2383. doi: 10.1101/gad.1127103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sonnleitner E, Prindl K, Blasi U. 2017. The Pseudomonas aeruginosa CrcZ RNA interferes with Hfq-mediated riboregulation. PLoS One 12:e0180887. doi: 10.1371/journal.pone.0180887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osborne J, Djapgne L, Tran BQ, Goo YA, Oglesby-Sherrouse AG. 2014. A method for in vivo identification of bacterial small RNA-binding proteins. Microbiologyopen 3:950–960. doi: 10.1002/mbo3.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peng Y, Soper TJ, Woodson SA. 2014. Positional effects of AAN motifs in rpoS regulation by sRNAs and Hfq. J Mol Biol 426:275–285. doi: 10.1016/j.jmb.2013.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Panja S, Schu DJ, Woodson SA. 2013. Conserved arginines on the rim of Hfq catalyze base pair formation and exchange. Nucleic Acids Res 41:7536–7546. doi: 10.1093/nar/gkt521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen JS, Lei LK, Ebersbach T, Olsen AS, Klitgaard JK, Valentin-Hansen P, Kallipolitis BH. 2010. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res 38:907–919. doi: 10.1093/nar/gkp1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Metruccio MM, Fantappie L, Serruto D, Muzzi A, Roncarati D, Donati C, Scarlato V, Delany I. 2009. The Hfq-dependent small noncoding RNA NrrF directly mediates Fur-dependent positive regulation of succinate dehydrogenase in Neisseria meningitidis. J Bacteriol 191:1330–1342. doi: 10.1128/JB.00849-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Papenfort K, Said N, Welsink T, Lucchini S, Hinton JC, Vogel J. 2009. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol Microbiol 74:139–158. doi: 10.1111/j.1365-2958.2009.06857.x. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen AT, Jones JW, Camara M, Williams P, Kane MA, Oglesby-Sherrouse AG. 2016. Cystic fibrosis isolates of Pseudomonas aeruginosa retain iron-regulated antimicrobial activity against Staphylococcus aureus through the action of multiple alkylquinolones. Front Microbiol 7:1171–1183. doi: 10.3389/fmicb.2016.01171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nguyen AT, Jones JW, Ruge MA, Kane MA, Oglesby-Sherrouse AG. 2015. Iron depletion enhances production of antimicrobials by Pseudomonas aeruginosa. J Bacteriol 197:2265–2275. doi: 10.1128/JB.00072-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nguyen AT, O'Neill MJ, Watts AM, Robson CL, Lamont IL, Wilks A, Oglesby-Sherrouse AG. 2014. Adaptation of iron homeostasis pathways by a Pseudomonas aeruginosa pyoverdine mutant in the cystic fibrosis lung. J Bacteriol 196:2265–2276. doi: 10.1128/JB.01491-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lepine F, Milot S, Deziel E, He J, Rahme LG. 2004. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J Am Soc Mass Spectrom 15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 44.Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S. 2012. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog 8:e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morita T, Nishino R, Aiba H. 2017. Role of the terminator hairpin in the biogenesis of functional Hfq-binding sRNAs. RNA 23:1419–1431. doi: 10.1261/rna.060756.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sauer E, Weichenrieder O. 2011. Structural basis for RNA 3′-end recognition by Hfq. Proc Natl Acad Sci U S A 108:13065–13070. doi: 10.1073/pnas.1103420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sauer E, Schmidt S, Weichenrieder O. 2012. Small RNA binding to the lateral surface of Hfq hexamers and structural rearrangements upon mRNA target recognition. Proc Natl Acad Sci U S A 109:9396–9401. doi: 10.1073/pnas.1202521109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang A, Schu DJ, Tjaden BC, Storz G, Gottesman S. 2013. Mutations in interaction surfaces differentially impact E. coli Hfq association with small RNAs and their mRNA targets. J Mol Biol 425:3678–3697. doi: 10.1016/j.jmb.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santiago-Frangos A, Kavita K, Schu DJ, Gottesman S, Woodson SA. 2016. C-terminal domain of the RNA chaperone Hfq drives sRNA competition and release of target RNA. Proc Natl Acad Sci U S A 113:E6089–E6096. doi: 10.1073/pnas.1613053113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sonnleitner E, Blasi U. 2014. Regulation of Hfq by the RNA CrcZ in Pseudomonas aeruginosa carbon catabolite repression. PLoS Genet 10:e1004440. doi: 10.1371/journal.pgen.1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sonnleitner E, Abdou L, Haas D. 2009. Small RNA as global regulator of carbon catabolite repression in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 106:21866–21871. doi: 10.1073/pnas.0910308106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lease RA, Woodson SA. 2004. Cycling of the Sm-like protein Hfq on the DsrA small regulatory RNA. J Mol Biol 344:1211–1223. doi: 10.1016/j.jmb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 53.Zheng A, Panja S, Woodson SA. 2016. Arginine patch predicts the RNA annealing activity of Hfq from Gram-negative and Gram-positive bacteria. J Mol Biol 428:2259–2264. doi: 10.1016/j.jmb.2016.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Filkins LM, Graber JA, Olson DG, Dolben EL, Lynd LR, Bhuju S, O'Toole GA. 2015. Coculture of Staphylococcus aureus with Pseudomonas aeruginosa drives S. aureus towards fermentative metabolism and reduced viability in a cystic fibrosis model. J Bacteriol 197:2252–2264. doi: 10.1128/JB.00059-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Korgaonkar A, Trivedi U, Rumbaugh KP, Whiteley M. 2013. Community surveillance enhances Pseudomonas aeruginosa virulence during polymicrobial infection. Proc Natl Acad Sci U S A 110:1059–1064. doi: 10.1073/pnas.1214550110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hazan R, Que YA, Maura D, Strobel B, Majcherczyk PA, Hopper LR, Wilbur DJ, Hreha TN, Barquera B, Rahme LG. 2016. Auto poisoning of the respiratory chain by a quorum-sensing-regulated molecule favors biofilm formation and antibiotic tolerance. Curr Biol 26:195–206. doi: 10.1016/j.cub.2015.11.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng Y, Curtis JE, Fang X, Woodson SA. 2014. Structural model of an mRNA in complex with the bacterial chaperone Hfq. Proc Natl Acad Sci U S A 111:17134–17139. doi: 10.1073/pnas.1410114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Herrmann KM, Weaver LM. 1999. The shikimate pathway. Annu Rev Plant Physiol Plant Mol Biol 50:473–503. doi: 10.1146/annurev.arplant.50.1.473. [DOI] [PubMed] [Google Scholar]
- 59.Palmer GC, Jorth PA, Whiteley M. 2013. The role of two Pseudomonas aeruginosa anthranilate synthases in tryptophan and quorum signal production. Microbiology 159:959–969. doi: 10.1099/mic.0.063065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eby DM, Beharry ZM, Coulter ED, Kurtz DM Jr, Neidle EL. 2001. Characterization and evolution of anthranilate 1,2-dioxygenase from Acinetobacter sp. strain ADP1. J Bacteriol 183:109–118. doi: 10.1128/JB.183-1.109-118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urata M, Miyakoshi M, Kai S, Maeda K, Habe H, Omori T, Yamane H, Nojiri H. 2004. Transcriptional regulation of the ant operon, encoding two-component anthranilate 1,2-dioxygenase, on the carbazole-degradative plasmid pCAR1 of Pseudomonas resinovorans strain CA10. J Bacteriol 186:6815–6823. doi: 10.1128/JB.186.20.6815-6823.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim SK, Im SJ, Yeom DH, Lee JH. 2012. AntR-mediated bidirectional activation of antA and antR, anthranilate degradative genes in Pseudomonas aeruginosa. Gene 505:146–152. doi: 10.1016/j.gene.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 63.Miyakoshi M, Shintani M, Terabayashi T, Kai S, Yamane H, Nojiri H. 2007. Transcriptome analysis of Pseudomonas putida KT2440 harboring the completely sequenced IncP-7 plasmid pCAR1. J Bacteriol 189:6849–6860. doi: 10.1128/JB.00684-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dulcey CE, Dekimpe V, Fauvelle DA, Milot S, Groleau MC, Doucet N, Rahme LG, Lepine F, Deziel E. 2013. The end of an old hypothesis: the pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem Biol 20:1481–1491. doi: 10.1016/j.chembiol.2013.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Little AS, Okkotsu Y, Reinhart AA, Damron FH, Barbier M, Barrett B, Oglesby-Sherrouse AG, Goldberg JB, Cody WL, Schurr MJ, Vasil ML, Schurr MJ. 2018. Pseudomonas aeruginosa AlgR phosphorylation status differentially regulates pyocyanin and pyoverdine production. mBio 30:e02318-. doi: 10.1128/mBio.02318-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oglesby-Sherrouse AG, Vasil ML. 2010. Characterization of a heme-regulated non-coding RNA encoded by the prrF locus of Pseudomonas aeruginosa. PLoS One 5:e9930. doi: 10.1371/journal.pone.0009930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 68.Hoang TT, Kutchma AJ, Becher A, Schweizer HP. 2000. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid 43:59–72. doi: 10.1006/plas.1999.1441. [DOI] [PubMed] [Google Scholar]
- 69.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 70.Donis-Keller H, Maxam AM, Gilbert W. 1977. Mapping adenines, guanines, and pyrimidines in RNA. Nucleic Acids Res 4:2527–2538. doi: 10.1093/nar/4.8.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. 2005. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. RNA 11:344–354. doi: 10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soper TJ, Woodson SA. 2008. The rpoS mRNA leader recruits Hfq to facilitate annealing with DsrA sRNA. RNA 14:1907–1917. doi: 10.1261/rna.1110608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuker M. 2003. Mfold Web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.