

Summary
The BCL-2 family proteins are central regulators of apoptosis. However, cells deficient for BAX and BAK or overexpressing BCL-2 still succumb to oxidative stress upon DNA damage or matrix detachment. Here, we show that ΔNp63α overexpression protects cells from oxidative stress induced by oxidants, DNA damage, anoikis, or ferroptosis-inducing agents. Conversely, ΔNp63α deficiency increases oxidative stress. Mechanistically, ΔNp63α orchestrates redox homeostasis through transcriptional control of glutathione biogenesis, utilization, and regeneration. Analysis of The Cancer Genome Atlas (TCGA) lung squamous cell carcinoma dataset reveals that TP63 amplification/overexpression upregulates the glutathione metabolism pathway in primary human tumors. Strikingly, overexpression of ΔNp63α promotes clonogenic survival of p53-/-Bax-/-Bak-/- cells against DNA damage. Furthermore, co-expression of BCL-2 and ΔNp63α confers clonogenic survival against matrix detachment, disrupts the luminal clearance of mammary acini, and promotes cancer metastasis. Our findings highlight the need for a simultaneous blockade of apoptosis and oxidative stress to promote long-term cellular wellbeing.
Graphical abstract
Apoptosis-defective cells remain vulnerable to oxidative stress that limits long-term survival. Wang et al. identify ΔNp63α as a central regulator of redox homeostasis through transcriptional control of a tightly-coupled glutathione metabolic circuit. ΔNp63α alleviates oxidative stress and cooperates with BCL-2 family to promote both long-term cellular wellbeing and cancer metastasis.
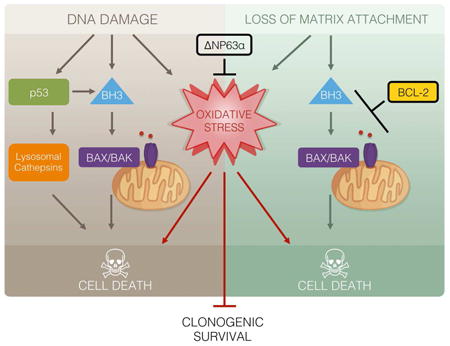
Introduction
Proper execution of cell death ensures normal biological processes, and its deregulation causes human diseases, ranging from cancer to neurodegenerative disorders (Thompson, 1995). The evolutionarily conserved signaling cascade, consisting of the BCL-2 family, the adaptor protein Apaf-1, and the caspase family, outlines the quintessential apoptotic network (Wang, 2001). In response to apoptotic signals, the “activator” BH3-only molecules, including BID, BIM, PUMA, and NOXA, trigger the homo-oligomerization of BAX and BAK to permeabilize mitochondria, leading to the efflux of cytochrome c for caspase activation (Chen et al., 2015; Cheng et al., 2001; Inoue-Yamauchi et al., 2017; Kim et al., 2006; Kim et al., 2009; Ren et al., 2010; Wang, 2001; Wei et al., 2001). Although apoptosis has long been considered as the major cell death mechanism required for the successful development and maintenance of tissue homeostasis in metazoans, double deficiency of Bax and Bak only disrupts the development and homeostasis in restricted sets of tissues (Lindsten et al., 2000), suggesting the existence of BAX/BAK-independent cell death mechanism(s) in maintaining tissue homeostatic state.
In exploring this cell death conundrum, we discovered that apoptosis-deficient Bax-/-Bak-/- double-knockout (DKO) mouse embryonic fibroblasts (MEFs) underwent a regulated form of necrotic cell death in response to DNA damage induced by topoisomerase inhibitors (Tu et al., 2009). Surprisingly, this type of necrotic cell death requires active transcription/translation (Tu et al., 2009). As this DNA damage-induced “programmed necrotic death” (PND) does not require RIP1 and is not blocked by inhibitors of RIP1 and RIP3, it is distinct from TNF-induced “necroptosis” (Pasparakis and Vandenabeele, 2015; Sun and Wang, 2014; Yuan and Kroemer, 2010) (Figure S1). Furthermore, we have reported that this death is independent of caspases, mitochondrial permeability transition pore (PTP), autophagy, or poly (ADP-ribose) polymerase (PARP) (Tu et al., 2009). Notably, DNA alkylation induces PARP-dependent necrotic death whereas double-strand DNA breaks induce PARP-independent necrotic death in DKO cells (Tu et al., 2009; Zong et al., 2004). Mechanistically, we have delineated a p53-cathepsin axis that cooperates with ROS (reactive oxygen species) to activate PND in DKO cells undergoing double-strand DNA breaks (Tu et al., 2009).
Similar to DNA damage-induced cell death, it was reported that inhibition of apoptosis by BCL-2 is insufficient to provide long-term clonogenic survival against anoikis (Schafer et al., 2009), a form of cell death that is induced by detachment from extracellular matrix in anchorage-dependent cells. Interestingly, antioxidant Trolox cooperates with BCL-2 to enhance clonogenic survival and prevent the luminal clearance of acini in three-dimensional (3D) culture of mammary epithelial cells (Schafer et al., 2009). Hence, although apoptosis is the fastest mechanism for eliminating cells upon death stimuli, inhibition of apoptosis is insufficient to confer long-term clonogenic survival that is required to prevent the pathological loss of cells during disease processes. ROS appears to play a critical role in abrogating long-term clonogenic survival. The importance of ROS in regulating cell death is further exemplified by the recent characterization of ferroptosis, an iron-dependent, oxidative form of PND that is triggered by the depletion of intracellular glutathione or inhibition of GPX4, leading to accumulation of lipid hydroperoxides (Conrad et al., 2016). Of note, ferroptosis is not involved in DNA damage-induced death of Bax-/-Bak-/- cells because ferrostatin-1, an inhibitor of ferroptosis, failed to protect DKO cells from etoposide (Figure S1). In contrast, iron chelators protected Bax-/-Bak-/- cells from etoposide-induced death (Figure S1). In this study, we sought to identify a master regulator of ROS and determine whether the identified guardian of oxidative stress can cooperate with the gatekeepers of mitochondrial apoptosis, i.e. BCL-2 family proteins, to promote clonogenic survival against intrinsic cell death signals.
Here, we demonstrate that long-term clonogenic survival of apoptosis-defective cells in response to DNA damage or loss of matrix attachment could be achieved through overexpression of the putative oncogene ΔNp63α, the major isoform of p63. ΔNp63α increases cellular resistance against oxidative stress and ferroptosis by regulating the synthesis, utilization, and regeneration of glutathione, a central component of the cellular antioxidant defense system. The regulation of the glutathione metabolic pathway by ΔNp63α was further demonstrated in human squamous cell lung cancer in which TP63 is frequently amplified. Amplification of TP63 appears to serve as a mechanism by which squamous cell lung cancer evades oxidative stress and promotes tumorigenesis. In summary, simultaneous inhibition of mitochondrion-dependent apoptosis and ROS-induced cell death can confer long-term cellular survival, a strategy likely hijacked by cancer.
Results
ΔNp63α confers clonogenic survival of p53-/-Bax-/-Bak-/- TKO cells against DNA damage through inhibition of oxidative stress
Our prior studies of Bax-/-Bak-/- cells showed that genotoxic stress-induced, ROS-dependent necrotic death requires active transcription (Tu et al., 2009), suggesting that genotoxic stress-induced ROS can be regulated by specific transcription factor(s). To search for such candidates, we examined several transcription factors known to regulate cell death or the DNA damage response, which led to the discovery of ΔNp63α as a key transcriptional regulator of ROS. Overexpression of ΔNp63α prevented etoposide-induced ROS accumulation in transformed Bax-/-Bak-/- MEFs (Figure 1A). Three ROS-sensitive dyes were employed to interrogate intracellular ROS levels—H2DCFDA, a broad-spectrum ROS indicator; dihydroethidium (DHE), a specific indicator for superoxide; and MitoSOX Red, a specific indicator for mitochondrial matrix superoxide. Overexpression of ΔNp63α also reduced etoposide-induced lipid peroxidation (Figure 1B). Consistent with our previous characterization of p53 and ROS as two independent effectors in executing necrotic death (Tu et al., 2009), overexpression of a dominant negative mutant of p53 (p53 DN) did not prevent ROS induced by genotoxic stress (Figure 1A). Furthermore, overexpression of either ΔNp63α or p53 DN provided only a short-term survival advantage against etoposide (Figure 1C), whereas co-expression of both ΔNp63α and p53 DN enhanced clonogenic survival of DKO cells (Figure 1D). Of note, we have previously shown that transformation of MEFs by SV40 genome does not inactivate p53 (Tu et al., 2009).
Figure 1. ΔNp63α alleviates DNA damage-induced oxidative stress and enhances clonogenic survival of transformed p53-/-Bax-/-Bak-/- TKO MEFs.
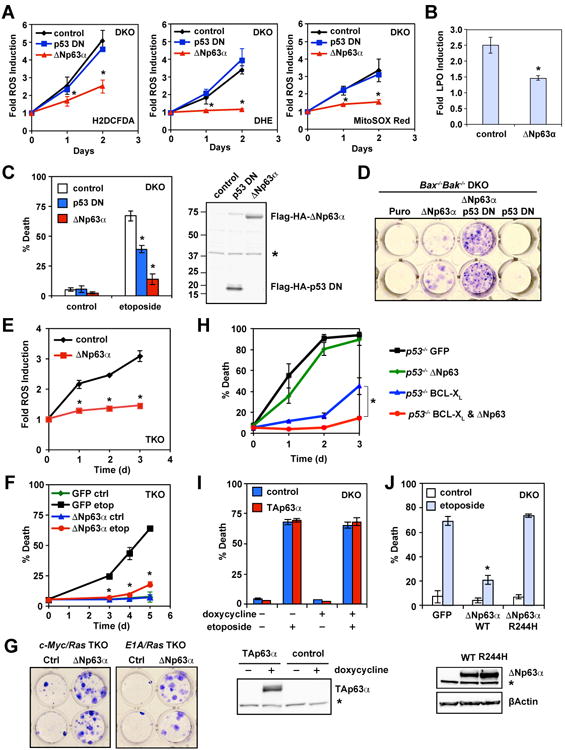
(A) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were untreated or treated with etoposide (10 μg/ml). Oxidation of the ROS-sensitive dyes H2DCFDA, DHE, or MitoSOX Red was quantified by flow cytometry. Data shown are fold increase of ROS after etoposide treatment (mean ± s.d., n = 3). (B) Cells as described in (A) were subjected to staining using the lipid peroxidation probe C11-BODIPY665/676. Oxidation of C11-BODIPY was quantified by flow cytometry. Data shown are fold increase of lipid peroxidation (LPO) after etoposide treatment (mean ± s.d., n = 3). (C) Cell death was quantified by propidium iodide staining at 3 days after etoposide treatment (mean ± s.d., n = 3). The expression of N-terminal Flag-HA-tagged ΔNp63α and dominant negative p53 was assessed by an anti-Flag immunoblot. Asterisk denotes a cross-reactive band serving as a loading control. (D) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were treated with etoposide for 14 hours. Colonies were stained with crystal violet after 12 days. The data shown are representative of three independent experiments. (E) SV40-transformed p53-/-Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were untreated or treated with etoposide. Oxidation of the ROS-sensitive dye DHE was quantified by flow cytometry. Data shown are fold increase of ROS after etoposide treatment (mean ± s.d., n = 3). (F) Cell death was quantified by propidium iodide staining at the indicated times after etoposide treatment (mean ± s.d., n = 3). (G) c-Myc/Ras or E1A/Ras transformed p53-/-Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were treated with etoposide for 9 hours. Colonies were stained with crystal violet after 10 days. (H) SV40-transformed p53-/- MEFs transduced with the indicated retrovirus were untreated or treated with etoposide. Cell death was quantified by propidium iodide staining at the indicated times (mean ± s.d., n = 3). (I) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were untreated or treated with doxycycline for 2 days, followed by etoposide treatment for 3 days. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). The expression of TAp63α was assessed by an anti-p63 immunoblot. Asterisk denotes a cross-reactive band serving as a loading control. (J) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were untreated or treated with etoposide. Cell death was quantified by propidium iodide staining at 3 days after etoposide treatment (mean ± s.d., n = 3). The expression of WT or mutant ΔNp63α was assessed by an anti-p63 immunoblot. Asterisk denotes a cross-reactive band serving as a loading control. * P < 0.05 (Student's t-test).
To evaluate whether ΔNp63α regulates ROS independently of p53, p53-/-Bax-/-Bak-/- triple-knockout (TKO) MEFs were generated. Although transformed p53-/-Bax-/-Bak-/- TKO MEFs were more resistant to DNA damage than Bax-/-Bak-/- DKO MEFs (Figure S2), TKO cells were eventually killed by DNA damage due to oxidative stress that could be alleviated by antioxidants N-acetyl-L-cysteine (NAC) and diphenyleneiodonium (DPI) (Figure S2). Importantly, overexpression of ΔNp63α protected TKO from etoposide-induced ROS accumulation and cell death (Figures 1E, 1F, and S2). Furthermore, inhibition of oxidative stress by ΔNp63α conferred clonogenic survival of TKO cells against DNA damage (Figure 1G). Because BCL-XL amplification/overexpression and p53 mutations can co-occur in human cancers, we next examined whether ΔNp63α can inhibit etoposide-induced death in BCL-XL-overexpressing p53-deficient MEFs that resemble p53-/-Bax-/-Bak-/- TKO MEFs. Indeed, overexpression of both BCL-XL and ΔNp63α but not alone completely blocked etoposide-induced death of p53-/- MEFs (Figure 1H). Notably, overexpression of BCL-XL provided more protection than ΔNp63α because BIM-mediated activation of BAX/BAK occurs in p53-deficient cells.
It was reported that another p63 isoform, TAp63, is induced by DNA damage in MEFs (Flores et al., 2002) and deficiency of TAp63 but not p53 protects oocytes from ionizing irradiation (Suh et al., 2006). Of note, MEFs only express TAp63 but not ΔNp63 as determined by qRT-PCR (Figure S3). However, TAp63 protein was below the immunoblot detection limit. Knockdown of TAp63 in DKO or TKO MEFs had no effect on etoposide-induced cell death (Figure S3), indicating that endogenous TAp63 is not involved in DNA damage-induced necrotic death and ΔNp63α does not inhibit oxidative stress through antagonizing endogenous TAp63 in MEFs. Consistent with the reported pro-death activity of TAp63α (Suh et al., 2006), we were unable to stably express TAp63α in DKO cells. However, when the expression of TAp63α was induced in DKO cells using a lentiviral tetracycline-inducible system, it neither enhanced nor blocked DNA damage-induced PND in DKO cells (Figure 1I). Moreover, expression of a human EEC syndrome (ectrodactyly, ectodermal dysplasia, and cleft lip/palate)-derived R244H ΔNp63α mutant (Dotsch et al., 2010; van Bokhoven et al., 2001) that loses DNA binding activity failed to protect DKO cells from DNA damage-induce cell death (Figure 1J). These results indicate that binding to DNA but not p53 family members is critical for ΔNp63α to prevent DNA damage-induced necrotic death.
ΔNp63α orchestrates redox homeostasis through transcriptional control of glutathione metabolism genes
We envisioned that if ΔNp63α prevents DNA damage-induced oxidative stress by increasing antioxidant capacity, ΔNp63α could protect cells against exogenous oxidants. Indeed, overexpression of ΔNp63α protected wild-type (WT) MEFs from thiol oxidant diamide and lipid oxidant tert-butyl hydroperoxide (TBH) (Figure 2A). Given that glutathione is the major antioxidant produced by the cell and plays a central role in regulating the intracellular redox state (Meister, 1995; Trachootham et al., 2009), one testable thesis is that ΔNp63α mitigates oxidative stress through transcriptional regulation of glutathione metabolism genes. Glutathione is synthesized in two sequential steps that are catalyzed by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS), respectively (Meister, 1995) (Figure 2B). It exists in both reduced (GSH) and oxidized (GSSG) states (Meister, 1995). GSH participates in the detoxification of hydrogen peroxide and lipid hydroperoxides through various glutathione peroxidases (GPX) (Figure 2B). During this process, GSH is oxidized to GSSG, which can then be recycled back to GSH through the action of glutathione reductase (GSR) at the expense of reduced nicotinamide adenine dinucleotide phosphate (NADPH) (Figure 2B). NADPH can be generated by several enzymes, including isocitrate dehydrogenase 2 (IDH2). Significantly, gene expression profiling revealed that genes involved in the glutathione redox pathway were upregulated in ΔNp63α-overexpressing cells both before and after DNA damage (Figure S4). qRT-PCR confirmed that GCLC (the catalytic subunit of GCL), GSS, IDH2, and GPX2 were upregulated by ΔNp63α (Figure 2C). Hence, it appears that ΔNp63α orchestrates a transcription program to coordinate the biosynthesis of glutathione, the utilization of GSH as an antioxidant, and the regeneration of GSH from GSSG (Figure 2B), the outcome of which would be an increased GSH to GSSG ratio, indicating a more reduced redox state. Indeed, overexpression of ΔNp63α significantly increased the GSH/GSSG ratio in cells both before and after etoposide treatment (Figure 2D). The functional significance of these ΔNp63α-induced glutathione metabolism genes in regulating ROS-induced cell death was interrogated by siRNA-mediated knockdown (Figure S3). Knockdown of GCLC, GSS, GPX2, or IDH2 compromised the ability of ΔNp63α to protect Bax-/-Bak-/- DKO cells against DNA damage to varying extents (Figures 2E-2H). The protective effect of ΔNp63α was also mitigated by the GCLC inhibitor buthionine sulfoximine (BSO) (Figure S4). Of note, knockdown of these genes also enhanced etoposide-induced cell death in control DKO cells, but to a lesser degree (Figures 2E-2H), supporting the involvement of oxidative stress in this form of cell death. Collectively, our data show that ΔNp63α orchestrates a tightly coupled glutathione metabolic circuit to alleviate oxidative stress (Figure 2B), protecting apoptosis-defective cells from ROS-mediated necrotic cell death.
Figure 2. ΔNp63α controls the homeostasis of intracellular redox through transcriptional control of glutathione metabolism genes.

(A) SV40-transformed WT MEFs transduced with the indicated retrovirus were untreated or treated with diamide (100 μM) for 4 hours or tert-butyl hydroperoxide (TBH, 50 μM) for 7 hours. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (B) A schematic of the effect of ΔNp63α on glutathione metabolism. Overexpression of ΔNp63α in cells generates a more reduced intracellular redox state and increases resistance to oxidative damage through a coordinated regulation of glutathione metabolism genes (highlighted in red). Specifically, ΔNp63α upregulates enzymes responsible for (1) glutathione biosynthesis, (2) ROS detoxification by glutathione, and (3) regeneration of GSH from GSSG. (C) SV40-transformed Bax-/-Bak-/- cells transduced with the indicated retrovirus were untreated or treated with etoposide for 6 hours. The mRNA levels of the indicated genes were analyzed by qRT-PCR (mean ± s.d., n = 3). (D) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were untreated or treated with etoposide for 24 hours. Intracellular reduced (GSH) and oxidized (GSSG) glutathione concentrations were determined using an enzyme-cycling assay and normalized against protein concentration. Normalized [GSH] and [GSSG] were used to calculate the GSH/GSSG ratio (mean ± s.d., n = 3). (E-H) SV40-transformed Bax-/-Bak-/- cells stably expressing GFP or ΔNp63α were transfected with scrambled siRNA (siScr) or siRNA against Gclc (E), Gss (F), Gpx2 (G), or Idh2 (H). Cells were subsequently untreated or treated with etoposide for 3 days. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). *, P < 0.05 (Student's t-test).
As the DNA binding activity is required for ΔNp63α to alleviate oxidative stress, we searched for the p63 response element in glutathione metabolism genes (Hoh et al., 2002; Perez et al., 2007). Potential p63 binding sites were identified in the intron 1 of GCLC and GPX2. Direct targeting of ΔNp63α to these sites was demonstrated by chromatin immunoprecipitation (ChIP) assays (Figure 3A), which is consistent with a previous report showing direct regulation of GPX2 by p63 (Yan and Chen, 2006). Notably, overexpression of GCLC or GPX2 alone was insufficient to protect DKO cells from DNA damage-induced PND because neither measure would sustain the regeneration of GSH from GSSG (Figures 3B and 2B). These results highlight the importance of ΔNp63α in orchestrating a tightly coupled glutathione metabolic circuit in maintaining redox homeostasis. As NRF2 (NFE2L2) is the most studied transcription factor that controls the expression of antioxidant genes, including GCLC (Motohashi and Yamamoto, 2004), we investigated whether ΔNp63α regulates GCLC through NRF2. Knockdown of Nrf2 (Nfe2l2) had minimal effect on ΔNp63α-mediated upregulation of GCLC even though it partially reduced GCLC in control cells (Figures 3C and 3D). Consequently, the protective effect of ΔNp63α against DNA damage-induced necrotic death was not affected by NRF2 loss (Figure 3E). Furthermore, the DNA-binding incompetent R244H mutant of ΔNp63α failed to upregulate GCLC (Figure 3F). In summary, these data not only illustrate an NRF2-independent regulation of GCLC by ΔNp63α but also indicate that ΔNp63α is a central regulator of glutathione biogenesis.
Figure 3. ΔNp63α regulates glutathione metabolism and oxidative stress-induced cell death independently of NRF2.
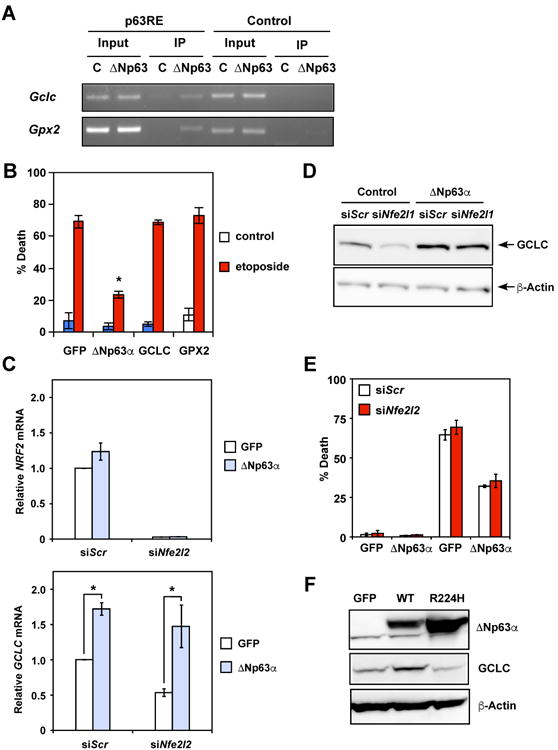
(A) SV40-transformed Bax-/-Bak-/- MEFs transduced with control or ΔNp63α-expressing retrovirus were subjected to chromatin immunoprecipitation using the anti-p63 antibody, followed by PCR amplification of the intron 1 of Gclc and Gpx2, respectively. A 5′ sequence located ∼3-4 kb upstream of the transcription start site of Gclc or Gpx2 served as a negative control. (B) SV40-transformed Bax-/-Bak-/- DKO MEFs transduced with the indicated retrovirus were untreated or treated with etoposide. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (C) SV40-transformed Bax-/-Bak-/- MEFs transduced with retrovirus expressing GFP or ΔNp63α were transfected with scrambled siRNA (siScr) or siRNA against Nrf2 (Nfe2l2). The mRNA levels of Nrf2 (Nfe2l2) and Gclc were analyzed by qRT-PCR (mean ± s.d., n = 3). (D) Protein lysates from cells described in (C) were analyzed by immunoblots using the indicated antibodies. (E) SV40-transformed Bax-/-Bak-/- MEFs stably expressing GFP or ΔNp63α were transfected with scrambled siRNA or siRNA against Nrf2 (Nfe2l2). Cells were subsequently untreated or treated with etoposide for 3 days. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (F) SV40-transformed Bax-/-Bak-/- MEFs transduced with the indicated retrovirus were analyzed by immunoblots using the indicated antibodies. * P < 0.05 (Student's t-test).
Deficiency of ΔNp63α generates a more oxidized intracellular redox state and sensitizes cells to oxidative stress
To investigate the role of endogenous ΔNp63α in regulating redox homeostasis, knockdown of p63 using a validated shRNA that target all p63 isoforms (Godar et al., 2008) was performed in cell lines where ΔNp63α but not TAp63 protein was detected by immunoblots (Figure 4A). Consistent with our gain-of-function studies, knockdown of endogenous ΔNp63α led to increased intracellular ROS in human mammary epithelial cell line MCF-10A and cervical carcinoma cell line ME-180 (Figure 4B). Knockdown of ΔNp63α using miR30-based shRNA or siRNA specific for ΔNp63 also increased ROS (Figure S5). As MCF-10A was reported to express low levels of TAp63 that is below the immunoblot detection limit (Figure 4A), knockdown of either TAp63 or ΔNp63α was performed in MCF-10A using isoform-specific siRNA. Knockdown of ΔNp63α increased ROS whereas knockdown of TAp63 increased ΔNp63 expression and slightly reduced ROS (Figure S5). Together, these results support that ΔNp63 suppresses ROS accumulation. Importantly, qRT-PCR showed that ΔNp63α knockdown downregulated GCLC, GSS, IDH2, and GPX2 (Figure 4C)—the same set of genes that was upregulated by ΔNp63α (Figure 2C). Of note, GSR was suppressed by loss of endogenous ΔNp63α in human cells but not induced by ΔNp63α overexpression in MEFs (Figure 4C and data not shown), which might reflect cell type-specific regulation. Consistent with downregulation of glutathione metabolism genes by ΔNp63α knockdown, the GSH/GSSG ratio was reduced in cells deficient for ΔNp63α (Figure 4D), indicating a more oxidized redox state. Consequently, ΔNp63α knockdown enhanced diamide-induced cell death, which was blocked by NAC (Figure 4E). Furthermore, ΔNp63α knockdown slightly increased baseline cell death over time and sensitized cells to chemotherapeutic agent-induced cell death (Figure S5). Altogether, our loss-of-function studies revealed an important role of endogenous ΔNp63α in maintaining redox homeostasis.
Figure 4. Deficiency of ΔNp63α generates a more oxidized intracellular redox state and sensitizes cells to oxidative stress.
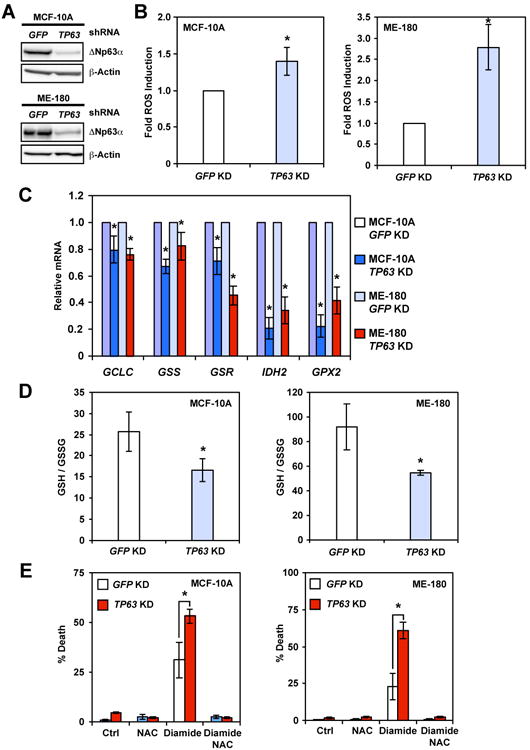
(A) MCF-10A or ME-180 cells transduced with lentivirus expressing shRNA against GFP or TP63 were analyzed by immunoblots using the indicated antibodies. (B) MCF-10A or ME-180 cells transduced with lentivirus expressing shRNA against GFP or TP63 were stained with CM-H2DCFDA or H2DCFDA. Oxidation of the respective dyes was quantified by flow cytometry. Data shown are fold increase of ROS induced by knockdown of p63 (mean ± s.d., n = 3). (C) MCF-10A or ME-180 cells were transduced with lentivirus expressing shRNA against GFP or TP63. The mRNA levels of the indicated genes were analyzed by qRT-PCR (mean ± s.d., n = 3). (D) MCF-10A or ME-180 cells transduced with lentivirus expressing shRNA against GFP or TP63 were analyzed for intracellular reduced (GSH) and oxidized (GSSG) glutathione concentrations using an enzyme-cycling assay. Normalized [GSH] and [GSSG] were used to calculate the GSH/GSSG ratio (mean ± s.d., n = 3). (E) MCF-10A or ME-180 cells transduced with lentivirus expressing shRNA against GFP or TP63 were untreated or treated with oxidant diamide in combination with antioxidant N-acetyl-L-cysteine (NAC) for 6 hours (MCF-10A) or 4 hours (ME-180). Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). *, P < 0.05 (Student's t-test).
ΔNp63α can inhibit ferroptosis independently of p53
Given that ΔNp63α orchestrates glutathione homeostasis and glutathione depletion induces ferroptosis, we envisioned that ΔNp63α might inhibit ferroptosis. As p53 is reported to positively regulate ferroptosis through transcriptional repression of Slc7a11 (Jiang et al., 2015), a central component of the cystine-glutamate antiporter (System Xc-), and ΔNp63α can potentially function as a dominant negative regulator of p53, we compared the effect of ΔNp63α on ferroptosis in both WT and p53-/- MEFs. Consistent with the published results (Jiang et al., 2015), p53 deficiency protected MEFs from ferroptosis induced by erastin but only at early time point (Figures 5A and 5B). Overexpression of ΔNp63α protected both WT and p53-/- MEFs from erastin-induced death, suggesting that the anti-ferroptotic activity of ΔNp63α is independent of p53. Similar results were observed in RSL3-induced ferroptosis (Figure 5C). Although the effect of ΔNp63α on glutathione homeostasis is probably sufficient to inhibit ferroptosis, ΔNp63α may also regulate the expression of Slc7a11 directly or indirectly through p53. Interestingly, ΔNp63α upregulated Slc7a11 expression in p53-/- but not in WT MEFs where p53 suppressed Slc7a11 expression (Figure 5D), suggesting that ΔNp63α is unable to abrogate p53-mediated transcriptional repression of Slc7a11. Together, these findings suggest that the protective effect of ΔNp63α against ferroptosis is probably due to its transcriptional control of glutathione metabolism genes in WT MEFs whereas its regulation of Slc7a11 further contributes to its anti-ferroptotic activity in p53-deficient MEFs. We subsequently performed loss-of-function studies of ΔNp63α in ME-180 cells that are HPV-positive. ΔNp63α knockdown enhanced ferroptosis and downregulated SLC7A11 expression in ME-180 (Figures 5E-5G), which is consistent with our gain-of-function studies in MEFs. In contrast, TAp63 knockdown in ME-180 neither reduced SLC7A11 expression nor enhanced ferroptosis (Figure S5). Overall, our data suggest that ΔNp63α can inhibit ferroptosis independently of p53.
Figure 5. ΔNp63α can inhibit ferroptosis independently of p53.

(A) SV40-transformed WT MEFs transduced with GFP or ΔNp63-expressing retrovirus were untreated or treated with erastin (4 μM) for the indicated times. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (B) SV40-transformed p53-/- MEFs transduced with GFP or ΔNp63-expressing retrovirus were untreated or treated with erastin (4 μM) for the indicated times. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (C) SV40-transformed WT or p53-/- MEFs transduced with GFP or ΔNp63-expressing retrovirus were untreated or treated with RSL3 (1 μM) for 8 h. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (D) SV40-transformed WT or p53-/- MEFs transduced with GFP or ΔNp63-expressing were subjected to qRT-PCR analysis of Slc7a11 mRNA. Data were normalized against β-Actin (mean ± s.d., n = 3). (E) ME-180 cells, transfected with scrambled siRNA or siRNA against ΔNp63, were untreated or treated with erastin (20 μM) for 72 h. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (F) ME-180 cells, transfected with scrambled siRNA or siRNA against ΔNp63 were untreated or treated with RSL3 (10 μM) for 48 h. Cell death was quantified by propidium iodide staining (mean ± s.d., n = 3). (G) ME-180 cells, transfected with scrambled siRNA or siRNA against ΔNp63, were subjected to qRT-PCR analysis of the indicated genes (mean ± s.d., n = 3). *, P < 0.05; **, P < 0.01; ***, P < 0.001 (Student's t-test).
ΔNp63α alleviates anoikis-induced oxidative stress and cooperates with BCL-2 to disrupt the luminal clearance of mammary acini and promote metastasis
The observation that antioxidant and anti-apoptotic BCL-2 work in concert to promote long-term clonogenic survival against matrix detachment (Schafer et al., 2009) prompted us to investigate whether ΔNp63α can mitigate oxidative stress associated with anoikis and whether ΔNp63α can cooperate with BCL-2 to prevent the luminal clearance of acini in 3D culture of MCF-10A. Interestingly, loss of matrix attachment led to downregulation of ΔNp63α in both MCF-10A and ME-180 where ΔNp63α knockdown induced oxidative stress (Figures 6A and 4B). Notably, overexpression of ΔNp63α but not BCL-2 prevented matrix detachment-induced ROS accumulation in MCF-10A even though ΔNp63α provided less protection against cell death than BCL-2 (Figures 6B and 6C). To investigate long-term survival upon loss of matrix attachment, MCF-10A cells were seeded onto reconstituted basement membrane under conditions that enable the formation of 3D acini where the inner, matrix-deprived cells are eliminated to facilitate lumen formation (Schafer et al., 2009). Overexpression of either BCL-2 or ΔNp63α only weakly inhibited luminal clearance whereas co-expression of BCL-2 and ΔNp63α markedly blocked luminal clearance (Figures 6D and 6E). Luminal clearance of MCF-10A acini was evident within two weeks after seeding, whereas co-expression of BCL-2 and ΔNp63α potently blocked luminal clearance for up to a month (Figures 6D and 6E). Importantly, overexpression of ΔNp63α but not BCL-2 suppressed ROS accumulation in the matrix-deprived luminal cells at early time points before these cells were eliminated (Figure 6F). Both TAp63 and ΔNp63 have been shown to regulate the expression of adhesion molecules and adenovirus-mediated knockdown of p63 was reported to induce anoikis (Carroll et al., 2006). However, we did not observe obvious adhesion problems when p63 was silenced, which is in accordance with a previous report (Senoo et al., 2007). Notably, only ΔNp63 but not TAp63 is capable of regulating oxidative stress (Figures 1I and S5). Ferrostatin-1 had minimal effect on the luminal clearance of mammary acini in 3D culture (Figure S6).
Figure 6. ΔNp63α mitigates anoikis-induced oxidative stress and cooperates with anti-apoptotic BCL-2 to disrupt the luminal clearance of mammary acini and promote cancer metastasis.
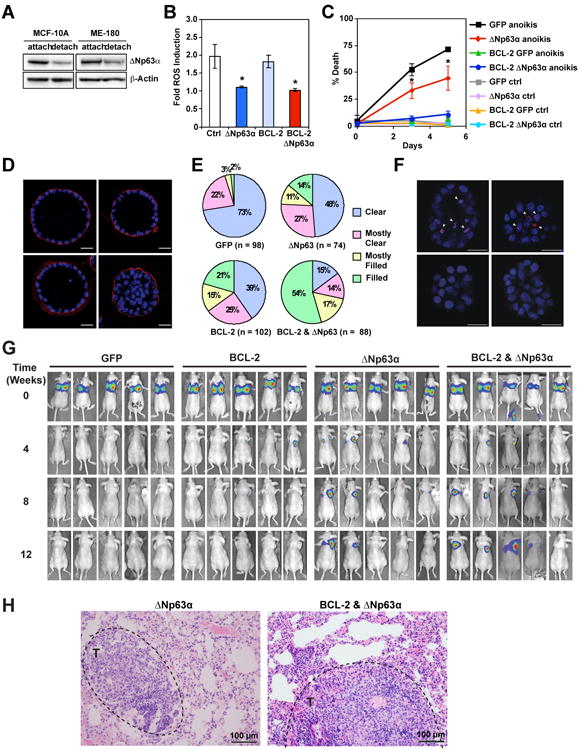
(A) MCF-10A or ME-180 cells were grown on poly-HEMA-coated plates in the presence of 1% methylcellulose for 12 hours to induce anoikis. Paired samples grown in either regular or detachment condition were assessed by anti-p63 and anti-Actin immunoblots. (B) MCF-10A cells transduced with the indicated retrovirus were grown on poly-HEMA-coated plates in the presence of 1% methylcellulose to induce anoikis. Paired samples grown in either regular or detachment condition were stained with DHE at 1.5 days. Oxidation of DHE was quantified by flow cytometry. Data shown are fold increase of ROS induced by anoikis (mean ± s.d., n = 3). (C) MCF-10A cells transduced with the indicated retrovirus were grown on poly-HEMA-coated plates in the presence of 1% methylcellulose to induce anoikis. Cell death was quantified by annexin-V staining at the indicated times (mean ± s.d., n = 3). (D and E) MCF-10A cells transduced with the indicated retrovirus were cultured in reconstituted basement membrane (Matrigel) for 31 days. Acini were fixed and stained for Laminin 5 (red) and nuclear stain 4′,6-diamidino-2-phenylindole (DAPI, blue). Luminal cellularity of acini was scored using confocal microscopy. Clear, 0-25% of the luminal space filled with cell nuclei; mostly clear, 25-50%; mostly filled, 50-75%; filled, 75-100%. Representative confocal microscopy images from three independent experiments are shown in (D). Scale bar, 25 μm. Data shown in (E) are average of three independent experiments. (F) MCF-10A cells transduced with the indicated retrovirus were cultured in Matrigel for 6-8 days. Acini were stained with DHE (red) and Hoechst 33342 (blue). Representative confocal microscopy images are shown. Arrowheads indicate DHE-high cells. Scale bar, 25 μm. (G) Luciferase-transduced MDA-MB-231 cells overexpressing GFP, BCL-2, ΔNp63α, or both ΔNp63α and BCL-2 were injected into the tail vein of immunodeficient mice, and lung colonization was analyzed by in vivo bioluminescence imaging (n = 5 for each group). (H) Representative histopathological images of lung with metastatic tumors (black dashed circle) from mice injected with luciferase-transduced MDA-MB-231 cells overexpressing ΔNp63α or both ΔNp63α and BCL-2. Scale bar, 100 μm. * P < 0.05 (Student's t-test).
As resistance to anoikis has been proposed to promote cancer metastasis and oxidative stress has been shown to limit cancer metastasis (Buchheit et al., 2014; Piskounova et al., 2015), we next examined whether overexpression of ΔNp63α and/or BCL-2 promotes lung metastasis of a triple-negative breast cancer cell line MDA-MB-231 that is transduced with luciferase for bioluminescence imaging. Tail vein injection of ΔNp63α overexpressing, luciferase-transduced MDA-MB-231 cells into immunodeficient mice resulted in lung metastases in 2 out of 5 mice whereas GFP-expressing control cells failed to metastasize to lung (Figures 6G and 6H). These findings are consistent with the report showing that NAC increases metastatic melanoma burden due to increased ROS and reduced GSH/GSSG ratios in metastatic tumors (Piskounova et al., 2015). Although overexpression of BCL-2 alone did not increase lung metastasis, co-expression of BCL-2 and ΔNp63α further increased lung metastases and 4 out of 5 mice following injection developed lung metastases (Figures 6G and 6H). In summary, combined inhibition of apoptosis and oxidative stress through co-expression of BCL-2 and ΔNp63α confers clonogenic survival against anoikis and promotes lung metastasis.
Amplification of TP63 in human squamous cell lung cancer upregulates glutathione metabolism genes to promote anchorage-independent growth
In contrast to the tumor suppressor function of p53, ΔNp63α has been implicated as an oncogene and is frequently amplified and/or overexpressed in human squamous cell carcinoma of the lung and of the head and neck (Dotsch et al., 2010; Keyes et al., 2011; Tonon et al., 2005). As cancers often have an increased demand for antioxidant capacity compared to normal tissues likely due to inherent metabolic derangements that increase ROS production (Trachootham et al., 2009), ΔNp63α might promote tumorigenesis by reducing oxidative stress. To explore this possibility, we analyzed The Cancer Genome Atlas (TCGA) lung squamous cell carcinoma dataset (Cancer Genome Atlas Research, 2012) to determine whether amplification/overexpression of p63 upregulates the glutathione metabolism pathway in primary human tumors. Consistent with previously published reports (Dotsch et al., 2010; Tonon et al., 2005), TP63 amplification is a frequent event in human lung squamous cell carcinoma (∼30%) and the majority of TP63 amplified tumors overexpress ΔNp63. We further interrogated the database using Gene Set Enrichment Analysis (GSEA) and found that tumor samples harboring TP63 amplification and overexpressing ΔNp63 had significant enrichment of glutathione metabolic pathway genes when compared to diploid tumor samples (Figures 7A and 7B). Furthermore, within the TP63-amplified subset, ΔNp63 expression levels positively correlated with the expression of several genes that were discovered through our mechanistic studies, including GCLC (r = 0.49, p = 8.49e-07), GPX2 (r = 0.52, p = 1.48e-07), and GSR (r = 0.46, p = 7.29e-06) (Figures 7C-7E).
Figure 7. Amplification of TP63 in human squamous cell lung cancer upregulates glutathione metabolism genes and promotes anchorage-independent growth.
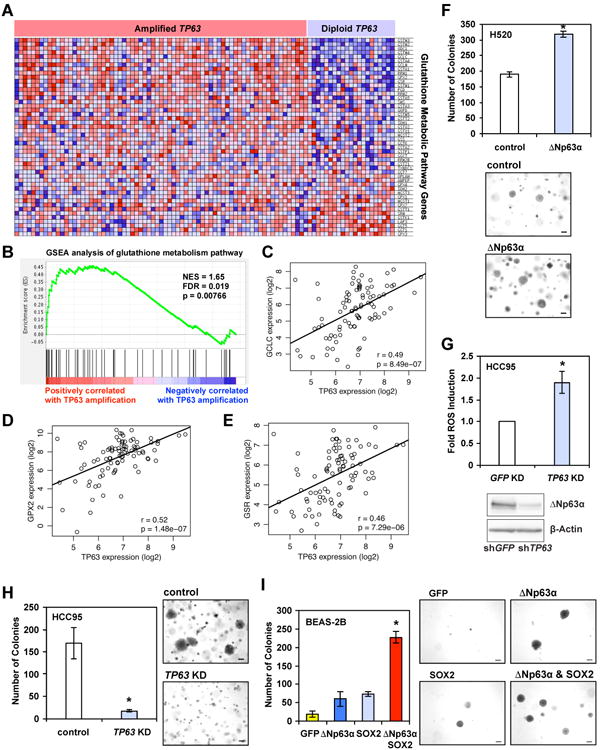
(A) Heat map of expression levels of KEGG Glutathione Metabolism Pathway genes in the TCGA lung squamous cell carcinoma samples harboring amplified or diploid TP63 as indicated. The expression levels are plotted in red-blue color scale with red indicating high expression and blue indicating low expression. (B) GSEA enrichment plot demonstrates enrichment of the KEGG Glutathione Metabolism Pathway in the TCGA lung squamous cell carcinoma samples with amplified TP63 compared to diploid samples. Nominal p-value = 0.00766. FDR q-value = 0.019. (C-E) Pairwise comparison of mRNA expression of TP63 versus GCLC (C), GPX2 (D), or GSR (E) in the TCGA lung squamous cell carcinoma samples harboring amplified TP63 with Pearson correlation and p-values shown. (F) H520 cells transduced with control retrovirus or retrovirus expressing ΔNp63α were plated in soft agar for 18 days. The colonies with diameters larger than 200 μm were counted (mean ± s.d., n = 3). Representative images are shown (scale bar = 200 μm). (G) HCC95 cells transduced with lentivirus expressing shRNA against GFP or TP63 were stained with CM-H2DCFDA or subjected to immunoblot analysis using the indicated antibodies. Oxidation of CM-H2DCFDA was quantified by flow cytometry. Data shown are fold increase of ROS induced by knockdown of TP63 (mean ± s.d., n = 3). (H) HCC95 cells transduced with lentivirus expressing control or shRNA against TP63 were plated in soft agar for 12 days. The colonies with diameters larger than 200 μm were counted (mean ± s.d., n = 3). Representative images are shown (scale bar = 200 μm). (I) BEAS-2B cells transduced with the indicated retrovirus were plated in soft agar for 18 days. The colonies with diameters larger than 200 μm were counted (mean ± s.d., n = 3). Representative images are shown (scale bar = 200 μm). *, P < 0.05 (Student's t-test).
Given that oxidative stress is induced in response to matrix detachment, the ability of cells to form colonies in soft agar is not only determined by their proliferative potential but also their resistance to oxidative stress. We next examined whether ΔNp63α promotes anchorage-independent growth by inhibiting oxidative stress. Indeed, overexpression of ΔNp63α in a squamous cell lung cancer cell line H520 with low ΔNp63α expression increased soft agar colony formation without enhancing cell proliferation (Figure 7F and data not shown). In contrast, ΔNp63α knockdown in a squamous cell lung cancer cell line HCC95 that highly expresses ΔNp63α led to increased ROS accumulation and reduced soft agar colony formation (Figures 7G and 7H). Because genomic amplification on chromosome 3q26.33 harbors an oncogene SOX2 in additional to TP63 (Bass et al., 2009), we envisioned that SOX2 and ΔNp63α might cooperate to promote oncogenesis. Consistent with a previous report (Bass et al., 2009), overexpression of SOX2 alone only slightly increased colony formation in soft agar (Fig. 7I). Significantly, SOX2 and ΔNp63α synergized to transform normal human bronchial epithelial cells BEAS-2B (Fig. 7I). Collectively, these data support a notion that amplification of TP63 may be a strategy by which squamous cell lung cancer evades oxidative stress and thereby promotes tumorigenesis.
Discussion
Here, we report that ΔNp63α prevents oxidative stress and cooperates with apoptotic inhibition to promote long-term cellular survival against DNA damage and extracellular matrix detachment, highlighting the quintessence of suppressing oxidative stress in promoting long-term survival (Figure S7). Caspase-dependent apoptosis has long been considered as the predominant pathway for cell death. Apoptosis can be initiated through either intrinsic death signals, such as DNA damage, loss of matrix attachment, cytokine/growth factor deprivation, and ER stress, or extrinsic death signals that are mediated by the ligand engagement of death receptors including FAS and TNF-R1. Studies of extrinsic cell death discovered a necrotic program “necroptosis” when apoptosis is inhibited (Pasparakis and Vandenabeele, 2015; Sun and Wang, 2014; Yuan and Kroemer, 2010). The discovery of programmed forms of necrotic death represents a significant conceptual advance for cell death research. However, it remains unsettled whether intrinsic death signals also activate necrosis; and whether intrinsic necrosis, if exists, is also programmed. We and others have previously shown that cells incompetent of undergoing apoptosis still succumb to oxidative stress upon DNA damage or loss of matrix attachment and display morphological features of necrosis (Schafer et al., 2009; Tu et al., 2009). Current study showed that oxidative stress-induced necrotic death in these settings could be blocked by overexpression of ΔNp63α through coordinated regulation of glutathione biosynthesis, utilization, and regeneration. In fact, ΔNp63α guards against oxidative stress triggered by a wide array of death stimuli in several different cell lines, including intrinsic apoptotic signals, thiol or lipid oxidants, and ferroptosis-inducing agents. These data support that ΔNp63α is a key cellular guardian against oxidative stress, which is analogous to antiapoptotic BCL-2 family proteins that act at the convergence of diverse apoptosis-inducing signaling pathways. Overall, our data support that intrinsic death signals can trigger necrosis when apoptosis is inhibited and intrinsic necrosis is also programmed.
Inhibition of oxidative stress may be especially critical to the wellbeing of long-lived cells such as stem cells (Holmstrom and Finkel, 2014; Kobayashi and Suda, 2012). Because stem cells must remain capable of both self-renewal and repletion of lost progeny throughout an organism's lifespan, they are expected to be more tolerant to the long-term exposure to ROS and oxidative stress. In fact, ΔNp63α is important in maintaining the stem cell populations of stratified epithelia (Dotsch et al., 2010; Keyes et al., 2011; Senoo et al., 2007). In addition to sustaining the proliferative capacity of epithelial stem cells as previously proposed (Keyes et al., 2011; Senoo et al., 2007), our data implicate that ΔNp63α may also promote stem cell survival through its antioxidant function. Intriguingly, p63+/- mice have a shortened life span and display features of accelerated aging, and p63-deficient cells exhibit heightened cellular senescence (Keyes et al., 2005), all of which could be caused by oxidative damage (Balaban et al., 2005) and may be linked to oxidative stress resulting from the loss of ΔNp63α.
Our findings highlight the need for a simultaneous blockade of apoptosis and oxidative stress to promote long-term cellular wellbeing, a strategy likely hijacked by cancer. To evade apoptotic checkpoints, cancer cells often overexpress anti-apoptotic BCL-2 family proteins through chromosome translocation involving BCL-2 (Korsmeyer, 1992) or amplification of BCL-XL and MCL-1 (Beroukhim et al., 2010). Likewise, amplification of TP63 can be a strategy for cancer cells to evade oxidative stress-induced cell death. The importance of antioxidant defense in tumorigenesis is also supported by the discovery of frequent loss-of-function mutations of KEAP1 and gain-of-function mutations of NRF2 in human cancers (Hayes and McMahon, 2009). NRF2 controls cellular adaptation to oxidative stress by inducing antioxidant and detoxification genes, whereas KEAP1 sequesters NRF2 in cytoplasm and promotes the degradation of NRF2. Here, we showed that ΔNp63α could regulate GCLC, the rate-limiting GSH biosynthetic enzyme, through an NRF2-independent manner, supporting a central role of ΔNp63α in glutathione biogenesis. In addition, we showed that ΔNp63α could inhibit ferroptosis independently of p53. Given the recent discovery of ferroptosis regulation as one of the tumor suppressor mechanisms of p53 (Jiang et al., 2015), suppression of ferroptosis by ΔNp63α may contribute to its oncogenic properties. Our study highlights the importance of evading two cell death mechanisms in tumorigenesis, which may provide a selection pressure for genetic/epigenetic alterations with oncogenic advantages.
Although a detailed blueprint of the core apoptotic pathway has been constructed through biochemical and genetic studies over the past two decades, we are still unable to fully rescue cells from most death stimuli. The identification of oxidative stress as a critical mechanism compromising long-term survival of apoptosis-defective cells may open new avenues for therapeutic interventions aimed at preventing excessive cell loss during disease processes. Conversely, targeting the antioxidant pathway may hold promise for the future development of anticancer therapeutics by eliminating cancer cells that evade apoptotic checkpoints.
Experimental Procedures
Plasmid construction, retrovirus production, and siRNA
Murine ΔNp63α was dually tagged with FLAG and HA at the N-terminus and cloned into MSCV-IRES-Puro or tagged with HA at the N-terminus and cloned into MSCV-IRES-GFP. The target sequence of shRNA against mouse p63 is 5′-AGCACACGATCGAAACGTA-3′. The target sequence of scramble shRNA is 5′-GCGCGCTTTGTAGGATTCG-3′. Lentivirus-mediated knockdown constructs for human p63 and GFP were previously described (Godar et al., 2008; Sancak et al., 2008). Lentiviral tetracycline-inducible miR30-based shRNA against human p63 was obtained from Open Biosystems. The target sequence is 5′-TCCGAGCTATGTCAGTACTATT-3′. The siRNA oligos were purchased from Ambion Silencer Select oligos (Applied Biosystems) or Dharmacon and summarized in Table S1.
Cell culture and viability assays
Generation and transformation of MEFs were performed as described (Chen et al., 2015; Cheng et al., 2001). HCC95 cell line was provided by Dr. John Minna at University of Texas Southwestern Medical Center. To induce anoikis, cells were grown on poly-HEMA (Sigma)-coated plates in the presence of 1% methylcellulose (Sigma). Cell death was quantified by annexin-V (BioVision) or propidium iodide (Sigma) staining, followed by flow cytometric analysis using either a FACSCalibur (BD Biosciences) or an LSRFortessa (BD Biosciences). Data were analyzed using CellQuest Pro (BD Biosciences) or FACSDiva (BD Biosciences).
Measurement of ROS
Production of ROS was monitored by flow cytometry using the redox-sensitive dyes (Invitrogen), including 2′,7′-dichlorofluorescein diacetate (H2DCFDA), 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), dihydroethidium (DHE), C11-BODIPY665/676, or MitoSOX Red. Oxidation of the ROS-sensitive dyes was quantified by flow cytometry. Median fluorescence in the appropriate detection channels was assessed by FlowJo (Tree Star). Fold induction of ROS was obtained by dividing the median fluorescence of experimental samples with that of control samples.
Reverse transcription and quantitative PCR
Total RNA was extracted from cells using TRIZOL (Invitrogen) and reverse transcription was performed with oligo-dT plus random decamer primers (Ambion) using Superscript II (Invitrogen). Quantitative PCR was performed with SYBR green master mix (Applied Biosystems) using gene-specific primers, summarized in Table S2. For TaqMan probes, quantitative PCR was performed with TaqMan PCR Master Mix (Applied Biosystems). Quantitative PCR was performed on an ABI Prism 7300 sequence detection system (Applied Biosystems) or a ViiA 7 Real-Time PCR System (Applied Biosystems). Data were analyzed by normalization against GAPDH, β-Actin, or 18S rRNA as described (Tu et al., 2009). The TaqMan probes for mouse GPX2 (TaqMan Assay ID Mm00850074_g1) and human GCLC (TaqMan Assay ID Hs00155249_m1) were obtained from Invitrogen.
Three-dimensional culture of MCF-10A and indirect immunofluorescence microscopy
3D culture of MCF-10A cells was performed as described (Debnath et al., 2003). Briefly, 104 cells were seeded onto one chamber of a 4-well chamber slide pre-coated with Matrigel (BD Biosciences). After 31 days, cells were fixed using 4% paraformaldehyde (Fisher) and permeabilized using 0.5% Triton X-100, and sequentially incubated with anti-Laminin-5 (D4B5, EMD Millipore), Alexa Fluor 568 conjugated goat anti-mouse IgG (Invitrogen), anti-GFP (ab13970, Abcam), Alexa Fluor 488 conjugated goat anti-chicken IgG (Invitrogen), and nuclear stain 4,6-diamidino-2-phenylindole (DAPI, Sigma). Images were acquired using the Leica TCS SP5-II Confocal Upright and the PerkinElmer UltraVIEW ERS confocal microscopes at the Molecular Cytology Core Facility at MSKCC and analyzed by MetaMorph software (Molecular Devices). To measure ROS levels, 3D culture of MCF-10A cells after 6-8 days were incubated with Hank's Balanced Salt Solution (HBSS, Invitrogen) containing 5 μM dihydroethidium (DHE, Invitrogen) and 3 μg/ml Hoechst 33342 (Invitrogen) at 37°C for 30 minutes in a humidified 5% CO2 incubator, washed once with HBSS, then immediately imaged.
Reagents, antibodies, and immunoblot analysis
Chemicals used are listed as follows: etoposide (Sigma), diamide (Sigma), tert-butyl hydroperoxide (Sigma), diphenyleneiodonium (Sigma), N-acetyl-L-cysteine (Sigma), ferrostatin-1 (Sigma), necrostatin-1 (Sigma), GSK'872 (EMD Millipore), deferoxamine (Sigma), and Tiron (Sigma). Antibodies used are listed as follows: anti-FLAG (M2, Sigma), anti-p53 (FL393, Santa Cruz Biotechnology), anti-p63 (4A4, Santa Cruz Biotechnology), anti-GCLC (HPA036359, Sigma), anti-IDH2 (ab55271, Abcam), anti-RIP1 (610458, BD Biosciences), and anti-β-Actin (A1978, Sigma). Cell lysates were resolved by NuPAGE gels (Invitrogen), transferred onto PVDF membrane (Immobilon-P, Millipore). Antibody detection was accomplished using enhanced chemiluminescence method (Western Lightning, PerkinElmer) and LAS-3000 Imaging system (FUJIFILM).
Glutathione assays
Measurement of intracellular GSH and GSSG was performed using the 5,5′-dithio-bis(2-nitrobenzoic acid)-glutathione reductase recycling method as described (Rahman et al., 2006). Briefly, the rate of 5-thio-2-nitrobenzoic acid (TNB) formation was determined by measuring the rate of change of absorbance at 412 nm. Linear regressions based on values obtained from standard curves were used to calculate concentrations of total and oxidized glutathione, which were then used to calculate concentration of reduced glutathione. Glutathione concentrations were normalized based on protein concentration determined using the BCA Kit (Pierce).
Gene expression profiling and heatmap plot
Total RNA was extracted from cells using TRIZOL (Invitrogen) and subjected to cleanup using the RNeasy Mini Kit (Qiagen). RNA samples were submitted to the Laboratory for Clinical Genomics at the Washington University in St. Louis for microarray analysis using the GeneChip Mouse Gene 1.0 ST array (Affymetrix). Heatmap plot was generated using Partek Genomics Suite (Partek).
Gene set enrichment and mRNA expression correlation analysis
Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was used to statistically evaluate the extent that genes in the glutathione metabolism pathway (KEGG_GLUTATHIONE_METABOLISM, MSigDB v3.0) were dysregulated in TCGA lung squamous cell carcinoma tumor samples with TP63 amplification. We first identified tumor samples with diploid (N = 25) and amplified TP63 (inferred gain of >=2 copies, N = 91). All genes expressed in tumor samples (N = 107) were sorted by mRNA expression change in TP63 amplified versus diploid samples (sorting from down to up-regulation, Wilcoxon rank-sum test), and GSEA was used to evaluate the null hypothesis that genes in glutathione metabolism gene set were not differentially expressed in amplified versus diploid samples (using 1000 permutations). Pairwise correlations between TP63 mRNA expression and mRNA expression levels of genes in the glutathione metabolism pathway in samples with TP63 amplification were evaluated using the Pearson correlation coefficient and regression lines were estimated using the ordinary least squares method.
Xenograft Studies
Animal experiments were performed in accordance to the MSKCC Institutional Animal Care and Use Committee. Tail vein injection was performed as described (Minn et al., 2005). Briefly, 2 × 105 cells were injected into the lateral tail vein of athymic nude mice. Successful injections were confirmed by bioluminescence imaging. For the imaging, 75 mg/kg of D-Luciferin (Xenogen) in PBS was injected retro-orbitally into anesthetized mice. Bioluminescence images were obtained with the IVIS Imaging System (Xenogen) and analyzed using Living Image software (Xenogen).
Statistical Analysis
Cell death, ROS induction, qRT-PCR, and glutathione assays were analyzed for statistical significance using unpaired Student's t-test with alpha = 0.05. Statistical analysis for GSEA is described above.
Supplementary Material
Highlights.
ΔNp63α is a key cellular guardian against oxidative stress including ferroptosis
ΔNp63α inhibits oxidative stress through regulation of glutathione metabolism
ΔNp63α cooperates with the BCL-2 family to promote long term clonogenic survival
TP63 amplification upregulates glutathione metabolism to promote tumorigenesis
Acknowledgments
We apologize to all the investigators whose research could not be appropriately cited due to space limitations. We thank Hsiu-Fang Chen and Po Chan for technical assistance, and Paul Jeng for graphic design and editorial assistance. This work was supported by grants to E. Cheng from the NIH (R01CA125562) and the American Cancer Society (118518-RSG-10-030-01-CCG).
Footnotes
Accession Number: The accession number for the microarray data reported in this paper is GEO: GSE106214.
Author Contributions: G.X.W. designed and conducted experiments, and analyzed data. E.H.C. designed research, analyzed data, and supervised the project. H.C.T., Y.D., Y.W., S.T., Y.T.G., S.H., and H.L. conducted some experiments. A.J.S. analyzed data. J.J.H supervised some experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bass AJ, Watanabe H, Mermel CH, Yu S, Perner S, Verhaak RG, Kim SY, Wardwell L, Tamayo P, Gat-Viks I, et al. SOX2 is an amplified lineagesurvival oncogene in lung and esophageal squamous cell carcinomas. Nature genetics. 2009;41:1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, Barretina J, Boehm JS, Dobson J, Urashima M, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nature Reviews Cancer. 2014;14:632–641. doi: 10.1038/nrc3789. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, Brugge JS, Ellisen LW. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol. 2006;8:551–561. doi: 10.1038/ncb1420. [DOI] [PubMed] [Google Scholar]
- Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, Kim H, Takeda S, Reyna DE, Chan PM, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–1281. doi: 10.1038/ncb3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell. 2001;8:705–711. doi: 10.1016/s1097-2765(01)00320-3. [DOI] [PubMed] [Google Scholar]
- Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15:348–366. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G. p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol. 2010;2:a004887. doi: 10.1101/cshperspect.a004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- Godar S, Ince TA, Bell GW, Feldser D, Donaher JL, Bergh J, Liu A, Miu K, Watnick RS, Reinhardt F, et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell. 2008;134:62–73. doi: 10.1016/j.cell.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, McMahon M. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci. 2009;34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- Hoh J, Jin S, Parrado T, Edington J, Levine AJ, Ott J. The p53MH algorithm and its application in detecting p53-responsive genes. Proc Natl Acad Sci U S A. 2002;99:8467–8472. doi: 10.1073/pnas.132268899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nature reviews Molecular cell biology. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, Ishizawa K, Jebiwott S, Dong Y, Pietanza MC. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nature Communications. 2017;8 doi: 10.1038/ncomms16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes WM, Pecoraro M, Aranda V, Vernersson-Lindahl E, Li W, Vogel H, Guo X, Garcia EL, Michurina TV, Enikolopov G, et al. DeltΔNp63αlpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell. 2011;8:164–176. doi: 10.1016/j.stem.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes WM, Wu Y, Vogel H, Guo X, Lowe SW, Mills AA. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005;19:1986–1999. doi: 10.1101/gad.342305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kim H, Tu HC, Ren D, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol Cell. 2009;36:487–499. doi: 10.1016/j.molcel.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi CI, Suda T. Regulation of reactive oxygen species in stem cells and cancer stem cells. Journal of cellular physiology. 2012;227:421–430. doi: 10.1002/jcp.22764. [DOI] [PubMed] [Google Scholar]
- Korsmeyer SJ. Bcl-2 initiates a new category of oncogenes: regulators of cell death. Blood. 1992;80:879–886. [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A. Glutathione metabolism. Methods in enzymology. 1995;251:3–7. doi: 10.1016/0076-6879(95)51106-7. [DOI] [PubMed] [Google Scholar]
- Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, Ponomarev V, Gerald WL, Blasberg R, Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. The Journal of clinical investigation. 2005;115:44–55. doi: 10.1172/JCI22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med. 2004;10:549–557. doi: 10.1016/j.molmed.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Perez CA, Ott J, Mays DJ, Pietenpol JA. p63 consensus DNA-binding site: identification, analysis and application into a p63MH algorithm. Oncogene. 2007;26:7363–7370. doi: 10.1038/sj.onc.1210561. [DOI] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. BID, BIM, and PUMA are essential for activation of the BAX- and BAK-dependent cell death program. Science. 2010;330:1390–1393. doi: 10.1126/science.1190217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senoo M, Pinto F, Crum CP, McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh EK, Yang A, Kettenbach A, Bamberger C, Michaelis AH, Zhu Z, Elvin JA, Bronson RT, Crum CP, McKeon F. p63 protects the female germ line during meiotic arrest. Nature. 2006;444:624–628. doi: 10.1038/nature05337. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang X. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends in biochemical sciences. 2014;39:587–593. doi: 10.1016/j.tibs.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- Tonon G, Wong KK, Maulik G, Brennan C, Feng B, Zhang Y, Khatry DB, Protopopov A, You MJ, Aguirre AJ, et al. High-resolution genomic profiles of human lung cancer. Proc Natl Acad Sci U S A. 2005;102:9625–9630. doi: 10.1073/pnas.0504126102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Tu HC, Ren D, Wang GX, Chen DY, Westergard TD, Kim H, Sasagawa S, Hsieh JJ, Cheng EH. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci U S A. 2009;106:1093–1098. doi: 10.1073/pnas.0808173106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, et al. p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. American journal of human genetics. 2001;69:481–492. doi: 10.1086/323123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–2933. [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem. 2006;281:7856–7862. doi: 10.1074/jbc.M512655200. [DOI] [PubMed] [Google Scholar]
- Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004;18:1272–1282. doi: 10.1101/gad.1199904. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.