

Abstract
Recent studies have demonstrated that enzyme-instructed self-assembly (EISA) in extra- or intracellular environments can serve as a multistep process for controlling cell fate. There is little knowledge, however, about the kinetics of EISA in the complex environments in or around cells. Here we design and synthesize three dipeptidic precursors (LD-1-SO3, DL-1-SO3, DD-1-SO3), consisting of diphenylalanine (L-Phe-D-Phe, D-Phe-L-Phe, D-Phe-D-Phe, respectively) as the backbone, which are capped by 2-(naphthalen-2-yl)acetic acid at the N-terminal and by 2-(4-(2-aminoethoxy)-4-oxobutanamido)ethane-1-sulfonic acid at the C-terminal. On hydrolysis by carboxylesterases (CES), these precursors result in hydrogelators, which self-assemble in water at different rates. While all three precursors selectively kill cancer cells, especially high-grade serous ovarian carcinoma cells (HGSC), by undergoing intracellular EISA, DL-1-SO3 and DD-1-SO3 exhibit the lowest and the highest activities, respectively, against the cancer cells. This trend inversely correlates with the rates of converting the precursors to the hydrogelators in PBS buffer. Because CES exists both extra- and intracellularly, we use kinetic modeling to analyze the kinetics of EISA inside cells and to calculate the cytotoxicity of each precursor for killing cancer cells. Our results indicate that (i) the stereochemistry of the precursors affects the morphology of the nanostructures formed by the hydrogelators, as well as the rate of enzymatic conversion; (ii) decreased extracellular hydrolysis of precursors favors intracellular EISA inside the cells; (iii) the inherent features (e.g., self-assembling ability and morphology) of the EISA molecules largely dictate the cytotoxicity of intracellular EISA. As the kinetic analysis of intracellular EISA, this work elucidates how the stereochemistry modulates EISA in the complex extra- and/or intracellular environment for developing anticancer molecular processes. Moreover, it provides insights for understanding the kinetics and cytotoxicity of aggregates of aberrant proteins or peptides formed inside and outside cells.
Keywords: nanostructure, self-assembly, enzyme, stereochemistry, anti-cancer
Graphical Abstract
Despite advances in cancer treatments such as surgery, chemotherapy and immunotherapy, cancer, as a class of diseases that induce abnormal cell growth, remains a public health threat and is the number two cause of death globally.1 Such a serious health problem requires the further exploration of approaches for cancer treatment. Recognizing tight ligand-receptor binding in the development of molecule-targeted chemotherapy as a likely cause of the emergence of multidrug resistance in cancer therapy, we2–11 and several other labs12–16 have been exploring molecular processes (e.g., peptidic self-assembly12, 17–42) for cancer therapy by using enzymatic reactions to generate self-assembled molecular nanostructures (e.g., nanofibers and nanoparticles) for killing cancer cells. Our previous study revealed that the intracellular enzyme-instructed self-assembly (EISA)43,44 of dipeptidic derivatives, by interacting with actin, significantly boosts the activity of cisplatin, a widely used anticancer drug, against drug-resistant ovarian cancer cells.6 However, there is little information on the kinetics of intracellular EISA and the effects of precursor stereochemistry on intracellular EISA. In this work, we examine the roles of the stereochemistry of dipeptidic precursors on the efficacy of intracellular EISA, without involving cisplatin, for killing cancer cells. We expect dipeptides of different stereochemistry to result in nanofibers with different morphologies and different rates of formation, factors that should contribute to the cytotoxicity of the nanofibers to cancer cells.
To test the above assumption, we design and synthesize three stereoisomers of diphenylalanine precursors that are capped by 2-(naphthalen-2-yl)acetic acid at the N-terminal and by 2-(4-(2-aminoethoxy)-4-oxobutanamido)ethane-1-sulfonic acid at the C-terminal. These dipeptide precursors (LD-1-SO3, DL-1-SO3, DD-1-SO3) (Scheme 1), as the substrates of carboxylesterases (CES),45–48 lose the hydrophilic 4-oxo-4-((2-sulfoethyl)amino)butanoic acid motif to form hydrogelators (LD-1, DL-1, DD-1) (Scheme 1) upon addition of CES. At certain concentrations of the precursors, enzymatic formation of LD-1, DL-1 or DD-1 results in supramolecular hydrogels, confirming that these precursors/hydrogelators are suitable molecules for EISA. While the products, formed by cleaving ester bonds, self-assemble to generate molecular nanofibers that exhibit slightly different morphologies as the result of the different stereochemistries of LD-1, DL-1, and DD-1, the cytotoxicity of the precursors is inversely correlated with the rates of converting the precursors to the hydrogelators in PBS buffer. This trend is counterintuitive and differs from the results of pericellular self-assembly,5, 9, 49 thus warranting further elucidation kinetic analysis for understanding the reaction-transport characteristics of intracellular EISA in the cellular environment, an important feature of molecular processes that has yet to be elucidated.50
Scheme 1.

Chemical structures of the precursors and the corresponding hydrogelators.
We use multivariate data fitting to elucidate the kinetics of ester hydrolysis outside and EISA inside the cells in order to evaluate the cytotoxicity of each precursor against cancer cells in a quantitative manner. Since the interaction between the drug and the cell is a complex process involving many different kinetic steps and parameters, we identify three key constitutive processes: (a) the exchange of the EISA molecules between the inside and outside of the cells, (b) the chemical mechanism that leads to supramolecular assembly through the action of the enzyme (e.g., CES), and (c) the interaction of the supramolecular assemblies with the cancer cells, resulting in cell death. Fitting appropriate subsets of a mathematical model on three separate sets of experiments allows the estimation of the kinetic parameters (i.e., the transport coefficients of molecules in and out the cells (kin/kout), the Michaelis-Menten constants (KM), the hydrogelation rate constant (Kgs), and the killing power of the assemblies (kk0, [GC]tr, ζ) associated with each of these precursors and processes (Scheme 2)). Our results indicate that the stereochemistry of the precursors affects the morphology of the nanostructures formed by the hydrogelators and the inherent features of the EISA molecules largely dictate the cytotoxicity of intracellular EISA. As the kinetic analysis of intracellular EISA, this work not only provides useful insights for understanding the kinetics and cytotoxicity of pathogenic molecular aggregates formed during biological processes, but also illustrates that the use of stereochemistry to enhance self-assembling ability and to slow down extracellular hydrolysis of small molecules is an effective approach for optimizing intracellular EISA.
Scheme 2.

Extracellular hydrolysis and cancer cell death caused by intracellular EISA.
RESULTS AND DISCUSSION
Molecular design
We choose diphenylalanine (FF) as the dipeptide backbone because of the self-assembling ability of FF in water.51–52 We connect 2-(naphthalen-2-yl)acetic acid (Nap) at the N-terminal to increase the aromatic-aromatic interaction for further enhancing the self-assembly of the peptide derivatives. To understand the effect of stereochemistry on hydrolysis rate and self-assembly, we choose three stereoisomers of the diphenylalanine backbone, L-Phe-D-Phe, D-Phe-L-Phe, and D-Phe-D-Phe. We omit the use of L-Phe-L-Phe because proteases degrade Nap-L-Phe-L-Phe to Nap-L-Phe and Phe readily.53 We install an ester bond at the C-terminal of the dipeptide derivatives to make them suitable substrates for CES. To ensure that the peptides are taken up by the cell for intracellular EISA, we also further extend the C-terminal of the diphenylalanine by attaching a taurine group (2-aminoethanesulfonic acid) (Scheme 1), because the taurine motif promotes the cellular uptake of D-peptides.54
Synthesis
Based on the chemical structures of the precursors and the corresponding hydrogelators in Scheme 1, we apply both solid phase peptide synthesis (SPPS)55 and solution phase synthesis to generate the precursors (Scheme S1). After purification of the compounds by high-performance liquid chromatography (HPLC) with an overall yield about 60%, LC-MS and 1H-NMR spectra confirm the purities of these molecules (Figures S1–S3).
CES-catalyzed self-assembly
After we obtain LD-1-SO3, DL-1-SO3, and DD-1-SO3, we test their ability to serve as precursors for EISA in PBS buffer. The precursors dissolve in PBS buffer to form clear solutions (0.2 wt%). After the addition of 1 U/mL of CES, all of solutions turn into hydrogels in 24 h (Figure S4). From LC-MS analysis of the hydrogels, we find that, catalyzed by CES for 24 h, 87%, 91%, and 85% of the precursors LD-1-SO3, DL-1-SO3, DD-1-SO3, respectively, become the hydrogelators LD-1, DL-1, and DD-1. Transmission electron microscopy (TEM) of the hydrogels reveals that the nanofibers, acting as the matrices of the hydrogels, exhibit different morphologies. Generated from EISA, the hydrogel of LD-1 mainly contains helical nanoribbons with some straight nanofibers; the hydrogel of DL-1 contains straight ribbons or narrow nanosheet-like structures with different widths as matrices; and the hydrogel of DD-1 consists mainly of nanofibers with a diameter of 8 ± 2 nm.
After using enzymatic hydrogelation to establish that the molecules we designed serve as precursors/hydrogelators for EISA, we examine the self-assembly of the hydrogelators at lower concentrations after adding CES to solutions of the precursors, because the precursors exhibit cytotoxicity at concentrations much lower than the concentrations needed for gelation (vide infra). As shown in Figure 1, we choose three concentrations (200, 100 and 50 μM) for each of the precursors, which dissolve in PBS buffer to form transparent solutions. After addition of CES to each solution for 24 h to convert the precursors to the corresponding hydrogelators, we use TEM to examine the reaction mixtures. For the enzymatic conversion of LD-1-SO3 to LD-1, TEM shows a considerable quantity of twisted nanoribbons (with a pitch of 130 ± 10 nm) at the precursor concentration of 200 μM, plus some nanoparticles. When the precursor concentration drops to 100 μM, the nanoribbons become narrower to form twisted nanofibers with a new, shorter pitch (72 ± 4 nm). There are hardly any nanofibers, but rather nanoparticles (700–900 nm in diameter) in the TEM when the concentration of the precursor is 50 μM. While the addition of CES to DL-1-SO3 (200 μM) results in short nanoribbons, there are hardly any nanoribbons remaining, except some very short nanofibers mixed with many nanoparticles, when DL-1-SO3 is present at 100 μM. When the concentration of DL-1-SO3 drops further to 50 μM, TEM shows that the addition of CES generates nanoparticles of diameter 320–510 nm. Unlike the cases of LD-1-SO3 or DL-1-SO3, adding CES to DD-1-SO3 (200 μM) produces relatively high-density twisted nanofibers (10–12 nm diameter) with indeterminate pitch. At 100 μM of DD-1-SO3, the amount of EISA-generated nanofibers decreases, and the twisted nanofibers (11 ± 2 nm in width) exhibit a well-defined pitch (43 ± 2 nm). When the concentration of DD-1-SO3 is 50 μM, there are still some fiber-like structures, together with nanoparticles (about 60 nm diameter) after CES-catalyzed hydrolysis. Among the three hydrogelators, DD-1 appears to have the highest self-assembling ability. For example, at 100 μM, solutions of both LD-1 and DD-1 contain fiber-like structures, while DL-1 solution contains only aggregates. In addition, solutions of DD-1 contain fiber-like structures at concentrations as low as 50 μM. On the other hand, DL-1 shows the weakest self-assembling ability; its 200 μM solution contains only a few short nanoribbon like-structures. These results provide useful morphological information about the assemblies of these stereoisomers formed by the EISA process.
Figure 1.

TEM images of samples formed 24 h after adding CES (1 U/mL) to solutions of LD-1-SO3, DL-1-SO3 and DD-1-SO3 at 200 μM, 100 μM, and 50 μM. All are in PBS buffer, pH = 7.4. Scale bar = 100 nm.
Circular dichroism
To gain further understanding of the molecular conformations of the hydrogelators in the hydrogels, we measure the circular dichroism (CD) of the hydrogels. Peaks from 200 nm to 250 nm originate from π-π* and n-π* transitions of amide groups. The hydrogel formed by LD-1 shows a strong positive peak at 247 nm while the hydrogel formed by DL-1 exhibits a negative peak at 234 nm (Figure S5). LD-1 and DL-1 are enantiomers, but the CD spectra of the hydrogels are slightly unsymmetrical, which likely originates from inclusion of CES in the hydrogels, which makes the hydrogels diastereromeric. Additionally, the secondary structure formed by the self-assembly process can be different because the rates of the enzymatic reactions probably differ for LD-1-SO3 and DL-1-SO3 (vide infra). While the CD spectra of the hydrogels of LD-1 and DL-1 indicate the presence of alpha-helical-like structures, the CD spectrum of the hydrogel formed by DD-1 shows both positive (220 nm) and negative peaks (237 nm), suggesting beta-sheet like structures. These results indicate that the stereochemistry of the hydrogelators indeed dictates their secondary structure in the assembled state.
Rheometry
From rheological measurements on the hydrogels formed by the three precursors after the addition of 1 U/mL of CES for 24 h, we compare the viscoelastic properties of the hydrogels. The storage moduli (G′) of all three hydrogels are larger than their loss moduli (G″), and both G′ and G″ are independent of frequency, which confirms the viscoelastic features of the hydrogels (Figure S6). In addition, we obtain the maximum storage moduli from a strain sweep. As shown in Figure S6A, the hydrogel of DL-1 exhibits the highest maximum storage modulus at 58 Pa. The maximum storage modulus of the LD-1 hydrogel is 51 Pa, slightly lower than the DL-1 hydrogel. The maximum storage modulus of the hydrogel of DD-1 is the smallest among the three hydrogels (13 Pa). These maximum storage moduli are consistent with the extent of the hydrolysis catalyzed by CES. We also measure the critical strain values at which the storage modulus G′ starts to drop drastically due to breakdown of the matrices of the hydrogels. The values of critical strain of the hydrogels formed by LD-1, DL-1 and DD-1 are 3.2%, 2.6% and 4.8%, respectively. The DD-1 hydrogel shows the highest critical strain, indicating that it is the most resilient among the three hydrogels. The critical strain value of the DL-1 hydrogel is the lowest, suggesting that this hydrogel is more rigid than the other two. These results imply that the stereochemistry of hydrogelators affects the viscoelasticity of the hydrogels. The resilience of the hydrogels also reflects the self-assembling ability of the hydrogelators.
Cytotoxicity
We test the cytotoxicity of these precursors on high-grade serous ovarian cancer (HGSC) cell lines (KURAMOCHI, OVSAHO, JHOS-2 and JHOS-4),56 cervical cancer cells (HeLa), as well as bone marrow stromal cells (HS-5, a normal cell line with low expression of CES57). We summarize the IC50 and IC90 values of these molecules in Figure 2A and 2B, respectively. The IC50 values of LD-1-SO3 on the cancer cell lines are around 50 μg/mL while its IC50 on a normal cell line (HS-5) is above 80 μg/mL. Similarly, the IC90 values of LD-1-SO3 on cancer cells are around 73 μg/mL, much lower than the IC90 for HS-5 cells, 136 μg/mL. The IC50 and IC90 values of DL-1-SO3 on the cancer cell lines are about 80 and 110 μg/mL, respectively, while the IC50 and IC90 values on HS-5 are 117 and 172 μg/mL, respectively. DD-1-SO3 shows the highest cytotoxicity, with IC50 and IC90 values on cancer cells of 24 and 29 μg/mL, respectively. DD-1-SO3 also shows excellent selectivity—IC50 and IC90 against HS-5 cells are 58 and 68 μg/mL, respectively. Figure 2C summarizes the viability of cancer cells and normal cells treated with 73 μg/mL of LD-1-SO3, DL-1-SO3 or 37 μg/mL of DD-1-SO3 for 48 h. After treatment with LD-1-SO3, all the cancer cell lines show cell viability below 30%, while HS-5 shows cell viability above 60%. DL-1-SO3 kills more than 40% of cancer cells; notably about 80% of HeLa cells are dead. On the other hand, more than 90% of HS-5 cells are still alive after treatment with 73 μg/mL of DL-1-SO3. DD-1-SO3 thus shows even better selectivity at 37 μg/mL dosage. While the viabilities of HGSC cells are less than 10%, more than 70% of HS-5 cells remain viable after the treatment with DD-1-SO3. The overall trend is that DD-1-SO3 is the most cytotoxic molecule against the ovarian cancer cells among the three precursors, and DL-1-SO3 shows the lowest cytotoxicity. These results also confirm that all three precursors selectively kill cancer cells, particularly HGSC cells.
Figure 2.

(A) IC50 and (B) IC90 values of the precursors LD-1-SO3, DL-1-SO3 and DD-1-SO3 against KURAMOCHI, OVSAHO, JHOS-2, JHOS-4, HeLa and HS-5 cells on the second day; (C) Cell viability of KURAMOCHI, OVSAHO, JHOS-2, JHOS-4, HeLa and HS-5 cells treated with 100 μM (73 μg/mL) of LD-1-SO3, DL-1-SO3 or 50 μM (37 μg/mL) of DD-1-SO3 for 48 h.
Inverse correlation of hydrolysis rate and cytotoxicity of the precursors
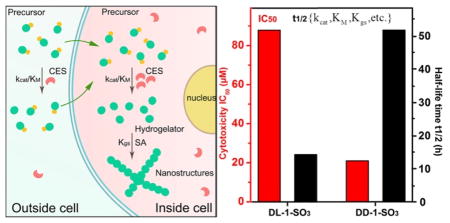
After we evaluate the cytotoxicity of the precursors, we test the enzyme catalytic rate by adding 0.1 U/mL of esterase to 500 μM solutions of the three precursors. We use HPLC to quantify the compositions of the solutions during 36 h incubation at 37 °C, and summarize the decrease of precursors with time (Figure 3A). Surprisingly, more than 60% of DD-1-SO3 remains, while only about 34% of DL-1-SO3 remains in the solution after 36 h. About 55% of LD-1-SO3 is converted to LD-1 by the esterase after 36 h. These results confirm that DD-1-SO3 is the most stable, while DL-1-SO3 is the most easily hydrolyzed by esterase among the three precursors. To examine the relationship between the hydrolysis rate and the cytotoxicity of these precursors, we plot the IC50 values (against JHOS-4 cells) and half-life time (t1/2) of the precursors (Figure 3B). Notably, the precursor having a longer half-life shows a smaller IC50. DD-1-SO3 is the most cytotoxic molecule among the three, though its hydrolysis catalyzed by esterases is the slowest. DL-1-SO3 shows the highest hydrolysis rate and the lowest cytotoxicity.
Figure 3.

(A) Decrease of the amount of precursors with time after their incubation with 0.1 U/mL of esterase at 37 °C. The precursors were dissolved in pH 7.4 PBS buffer at a concentration of 500 μM; (B) IC50 values (against JHOS-4) and t1/2 values of LD-1-SO3, DL-1-SO3 and DD-1-SO3.
Esterase inhibitors
The trend shown in Figure 3B for intracellular EISA is opposite to that for extracellular EISA,58 implying that these three precursors start hydrolyzing before they enter the cells. To examine whether CES is present in the culture medium, we dissolve 6-carboxyfluorescein diacetate (6-CFDA), which is a substrate for esterase, in the fresh culture medium or PBS buffer. As shown in Figure 4A, without addition of 6-CFDA, all of the culture media and PBS buffer barely show any fluorescence. However, after being incubated with 6-CFDA, the culture media (DMEM, DMEM/F12 and MEM) show about a 1000-fold increase in fluorescence, while the PBS buffer hardly shows any increase. This result confirms that the culture media used for the cell viability experiment contain soluble esterases. The esterases in the culture medium certainly catalyze the hydrolysis of the precursors, which not only explains the effectiveness of DD-1-SO3, but also underscores the unique advantage of using D-peptide-based precursors to slow down extracellular hydrolysis, thereby promoting effective intracellular EISA.
Figure 4.

(A) Fluorescence of medium and PBS buffer with and without addition of 6-CFDA (final concentration of 6-CFDA is 25 μM). (B, C) IC50 (48 h) of LD-1-SO3, DL-1-SO3 and DD-1-SO3 alone or together with troglitazone (10 μM), loperamide (10 μM), BNPP (100 μM) or mixture of troglitazone (10 μM) and loperamide (10 μM) (B) on OVSAHO cells and (C) on HeLa cells.
The esterases in the culture medium are analogous to the esterases in the blood, so our preparation offers a simple model system to explore approaches for enhancing intracellular EISA. Considering that there are more esterases inside cells than in serum, it would be reasonable to use esterase inhibitors to minimize the premature hydrolysis of the precursors before they enter the cells. Thus, we check the cytotoxicity of the precursors together with several CES inhibitors on OVSAHO cells and HeLa cells. We use the inhibitors at concentrations that are innocuous to the cells. As shown in Figure 4B and Figure S7, after treatment with LD-1-SO3 and the CES1 inhibitor troglitazone (10 μM), the viability of OVSAHO cells changes little. When BNPP (100 μM), an irreversible CES inhibitor, is used to treat OVSAHO cells together with LD-1-SO3, cell viability is decreased. When OVSAHO cells are treated with LD-1-SO3 and the CES2 inhibitor loperamide (10 μM), the viability of OVSAHO decreases considerably. When we use LD-1-SO3 combined with both troglitazone and loperamide (10 μM each) to treat OVSAHO cells, the cells show much lower viability. The use of the above-mentioned inhibitors to combine with DL-1-SO3 or DD-1-SO3 results in an overall trend that the addition of CES2 inhibitors alone or together with CES1 inhibitors causes a significant decrease of the viability of OVSAHO cells. Co-incubation of BNPP (a non-specific CES inhibitor) with the precursors also decreases cell viability, although it is less effective than the CES2 inhibitors. The addition of CES1 inhibitors barely changes the viability of OVSAHO cells, indicating that CES1 inhibitors are unable to rescue OVSAHO cells. In Figure 4C and Figure S8, we show the results of treating HeLa cells in the same way as the OVSAHO cells. The CES2 inhibitor causes the highest decrease in cell viability. The nonspecific CES inhibitor (BNPP) also helps decrease the viability of HeLa cells, while addition of a CES1 inhibitor has little effect on HeLa cells. The results in Figure 4 confirm that CES inhibitors help inhibit the esterases in culture medium, increasing the amount of precursors in the culture medium, thus allowing more molecular precursors to enter the cells for intracellular EISA.
ELISA
To gain insight into the mechanism of cell death caused by intracellular EISA, we use ELISA59,60 to detect the expression levels of six key signaling molecules of the cell death pathway61,62 in HeLa cells treated with the three precursors at 50 μM for up to 36 h. As shown in Figure 5A, after treatment with 50 μM LD-1-SO3, there is about a 2.5-fold increase in the amount of active-PARP63 in HeLa cells after 36 h of incubation. Also, the amount of active-caspase3 increases slightly after 24 and 36 h of incubation. For HeLa cells treated with DL-1-SO3, the level of active-PARP in HeLa cells starts to increase after 24 h of incubation and shows about a 3.5-fold increase after 36 h (Figure 5B). When HeLa cells are treated with 50 μM DD-1-SO3, the relative amounts of active-caspase3 and active-PARP increase significantly (Figure 5C). Active-PARP shows about a 7-fold increase and active-caspase3 shows about a 5.5-fold increase, which are higher than the increase of these two signaling molecules in HeLa cells treated with LD-1-SO3 and DL-1-SO3. The level of phospho-Bad also increases slightly after 24 h treatment with DD-1-SO3. These results suggest that HeLa cells treated with LD-1-SO3 undergo PARP- and caspase-dependent cell death, and that DL-1-SO3-treated cells undergo PARP-dependent cell death. Besides the activation of PARP and caspase364 in them, the HeLa cells treated with DD-1-SO3 also exhibit an increase in phospho-Bad,65 suggesting that DD-1-SO3 likely results in cell death via an intrinsic cell death signaling pathway.
Figure 5.

Change of relative amount of apoptosis signal molecules over time in HeLa cells treated with 50 μM of (A) LD-1-SO3, (B) DL-1-SO3, (C) DD-1-SO3.
F-actin staining
In order to examine the effect of intracellular self-assembly on the dynamics of actin filaments, we use Alexa fluor 633 phalloidin66 to stain the actin filaments of HeLa cells treated with the precursors. As shown in Figure 6, we treat the HeLa cells with 50 μM LD-1-SO3, DL-1-SO3, and DD-1-SO3 or culture medium for 20 h. Compared with the HeLa cells in the control group, HeLa cells treated with LD-1-SO3 show much less well-defined long actin filaments that extend throughout the cytoplasm. There are some short actin filaments and some red dots inside the cells. Similarly, after treatment with DL-1-SO3, actin filaments inside the HeLa cells also change. There are hardly any long actin filaments that can form the network of the cytoskeleton, but the actin filaments are slightly shorter than those in the HeLa cells treated with LD-1-SO3. Similar to HeLa cells treated with LD-1-SO3 and DL-1-SO3, DD-1-SO3-treated HeLa cells exhibit few well-defined actin filaments. Here there are more red dots and many fewer short actin filaments present inside the cells compared to the LD-1-SO3-treated cells. These results confirm that intracellular assemblies of these hydrogelators disrupt actin filaments in the cells, which likely contributes to cell death. DD-1-SO3 and DL-1-SO3 show the most and least obvious actin disruption, respectively, in agreement with the relative cytotoxicities of the precursors and further supporting the notion that intracellular EISA is a powerful process to interfere with actin dynamics, thereby killing cancer cells.57
Figure 6.

Fluorescence images of HeLa cells stained with Alexa Fluor 633 Phalloidin (F-actin) and Hoechst (nuclei) after treatment with 50 μM LD-1-SO3, DL-1-SO3, and DD-1-SO3 or culture medium (control) for 20 h. Scale bar = 10 μm.
Kinetic analysis
To quantitatively understand the kinetics of the EISA process, we fit the key reaction parameters for EISA occurring outside and inside the cells.67–73 To this end, we developed a mathematical model to simulate the mean-field kinetics of the physical, chemical and biological processes involved in our system and summarized in Scheme 3. The general set of ordinary differential equations constituting our model in dimensionless form reads:
Scheme 3.

Schematic view of the network of physical, chemical and biological interactions between the molecules and cells in the system.
Note: kin and kout: transport rate constants; kd: HeLa cell doubling rate constant, which is 4.81×10−4 min−1; kg: gelation rate constant which is 105 min−1; ks: dissociation rate constant of a gel; ktrap: rate constant of trapping process; kcat: catalytic rate constant; kk([GC]): killing rate constant; kf: forward rate constant; kr: reverse rate constant. S stands for precursor, P stands for free hydrogelators in solution; G stands for hydrogelators in the assemblies. The superscripts M and C designate species in the extracellular medium and inside the cells, respectively.
Here c, sM, pM, gM, sC, pC and gC are dimensionless variables respectively representing the population of cancer cells and the concentrations of precursors, free hydrogelators in solution and hydrogelators in the assemblies (gel). The superscripts M and C designate species in the extracellular medium (M) and inside the cells (C), respectively. The model contains 15 parameters, which we divide into 4 groups:
Group 0: S0, C0, kd, kg, Kgs. Parameters that we can directly measure or estimate from the literature.
Group 1: k̃cat, KM, ζ. Parameters related to the chemical reactions occurring within each compartment.
Group 2: δM, δC, kin, kout. Parameters related to the transport between the media (M) and cells (C) and to the different enzymatic concentrations in the two compartments.
Group 3: kk0, σ, [GC]tr. Parameters characterizing the cytotoxicity of the intracellular gels.
The parameters S0, C0, kd, and kg respectively are: the initial concentration of precursor; the volume fraction of tumor cells to the total volume; the rate constant of cell doubling; and the rate constant of gelation. The definition of the other parameters is given in the caption of Table 1. The precise definition of these quantities, as well as the systematic derivation of the above equations can be found in Section 3 of the Supporting Information.
Table 1.
Rate and equilibrium constants calculated by multivariate data fitting.
| Constant | DL-1-SO3 | LD-1-SO3 | DD-1-SO3 |
|---|---|---|---|
|
| |||
| Kgs (mM−1) | 1.31×101 | 2.19×101 | 4.52×101 |
|
| |||
| k̃cat (mM·min−1) | 2.63×10−2 | 2.78×10−1 | 6.38×10−3 |
| KM (mM) | 20.3 | 31.1 | 6.58 |
| ζ | 0.96 | 1.13 | 1.98 |
|
| |||
| kin (min−1) | 5.06×10−2 | 5.06×10−2 | 5.06×10−2 |
| kout (min−1) | 0.79×10−2 | 0.79×10−2 | 0.79×10−2 |
| δM | 0.83 | 0. 83 | 0. 83 |
| δC | 15.7 | 15.7 | 15.7 |
|
| |||
| kk0 (min−1) | 2.11×10−3 | 2.97×10−3 | 6.29×10−3 |
| σ (mM−1) | 0.48 | 1.24 | 3.69 |
| [GC]tr (mM) | 2.99 | 2.29 | 1.30 |
Kgs: equilibrium constant for the gelation process; k̃cat: catalytic rate constant; KM: Michaelis-Menten constant; ζ: gel-specific constant representing the average number of empty sites in the gel structure per molecule of hydrogelator available for the inclusion of one precursor; kin and kout: transport rate constants; δM: ratio of enzyme concentration in the culture medium to enzyme concentration used in the hydrolysis experiment; δC: ratio of enzyme concentration in the cell to enzyme concentration used in the hydrolysis experiment. [GC]tr (a threshold value), kk0 (a plateau value) and σ (slope) of the killing efficacy curve. Known parameters: kd: HeLa cell doubling rate constant which is 4.81×10−4 min−1; kg: gelation rate constant which is 105 min−1.
We use the above model to estimate the values of the control parameters from our experimental data. Our aim is to compare the values for the DL-1-SO3, LD-1-SO3 and DD-1-SO3 stereoisomers and to understand the reason behind their different cytotoxicity (see Table 1). Whereas the parameters in Group 0 are derived from our experiments or the literature, we use a numerical fitting procedure to obtain the values of the other parameters. Our approach is to fit Group 1, Group 2 and Group 3 in this order, on datasets coming from ad hoc experiments.
In order to fit group 1 we measure and analyze the kinetics of assembly in the absence of cancer cells, by measuring the hydrolysis rates of the three precursors. We choose three initial concentrations for each compound (500, 200, and 100 μM) to incubate together with esterase (0.1 U/mL) at 37 °C for 36 h, and analyze and plot the percentages of the remaining precursors. To understand at which concentration the compounds start to self-assemble, we determine the CMC (critical micelle concentration) of the three precursors and the hydrogelators (Table S1). We also measure the CMC of mixtures of precursors and their corresponding hydrogelators at different ratios (1:3, 1:1 and 3:1) to check if the coexistence of precursor and hydrogelator affects the CMC values (Figure S9). The CMC measurement shows that the precursors always have higher CMCs than their corresponding hydrogelators. The existence of precursors in the solution has little effect on the overall CMC (i.e., CMC values of the mixtures of precursors and hydrogelators). Though it is hard to test intracellular CMCs in live cells, previous study suggests that CMC in cell lysates is about 10 times lower than that in PBS buffer.74,75 To clarify if the hydrolysis of the three precursors by esterase is reversible and to check if the esterase catalyzes ester bond formation, we co-incubate the hydrogelators (initial concentration 500 μM) with 4-oxo-4-((2-sulfoethyl)amino)butanoic acid as well as esterase (0.1 U/mL). We check the composition of the mixtures by LC-MS after 48 h incubation. The results (Table S2) indicate that the esterase appears not to catalyze the reverse reaction of hydrolysis, and the hydrolysis process of the three precursors by the esterase is irreversible. Based on this fact, the deviations of the trend of the hydrolysis curves from simple Michaelis-Menten kinetics can be explained by assuming that there are some precursor molecules trapped in the assemblies of the hydrogelators.
For group 2, we quantify the intracellular and extracellular concentrations of the precursors as well as the hydrogelators after incubating 100 μM precursor with HeLa cells for 1, 2, 4, 8 and 12 h. These time intervals are smaller than the doubling time of the cells (about 24 h): this treatment allows us to assume constancy of the cell population, so that we can neglect the contribution of terms associated with cell proliferation and death and use the chemical constants previously found for group 1 for the cell-free process. After collecting cell lysates and culture medium following incubation, we obtain intracellular and extracellular concentrations of the precursors and hydrogelators by analyzing the cell lysates as well as the culture media using LC-MS (Table S3 and S4). All of the measurements of CMCs, intracellular concentrations, and extracellular concentrations are repeated for three times. The calculated standard deviations indicate that the measurements are reproducible.
Finally we fit the only remaining unknown kinetic constants in group 3 (i.e., kk0, σ and [GC]tr) — the ones associated with the killing power of each assembly — from viability data of cell cultures exposed to different concentrations of precursor during 72 h.
Detailed information about the fittings is also provided in section 3 of the supporting information (Figure S9–S14, Table S7). The model is quite successful in reproducing the trends in the experimental data. One interesting thing to notice is that although the DD-1-SO3 stereoisomer produces the smallest concentrations of intracellular gel (see center row of Figure S14), it is nevertheless the quickest and most effective in killing the tumor cells. This apparently counterintuitive result is due to the fact that the killing efficacy is higher for DD-1-SO3 than LD-1-SO3 than DL-1-SO3 (see bottom row of Figure S14 or Figure 7). The overall killing efficacy of a gel can be estimated by considering the shape of kk([GC]) or, alternatively, at the values of the three parameters defining it: the maximum killing power (or the top plateau of the S-shaped curve), kk0; the slope at the jump from 0 to maximum killing power, σ; and the threshold at which the jump happens, [GC]tr. The overall killing power is greater when the [GC]tr is small and the value of kk0 is large. The role played by the slope σ depends on the location of the jump [GC]tr. If the jump occurs at a high value of [GC], a smaller σ will ensure a higher killing efficacy for a wider range of gel concentrations before the jump; if, however, the jump occurs at a low value of [GC], a larger σ will ensure that the maximum killing power is achieved sooner after the jump. Figure 7 and Table 1 demonstrate that the maximum killing power is larger for DD-1-SO3 than for LD-1-SO3 than for DL-1-SO3. The jump in kk([GC]) occurs for DD-1-SO3 first, for LD-1-SO3 second and for DL-1-SO3 third, while the slopes of the DD-1-SO3 and LD-1-SO3 curves are comparable. This makes the DD-1-SO3 stereoisomer the most potent among the three, followed by LD-1-SO3 and then DL-1-SO3, which is likely because of the different morphologies formed by the corresponding hydrogelators. The high killing efficacy of DD-1-SO3 is also likely due to that self-assembling ability of the hydrogelators also play critical role for intracellular EISA killing cancer cells, in addition to the contribution of morphological differences of the nanofibers.
Figure 7.

Values of the killing rate constants of the three stereoisomers DL-1-SO3 (dark blue), LD-1-SO3 (dark red) and DD-1-SO3 (dark green) as a function of the intracellular gelator concentration [GC].
CONCLUSION
In summary, the investigation of stereochemical isomers of the building block for intracellular self-assembly supports the notion that intracellular self-assembly catalyzed by intracellular CES is an effective approach for killing HGSC cells (Scheme 4). All of the three precursors show excellent selectivity against HGSC cells and HeLa cells. It is noteworthy that DD-1-SO3 is the slowest to be converted into the corresponding hydrogelator DD-1 among the three molecules, but exhibits the highest cytotoxicity. This result highlights the complexity of the extracellular as well as intracellular environment when the enzymes are unequally present outside and inside cells. Besides confirm the importance of self-assembling ability, this work underscores the advantages of using D-peptides for intramolecular EISA, since D-peptides have excellent stability in the cellular environment, which is a critical factor for the development of potent precursors of intracellular EISA against cancer cells. More importantly, the use of a CES2 inhibitor drastically increases the inhibitory activity of DL-1-SO3, not only suggesting the combination of enzyme inhibition and enzyme reaction for precisely targeting cancer cells, but also implying that regulating the activity of CES2 in serum may become part of the treatment. The intracellular EISA also has potential applications such as being applied in combination therapy. Our previous research indicated that DD-1-SO3 could significantly boost the activity of an anti-cancer drug, cisplatin.6 Moreover, this work provides useful insights for understanding the cytotoxicity of the molecular aggregates of aberrant peptides that cause neurodegenerative diseases. For example, cytotoxic peptides usually result from extracellular and intracellular hydrolysis of proteins. Results summarized in Table S5 indicate that the concentrations of the esterase inhibitors inside cells are too low to be detectable. The use of an inhibitor of proteolytic enzymes that is unable to enter the cell rapidly may aggravate the disease. Though proteases may play a role in the hydrolysis of the precursors, the incubation of proteinase K with the precursors is unable to produce the hydrogelators (Table S6). This result also agrees with the previous report that D-amino acids reduce the susceptibility of small peptides to proteolysis.76 Thus, this work underscores the need for perspectives and approaches to develop drugs for treating neurodegenerative diseases.
Scheme 4.

Relationship between the rates of enzymatic conversion of the precursors and the cancer cell inhibitions caused by intracellular EISA.
METHODS
TEM measurement
We first placed 5 μL of sample solutions on glow discharge thin carbon-coated copper grids (400 meshes, Pacific Grid-Tech) and incubated for 30s at room temperature. Then we washed the grid with ddH2O for three times. Finally we stained the sample-loaded grid with three successive drops of UA (uranyl acetate) and allowed it to dry in air.
MTT assay
The cytotoxicity was determined by the viability of cells by MTT assay. The cells were seeded in exponential growth phase in a 96-well plate at a concentration of 1 × 104 cell/well with 100 μL of culture medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/ml streptomycin. The cells were allowed to attach to the wells for 24 h at 37 ºC, 5% CO2. The culture medium was removed, and 100 μL culture medium containing compounds (immediately diluted from fresh prepared stock solution of 10 mM) at gradient concentrations (0 μM as the control) was placed into each well. Culture medium was regarded as the control. After culturing at 37 ºC, 5% CO2 for 24 h, 48 h and 72 h, 10 μL of 5 mg/mL MTT ((3-(4,5-DimethylthiazoL-2-yl)-2,5-diphenyltetrazolium bromide) was added to each well, and the plated cells were incubated in the dark for another 4 h. 100 μL of SDS-HCl solution was added to each well to stop the reduction reaction and to dissolve the purple formazan. After incubation of the wells at 37 ºC overnight, the absorbance of each well at 595 nm was measured by a microplate reader.
Measurement of CES activity
To examine whether CES is present in the culture medium, we dissolved 6-carboxyfluorescein diacetate (6-CFDA), which is a substrate of esterase, in the fresh culture medium or PBS buffer. After 30 min incubation of 6-CFDA in the culture medium at 37 °C, we determined the fluorescence by using a microplate reader equipped with 485 nm excitation and 535 nm emission filters.
CMC measurements
A series of precursor or hydrogelator solutions were prepared in pH 7.4 PBS buffer and incubated together with Rhodamine 6G (5 μM). The absorbance from 520 to 540 nm was measured by using a Biotek Synergy 4 hybrid multi-mode microplate reader, and λmax was determined.
Supplementary Material
Acknowledgments
This work was partially supported by NIH (R01CA142746), NSF (MRSEC-1420382 and CHE-1362477), and the W.M. Keck Foundation. We thank the Brandeis EM and Optical Imaging facilities for TEM.
Footnotes
The supporting information contains details of the synthesis, NMR Spectra and LC-MS data for all compounds, rheological data, and details of the kinetic model. This material is available free of charge on the ACS Publications website.
References
- 1.Cancer Facts & Figures 2015. American Cancer Society; Atlanta: 2015. [Google Scholar]
- 2.Yang Z, Xu K, Guo Z, Guo Z, Xu B. Intracellular enzymatic formation of nanofibers results in hydrogelation and regulated cell death. Adv Mater. 2007;19:3152–3156. [Google Scholar]
- 3.Gao Y, Kuang Y, Guo Z-F, Guo Z, Krauss IJ, Xu B. Enzyme-Instructed Molecular Self-assembly Confers Nanofibers and a Supramolecular Hydrogel of Taxol Derivative. J Am Chem Soc. 2009;131:13576–13577. doi: 10.1021/ja904411z. [DOI] [PubMed] [Google Scholar]
- 4.Li J, Gao Y, Kuang Y, Shi J, Du X, Zhou J, Wang H, Yang Z, Xu B. Dephosphorylation of D-Peptide Derivatives to Form Biofunctional, Supramolecular Nanofibers/Hydrogels and Their Potential Applications for Intracellular Imaging and Intratumoral Chemotherapy. J Am Chem Soc. 2013;135:9907–9914. doi: 10.1021/ja404215g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q, Xu B. Pericellular Hydrogel/Nanonets Inhibit Cancer Cells. Angew Chem Int Ed. 2014;53:8104–8107. doi: 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z, Liu J, Dinulescu DM, Xu B. Enzyme-Instructed Intracellular Molecular Self-Assembly to Boost Activity of Cisplatin against Drug-Resistant Ovarian Cancer Cells. Angew Chem Int Ed. 2015;54:13307–13311. doi: 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou J, Du X, Yamagata N, Xu B. Enzyme-Instructed Self-Assembly of Small D-Peptides as a Multiple-Step Process for Selectively Killing Cancer Cells. J Am Chem Soc. 2016;138:3813–3823. doi: 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Z, Wang H, Du X, Shi J, Li J, Xu B. Minimal C-terminal modification boosts peptide self-assembling ability for necroptosis of cancer cells. Chem Commun. 2016;52:6332–6335. doi: 10.1039/c6cc02282k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B. Enzyme-Instructed Self-Assembly for Spatiotemporal Profiling of the Activities of Alkaline Phosphatases on Live Cells. Chem. 2016;1:246–263. doi: 10.1016/j.chempr.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Feng Z, Wu D, Fritzsching KJ, Rigney M, Zhou J, Jiang Y, Schmidt-Rohr K, Xu B. Enzyme-Regulated Supramolecular Assemblies of Cholesterol Conjugates against Drug-Resistant Ovarian Cancer Cells. J Am Chem Soc. 2016;138:10758–10761. doi: 10.1021/jacs.6b06075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du XW, Zhou J, Xu B. Ectoenzyme switches the surface of magnetic nanoparticles for selective binding of cancer cells. J Colloid Interface Sci. 2015;447:273–277. doi: 10.1016/j.jcis.2014.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I. Controlling Cancer Cell Fate Using Localized Biocatalytic Self-Assembly of an Aromatic Carbohydrate Amphiphile. J Am Chem Soc. 2015;137:576–579. doi: 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka A, Fukuoka Y, Morimoto Y, Honjo T, Koda D, Goto M, Maruyama T. Cancer Cell Death Induced by the Intracellular Self-Assembly of an Enzyme-Responsive Supramolecular Gelator. J Am Chem Soc. 2015;137:770–775. doi: 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]
- 14.Wang H, Yang Z. Molecular hydrogels of hydrophobic compounds: a novel self-delivery system for anti-cancer drugs. Soft Matter. 2012;8:2344–2347. [Google Scholar]
- 15.Huang P, Gao Y, Lin J, Hu H, Liao H-S, Yan X, Tang Y, Jin A, Song J, Niu G, Zhang G, Horkay F, Chen X. Tumor-Specific Formation of Enzyme-Instructed Supramolecular Self-Assemblies as Cancer Theranostics. Acs Nano. 2015;9:9517–9527. doi: 10.1021/acsnano.5b03874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kalafatovic D, Nobis M, Son JY, Anderson KI, Ulijn RV. MMP-9 triggered self-assembly of doxorubicin nanofiber depots halts tumor growth. Biomaterials. 2016;98:192–202. doi: 10.1016/j.biomaterials.2016.04.039. [DOI] [PubMed] [Google Scholar]
- 17.Fry HC, Garcia JM, Medina MJ, Ricoy UM, Gosztola DJ, Nikiforov MP, Palmer LC, Stupp SI. Self-Assembly of Highly Ordered Peptide Amphiphile Metalloporphyrin Arrays. J Am Chem Soc. 2012;134:14646–14649. doi: 10.1021/ja304674d. [DOI] [PubMed] [Google Scholar]
- 18.Lock LL, Reyes CD, Zhang PC, Cui HG. Tuning Cellular Uptake of Molecular Probes by Rational Design of Their Assembly into Supramolecular Nanoprobes. J Am Chem Soc. 2016;138:3533–3540. doi: 10.1021/jacs.6b00073. [DOI] [PubMed] [Google Scholar]
- 19.Nagy KJ, Giano MC, Jin A, Pochan DJ, Schneider JP. Enhanced Mechanical Rigidity of Hydrogels Formed from Enantiomeric Peptide Assemblies. J Am Chem Soc. 2011;133:14975–14977. doi: 10.1021/ja206742m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dong JJ, Shokes JE, Scott RA, Lynn DG. Modulating amyloid self-assembly and fibril morphology with Zn(II) J Am Chem Soc. 2006;128:3540–3542. doi: 10.1021/ja055973j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagarkar RP, Hule RA, Pochan DJ, Schneider JP. De novo design of strand-swapped beta-hairpin hydrogels. J Am Chem Soc. 2008;130:4466–4474. doi: 10.1021/ja710295t. [DOI] [PubMed] [Google Scholar]
- 22.Huang BQ, Hirst AR, Smith DK, Castelletto V, Hamley IW. A direct comparison of one- and two-component dendritic self-assembled materials: Elucidating molecular recognition pathways. J Am Chem Soc. 2005;127:7130–7139. doi: 10.1021/ja050412d. [DOI] [PubMed] [Google Scholar]
- 23.Priftis D, Leon L, Song ZY, Perry SL, Margossian KO, Tropnikova A, Cheng JJ, Tirrell M. Self-Assembly of alpha-Helical Polypeptides Driven by Complex Coacervation. Angew Chem Int Ed. 2015;54:11128–11132. doi: 10.1002/anie.201504861. [DOI] [PubMed] [Google Scholar]
- 24.Cai KM, He X, Song ZY, Yin Q, Zhang YF, Uckun FM, Jiang C, Cheng JJ. Dimeric Drug Polymeric Nanoparticles with Exceptionally High Drug Loading and Quantitative Loading Efficiency. J Am Chem Soc. 2015;137:3458–3461. doi: 10.1021/ja513034e. [DOI] [PubMed] [Google Scholar]
- 25.Zheng Z, Chen PY, Xie ML, Wu CF, Luo YF, Wang WT, Jiang J, Liang GL. Cell Environment-Differentiated Self-Assembly of Nanofibers. J Am Chem Soc. 2016;138:11128–11131. doi: 10.1021/jacs.6b06903. [DOI] [PubMed] [Google Scholar]
- 26.Wang HM, Wei J, Yang CB, Zhao HY, Li DX, Yin ZN, Yang ZM. The inhibition of tumor growth and metastasis by self-assembled nanofibers of taxol. Biomaterials. 2012;33:5848–5853. doi: 10.1016/j.biomaterials.2012.04.047. [DOI] [PubMed] [Google Scholar]
- 27.Galler KM, Aulisa L, Regan KR, D’Souza RN, Hartgerink JD. Self-Assembling Multidomain Peptide Hydrogels: Designed Susceptibility to Enzymatic Cleavage Allows Enhanced Cell Migration and Spreading. J Am Chem Soc. 2010;132:3217–3223. doi: 10.1021/ja910481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Debnath S, Roy S, Ulijn RV. Peptide Nanofibers with Dynamic Instability through Nonequilibrium Biocatalytic Assembly. J Am Chem Soc. 2013;135:16789–16792. doi: 10.1021/ja4086353. [DOI] [PubMed] [Google Scholar]
- 29.Yan N, Xu Z, Diehn KK, Raghavan SR, Fang Y, Weiss RG. How Do Liquid Mixtures Solubilize Insoluble Gelators? Self-Assembly Properties of Pyrenyl-Linker-Glucono Gelators in Tetrahydrofuran–Water Mixtures. J Am Chem Soc. 2013;135:8989–8999. doi: 10.1021/ja402560n. [DOI] [PubMed] [Google Scholar]
- 30.Mallia VA, Butler PD, Sarkar B, Holman KT, Weiss RG. Reversible Phase Transitions within Self-Assembled Fibrillar Networks of (R)-18-(n-Alkylamino)octadecan-7-ols in Their Carbon Tetrachloride Gels. J Am Chem Soc. 2011;133:15045–15054. doi: 10.1021/ja204371b. [DOI] [PubMed] [Google Scholar]
- 31.Li C, Numata M, Bae A-H, Sakurai K, Shinkai S. Self-Assembly of Supramolecular Chiral Insulated Molecular Wire. J Am Chem Soc. 2005;127:4548–4549. doi: 10.1021/ja050168q. [DOI] [PubMed] [Google Scholar]
- 32.Kitahara T, Shirakawa M, Kawano S, Beginn U, Fujita N, Shinkai S. Creation of a mixed-valence state from one-dimensionally aligned TTF utilizing the self-assembling nature of a low molecular-weight gel. J Am Chem Soc. 2005;127:14980–14981. doi: 10.1021/ja0552038. [DOI] [PubMed] [Google Scholar]
- 33.Pashuck ET, Duchet BJR, Hansel CS, Maynard SA, Chow LW, Stevens MM. Controlled Sub-Nanometer Epitope Spacing in a Three-Dimensional Self-Assembled Peptide Hydrogel. ACS Nano. 2016;10:11096–11104. doi: 10.1021/acsnano.6b05975. [DOI] [PubMed] [Google Scholar]
- 34.Wang ST, Lin YY, Spencer RK, Thomas MR, Nguyen AI, Amdursky N, Pashuck ET, Skaalure SC, Song CY, Parmar PA, Morgan RM, Ercius P, Aloni S, Zuckermann RN, Stevens MM. Sequence-Dependent Self-Assembly and Structural Diversity of Islet Amyloid Polypeptide-Derived beta-Sheet Fibrils. ACS Nano. 2017;11:8579–8589. doi: 10.1021/acsnano.7b02325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clarke DE, Pashuck ET, Bertazzo S, Weaver JVM, Stevens MM. Self-Healing, Self-Assembled beta-Sheet Peptide Poly(gamma-glutamic acid) Hybrid Hydrogels. J Am Chem Soc. 2017;139:7250–7255. doi: 10.1021/jacs.7b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HM, Liu J, Han AT, Xiao NN, Xue ZS, Wang G, Long JF, Kong DL, Liu B, Yang ZM, Ding D. Self-Assembly-Induced Far-Red/Near-Infrared Fluorescence Light-Up for Detecting and Visualizing Specific Protein-Peptide Interactions. ACS Nano. 2014;8:1475–1484. doi: 10.1021/nn4054914. [DOI] [PubMed] [Google Scholar]
- 37.Yuan Y, Wang FQ, Tang W, Ding ZL, Wang L, Liang LL, Zheng Z, Zhang HF, Liang GL. Intracellular Self-Assembly of Cyclic D-Luciferin Nanoparticles for Persistent Bioluminescence Imaging of Fatty Acid Amide Hydrolase. ACS Nano. 2016;10:7147–7153. doi: 10.1021/acsnano.6b03412. [DOI] [PubMed] [Google Scholar]
- 38.Yuan Y, Sun H, Ge S, Wang M, Zhao H, Wang L, An L, Zhang J, Zhang H, Hu B, Wang J, Liang G. Controlled Intracellular Self-Assembly and Disassembly of 19F Nanoparticles for MR Imaging of Caspase 3/7 in Zebrafish. ACS Nano. 2015;9:761–768. doi: 10.1021/nn5062657. [DOI] [PubMed] [Google Scholar]
- 39.Leckie J, Hope A, Hughes M, Debnath S, Fleming S, Wark AW, Ulijn RV, Haw MD. Nanopropulsion by Biocatalytic Self-Assembly. ACS Nano. 2014;8:9580–9589. doi: 10.1021/nn503875y. [DOI] [PubMed] [Google Scholar]
- 40.Bai S, Pappas C, Debnath S, Frederix PWJM, Leckie J, Fleming S, Ulijn RV. Stable Emulsions Formed by Self-Assembly of Interfacial Networks of Dipeptide Derivatives. ACS Nano. 2014;8:7005–7013. doi: 10.1021/nn501909j. [DOI] [PubMed] [Google Scholar]
- 41.Nagy-Smith K, Beltramo PJ, Moore E, Tycko R, Furst EM, Schneider JP. Molecular Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey. ACS Cent Sci. 2017;3:586–597. doi: 10.1021/acscentsci.7b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cohen E, Weissman H, Pinkas I, Shimoni E, Rehak P, Král P, Rybtchinski B. Controlled Self-Assembly of Photofunctional Supramolecular Nanotubes. ACS Nano. 2018;12:317–326. doi: 10.1021/acsnano.7b06376. [DOI] [PubMed] [Google Scholar]
- 43.Yang ZM, Gu HW, Fu DG, Gao P, Lam JK, Xu B. Enzymatic formation of supramolecular hydrogels. Adv Mater. 2004;16:1440–1444. [Google Scholar]
- 44.Yang Z, Liang G, Xu B. Enzymatic hydrogelation of small molecules. Acc Chem Res. 2008;41:315–326. doi: 10.1021/ar7001914. [DOI] [PubMed] [Google Scholar]
- 45.Ohtsuka K, Inoue S, Kameyama M, Kanetoshi A, Fujimoto T, Takaoka K, Araya Y, Shida A. Intracellular conversion of irinotecan to its active form SN-38, by native carboxylesterase in human non-small cell lung cancer. Lung Cancer. 2003;41:187–198. doi: 10.1016/s0169-5002(03)00223-x. [DOI] [PubMed] [Google Scholar]
- 46.Guichard S, Terret C, Hennebelle I, Lochon I, Chevreau P, Fretigny E, Selves J, Chatelut E, Bugat R, Canal P. CPT-11 converting carboxylesterase and topoisomerase I activities in tumour and normal colon and liver tissues. Br J Cancer. 1999;80:364–370. doi: 10.1038/sj.bjc.6690364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanghani SP, Quinney SK, Fredenburg TB, Sun ZJ, Davis WI, Murry DJ, Cummings OW, Seitz DE, Bosron WF. Carboxylesterases expressed in human colon tumor tissue and their role in CPT-11 hydrolysis. Clin Cancer Res. 2003;9:4983–4991. [PubMed] [Google Scholar]
- 48.Satoh T, Hosokawa M. The mammalian carboxylesterases: From molecules to functions. Annu Rev Pharmacol Toxicol. 1998;38:257–288. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 49.Zhou J, Du XW, Yamagata N, Xu B. Enzyme-Instructed Self-Assembly of Small D-Peptides as a Multiple-Step Process for Selectively Killing Cancer Cells. J Am Chem Soc. 2016;138:3813–3823. doi: 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Epstein IR, Xu B. Reaction-diffusion processes at the nano- and microscales. Nat Nanotechnol. 2016;11:312–319. doi: 10.1038/nnano.2016.41. [DOI] [PubMed] [Google Scholar]
- 51.Reches M, Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Kuang Y, Gao YA, Xu B. Versatile Small-Molecule Motifs for Self-Assembly in Water and the Formation of Biofunctional Supramolecular Hydrogels. Langmuir. 2011;27:529–537. doi: 10.1021/la1020324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuang Y, Xu B. Disruption of the Dynamics of Microtubules and Selective Inhibition of Glioblastoma Cells by Nanofibers of Small Hydrophobic Molecules. Angew Chem Int Ed. 2013;52:6944–6948. doi: 10.1002/anie.201302658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou J, Du X, Li J, Yamagata N, Xu B. Taurine Boosts Cellular Uptake of Small D-Peptides for Enzyme-Instructed Intracellular Molecular Self-Assembly. J Am Chem Soc. 2015;137:10040–10043. doi: 10.1021/jacs.5b06181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chan WC, White PD. Fmoc solid phase peptide synthesis: A Practical Approach. Oxford University Press Inc; New York: 2000. [Google Scholar]
- 56.Domcke S, Sinha R, Levine DA, Sander C, Schultz N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun. 2013;4 doi: 10.1038/ncomms3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li J, Shi JF, Medina JE, Zhou J, Du XW, Wang HM, Yang CH, Liu JF, Yang ZM, Dinulescu DM, Xu B. Selectively Inducing Cancer Cell Death by Intracellular Enzyme-Instructed Self-Assembly (EISA) of Dipeptide Derivatives. Adv Healthc Mater. 2017;6 doi: 10.1002/adhm.201601400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou J, Du XW, Xu B. Regulating the Rate of Molecular Self-Assembly for Targeting Cancer Cells. Angew Chem Int Ed. 2016;55:5770–5775. doi: 10.1002/anie.201600753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–3270. doi: 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- 60.Li FZ, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature. 1998;396:580–584. doi: 10.1038/25141. [DOI] [PubMed] [Google Scholar]
- 61.Elmore S. Apoptosis: A review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 63.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 64.Fernandesalnemri T, Litwack G, Alnemri ES. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J Biol Chem. 1994;269:30761–30764. [PubMed] [Google Scholar]
- 65.Datta SR, Dudek H, Tao X, Masters S, Fu HA, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 66.Chazotte B. Labeling Cytoskeletal F-Actin with Rhodamine Phalloidin or Fluorescein Phalloidin for Imaging. Cold Spring Harbor Protocols. 2010;2010 doi: 10.1101/pdb.prot4947. pdb.prot4947. [DOI] [PubMed] [Google Scholar]
- 67.Epstein IR, Xu B. Reaction–diffusion processes at the nano- and microscales. Nat Nanotechnol. 2016;11:312. doi: 10.1038/nnano.2016.41. [DOI] [PubMed] [Google Scholar]
- 68.Serizawa T, Fukaya Y, Sawada T. Self-Assembly of Cellulose Oligomers into Nanoribbon Network Structures Based on Kinetic Control of Enzymatic Oligomerization. Langmuir. 2017;33:13415–13422. doi: 10.1021/acs.langmuir.7b03653. [DOI] [PubMed] [Google Scholar]
- 69.Sahoo JK, Pappas CG, Sasselli IR, Abul-Haija YM, Ulijn RV. Biocatalytic Self-Assembly Cascades. Angew Chem Int Ed. 2017;56:6828–6832. doi: 10.1002/anie.201701870. [DOI] [PubMed] [Google Scholar]
- 70.Kapur A, Aldeek F, Ji X, Safi M, Wang WT, Del Cid A, Steinbock O, Mattoussi H. Self-Assembled Gold Nanoparticle-Fluorescent Protein Conjugates as Platforms for Sensing Thiolate Compounds via Modulation of Energy Transfer Quenching. Bioconj Chem. 2017;28:678–687. doi: 10.1021/acs.bioconjchem.7b00006. [DOI] [PubMed] [Google Scholar]
- 71.Nakouzi E, Steinbock O. Self-organization in precipitation reactions far from the equilibrium. Sci Adv. 2016;2:e1601144–e1601144. doi: 10.1126/sciadv.1601144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Whitesell JK, Pojman JA. Homochiral and heterochiral polyesters: polymers derived from mandelic acid. Chem Mater. 1990;2:248–254. [Google Scholar]
- 73.Zhou H, Jimenez Z, Pojman JA, Paley MS, Hoyle CE. Photopolymerization kinetics of tributylmethylammonium-based (meth)acrylate ionic liquids and the effect of water. J Polym Sci A Polym Chem. 2008;46:3766–3773. [Google Scholar]
- 74.Yang Z, Liang G, Guo Z, Guo Z, Xu B. Intracellular hydrogelation of small molecules inhibits bacterial growth. Angew Chem Int Ed. 2007;46:8216–8219. doi: 10.1002/anie.200701697. [DOI] [PubMed] [Google Scholar]
- 75.Yang ZM, Xu KM, Guo ZF, Guo ZH, Xu B. Intracellular enzymatic formation of nanofibers results in hydrogelation and regulated cell death. Adv Mater. 2007;19:3152–3156. [Google Scholar]
- 76.Li XM, Du XW, Li JY, Gao Y, Pan Y, Shi JF, Zhou N, Xu B. Introducing D-Amino Acid or Simple Glycoside into Small Peptides to Enable Supramolecular Hydrogelators to Resist Proteolysis. Langmuir. 2012;28:13512–13517. doi: 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.