

Abstract
Excess telomere shortening has been observed in most cancer cells. The telomere quantitative polymerase chain reaction (qPCR) assay has become an important tool for epidemiological studies examining the effects of aging, stress, and other factors on the length of telomeres. Current telomere qPCR methods analyze the relative length of telomeres by amplifying telomere sequence products and normalizing with single-copy gene products. However, the current telomere qPCR does not always reflect absolute telomere length in cancer DNA. Because of genomic instability in cancer cells, we hypothesized that the use of single-copy genes (scg) is less accurate for normalizing data in cancer DNA and that new primer sets are required to better represent relative telomere length in cancer DNA. We first confirmed that cancer cells had a different copy ratio among different scg, implying that DNA is aneuploid. By using the new primer sets that amplify multiple-copy sequences (mcs) throughout the genome, the telomere qPCR results showed that the mcs primers were interchangeable with the scg primers as reference primers in normal DNA. By comparing results from the traditional southern blotting method (as kilobases) and results from monochrome multiplex qPCR using the mcs primers (as T/M ratios), we verified that the T/M ratio is highly correlated with absolute telomere length from the southern blot analysis. Together, the mcs primers were able to represent the telomere lengths accurately in cancer DNA samples. These results would allow for analyses of telomeres within cancerous DNA and the development of new, less invasive diagnostic tools for cancer.
Abbreviations: scg, single-copy genes; mcs, multiple-copy sequences; T, telomere quantity; S, scg quantity; M, mcs quantity; DNA, deoxyribonucleic acid; PCR, polymerase chain reaction; MMQPCR, monochrome multiplex quantitative PCR; EDTA, ethylenediaminetetraacetic acid; SDS, sodium dodecyl sulfate; TE, Tris-EDTA; DIG, digoxigenin; CV, coefficient of variation; UCSC, University of California, Santa Cruz; Tm, melting temperature; Eff, PCR efficiency; Ct, cycle threshold; ALT, alternative lengthening of telomeres; IDC, invasive ductal carcinoma; DCIS, ductal carcinoma in situ
Introduction
The telomere quantitative polymerase chain reaction (qPCR) assay was originally developed by R.M. Cawthon for measuring relative average telomere length [1], [2]. This telomere qPCR assay has been widely distributed because of its time efficiency and low DNA requirement (theoretically <100 ng); therefore, the technique is advantageous for studies using limited resources such as large-scale epidemiological studies in a variety of fields including aging, metabolic, and psychosocial research [3], [4], [5], [6], [7], [8]. In this qPCR assay, DNA quantities (in nanograms) both from telomere and reference amplifications are determined by each standard curve created using a serial dilution of standard DNA (e.g., normal genomic DNA with known telomere length). The PCR primers for the reference are typically designed within single-copy gene (scg) sequences such as albumin (ALB) or gamma globin (HBG) genes [1], [2]. The relative telomere length is expressed as the T/S ratio that is calculated by dividing the telomere quantity (T) by the reference quantity of a single-copy gene (S) to normalize the data. Each quantity is calculated by standard curves created using standard DNA with known concentrations. The telomere length of the standard DNA always indicates the T/S ratio is equal to 1.0.
The latest version of telomere qPCR being performed is monochrome multiplex quantitative PCR (MMQPCR) which uses a single fluorescent DNA-intercalating dye (i.e., SYBR green I) and adds both target and reference primer sets in a single reaction tube.1 The MMQPCR method is feasible only when the number of target sequences is different from the number of reference sequences in a PCR template; for example, telomere repeat sequences are more abundant than single-copy genes such as ALB and HBG in genome. This difference in target sequences allows for the distinguishing between the two signals created by the separate primer sets. The multiplexing method further increases throughput and improves the accuracy by minimizing sample handling error while also saving the amount of starting materials and the cost of reagents, which are sometimes of critical value.
Regardless of whether the telomere qPCR is singleplexing or multiplexing, it is worth remembering that the current telomere qPCR method is optimized for diploid DNA samples. To normalize the data, the single-copy genes (scg) are used because diploid cells, in theory, always contain two copies (or alleles) of the single-copy genes, one per chromosome. The current telomere qPCR method is beneficial for most epidemiological studies which use peripheral blood DNA samples as a PCR template. However, when nondiploid DNA is the study subject, such as is the case in cancerous tissues, the current telomere qPCR may mislead the interpretations by reducing the data accuracy and integrity [9], [10]. In other words, since tumor tissues or cells often form aneuploidy which represents an abnormal number of chromosomes in a cell, the scg may not reflect the amount of input DNA if the aneuploidy exists within the scg sequences.
To eliminate the abovementioned concern, we introduce an alternative telomere MMQPCR method which is applicable for cancer DNA. Using cancerous DNA, the results from our newly developed MMQPCR method is well correlated with those from traditional southern blot analysis, suggesting that the refined MMQPCR method is useful for human genomic DNA samples with abnormal ploidy levels.
Materials and Methods
Tissue and Blood Specimens
Tumor tissue specimens were obtained at the time of surgery, immediately frozen in liquid nitrogen, and stored in liquid nitrogen until use. Frozen tissues (100-150 mg) from colon cancer (n = 8), renal cell carcinoma (n = 7), and breast cancer (n = 2) were obtained from the tissue bank at the Indiana University Simon Cancer Center. All cases were reviewed by pathologists to assess tumor histology (Supplemental Table S1). The purity of each specimen was shown as at least 50% tumor. Whole blood DNA samples (n = 18, 23 ≤ age ≤ 70, mean age = 48.8) from healthy women were obtained from the Susan Komen Tissue Bank. Additional frozen breast tissues of ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC) were also obtained from the tissue bank at the Indiana University Simon Cancer Center (DCIS, 30-80 years, mean, 55 ± 12 years; IDC, 31-86 years, mean, 47.6 ± 13.3 years) [11]. The study was approved by the institutional review board of the Indiana University.
Cell Lines
A total of 21 human cell lines were used to isolate genomic DNA as follows: cervical carcinoma (HeLa), breast carcinoma (MCF7, T47D), renal cell carcinoma (RCC23) [12], lung adenocarcinoma (H1299), colorectal carcinoma (HT29, SW620, HCT116, and DLD-1), pancreatic carcinoma (PANC1, MIA PaCa-2, and AsPc-1), ovarian carcinoma (A2780, SKOV3), prostate carcinoma (PC-3), giant cell tumor (GCT), fibrosarcoma, (HT1080), preneoplastic mammary epithelial cells (MCF10A), and three ALT-positive cells (U-2 OS, Saos-2, and VA13). All cell lines were cultured in ambient oxygen, 5% CO2 and maintained in appropriate medium according to ATCC (www.atcc.org).
DNA Extraction
Genomic DNA from both cell lines and tumors was extracted by a salt precipitation method as previously described [13]. Briefly, about 20 mg of each frozen tissue fragment was minced quickly in cold PBS(−) (which is PBS without divalent cations) or cells from 10-cm dish were trypsinized and washed with PBS(−). The PBS(−) was removed by centrifuging at 3500×g, and the pellets were resolved in lysis buffer (20 mM Tri-HCl/pH 8.2, 10 mM EDTA, 400 mM NaCl, 0.5% SDS, 0.05 μg/μl proteinase K) and incubated overnight at 56°C. The DNA solution was centrifuged for 15 minutes at 9600×g after adding one-fourth volume of saturated NaCl. The supernatant was transferred to a new tube, and the DNA was precipitated by adding the equal volume of isopropanol. After rinsing with 70% ethanol, the DNA was resolved in TE buffer.
DNA Quantitation
The DNA concentration was quantitated by Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific) as well as picogreen assay. Quant-iT TM PicoGreen dsDNA Assay Kit (Life Technologies) was used according to the manufacturer's instruction. Fluorescence intensity was measured with a Synergy 2 Multi-Mode Reader at emission wavelength of 520 nm and excitation wavelength of 480 nm.
Telomere Length Measurement: Southern Blot Analysis
Absolute mean telomere length was measured by TeloTAGGG Telomere Length Assay (Roche). Briefly, 4 μg of each DNA sample were digested overnight with two tetra-cutter restriction enzymes, RsaI and HinfI. Digested DNA samples were resolved on a 0.8% agarose gel, blotted to a hybond-N+ membrane (GE Healthcare) using capillary transfer and 20× SSC buffer. A digoxigenin-labeled probe was used for the hybridization. The results were scanned from Amersham Imager 600, and mean telomere lengths were calculated by ImageQuant and the Excel spreadsheet program TELORUN as described [14], [15].
Telomere Length Measurement: qPCR
Telomere length qPCR was performed with a QuantStudio 6 Flex Real-Time PCR System and analyzed under either singleplex or multiplex conditions. The telomere primers were used for the telomere signal (telg and telc, final concentrations 900 nM) [1]. A set of single-copy gene (scg) signal primers within albumin or β-globin genes was used as reference (final concentrations 200 nM each) [1], [2]. In the case of the new multiple-copy sequence (mcs) primer sets (final concentrations 800 nM each) [16], the telomere primers contained a GC clamp and were used for the telomere signal (GC-Telg and GC-Telc, final concentrations 150 nM). The PCR cycle for MMQPCR is as follows: Stage 1: 2 minutes at 95°C; Stage 2: 2 cycles of 15 seconds at 94°C, 15 seconds at 49°C; and Stage 3: 35 cycles of 15 seconds at 94°C, 10 seconds at 60°C, 20 seconds at 74°C with signal acquisition (for mcs amplification), 10 seconds at 84°C, 20 seconds at 88°C with signal acquisition (for telomere amplification). Each DNA standard curve was generated using a pooled female genomic DNA (Promega, G152A) and used for calculation of the T/S ratios (= ratios of “telomere signals per scg signals”) or the T/M ratios (= ratios of “telomere signals per mcs signals”). The same control DNA was included in every run, and intra- and interassay coefficients of variation (CVs) were carefully monitored. All data were collected from three independent experiments with duplicate or triplicate reactions. The values were accepted when the standard deviation of Ct was below 0.5 among replications.
Other qPCR
Singleplex qPCR was performed with a QuantStudio 6 Flex Real-Time PCR System and analyzed. Three sets of scg primers (e.g., HBG, CDK6, and ALB) were used to quantify ratios among three amplifications. Additionally, the sets of mcs primers (MRef 1 and MRef 2) were used to compare DNA quantifications between the mcs primers and the scg primers. Each DNA standard curve was generated using a pooled female genomic DNA (Promega, G152A) and used to calculate each quantity from unknown DNA. All primer information is shown in Supplemental Table S2.
Statistical Analysis
Student’s t test was applied for calculation of statistical differences, and two-tailed P values of less than .05 were considered to be statistically significant. Regression analysis represented correlation by the coefficient of determination (R2) and P value.
Results
Revalidation of Single-Copy Gene Primers
To determine whether T/S ratios are reliable for measuring relative telomere length in cancer DNA samples, we compared T/S ratios of cancer DNA samples with their absolute mean telomere length of the same DNA by southern blot analysis. The results showed the correlation in mean telomere lengths measured by these two different techniques (R2 = 0.578, P = .0002, Figure 1, A & B); however, in some cell lines (e.g., PANC-1, HeLa, and RCC23), the T/S ratios were abnormally far from their absolute mean telomere length by southern blotting method (Figure 1B and Supplemental Table S3). Based on the ploidy status collected from publically available information (e.g., www.atcc.org/),[17], [18] all cancer/immortalized cell lines used in this study indicated a lack of diploid status (Supplemental Table S3). Therefore, we investigated whether scg primers might be less accurate for normalizing telomere amplification signals in nondiploid DNA. To assess the DNA ploidy level in each sample, we quantified ratios between two scg amplifications, ALB and HBG, using DNA extracted from 18 whole blood DNA from healthy individuals, 18 cancer and noncancer cell lines, and 17 tumor tissues from colon, breast, and renal cell carcinoma using qPCR. In principle, we would expect that diploid DNA contains the same amount of scg; therefore, the quantity ratio between one scg and another should be close to 1.0 using the qPCR analysis. We confirmed that the amplification performance of normal whole blood DNA samples was indeed close to 1.0 when either HBG or CDK6 gene quantity was divided by ALB gene quantity (Figure 1C and Supplemental Figure S1). These results indicate that the HBG, CDK6, and ALB primers substitute each other to normalize the telomere qPCR results for normal DNA, while no such correlation existed in DNA samples from both cancer cell lines and tumor tissue specimens. Indeed, when DNA samples from cancer cell lines were used, the ratios of quantities (HBG/ALB) showed significant spread with ratios between 0.70 and 2.65 (mean ± S.D. = 1.27 ± 0.40). Because ALB, CDK6, and HBG genes are located on chromosomes 4, 7, and 11, respectively, it is possible that those alleles changed structurally or numerically, and thereby, these genes no longer existed as two copies per diploid. These findings suggest that gain or loss of certain loci indeed occurs in cancer DNA, and scg sequences are not always accurate for use to normalize the data when nondiploid DNA (e.g., cancerous DNA) is the subject for study.
Figure 1.

The current telomere qPCR method does not always correlate with absolute telomere length in cancer DNA because of its aneuploidy. (A) A representative southern blotting image is shown. The traditional southern blot analysis was used to measure the absolute length of the telomeres in the cell line cancer DNA samples. Commercialized pooled normal DNA was used as reference. TIG-3 is a normal diploid fibroblast cell [31]. (B) The relative telomere length (T/S ratio) calculated utilizing the qPCR method had a moderate correlation with the Southern blot measurements when using 18 cancer cell line DNA samples (R2 = 0.5784). The individual values were shown in Supplemental Table S3. (C) Cancer DNA from both cell lines and tumor tissue samples indeed exhibited aneuploidy (see details in the Results section). Singleplex qPCR was performed using two single-copy genes (HBG and ALB) primer sets in the same run, and the quantity from HBG amplification was divided by the quantity from ALB amplification. Normal DNA showed an approximate 1:1 ratio between the two genes. The results were generated from three independent experiments using DNA samples from 18 normal blood samples, 18 cancer cell lines, and 17 tumor tissue samples.
Supplemental Figure S1.
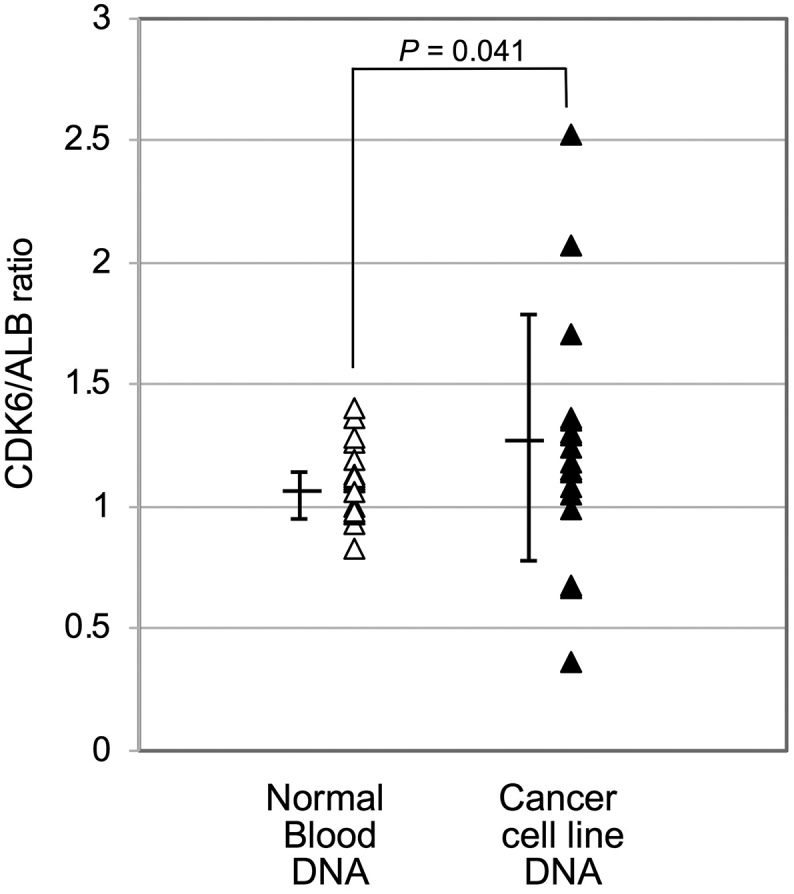
Additional examination for DNA ploidy status comparing two different single-copy genes (CDK6 and ALB) amplifications. The results revealed further evidence of aneuploidy within cancer cell line DNA. Singleplex qPCR was performed using two single-copy genes (CDK6 and ALB) primer sets in the same run, and the quantity from CDK6 amplification was divided by the quantity from ALB amplification. The average ± S.D. of CDK6/ALB ratio was 1.04 ± 0.09 and 1.30 ± 0.51 in normal DNA and cancer DNA, respectively. The results were generated from three independent repeats using DNA samples from 15 normal blood and 15 cancer cell lines.
Reference Primers for Nondiploid DNA
To refine and develop a new telomere qPCR assay suitable for cancer DNA, we tested two reference primer sets as we previously reported [16]. These two primer pairs are, in silico, able to amplify loci on multiple chromosomes. According to the UCSC in silico PCR database (http://genome.ucsc.edu/cgi-bin/hgPcr), the MRef1 primer set (MRef1F and MRef1R) amplifies 116 to 119–base pair products from at least 6 loci on chromosomes 1, 5, 8, 12, and 19; on the other hand, the MRef2 primer set (MRef2F and MRef2R) amplifies 104–base pair products from at least7 loci on chromosome 1, 4, 6, 7, 12, 13, and X. These primer sets can be amplified regions within glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene and its pseudogenes [19]. Thus, we named these primers “multicopy sequences (mcs)” primers. We first examined whether these mcs primer sets were interchangeable with the scg primer set in normal DNA samples. Using 19 normal blood DNA samples and 1 commercialized pooled DNA, we performed singleplex qPCR using three different primer sets (MRef1, MRef2, and HBG) in the same plate and calculated each quantity based on the standard curve generated using that primer set. The amplification signals using both MRef1 and MRef2 primer sets resulted in almost identical quantities to the one using the HBG primer set (R2 = 0.997, slope = 1.038, and intercept = -0.043 in Figure 2A; R2 = 0.996, slope = 0.996, and intercept = 0.010 in Figure 2B). The intraassay CV was very low: 1.59% between MRef1 and HBG as well as 1.49% between Mref2 and HBG. These findings suggest that the new mcs primers (both MRef1 and MRef2) are interchangeable with the scg primers when using normal DNA and function as reference primers under our condition. Because MRef2 primers amplify a product on chromosome X, we addressed the possible effects sex of the sample may have on the amplification of MRef2 compared to MRef1. The relative quantification of MRef1/ALB ratios and MRef2/ALB ratios was compared among genomic male (XY), female (XX), and mixed DNA (XY:XX = 1:1). The results of this comparison showed that while only minor differences were observed in amplifications of the female DNA signal compared to the male DNA signal, these differences are not significant enough to discount MRef2 as a potential internal control moving forward (Figure 2C). In addition, we found that the quantities were very similar between the MRef1 primers and the MRef2 primers, and the intraassay CV between MRef1 and MRef2 was 2.13% (R2 = 0.996, slope = 1.0325, and intercept = −0.0243 in Figure 2D). The average interassay CV of control DNA was 6.12% throughout the experiments. These findings suggest that all of three primer sets (Mref1, Mref2, and HBG) are interchangeable with each other.
Figure 2.

The new multiple copy sequence primer sets are interchangeable with the single-copy gene primer set in normal DNA. (A and B) The quantification of DNA utilizing either of the new multiple copy sequence primer sets (MRef1 or MRef2) was compared to the same amount of DNA quantified by the single-copy gene primer set (HBG). Each quantity was calculated based on the standard curves after singleplex qPCR. The results of this comparison illustrated an approximate 1:1 relationship between these two quantifications, indicating an interchangeable relationship (R2 = 0.9974 and 0.9965, respectively). (C) The qPCR was performed to compare the amplifications of MRef1 and MRef2 in pooled genomic male (XY), genomic female (XX), and genomic male/female mixed DNA (XX & XY). The results were analyzed by ΔΔCt method using ALB amplification. The effect of sex differences was not significant. (D) The quantification of DNA utilizing the two multiple-copy sequence primers (MRef1 and MRef2) was also compared using the same amount of DNA. Each quantity was calculated based on the standard curves after singleplex qPCR. The results of this comparison illustrated an approximate 1:1 relationship between these two quantifications, indicating an interchangeable relationship (R2 = 0.9960). All results in A, B, and D were generated using DNA from 18 normal blood samples.
Optimization of the MMQPCR Reaction Using mcs Primers
To perform monochrome qPCR in multiplex, the amount of target sequences amplified by one primer set must differ from the amount amplified by the other set such that the signals can be distinguished. In addition, differing melting temperatures (Tms) allow for separate amplification steps in the PCR cycle and help create two distinct signals by increasing the Tm of the more abundant target sequence, allowing for the recording of the two different target sequences at the two different temperatures within the PCR cycle. In the case of the traditional MMPCR,1 the telomere repeat sequences are more abundant than single-copy genes (scg) within the genome; therefore, the scg amplification signal was able to be separated with the telomere amplification signal by increasing the Tm of scg primers. In the new telomere qPCR assay presented here, multiple-copy sequences (mcs) are amplified earlier than telomere repeat sequences; therefore, the Tm of a new telomere primer set (GC-Telg and GC-Telc) was increased by adding a GC clamp to the telomere primer set (telc and telg) which was previously designed.1 DNA standard curves demonstrated that there were at least three cycle differences between mcs amplification curves and a telomere amplification curve (Figure 3A) under our MMQPCR condition. Each PCR plate always included standard curves and quality control DNA. Of note, we verified that both MMQPCR and singleplex qPCR produced the similar results under our condition (Supplemental Figure S2). To test our hypothesis that the newly developed MMQPCR could better represent relative telomere length in cancer DNA, we compared the T/M ratio of cancer DNA samples with their absolute mean telomere length by telomere southern blot analysis. The T/M ratio was calculated by dividing the telomere quantity (T) by the quantity of mcs (M) to normalize the data. The results showed that the relative ratios of T/M1 (using MRef1 primers) and T/M2 (using MRef2 primers) were correlated with absolute mean telomere length from southern blot analysis (P = 1.94E-6, R2 = 0.86 in Figure 3B; 1.94E-6, R2 = 0.87 in Figure 3C), a notable improvement from that seen in Figure 1C comparing T/S to the absolute telomere length from southern blot analysis. The average CV between T/M1 and T/M2 was 4.9%, indicating again that T/M1 and T/M2 were interchangeable. This correlation between relative T/M ratios measured by qPCR and mean telomere lengths by southern blotting supports the modified MMQPCR method as a useful for measuring relative telomere lengths in cancer DNA. The same trend was also observed in different telomere length analyses using 17 tumor tissue samples used in Figure 1C (Supplemental Table S1). It has been shown that telomeres are shorter in invasive tumors than in early stages of tumors.[20], [21] To further determine whether we can detect such relationship using the new MMQPCR method, T/M1 ratios were compared with T/S ratios between invasive breast tumor samples (IDC, n = 13) and noninvasive breast tumor samples (DCIS, n = 13). While the T/S ratios did not find a noticeable trend between two breast cancer groups, the T/M ratios showed a significant telomere shortening in invasive breast tumor samples compared to noninvasive DCIS tumor samples (P = 0.034, Figure 3D).
Figure 3.

Utility of the modified MMQPCR method for measuring relative telomere lengths in cancer DNA. (A) Representative standard curves of MMQPCR were shown along with R2 and PCR efficiency (Eff). Each qPCR run included all standard DNAs with five serial dilutions. The separation (about three cycle differences) between the amplification of the telomere primer set and the two mcs primer sets (MRef1 and MRef2) allows for use of the multiplexing method. (B and C) The relative telomere length measurements (T/M1 and T/M2) calculated utilizing the new MMQPCR methods were highly correlated with the southern blot measurements when using cell-line cancer DNA samples (n = 18). Correlation of absolute telomere length (kb) vs. T/M1 and absolute telomere length (kb) vs. T/M2 was shown in B and C, respectively. (D) Comparison of two qPCR methods (new MMQPCR as T/M1 vs. current telomere qPCR as T/S) using the same noninvasive and invasive breast cancer samples (n = 13 each). Each value was generated from three experimental repeats in duplicate.
Supplemental Figure S2.

Comparison of qPCR by singleplex and multiplex methods. The five serial dilutions of standard DNA showed similar amplifications curves between singleflex qPCR and MMQPCR using the primers of MRef1 in (A), MRef2 in (B), and GC-tel in (C). The graph for GC-tel includes amplifications of GC-tel gathered in runs with MRef 1 and MRef2.
In many cases of human cancers, telomerase activation is responsible for stable telomere maintenance; however, 5% to 15% of cases (which depend on cancer types) are associated with an alternative mechanism termed ALT (alternative lengthening of telomeres) [22]. ALT-positive cells are characterized by long and heterogeneous telomeres, ranging from 2 to > 20 kilobases (kb). To evaluate whether this MMQPCR is useful for ALT-positive cells, we measured T/M ratios using three ALT-positive cell lines (U-2 OS, Saos-2, and VA13). Although T/M ratios of U-2 OS and VA13 cells were indeed fairly higher than those of telomerase-positive cell lines (>3.5 vs. < 1.0), these ratios from the MMQPCR did not correlate very well with the telomere lengths measured via Southern blotting [23], [24]. This result is consistent with the fact that ALT-positive cells have extrachromosomal telomeric repeats [25], [26], and the telomere primers used in this study do not distinguish between chromosomal and extrachromosomal telomeric repeats. Moreover, Saos-2’s DNA was not amplified well using MRef1 and MRef2 primers. This result does not conflict with the fact that Saos-2 has a highly rearranged hyperdiploid karyotype [27], [28]. It is likely that intact DNA sequences of MRef1 and MRef2 primers no longer exist in Saos-2’s genome. Together, we conclude that the MMQPCR presented in this study might not be suitable for ALT-positive cells or/and cells which have highly rearranged chromosomes.
Discussion
In this study, we demonstrated the optimization of the telomere qPCR method in order to improve the accuracy of this assay for analyzing cancer DNA samples. Relative telomere lengths (T/M ratios) were measured in DNA from 21 cancer and immortalized cell lines including ALT-positive cells by the modified MMQPCR method as T/M ratios. The results described here were highly correlated with absolute mean telomere lengths measured by southern blotting in almost all cells and tissues (except for ALT-positive cells). On the other hand, the T/S ratios measured in these same DNA samples by the original telomere qPCR assay were not as highly correlated with the telomere lengths by southern blotting. These results highlighted the importance of choosing appropriate primers to normalize qPCR data such that the accuracy of the assay is maintained. Importantly, the T/M ratios obtained by the MMQPCR were reproducible in independent runs of the singleplex assay using both cancer and normal DNA. Other techniques outside of qPCR and Southern blotting exist for use for telomere length measurement, including a variety of quantitative fluorescence in situ hybridization (Q-FISH) methods [29], [30]. For example, metaphase Q-FISH has the benefit of being able to identify chromosome-specific telomeres, but it is only effective on mitotically active cells. Flow-FISH is another method to measure mean telomere length of interphase nuclei in combination with FISH and flow cytometry. These FISH methods usually require a high level of cytogenetic skills and are labor intensive. In addition, these assays cannot identify chromosomes with telomeres that are too short to hybridize with the probe. Recently, a new technique, Telomere Shortest Length Assay, was reported to detect telomeres from all chromosome ends from <1 kb to 18 kb using small amounts of input DNA [10]. This method has the advantage of providing more information about the shortest telomeres, while this method is still labor intensive and not applicable to ALT-positive cells. For epidemiologic studies of telomere length using a large sample size, we believe that the MMQPCR would have significant advantages of increased speed, reduced costs, reduced variability of the assay, and reduced amounts of DNA sample per assay. We also propose that this new MMQPCR method could have potential for the development of new, less invasive diagnostic tools for cancer and other diseases which have aneuploid DNA.
Conclusions
The aneuploidic nature of cancer DNA requires usage of primers that account for this aneuploidy when measuring relative telomere length. By utilizing mcs primers with telomere primers in a MMQPCR method, new diagnostic tools may be developed for cancer and other diseases with aneuploid DNA.
The following are the supplementary data related to this article.
Supplemental tables
Acknowledgments
Acknowledgement
We thank the Indiana University Simon Cancer Center for the use of the Tissue Procurement and Distribution Core and the Susan G. Komen Tissue Bank: both provided tissue specimen service. We also thank Rie Matern and Kayla Quirin for technical assistance and Xi Wu for helping initial experiments at an early stage of this project. P.N.D. was a fellow of the Student Research Program in Academic Medicine at Indiana University School of Medicine at Indianapolis. K.B. was a fellow of the Undergraduate Research Opportunity Program at Indiana University-Purdue University at Indianapolis.
Funding
This work is supported in part by National Institutes of Health under CA205434, an Indiana University Simon Cancer Center Breast Cancer Signature pilot grant, the Indiana Clinical and Translational Institute under grant #TR001107 from the NIH, National Center for Advancing Translational Sciences, an Indiana University School of Medicine Biomedical Research grant, the Showalter Foundation, the Friends for an Earlier Breast Cancer Test, the Susan G. Komen Foundation, and the American Cancer Society.
References
- 1.Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nucleic Acids Res. 2009;37:e21. doi: 10.1093/nar/gkn1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Needham BL, Adler N, Gregorich S, Rehkopf D, Lin J, Blackburn EH, Epel ES. Socioeconomic status, health behavior, and leukocyte telomere length in the National Health and Nutrition Examination Survey, 1999-2002. Soc Sci Med. 2013;85:1–8. doi: 10.1016/j.socscimed.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shalev I, Entringer S, Wadhwa PD, Wolkowitz OM, Puterman E, Lin J, Epel ES. Stress and telomere biology: a lifespan perspective. Psychoneuroendocrinology. 2013;38:1835–1842. doi: 10.1016/j.psyneuen.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Astuti Y, Wardhana A, Watkins J, Wulaningsih W. Cigarette smoking and telomere length: a systematic review of 84 studies and meta-analysis. Environ Res. 2017;158:480–489. doi: 10.1016/j.envres.2017.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu X, Han W, Xue W, Zou Y, Xie C, Du J, Jin G. The association between telomere length and cancer risk in population studies. Sci Rep. 2016;6:22243. doi: 10.1038/srep22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wentzensen IM, Mirabello L, Pfeiffer RM, Savage SA. The association of telomere length and cancer: a meta-analysis. Cancer Epidemiol Biomark Prev. 2011;20:1238–1250. doi: 10.1158/1055-9965.EPI-11-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muezzinler A, Zaineddin AK, Brenner H. Body mass index and leukocyte telomere length in adults: a systematic review and meta-analysis. Obes Rev. 2014;15:192–201. doi: 10.1111/obr.12126. [DOI] [PubMed] [Google Scholar]
- 9.Lustig AJ. Potential risks in the paradigm of basic to translational research: a critical evaluation of qPCR telomere size techniques. J Cancer Epidemiol Treat. 2015;1:28–37. doi: 10.24218/jcet.2015.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lai TP, Zhang N, Noh J, Mender I, Tedone E, Huang E, Wright WE, Danuser G, Shay JW. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun. 2017;8:1356. doi: 10.1038/s41467-017-01291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka H, Abe S, Huda N, Tu L, Beam MJ, Grimes B, Gilley D. Telomere fusions in early human breast carcinoma. Proc Natl Acad Sci U S A. 2012;109:14098–14103. doi: 10.1073/pnas.1120062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horikawa I, Suzuki M, Yoshida MA, Oshimura M. Frame-shift mutation and reduced transcript of p53 gene in a renal cell carcinoma cell line, RCC23. Hum Mol Genet. 1995;4:771–773. doi: 10.1093/hmg/4.4.771. [DOI] [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huda N, Tanaka H, Herbert BS, Reed T, Gilley D. Shared environmental factors associated with telomere length maintenance in elderly male twins. Aging Cell. 2007;6:709–713. doi: 10.1111/j.1474-9726.2007.00330.x. [DOI] [PubMed] [Google Scholar]
- 15.Herbert BS, Shay JW, Wright WE. Analysis of telomeres and telomerase. Curr Protoc Cell Biol. 2003 doi: 10.1002/0471143030.cb1806s20. [Chapter 18:Unit 18.16] [DOI] [PubMed] [Google Scholar]
- 16.Tanaka H, Beam MJ, Caruana K. The presence of telomere fusion in sporadic colon cancer independently of disease stage, TP53/KRAS mutation status, mean telomere length, and telomerase activity. Neoplasia. 2014;16:814–823. doi: 10.1016/j.neo.2014.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang ZQ, Yoshida MA, Fukuda Y, Kurihara N, Nakamura Y, Inazawa J. Molecular cytogenetic analysis of 17 renal cancer cell lines: increased copy number at 5q31-33 in cell lines from nonpapillary carcinomas. Jpn J Cancer Res. 2000;91:156–163. doi: 10.1111/j.1349-7006.2000.tb00927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giladi M, Schneiderman RS, Voloshin T, Porat Y, Munster M, Blat R, Sherbo S, Bomzon Z, Urman N, Itzhaki A. Mitotic spindle disruption by alternating electric fields leads to improper chromosome segregation and mitotic catastrophe in cancer cells. Sci Rep. 2015;5:18046. doi: 10.1038/srep18046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tso JY, Sun XH, Kao TH, Reece KS, Wu R. Isolation and characterization of rat and human glyceraldehyde-3-phosphate dehydrogenase cDNAs: genomic complexity and molecular evolution of the gene. Nucleic Acids Res. 1985;13:2485–2502. doi: 10.1093/nar/13.7.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chin K, de Solorzano CO, Knowles D, Jones A, Chou W, Rodriguez EG, Kuo WL, Ljung BM, Chew K, Myambo K. In situ analyses of genome instability in breast cancer. Nat Genet. 2004;36:984–988. doi: 10.1038/ng1409. [DOI] [PubMed] [Google Scholar]
- 21.Artandi SE, DePinho RA. Telomeres and telomerase in cancer. Carcinogenesis. 2010;31:9–18. doi: 10.1093/carcin/bgp268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bryan TM, Englezou A, Dalla-Pozza L, Dunham MA, Reddel RR. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med. 1997;3:1271–1274. doi: 10.1038/nm1197-1271. [DOI] [PubMed] [Google Scholar]
- 23.Lee M, Hills M, Conomos D, Stutz MD, Dagg RA, Lau LM, Reddel RR, Pickett HA. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014;42:1733–1746. doi: 10.1093/nar/gkt1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Scheel C, Schaefer KL, Jauch A, Keller M, Wai D, Brinkschmidt C, van Valen F, Boecker W, Dockhorn-Dworniczak B, Poremba C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene. 2001;20:3835–3844. doi: 10.1038/sj.onc.1204493. [DOI] [PubMed] [Google Scholar]
- 25.Cesare AJ, Griffith JD. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol. 2004;24:9948–9957. doi: 10.1128/MCB.24.22.9948-9957.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, Reddel RR. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27:1181–1185. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 27.Ozaki T, Neumann T, Wai D, Schafer KL, van Valen F, Lindner N, Scheel C, Bocker W, Winkelmann W, Dockhorn-Dworniczak B. Chromosomal alterations in osteosarcoma cell lines revealed by comparative genomic hybridization and multicolor karyotyping. Cancer Genet Cytogenet. 2003;140:145–152. doi: 10.1016/s0165-4608(02)00685-4. [DOI] [PubMed] [Google Scholar]
- 28.Hattinger CM, Reverter-Branchat G, Remondini D, Castellani GC, Benini S, Pasello M, Manara MC, Scotlandi K, Picci P, Serra M. Genomic imbalances associated with methotrexate resistance in human osteosarcoma cell lines detected by comparative genomic hybridization-based techniques. Eur J Cell Biol. 2003;82:483–493. doi: 10.1078/0171-9335-00336. [DOI] [PubMed] [Google Scholar]
- 29.Aubert G, Hills M, Lansdorp PM. Telomere length measurement-caveats and a critical assessment of the available technologies and tools. Mutat Res. 2012;730:59–67. doi: 10.1016/j.mrfmmm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montpetit AJ, Alhareeri AA, Montpetit M, Starkweather AR, Elmore LW, Filler K, Mohanraj L, Burton CW, Menzies VS, Lyon DE. Telomere length: a review of methods for measurement. Nurs Res. 2014;63:289–299. doi: 10.1097/NNR.0000000000000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsuo M, Kaji K, Utakoji T, Hosoda K. Ploidy of human embryonic fibroblasts during in vitro aging. J Gerontol. 1982;37:33–37. doi: 10.1093/geronj/37.1.33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental tables