

Abstract
We have identified a series of positive allosteric NMDA receptor (NMDAR) modulators derived from a known class of GluN2C/D-selective tetrahydroisoquinoline analogues that includes CIQ. The prototypical compound of this series contains a single isopropoxy moiety in place of the two methoxy substituents present in CIQ. Modifications of this isopropoxy-containing scaffold led to the identification of analogues with enhanced activity at the GluN2B subunit. We identified molecules that potentiate the response of GluN2B/GluN2C/GluN2D, GluN2B/GluN2C, and GluN2C/GluN2D-containing NMDARs to maximally effective concentrations of agonist. Multiple compounds potentiate the response of NMDARs with submicromolar EC50 values. Analysis of enantiomeric pairs revealed that the S-(−) enantiomer is active at the GluN2B, GluN2C, and/or GluN2D subunits, whereas the R-(+) enantiomer is only active at GluN2C/D subunits. These results provide a starting point for the development of selective positive allosteric modulators for GluN2B-containing receptors.
Graphical Abstract
Introduction
The NMDA (N-methyl-D-aspartic acid), AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid), and kainic acid receptors are ligand-gated ionotropic glutamate receptors expressed in the central nervous system (CNS) 1. NMDARs are involved in nervous system development, learning, memory,2,3 and synaptic plasticity,4,5 and these receptors mediate a slow Ca2+-permeable component of the excitatory postsynaptic current that is voltage-dependent by virtue of channel block by extracellular Mg2+ 6–9. The NMDAR is a heterotetrameric assembly of two GluN1 subunits and two GluN2 subunits, and the opening of the cation-selective pore is induced by simultaneous binding of the co-agonists glycine and glutamate1 to GluN1 and GluN2, respectively. Pharmacological and functional properties of the NMDAR, including different agonist potency10, open probability,11–14 and deactivation time course following removal of agonist,15 depend on GluN2 subunit composition. The four grin2 genes encoding GluN2A-D are differentially expressed throughout the brain and show distinct developmental profiles.16–18 Each GluN1 and GluN2 subunit comprises four semiautonomous domains: an amino terminal domain (ATD), an agonist binding domain (ABD), a transmembrane domain (TMD), and a carboxy terminal domain (CTD). Crystallographic analysis of GluN1/GluN2 receptors that contained the ATD-ABD-TMD domains revealed inter-subunit and inter-domain interactions that distinguish the NMDA receptor from other members of the glutamate receptor family. Notably, the ATD of the NMDA receptor is tightly packed and in close contact with the ABD, whereas in the AMPA receptor, the ATD and ABD are clearly separated. The close contact between these domains may account for ATD-regulation of channel function in the NMDA receptor, a property that is not observed in non-NMDA receptors.19,20
NMDARs have been discussed as both contributing factors and therapeutic targets in multiple neurological disease states, notably Alzheimer’s disease,21 brain ischemia,22,23 depression,24 Parkinson’s disease,25 epilepsy26, and schizophrenia27,28. For this reason, a number of NMDA-selective ligands with potential therapeutic use have been developed.29–31 Channel blockers show minimal subunit-selectivity as a result of their binding within the highly conserved channel pore.32,33 FDA-approved channel blockers include amantadine and memantine, which are used for the symptomatic treatment of Parkinson’s disease and Alzheimer’s disease, respectively,34 and the antitussive dextromethorphan, which has been approved for use in pseudobulbar affect35. The anesthetic ketamine appears effective in clinical trials for treatment-resistant depression,36 and also has had a positive signal in preliminary trials for obsessive compulsive disorder (OCD)37 and post-traumatic stress disorder (PTSD).38 Although these compounds do have useful clinical applications, channel blocker administration is often accompanied by side effects that include altered cardiovascular activity, decreased motor function, hallucinations, and delusions, perhaps due to the strong nonselective block of all NMDA receptors regardless of subunit composition.39
The first subunit-selective NMDAR modulator reported was the noncompetitive inhibitor ifenprodil (Figure 1), which is over a 100-fold more potent at NMDA receptors that contain the GluN2B subunit compared to the GluN2A, GluN2C, or GluN2D subunits.40,41 Ifenprodil and related derivatives have been evaluated in animal models of disease and clinical trials.42–45 A negative allosteric modulator that is selective for GluN2A-containing receptors,46 3-chloro-4-fluoro-N-[4-[[2-(phenylcarbonyl)hydrazino]carbonyl]benzyl]benzenesulfonamide (TCN-201, Figure 1), binds to the GluN1-GluN2 ligand binding domain heterodimer interface, resulting in a decrease in glycine potency. 46–48 Additional classes of subunit-selective negative allosteric NMDAR modulators include pyrazine-containing GluN2A-selective antagonists such as 5-(((3-fluoro-4-fluorophenyl)sulfonamido)methyl)-N-((2-methylthiazol-5-yl)methyl)methylpyrazine-2-carboxamide (MPX-007, Figure 1),49 GluN2C/GluN2D-selective quinazolin-4-ones (QNZ-46), and dihydroquinoline-pyrazolines (DQP-1105).50–53 Several studies have described naphthalene and phenanthrene compounds with varying potency and subunit-selectivity.54–57
Figure 1.

Structures of relevant NMDA receptor positive and negative allosteric modulators.
By contrast, less is known about subunit-selective positive allosteric NMDAR modulators. Tetrahydroisoquinoline-containing compounds selectively enhance the response of recombinant and native GluN2C- and GluN2D-containing NMDARs 58,59 to maximally effective concentrations of agonist with EC50 values between 5–10 μM60,61. The naphthalene derivative 9-cyclopylphenanthrene-3-carboxylic acid (UBP-710, Figure 1) potentiates GluN2A- and GluN2B-containing receptors at 100 μM and inhibits GluN2C and GluN2D receptors at higher concentrations.54 Pyrrolidinone-containing compounds are highly selective positive allosteric modulators of diheteromeric GluN1/GluN2C (EC50 values 5–20 μM) with minimal effects on NMDARs that contain GluN2A, GluN2B, or GluN2D.62,63 Recently, a class of positive allosteric modulators of GluN2A-containing NMDA receptors were developed by Genentech64,65. The best-in-class compound of this series has an EC50 value of 0.021 μM at GluN2A and is >200-fold more potent at GluN2A than the GluA2 AMPA receptor65.
Multiple endogenous compounds have been characterized as modulators of NMDA receptors that contain GluN2B. For example, polyamines, such as spermine, selectively potentiate the response of GluN1/GluN2B receptors with low potency (100–200 μM EC50) by reducing tonic proton inhibition that is present at physiological pH.66,67 A number of positively charged aminoglycoside antibiotics also selectively potentiate GluN2B-containg receptors with EC50 values typically between 30 and 150 μM.68 The neurosteroid pregnenolone sulfate potentiates GluN2A and GluN2B-containing receptors by enhancing open probability.69 However, each of these classes of molecules have characteristics that complicate their use as tool compounds as well as their development as therapeutic agents. Despite the lack of a potent and selective tool compounds that selectively potentiate NMDARs that contain the GluN2B, there is evidence supporting the therapeutic value of such a compound. For example, overexpression of GluN2B receptors in the forebrain of mice enhances activation of synaptic NMDA receptor.2 This activation has been shown to improve learning, long-term memory, spatial performance, and cued and contextual fear conditioning70–73.
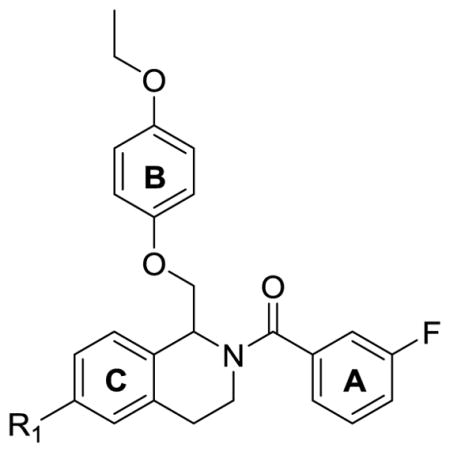
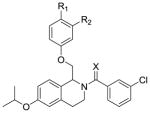
We have previously described a series of tetrahydroisoqinoline analogues based on the prototypical positive allosteric modulator CIQ (1).60,61 CIQ is selective for GluN2C- and GluN2D-containing NMDARs over GluN2A- and GluN2B-containing NMDARs and has no effect on AMPA or kainate receptors. An extensive SAR was described, and the most potent compounds in the series had EC50 values of 300 nM at GluN2C and GluN2D subunits.61 During our initial exploration of the SAR of this series of GluN2C/GluN2D-selective positive allosteric modulators (1), we discovered a single compound (2) that displayed activity not just at GluN2C and GluN2D subunits, but also at the GluN2B subunit (Figure 2). Compound 2 resulted from the replacement of the dimethoxy groups in CIQ (1) with a single isopropoxy group. Here we describe an extensive structure-activity relationship (SAR) around compound 2 with significant activity at the GluN2B subunit. We also have separated enantiomers for the most potent compounds in the class to show that the GluN2B activity is stereodependent. The modifications and stereodependence exhibited by the tetrahydroisoquinoline compounds reported here show that the CIQ scaffold, originally described as selective for GluN2C and GluN2D subunits, can be tuned to target the GluN2A and GluN2B subunits.
Figure 2.
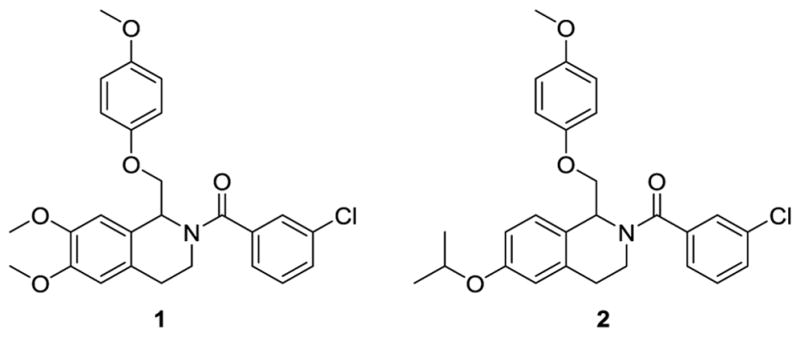
Scaffold of CIQ (1) and the structurally similar isopropoxy compound (2)
Results
Chemistry
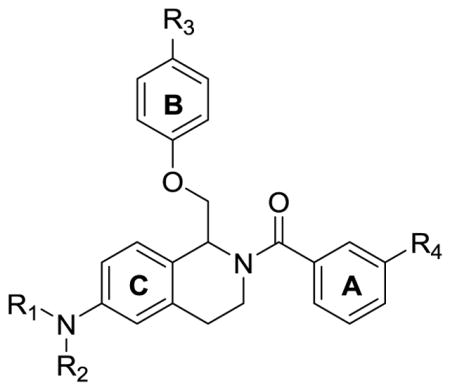
To probe the SAR around the new analogue that potentiates GluN2B-, GluN2C-, and GluN2D-containing NMDA receptors, each of the three aromatic rings (referred to as A, B, and C) of the tetrahydroisoquinoline were modified (Figure 3). The nonselective potentiation arose from modifications to the C-ring, and all functionality to the C-ring was installed via one of three methods (Scheme 1, Scheme 2, or Scheme 3). Compounds with an ether linkage in the R1 position were synthesized beginning with the previously described tert-butyl 3-hydroxyphenethylcarbamate 3,61 which was either subjected to alkylation conditions or Mitsunobu coupling conditions to yield phenethylcarbamate compounds 4–8. Under acidic conditions, the Boc group was removed to afford the functionalized phenethylamine compounds 9–13, which were acylated by chloroacetyl chloride to afford 2-chloro-N-phenethylacetamide compounds 14–19. Compounds 14–19 were then alkylated with ortho, meta, and para-substituted phenols to yield linear amides 20–29 (Scheme 1).
Figure 3.
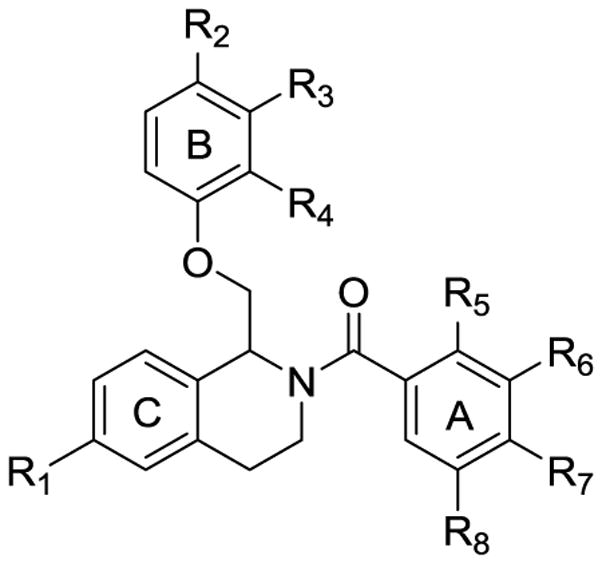
Labeled tetrahydroisoquinoline to differentiate the A, B, and C-rings.
Scheme 1.

a) alkyl halide, K2CO3, DMF, 24 h, 35–72% (procedure I); b) substituted alcohol, DIAD, PPh3, Et3N, 6 days, 23–34% (procedure II); c) HCl/ether, 1 h, quant. (procedure III); d) chloroacetyl chloride, Et3N, DCM, 3 h, 52–72% (procedure IV); e) substituted phenol, Cs2CO3, ACN, 15 h, 58–92% (procedure V)
Scheme 2.

a) chloroacetyl chloride, Et3N, DCM, 3 h, 65% (procedure IV); b) substituted phenol, Cs2CO3, ACN, 15 h, 83–92% (procedure V); c) NaI, CuI, N, N-dimethylethylenediamine, dioxane, sealed tube, 110°C, 24 h, 69–80% (procedure VI); d) substituted alcohol, CuI, 1,10-phen, sealed tube, 110°C, 24–48 h, 36–76% (procedure VII); e) Pd(OH)2, H2, MeOH/THF; 91%; f) 2-iodopropane, Cs2CO3, ACN/DMF; 24 h, 88%
Scheme 3.

a) cyclic amines or cyclohexylamine, CuI, L-proline, K2CO3, DMSO, sealed tube, 80°C, 43–87% (procedure VIII); b) Cu(I)O2, NH4OH, NMP, sealed tube, 85°C, 72%; c) paraformaldehyde, NaCNBH3, AcOH, 4–20 h, 57–70% (procedure IX); d) 2-propanone, NaCNBH3, AcOH/ACN, 2 h, 32%
Oxygen functionality on the C-ring could also be installed utilizing Scheme 2, which includes an Ullmann copper coupling sequence.74 Bromine-substituted phenethylamine 30 was subjected to acylation conditions to yield N-(3-bromophenethyl)-2-chloroacetamide 31, and the chlorine was then displaced by para-substituted phenols to give linear amides 32–33. The bromine in compounds 32 and 33 was then exchanged for an iodine in an aromatic Finkelstein reaction75 to give 34 and 35, respectively. The aryl iodine-containing compounds were then reacted with a variety of substituted alcohols in a copper-catalyzed coupling reaction74 to yield oxygen-containing linear amides 36–38, and this is an alternative method to develop the phenethylamine scaffold from that shown in Scheme 1. Since installing an isopropoxy is challenging via an Ullmann-type coupling, compound 40 was ultimately prepared by removing the benzyl group of compound 38 by hydrogenolysis and subjecting the resulting hydroxyphenethylacetamide (39) to alkylation conditions with 2-iodopropane.
The aryl iodine intermediates 34 and 35 can also be directly converted to nitrogen functionality by one of two methods shown in Scheme 3. Compounds 34 and 35 were subjected to Ullmann copper-catalyzed coupling76 with cyclohexylamine, piperidine, or morpholine to afford compounds 41–43. An alternate copper-catalyzed reaction77 of 34 and aqueous ammonia afforded aniline-containing amide 44, which underwent reductive amination conditions to yield either the dimethylamino acetamide 45 or the isopropylamino-substituted acetamide 46.78 Compound 46 was then subjected to a second reductive amination to afford di-N-substituted compound 47. Oxygen or nitrogen-containing intermediates (20–29, 36–37, 40–43, and 45–47) were then cyclized using Bischler-Napieralski conditions to form the dihydroisoquinolines 48–66, which were reduced with sodium borohydride to afford the corresponding tetrahydroisoquinolines 67–85 (Scheme 4).
Scheme 4.

a) POCl3, toluene, reflux, 1–5 h (procedure X or XI); b) NaBH4, MeOH, 20h, 8–55% over 2 steps (procedure XII)
Tetrahydroisoquinolines (67–85) were then either acylated with a variety of benzoyl chlorides or were subjected to typical EDCI coupling conditions with benzoic acids to afford final compounds (2, 87–128, 130, 133–137, 147). Nitro-containing 128 was reduced via hydrogenation to afford aniline-containing 132. Once TBDMS-protected intermediate 130 had been synthesized via typical EDCI coupling conditions, the protected phenol was deprotected using TBAF to afford analog 131 (Scheme 5). Amide-containing analogs were then converted to thioamide analogs (138–146, 148) using Lawesson’s reagent in refluxing toluene (Scheme 5).
Scheme 5.

a) substituted benzoyl chlorides, Et3N, DCM, 3 h, 23–81% (procedure XIII); b) substituted benzoic acids, EDCI, DMAP, 24 h, 15–69% (procedure XIV); c) Pd/C, H2, THF/2-propanol, 90%; d) TBAF, THF, 66%; e) Lawesson’s reagent, toluene, reflux, 33–69% (procedure XV); f) Lawesson’s reagent, toluene, MW, 35 min, 51–69% (procedure XVI)
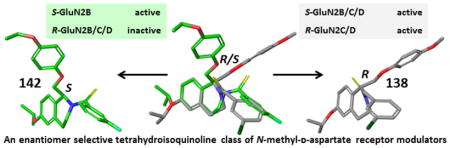
The enantiomers of compounds 2 and 97 were separated via a ChiralPakAD-H semi-preparatory chiral column chromatography (Scheme 6), and both enantiomers of compounds 2 and 97 were converted to their thioamide-containing counterparts, compounds 138 and 142, respectively. Crystal structures of S-(-)-138 (Figure 4) allowed for the assignment of the absolute stereochemistry of the enantiomers.
Scheme 6.

The separation of 2 and 97. Methods for separation are given in the experimental section. a) Lawesson’s reagent, toluene, reflux, 49–65% (procedure XV); b) Lawesson’s reagent, toluene, MW, 35 min, 56–65% (procedure XVI)
Figure 4.

Crystal structure of thioamide S-(−) enantiomer of 138, active at the GluN2B, GluN2C, and GluN2D subunit.
Potentiation of NMDA Receptors
All compounds synthesized were evaluated in two-electrode voltage clamp recordings of currents arising from recombinant GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, and GluN1/GluN2D NMDA receptors expressed in Xenopus oocytes and activated by saturating concentrations of glutamate (100 μM) and glycine (30 μM). Compound 2 served as the starting point for this study, and retained selectivity over AMPA and kainate receptors; the response of co-application of 30 μM of compound 2 with 100 μM glutamate and 30 μM glycine was 106+2% and 96+9% of the control response to glutamate alone for GluA1 (n=8) and GluK2 homomeric receptors (n=9), respectively (See Methods). In addition, 30 μM of compound 2 showed no agonist activity when co-applied with either 30 μM glutamate or 10 μM glycine onto oocytes expressing GluN1/GluN2B (n=6), GluN1/GluN2C (n=6–8), or GluN1/GluN2D (n=6–8). All analogues developed from compound 2 were tested in at least 3 oocytes from at least 2 different frogs at GluN2B, GluN2C, and GluN2D-containing NMDA receptors, and in at least 4 oocytes from 1–3 frogs at GluN2A-containing NMDA receptors. The average potentiation at 30 μM of drug is displayed in Tables 1–5, and is defined as the mean ratio of current in the presence of test compound to current response in the absence of test compound; the standard error of the mean is given. If the average potentiation exceeded 120% of control, concentration-effect curves were generated by co-applying increasing concentrations of drug with 100 μM glutamate and 30 μM glycine, and the EC50 value was calculated by fitting equation 1 to the data. Only ten compounds displayed modest potentiation (greater than 120%) at GluN2A-containing receptors, and this effect was not studied further (See Supplemental Table S1).
Table 1.
Optimization of potency based on substituent identification of C-ring.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (maximum) (μM, %)a | |||||||
| # | R1 | R2 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D |
| 86b | OMe | OMe | 90 ± 5.4 | 213 ± 13 | 215 ± 19 | -- | 1.3 (240%) | 1.0 (219%) |
| 87 | OEt | OMe | 158 ± 8.9 | 285 ± 23 | 329 ± 15 | 7.8 (167%) | 2.4 (294%) | 3.0 (344%) |
| 2 | OiPr | OMe | 195 ± 11 | 233 ± 18 | 240 ± 29 | 5.2 (202%) | 2.0 (243%) | 3.0 (245%) |
| 88c | OiBu | OMe | 313 ± 18 | 400 ± 58 | 472 ± 70 | 1.5 (333%)e | 0.9 (448%) | 1.4 (521%) |
| 89d | OCH(CH2CH3)2 | OMe | 331 ± 17 | 286 ± 26 | 381 ± 40 | 1.8 (344%) | 1.6 (294%) | 1.7 (390%) |
| 90 | OcHex | OMe | 259 ± 8.7 | 222 ± 19 | 309 ± 37 | 2.7 (265%) | 1.2 (225%) | 1.9 (317%) |
| 91 | OEt | OEt | 104 ± 2.7 | 91 ± 3.9 | 75 ± 2.7 | -- | -- | -- |
| 92c | OiPr | OEt | 145 ± 7.8 | 126 ± 2.8 | 102 ± 2.0 | 5.3 (158%) | 1.8 (128%) | -- |
| 93 | OiBu | OEt | 294 ± 33 | 238 ± 11 | 342 ± 21 | 1.0 (293%)f | 1.0 (242%) | 1.5 (351%) |
| 94c | OcPent | OEt | 204 ± 16 | 187 ± 11 | 193 ± 6.6 | 4.0 (221%) | 4.2 (210%) | 4.3 (188%) |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 7–26 oocytes from 2–5 frogs for each compound and receptor tested.
Previously published61 data for compound 86 were included for comparison.
Current ratio reported for 10 μM test compound due to visible precipitate at 30 μM.
What was d?
p value <0.05 when comparing 88 and 2 EC50 value and max. response; p value <0.05 when comparing 88 and 90 max. response.
p value <0.05 when comparing 93 and 94 EC50.
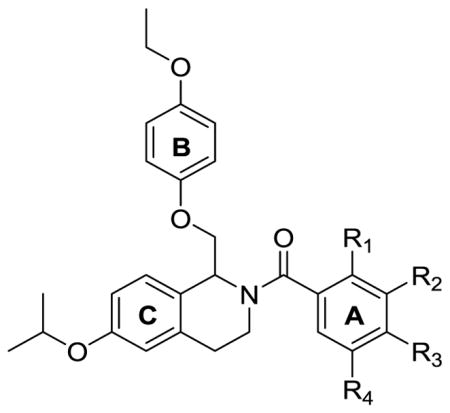
Table 5.
Optimization of potency based on substituent identification of C-ring.
| |||||||
|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (max.) (μM %)a | ||||||
| R1 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D | |
| 135 | OEt | 94 ± 1.3 | 77 ± 3.2 | 67 ± 2.0 | – | – | – |
| 97b | OiPr | 147 ± 3.7 | 104 ± 2.2 | 91 ± 1.7 | 7.2 (152%) | – | – |
| 136 | OiBu | 332 ± 13 | 309 ± 15 | 351 ± 17 | 2.5 (342%) | 3.1 (325%) | 3.4 (364%) |
| 137 | OcPent | 202 ± 13 | 169 ± 2.7 | 161 ± 4.4 | 4.4 (210%) | 4.5 (173%) | 4.9 (163%) |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 8–17 oocytes from 2–3 frogs for each compound and receptor tested.
Compound 97 was shown in previous tables and was included here for comparison.
Effect of Modifying the C-ring
In order to evaluate whether other modifications to the C-ring, in addition to the isopropoxy group in compound 2, would result in potentiation at the GluN2B subunit, a variety of ether-linked moieties were installed at that site (Table 1). Previously published compound 86 with a methoxy group in the R1 position61 was selective for GluN2C and GluN2D subunits. When the methoxy group on the C-ring was replaced with an ethoxy group as in compound 87, potentiation was observed at the GluN2B subunit in addition to GluN2C and GluN2D. Similarly to compound 2, all compounds with a methoxy group on the B-ring and a branched or cyclic ether moiety on the C-ring at R1 (88, 89, and 90) were active at GluN2B, GluN2C, and GluN2D subunits. When the methoxy substitution in the R2 position on the B-ring was exchanged for an ethoxy group, more subunit-selectivity began to emerge. Compound 91 was inactive at all subunits, but once an ethoxy group at R1 was replaced with branched functionality as in compounds 92, 93, and 94, activity at the GluN2B subunit was restored. Compound 92 with an isopropoxy group on the C-ring was only active at GluN2B and GluN2C subunits, whereas 93 and 94 were active at all three subunits.
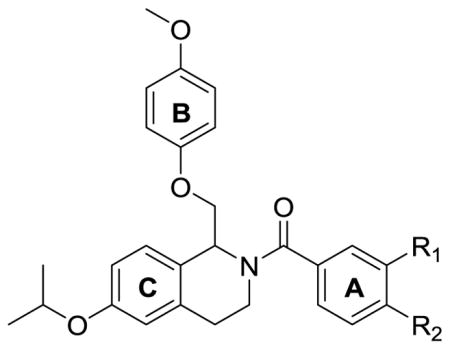
In addition to studying the effect of a variety of ether-linked moieties on the C-ring, nitrogen-containing functional groups were also installed and evaluated (Table 2). A number of mono- and di-substituted nitrogen-containing compounds were synthesized that were active at GluN2B, GluN2C, and GluN2D subunits, however, compounds with ether functionality at this position were generally more potent. After exploring a variety of changes to the C-ring, Compound 92 showed the strongest subunit-selectivity, with detectable potentiation of the response to maximal agonist at only the GluN2B and GluN2C subunit. For this reason, in moving forward with the SAR, the isopropoxy group on the Cring and the ethoxy group in the para-position of the B-ring were held constant.
Table 2.
Optimization of potency based on substituent identification of C-ring.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (max.) (μM %)a | |||||||||
| R1 | R2 | R3 | R4 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D | |
| 117 | Me | Me | OMe | Cl | 127 ± 4.1 | 288 ± 18 | 322 ± 20 | ND | 4.0 (315) | 4.4 (349%) |
| 118 | Me | Me | OMe | Br | 99 ± 3.6 | 285 ± 11 | 307 ± 18 | – | 2.4 (293%) | 3.8 (327%) |
| 119 | Me | iPr | OMe | Cl | 228 ± 12 | 404 ± 30 | 500 ± 56 | 5.9 (235%) | 2.0 (424%) | 4.0 (549%) |
| 120 | H | iPr | OMe | Cl | 138 ± 5.2 | 239 ± 14 | 333 ± 30 | 9.3 (141%) | 4.7 (261%) | 5.1 (363%) |
| 121 | piperidine | OMe | Cl | 213 ± 20 | 289 ± 14 | 418 ± 42 | 4.9 (221%) | 2.3 (313%) | 4.0 (464%) | |
| 122 | morpholine | OMe | Cl | 146 ± 4.3 | 273 ± 16 | 231 ± 40 | 9.3 (188%) | 4.7 (342%) | 22 (413%) | |
| 123 | morpholine | OMe | Br | 152 ± 4.4 | 279 ± 18 | 284 ± 50 | 4.8 (165%) | 3.0 (314%) | 4.3 (425%) | |
| 124 | morpholine | OEt | Cl | 127 ± 4.7 | 133 ± 4.4 | 104 ± 2.4 | ND | ND | – | |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 7–12 oocytes from 2 frogs for each compound and receptor tested. ND (not determined) indicates that we could not obtain adequate data to allow the concentration-effect curve to be fitted.
Effect of Modifying the A-ring
All of the compounds mentioned previously above were synthesized with a chlorine in the meta-position on the A-ring. We also synthesized compounds to determine optimal placement and identity of A-ring substitutions (Table 3). Specific substitution on the A-ring is required for activity as compound 95 was inactive at all subunits. Compounds with a phenyl group (99), a methyl group (100), and a methoxy group (101) were inactive as well, but compounds with halogens in the meta-position of the A-ring were active. Similar to compound 92, compound 96 with a meta-substituted bromine was active at GluN2B and GluN2C. Notably, compound 97 with fluorine substitution at that position was active at only GluN2B, but with modest potency (EC50 7.2 μM). Since 97 was selective for GluN2B, the ortho-and para-substituted fluorine and the chlorine analogues (102, 103, 104, 105) were also made to probe ideal substituent placement. A chlorine or fluorine in the ortho-position (102 and 103) or the para-position (104 and 105) caused a complete loss of activity, indicating that meta-position substitution is required for activity. Di-substituted compounds 106, 107, 108, and 109 were also synthesized, and with the exception of 2,4-dichloro compound 109, were active as well. Compounds were also synthesized that were similar to 2 with a methoxy group in the para-position of the B-ring (Table 4). These compounds exhibited potentiation at the GluN2B, GluN2C, and GluN2D subunits with occasional potentiation at the GluN2A subunit, although none showed selectivity for GluN2B. Thus, it appeared as if fluorine in the meta-position of the A-ring was optimal in terms of GluN2B selectivity, and for this reason a number of compounds were synthesized that maintained the fluorine on the A-ring and para-ethoxy functionality on the B-ring with various C-ring ether moieties (Table 5). Unfortunately, none of these compounds were more selective for GluN2B than 97, being either completely inactive or equally potent at GluN2B, GluN2C, and GluN2D. Compound 136, however represents a very efficacious potentiator of GluN2B, GluN2C, and GluN2D with maximum potentiation ranging from 325% to 364% at the three subunits.
Table 3.
Optimization of potency based on substituent placement and identity of A-ring.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (max.) (μM %)a | |||||||||
| # | R1 | R2 | R3 | R4 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D |
| 95 | H | H | H | H | 88 ± 1.1 | 83 ± 2.5 | 69 ± 2.3 | -- | -- | -- |
| 92b | H | Cl | H | H | 145 ± 7.8 | 126 ± 2.8 | 102 ± 2.0 | 5.3 (158%) | 1.8 (128%) | -- |
| 96 | H | Br | H | H | 136 ± 7.9 | 133 ± 5.1 | 98 ± 1.9 | 3.4 (137%) | 1.8 (135%) | -- |
| 97 | H | F | H | H | 147 ± 3.7 | 104 ± 2.2 | 91 ± 1.7 | 7.2 (152%) | -- | -- |
| 98 | H | CF3 | H | H | 124 ± 7.0 | 129 ± 3.8 | 95 ± 8.6 | 2.4 (124%) | 1.3 (130%) | -- |
| 99 | H | Ph | H | H | 112 ± 4.4 | 92 ± 3.7 | 92 ± 1.9 | -- | -- | -- |
| 100 | H | Me | H | H | 78 ± 1.2 | 67 ± 2.1 | 43 ± 2.3 | -- | -- | -- |
| 101 | H | OMe | H | H | 89 ± 5.1 | 65 ± 4.1 | 44 ± 4.5 | -- | -- | -- |
| 102 | F | H | H | H | 61 ± 2.4 | 68 ± 4.1 | 55 ± 4.5 | -- | -- | -- |
| 103 | Cl | H | H | H | 67 ± 4.1 | 93 ± 6.3 | 79 ± 3.4 | -- | -- | -- |
| 104 | H | H | F | H | 118 ± 5.8 | 107 ± 6.7 | 90 ± 3.7 | -- | -- | -- |
| 105 | H | H | Cl | H | 112 ± 2.1 | 101 ± 7.8 | 88 ± 7.7 | -- | -- | -- |
| 106 | H | F | F | H | 172 ± 6.0 | 145 ± 6.6 | 133 ± 3.7 | 3.7 (173%) | 1.8 (150%) | 2.9 (137%) |
| 107c | H | Cl | Cl | H | 133 ± 5.3 | 150 ± 8.1 | 122 ± 4.1 | ND | ND | ND |
| 108 | H | Cl | F | H | 169 ± 7.8 | 159 ± 6.3 | 133 ± 4.8 | 2.2 (170%) | 1.3 (162%) | 2.2 (137%) |
| 109c | H | Cl | H | Cl | 105 ± 3.3 | 97 ± 2.9 | 80 ± 3.0 | -- | -- | -- |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 6–17 oocytes from 2–3 frogs for each compound and receptor tested.
Compound 92 from Table 1 is included here for comparison.
Current ratio reported is for 3 μM (compound 107) or 10 μM (compounds 99, 109) because these compounds were not soluble at 10 or 30 μM, respectively.
ND (not determined) indicates that we could not obtain data at high enough concentrations to determine a fitted maximum value for the concentration-effect curve.
Table 4.
Optimization of potency based on substituent placement and identity of A-ring.
| ||||||||
|---|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (max.) (μM %)a | |||||||
| R1 | R2 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D | |
| 2b | Cl | H | 195 ± 11 | 233 ± 18 | 240 ± 29 | 5.2 (202%) | 2.0 (243%) | 3.0 (245%) |
| 125c | Br | H | 189 ± 8 | 270 ± 12 | 312 ± 17 | 3.9 (204%) | 1.8 (286%) | 2.6 (338%) |
| 126 | I | H | 159 ± 8.7 | 215 ± 15 | 408 ± 27 | 1.7 (172%) | 0.69 (310%) | 1.6 (414%) |
| 114b | F | H | 249 ± 7.6 | 271 ± 15 | 402 ± 19 | 15 (297%) | 5.9 (365%) | 7.4 (427%) |
| 127 | CF3 | H | 196 ± 13 | 363 ± 25 | 334 ± 16 | 6.1 (206%) | 1.7 (384%) | 2.7 (362%) |
| 128 | NO2 | H | 198 ± 7.7 | 337 ± 21 | 335 ± 52 | 5.3 (203%) | 2.8 (358%) | 3.4 (351%) |
| 131 | OH | H | 98 ± 3.4 | 117 ± 8.4 | 105 ± 6.2 | – | – | – |
| 132 | NH2 | H | 106 ± 3.4 | 105 ± 8.9 | 107 ± 9.1 | – | – | – |
| 133 | H | Cl | 163 ± 9.6 | 251 ± 9.5 | 244 ± 13 | 12 (182%) | 9.5 (290%) | 11 (288%) |
| 134 | H | OMe | 110 ± 1.8 | 138 ± 4.0 | 114 ± 3.0 | – | ND | – |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 7–17 oocytes from 2–3 frogs for each compound and receptor tested. ND (not determined) indicates that we could not obtain adequate data to allow the concentration-effect curve to be fitted.
Compounds 2 and 114 shown in previous tables were included here for comparison.
Current ratio reported for 10 μM due to precipitate at 30 μM.
Effect of Modifying the B-ring
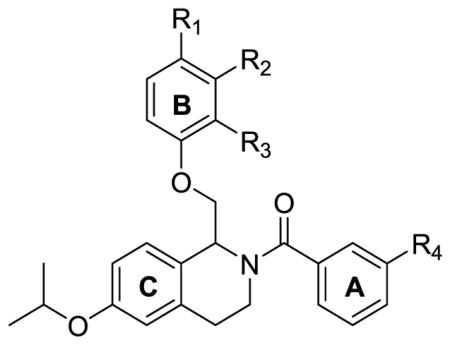
To first determine the optimal placement of substituents on the B-ring when an isopropoxy group was present on the C-ring, compounds 110 and 111 were synthesized (Table 6). When the methoxy functionality was moved from the para position to the meta position as in compound 110, the compound was selective for GluN2C and GluN2D. In 111, when the methoxy group was moved to the ortho position, all activity was lost. This data suggests that the para position is optimal for activity at GluN2B, and a number of compounds were synthesized with different substituents in the para-position, which resulted in varying degrees of selectivity. Compound 112 potentiated GluN2C and GluN2D with slight activity at GluN2B, while compound 92 was GluN2B- and GluN2C-selective. Compound 113 with an isopropoxy on the B-ring was moderately GluN2B-selective with maximum potentiation at GluN2B of 159% and only 134% at GluN2C. Since fluorine substitution on the A-ring (97) and isopropoxy substitution on the B-ring (113) both resulted in selectivity for the GluN2B subunit, these two modifications were combined in compound 116. This compound was active at GluN2C in addition to GluN2B, but the maximum potentiation at GluN2C was only 133%, suggesting that the isopropoxy group on the B-ring does indeed drive activity towards GluN2B (Table 6).
Table 6.
Optimization of potency based on substituent placement and identity of B-ring.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| I30μM / Icontrol (mean ± SEM, %) | EC50 (max.) (μM %)a | |||||||||
| # | R1 | R2 | R3 | R4 | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D |
| 2b | OMe | H | H | Cl | 195 ± 11 | 233 ± 18 | 240 ± 29 | 5.2 (202%) | 2.0 (243%) | 3.0 (245%) |
| 110 | H | OMe | H | Cl | 99 ± 5.6 | 132 ± 3.8 | 125 ± 2.4 | -- | 6.2 (132%) | 5.4 (125%) |
| 111c | H | H | OMe | Cl | 108 ± 1.9 | 110 ± 3.6 | 99 ± 1.6 | -- | -- | -- |
| 112 | OCF3 | H | H | Cl | 130 ± 2.7 | 189 ± 8.5 | 150 ± 5.9 | 1.4(130%) | 1.5 (192%) | 1.7 (152%) |
| 92b | OEt | H | H | Cl | 145 ± 7.8 | 126 ± 2.8 | 102 ± 2.0 | 5.3 (158%) | 1.8 (128%) | -- |
| 113 | OiPr | H | H | Cl | 159 ± 7.5 | 134 ± 8.0 | 111 ± 3.8 | 3.8 (160%) | 1.7 (138%) | -- |
| 114 | OMe | H | H | F | 249 ± 7.6 | 271 ± 15 | 402 ± 19 | 29 (391%) | 5.9 (365%) | 11 (493%) |
| 115 | OCF3 | H | H | F | 124 ± 3.8 | 154 ± 5.8 | 136 ± 3.2 | 3.2 (123%) | 2.5 (157%) | 4.1 (137%) |
| 97b | OEt | H | H | F | 147 ± 3.7 | 104 ± 2.2 | 91 ± 1.7 | 7.2 (152%) | -- | -- |
| 116 | OiPr | H | H | F | 179 ± 4.1 | 133 ± 4.5 | 103 ± 2.2 | 7.1 (185%) | 2.7 (135%) | -- |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 7–15 oocytes from 2–3 frogs for each compound and receptor tested.
Compounds 2, 92, and 97 were shown in previous tables and were included here for comparison.
Current ratio reported for 10 μM due to precipitate at 30 μM.
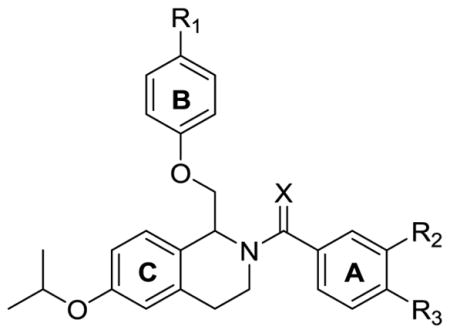
Effect of Modifying the A-ring Linker
Lastly, the linker between the tetrahydroisoquinoline core and the A-ring was modified by converting the amide to the thioamide (Table 7), a modification that yielded significant improvements in potency. When 2 was converted to thioamide 138, GluN2B, GluN2C, and GluN2D activity was observed, however there was approximately a 10-fold increase in potency at GluN2B and GluN2D with an approximate 4-fold increase at GluN2C potency compared to 2. Similarly, a significant increase in potency was observed when converting 114, which had an EC50 value at GluN2B of 36 μM, to thioamide 139, a compound with an EC50 at GluN2B of 1.4 μM. A number of compounds with an ethoxy group on the B-ring and an isopropoxy group on the C-ring were converted to thioamides, and these were consistently more potent at each of the subunits. Notably, compound 142, although not selective for GluN2B like its amide precursor 97, was 13-fold more potent at GluN2B than 97. The second GluN2B-selective compound, 113, was then converted to thioamide 145, and while activity was picked up at GluN2C and GluN2D, the EC50 values at each of the subunits was 0.48 μM and 0.41 μM, respectively, a substantial improvement in potency over the original GluN2B, GluN2C, and GluN2D potentiator, 2. Thioamide compounds 146 and 148 are the most potent GluN2B, GluN2C, and GluN2D potentiators in the class with EC50 values of approximately 0.3 μM, suggesting that the thioamide in combination with B-ring functionality is important, and enables compounds to simultaneously exhibit activity at these three subunits.
Table 7.
Optimization of potency based on A-ring linker and substituent identification of A-ring.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (max.) (μM, %)a | Response Doubling Concentration (μM)b | |||||||||
| # | R1 | R2 | R3 | X | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D |
| 2c | OMe | Cl | H | O | 5.2 (202%) | 2.0 (243%) | 3.0 (245%) | 64 | 4.6 | 4.9 |
| 138 | OMe | Cl | H | S | 0.49 (213%) d | 0.42 (225%) d | 0.43 (229%) d | 2.6 | 1.1 | 0.8 |
| 114c | OMe | F | H | O | 36 (391%) | 5.9 (365%) | 11 (493%) | 16 | 3.6 | 4.1 |
| 139 | OMe | F | H | S | 1.4 (259%) d | 0.97 (226%) d | 1.1 (208%) d | 1.9 | 2.1 | 4.2 |
| 92c | OEt | Cl | H | O | 5.3 (158%) | 1.8 (128%) | -- | -- | -- | -- |
| 140 | OEt | Cl | H | S | 0.40 (172%) d | 5.0 (278%) | 1.6 (192%) | -- | 7.6 | -- |
| 96c | OEt | Br | H | O | 3.4 (137%) | 1.8 (135%) | -- | -- | -- | -- |
| 141 | OEt | Br | H | S | 0.34 (174%) d | 0.94 (166%) d | 0.28 (135%) | -- | -- | -- |
| 97c | OEt | F | H | O | 7.2 (152%) | -- | -- | -- | -- | -- |
| 142 | OEt | F | H | S | 0.55 (184%) d | 1.4 (170%) | 1.2 (159%) | -- | -- | -- |
| 98c | OEt | CF3 | H | O | 2.4 (124%) | 1.3 (130%) | -- | -- | -- | -- |
| 143 | OEt | CF3 | H | S | 0.72 (190%) d | 0.35 (141%) d | 0.45 (128%) | -- | -- | -- |
| 108c | OEt | Cl | F | O | 2.2 (170%) | 1.3 (162%) | 2.2 (137%) | -- | -- | -- |
| 144 | OEt | Cl | F | S | 0.84 (222%) d | 0.35 (159%) d | 0.44 (128%) d | 17 | -- | -- |
| 113c | OiPr | Cl | H | O | 3.8 (160%) | 1.7 (138%) | -- | -- | -- | -- |
| 145 | OiPr | Cl | H | S | 0.55 (209%) d | 0.48 (141%) d | 0.41 (152%) | 4 | -- | -- |
| 116c | OiPr | F | H | O | 7.1 (185%) | 2.7 (135%) | -- | -- | -- | -- |
| 146 | OiPr | F | H | S | 0.67 (203%) d | 0.70 (158%) d | 1.2 (154%) | 9 | -- | -- |
| 147 | OiPr | CF3 | H | O | 1.5 (157%) | 0.92 (132%) | -- | -- | -- | -- |
| 148 | OiPr | CF3 | H | S | 0.27 (191%) d | 0.34 (162%) d | 0.33 (133%) | -- | -- | -- |
Fitted EC50 values are shown to two significant figures when potentiation at 30 μM exceeded 120%; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 3–26 oocytes from 2–5 frogs for each compound and receptor tested.
Concentration of test compound at which the response to maximally effective concentration of glutamate and glycine is increased 2-fold (see Methods)
Compounds with an amide linker (2, 114, 92, 96, 97, 98, 107, 108, 113, 116) shown in previous tables were included here for comparison to compounds with a thioamide linker.
The 95% confidence intervals for log fitted EC50 value determined from each oocyte were non-overlapping for the thioamide compound compared to its corresponding amide.
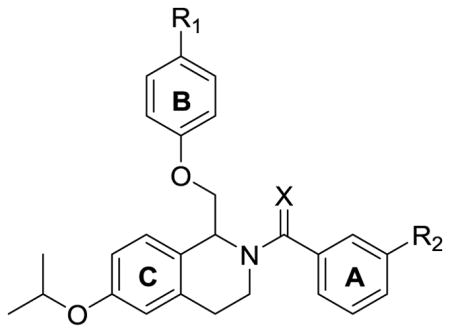
The increased potency when converting an amide to a thioamide was further investigated using computational methods. We hypothesized that there are two potential factors that contribute to the activity of these compounds. The first factor is the increase in lipophilicity when converting the amide to a thioamide (Table 8). A 1.42 log unit increase in the AlogP value upon conversion of the amide to the thioamide was calculated (Table 8) and could contribute to the increased potency of the thioamide. Second, we investigated the change in the cis/trans conformational preference of the molecules due steric and electronic properties of thioamides compared to amides. The cis/trans conformation is defined by carbonyl/thionyl orientation with respect to the chiral carbon (Supplemental Figure S1). We studied the cis/trans conformational preference by generating optimized geometries for both compounds S-(−)-138 and the S-(−)-2 in the cis and trans configuration using density functional theory (DFT) at the B3LYP/6–311-G(2d,2p) level. Implicit aqueous and oleaginous conditions were used to simulate the aqueous and protein environment. In the aqueous conditions the trans conformation of the thioamide compound is slightly favored (0.1 kcal/mol), while the cis conformation is moderately favored in the amide containing compound (0.3 kcal/mol; Supplemental Figure S1). In the oleaginous condition, representative of the diffusion of the compounds into the peptide core, both compounds 2 and 138 exhibit a shift away from the trans conformation (0.3 and 0.6 kcal/mol, respectively), although a greater portion of the thioamide remains in the trans conformation (Supplemental Figure S1). Given that the thioamide has a greater barrier to rotation with respect to the amide, it is possible that the peptide bound conformation is kinetically biased toward the aqueous equilibrium, while the amide more rapidly equilibrates toward the cis conformation within the binding pocket. We therefore postulate that the increased lipophilicity and trans population of the thioamide results in an increase potency of the thioamide-containing compounds.
Table 8.
Stereoselectivity of isopropoxy-containing tetrahydroisoquinoline compounds
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (max.) (μM, %)a | Doubling Concentration (μM)b | |||||||||
| # | R1 | R2 | X | AlogP | GluN2B | GluN2C | GluN2D | GluN2B | GluN2C | GluN2D |
| S-(−)-2c | OMe | Cl | O | 5.92 | 3.0 (276%) | 3.1 (277%) | 3.8 (267%) | 3.5 | 3.5 | 4.4 |
| S-(−)-138 | OMe | Cl | S | 7.34 | 0.32 (214%) d | 0.48 (307%) d | 0.48 (293%) d | 1.5 | 0.5 | 0.5 |
| R-(+)-2 | OMe | Cl | O | 5.92 | -- | 0.71 (252%) | 1.0 (297%) | -- | 1.3 | 1.0 |
| R-(+)-138 | OMe | Cl | S | 7.34 | -- | 1.7 (253%) d | 2.0 (250%) | -- | 3.8 | 4.9 |
| S-(−)-97c | OEt | F | O | 5.81 | 4.7 (204%) | 3.6 (128%) | -- | 40 | -- | -- |
| S-(−)-142 | OEt | F | S | 7.23 | 0.49 (213%) d | 0.38 (154%) d | 0.58 (179%) | 1.4 | -- | -- |
| R-(+)-97 | OEt | F | O | 5.81 | -- | -- | -- | -- | -- | -- |
| R-(+)-142 | OEt | F | S | 7.23 | -- | -- | -- | -- | -- | -- |
Fitted EC50 values are shown to two significant figures; values in parentheses are the fitted maximum response as a percentage of the initial glutamate (100 μM) and glycine (30 μM) current. Data are from between 12–27 oocytes from 2 frogs for each compound and receptor tested. Data for GluN1/GluN2A are given in Supplemental Table S1. 30 μM R-(+)-2 and R-(+)-138 had no significant effect on GluN1/GluN2B responses (101±5.9 and 105±3.5% of control, respectively, n=16–18). 30 μM S-(−)-97 had no significant effect on GluN1/GluN2D responses (110±4.4 of control, n=15). There was no detectable potentiation of any NMDA receptor subunit combinations by R-(+)-97 or R-(+)-142. AlogP values were calculated using Pipeline Pilot (Pipeline Pilot Server 17.1.0.115, Biovia)
Concentration of test compound co-applied at which the response to maximally effective concentration of co-applied glutamate and glycine is increased 2-fold over the response to glutamate and glycine alone (see Methods).
Compounds 2 and 97 were shown in previous tables and were included here for comparison.
The 95% confidence intervals for log fitted EC50 value determined from each oocyte were non-overlapping for the thioamide compound compared to its corresponding amide.
Potency of Enantiomers
The enantiomers of 2 were separated via chiral chromatography and both enantiomers were converted to thioamide-containing 138 enantiomers. It proved difficult to resolve the stereochemistry of either the R- or S-2 enantiomers with X-crystallography, but with the conversion of the amide to a thioamide, the absolute stereochemistry of one of the enantiomers was determined to be the S-(−) enantiomer by X-ray crystallography (see Supplemental Methods, Supplemental Table S2). In contrast to the CIQ (1) scaffold where only one enantiomer exhibited activity,61 evaluation of the 2 enantiomers revealed that both enantiomers were active, although they differed in potency and subunit selectivity. R-(+)-2 enantiomer was selective for GluN2C and GluN2D over GluN2A and GluN2B, with EC50 values of 0.71 μM and 1.0 μM, respectively (Table 8, Supplemental Table S1). The S-(−) enantiomer was less potent and showed different subunit selectivity, with EC50 values for potentiation of the response to maximal concentrations of glutamate and glycine at GluN2B-, GluN2C-, and GluN2D-containing NMDA receptors in the range of 3.0 – 3.8 μM. When both amide-containing enantiomers were converted to the thioamide, the same trend in subunit-selectivity was observed. R-(+)-138 was selective for GluN2C and GluN2D alone, while S-(−)-138 was active at GluN2B, GluN2C, and GluN2D subunits (Table 8, Supplemental Table S1). Interestingly, EC50 values were approximately 10-fold more potent (0.32–0.48 μM) at each subunit for the thioamide compared to the S-(−)-2 enantiomer, whereas the thioamide appears to have reduced potency for R-(+)-2 enantiomer at GluN2C and GluN2D. These data suggest that the GluN2B activity lies in the S-(−) enantiomer, while the R-(+) enantiomer is exclusively active at GluN2C and GluN2D.
Based on the results with enantiomers of 2 and 138, GluN2B-selective 97 was separated with the hypothesis that only the S-(−)-enantiomer would be active and the compound would exhibit exclusive activity at GluN2B. The potentiation by 30 μM S-(−)-97 at GluN2B was approximately 200%, whereas potentiation at GluN2C and GluN2D was only 127% and 110%, respectively. By contrast, no activity at any subunits was recorded for the R-(+)-enantiomer, further suggesting that GluN2B activity resides in the S-(−)-enantiomer (Table 8, Supplemental Table S1). When the 97 enantiomers were converted to the thioamide 142, only S-(−)-142 was active, whereas R-(+)-142 was inactive at all subunits (Table 8, Supplemental Table S1).
A significant proportion of NMDA receptors in adult brain contain two different GluN2 subunits, with triheteromeric receptors made up of GluN1/GluN2A/GluN2B present in principle cells79–82. We therefore tested GluN2B-preferring S-(−)-97 on triheteromeric receptors expressed in Xenopus oocytes coexpressing GluN1 with two different GluN2 subunits that contain modified intracellular domains that allow control of surface expression (see Methods). We found that 10 μM S-(−)-97 showed no effect on diheteromeric GluN1/GluN2A (103+1.2% of control, n=6) activated by maximally effective concentrations of glutamate and glycine, but potentiated diheteromeric GluN1/GluN2B (177+7.2% of control, n=14) and triheteromeric GluN1/GluN2A/GluN2B (136+2.7% of control, n=14). These data confirm that this series of positive allosteric modulators retains some activity at triheteromeric NMDA receptors that contain a single GluN2A and GluN2B subunit.
The development of an extensive SAR around this series with an isopropoxy group on the C-ring has allowed us to progress from compounds that exhibit activity as racemic mixtures to compounds that are only active as single enantiomers. This effect suggests that it may be possible to develop a unique class of compounds with stereodependency that can be exploited as potent and efficacious subunit–selective modulators. The thioamide-containing S-(−)-enantiomers of these two compounds, S-(−)-138 and S-(-)-142, are two of the most potent compounds in the class with EC50 values at GluN2B, GluN2C, and GluN2D subunits ranging from 0.32 μM – 0.58 μM.
Pharmacokinetic and metabolic analysis
We have previously shown that the GluN2C/GluN2D-selective tetrahydroisoquinoline CIQ was brain penetrant83 although CIQ has high protein binding and the free fraction in vivo is likely quite low (Jensen, Egebjerg, Bundgaard, unpublished data, personnel communication). We therefore determined the brain to plasma ratio for compound 2 of this new series of positive allosteric modulators. A dose of 10 mg/kg was administered IP in vehicle comprising 5% NMP/20% solutol/75% solution of 20% hydroxypropyl-β-cyclodextrin. Samples were evaluated by LC/MS/MS (See Methods) at 15, 30, 60, 120, 240 minutes. Administration of 10 mg/kg of compound 2 to C57Bl/6 mice produced a slow increase in plasma concentration that peaked at 2 hours (789 ng/mL, 1.7 μM) and slowly drops thereafter, and thus half-life could not be determined (n=3 mice). Brain concentration followed the plasma concentration, being 303 ng/mL (0.7 μM) at 15 and 30 min, and eventually dropping to 169 ng/mL (0.4 μM) at 4 hours. This corresponded to brain to plasma ratio of 0.7 at 15 minutes and 0.9 at 4 hours (n=3 mice). These data suggest that this class enters the brain, although more work is needed to define the pharmacokinetic properties and free fraction for members of this class.
Metabolic stability assays provide essential information on drug clearance, half-life and oral bioavailability, parameters that are useful during discovery/development. We therefore evaluated the metabolic stability in human liver microsomes (see Methods) of racemic 138, which is the thioamide analogue of prototypical compound 2. Compound 138 was found to have an in vitro microsome half-life of 29 minutes, being exclusively metabolized by cytochrome P450 enzymes and/or uridine-5-diphospho-glucuronosyltransferases. We also evaluated the stability of the thioamide, tracking conversion of compound 138 to compound 2, which represented conversion of the thioamide to an amide. We found that less than 1% of the thioamide was converted to an amide at 30 minutes, suggesting this functional group was relatively stable.
Conclusion
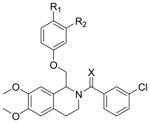
Whereas the previously published compound referred to as CIQ61 is highly selective for GluN2C- and GluN2D-containing NMDARs over GluN2A- and GluN2B-containing receptors, we have been able to target the GluN2B subunit with the replacement of dimethoxy groups on the C-ring with single isopropoxy functionality. Additionally, an extensive SAR has revealed that the tetrahydroisoquinoline series, once thought to be GluN2C/GluN2D selective, can be tuned to target the GluN2B, GluN2B/GluN2C, and GluN2B/GluN2C/GluN2D subunits. We have synthesized a number of compounds that are active at GluN2B, GluN2C, and GluN2D, and have shown that the isopropoxy-containing series has a distinct SAR from the series based around CIQ (several differences are summarized in Table 9). Changes to the scaffold of compound 2 induced different effects on NMDA potentiation than the same changes to the CIQ scaffold. Two important substituent changes were a methoxy group in the meta-position of the B-ring and an ethoxy group in the para-position of the B-ring. While these two functional groups were inactive when introduced into the CIQ scaffold61, they induced GluN2C/GluN2D activity (Compound 92) and GluN2B/GluN2C (Compound 110) when an isopropoxy on the Cring was present, suggesting the isopropoxy was able to revive some activity that had been eliminated in CIQ. Interestingly, the substitution of an ethoxy for methoxy in the para position of the B-ring in CIQ scaffold attenuated activity at GluN2C/GluN2D even for the isopropoxy-substituted C-ring, leaving GluN2B activity as the strongest effect of this analogue. Perhaps most significantly, the thioamide was inactive on the CIQ scaffold61, but was essential for potent activity when the tetrahydroisoquinoline contains an isopropoxy group as in compound 2.
Table 9.
Comparison of CIQ and Compound 2
|
|
|
|---|---|---|
| R1 = OEt, R2 = H | No activity at any subunits | GluN2B/C |
| X = S | No activity at any subunits | GluN2B/GluN2C/GluN2D |
| R1 = H, R2 = OMe | No activity at any subunits | GluN2C/GluN2D |
While the majority of compounds have EC50 values in the micromolar range, thioamide-containing compounds are significantly more potent with submicromolar EC50 values. Moreover, replacement of the para-methoxy on the B-ring with an ethoxy-group consistently yielded compounds that were selective for GluN2B and GluN2C over GluN2D. SAR studies around each of the rings led to varying degrees of selectivity, with significant shifts in potency arising from modifications to the A-ring linker. Compound 2, the first GluN2B, GluN2C, and GluN2D potentiator of maximally activated NMDARs, had EC50 values at all subunits in the low micromolar range. The most potent non-selective potentiators have EC50 values that are an order of magnitude lower (i.e., more potent) than 2; compound 148 had the highest potency at GluN2B (270 nM).
With the separation of the 2 and 138 enantiomers and the resolution of the S-(−) enantiomer by X-ray crystallography, we have determined that the GluN2B activity resides exclusively in the S-(−) enantiomer. While both enantiomers are active, the R-(+) enantiomer is selective for GluN2C and GluN2D-containing receptors and the S-(−) enantiomer causes potentiation at GluN2B, GluN2C, and GluN2D-containing receptors. This is a distinct difference from the GluN2C/GluN2D-selective CIQ enantiomers, where only one enantiomer was active (predicted to be the (R)-(+)-enantiomer based on a stereoselective reduction61,84). By exploring the tetrahydroisoquinoline scaffold, we have developed positive allosteric modulators of NMDAR function that are more active at the GluN2B subunit. This is demonstrated by compounds 97 and 142, which showed a significantly greater degree of potentiation at GluN2B than other GluN2 subunits. Racemic compound 97 exhibited a fitted maximal potentiation to 155+5.3% (mean + SEM) of control at GluN2B (n=17) compared to no discernable activity at GluN2C (104+2.2% of control at 30 μM, n=12) or at GluN2D (92+1.7% of control at 30 μM, n=15). Similarly, the (S)-( −)-enantiomer of 97 showed similar potencies at GluN2B and GluN2C, but had a greater maximal fitted response at GluN2B (208+7.3% of control, n=27) compared to GluN2C (135+4.8% of control, n=14, p<0.001 t-test). Potentiation of the GluN2B subunit has been observed with some endogenous molecules, such as polyamines, neurosteroids, as well as aminoglycoside antibiotics. However, small, drug-like molecules that selectively potentiate GluN2B-containing NMDARs have not been described. There is a need for potent compounds that can be utilized as probes to evaluate the ramifications and utility of GluN2B potentiation. Thus, the synthesis of compounds 97, 142, 147, 146, and 136 demonstrate a potential means for developing such tools.
Biology Experimental procedures
Unfertilized Xenopus laevis oocytes were obtained from Ecocyte (Austin TX). Two-electrode voltage-clamp recordings were performed on oocytes injected with mRNA to express recombinant rat GluN1/GluN2A, GluN1/GluN2B, GluN1/GluN2C, GluN1/GluN2D, GluA1, or GluK2 receptors. cDNAs for rat GluN1–1a (GenBank accession numbers U11418 and U08261; hereafter GluN1), GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), GluN2D (D13213), GluA1 (X17184), GluK2 (Z11548) were provided by Drs. S. Heinemann from the Salk Institute, S. Nakanishi from Kyoto University, and P. Seeburg from University of Heidelberg. GluN2C and GluN2D were modified as previously described16. Oocyte isolation, cRNA synthesis and cRNA were performed as previously described85. For the two-electrode voltage clamp recordings, oocytes were placed in a perfusion chamber and continually washed with a recording solution that contained (in mM) 90 NaCl, 1.0 KCl, 0.5 BaCl2, 0.005 EDTA, and 10 HEPES at pH 7.4 and a temperature of 23°C. The glass electrodes with a tip resistance of 0.5 – 2.5 MΩ were pulled from thin-walled glass capillary tubes and filled with 0.3–3.0 M KCl. The membrane potential of the oocytes was held at −40 mV by an OC-725C amplifier (Warner Instrument Co). All compounds were made as 20 mM stock solutions in DMSO and diluted to reach the desired final concentration in recording solution containing 100 μM glutamate and 30 μM glycine; final DMSO content was 0.05–0.5% (vol/vol). A gradual increase in current response over the course of the experiment is common with oocytes expressing GluN1/GluNA. To prevent this, oocytes expressing GluN1/GluN2A were either pretreated with 50 μM BAPTA-AM (1,2-bis(oaminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetraacetoxymethyl ester) for 10 min or injected with 20 nl of 100 mM K-BAPTA (potassium 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid). Compounds that had a modest effect on GluN1/GluN2A expressing oocytes were not studied further. We also evaluated the function of NMDA receptors that contained GluN2A and GluN2B subunits modified to contain the GluN2A intracellular C-terminal domain fused to either a C1 or C2 coiled-coil domain and an ER retention signal, as previously described86. Only receptors that contained GluN2 subunits with complementary C1 and C2 domains (GluN2AC1/GluN2AC2, GluN2AC1/GluN2BC2, GluN2BC1/GluN2BC2) will reach the cell surface, allowing us to evaluate a population of receptors with known subunit stoichiometry. Controls performed with GluN2 subunits inactivated by mutations in the agonist binding domain suggest that >95% of the current response arose from NMDA receptors that contained the desired GluN2 subunit combinations.86
Every test compound was recorded at 5–7 concentrations in at least 4 oocytes from at least 2 different frogs at GluN2B-, GluN2C-, and GluN2D-containing NMDA receptors and in 3–15 oocytes from 1–3 frogs at GluN2A-containing receptors. The potentiation of the test compounds at a concentration of 30 μM was averaged and reported as I30μM/Icontrol (mean + SEM, %), where I equals current. A few compounds with lower solubility were reported as I10μM/Icontrol or I3μM/Icontrol (mean + SEM, %). For test compounds with potentiation that exceeded 120% at 30 μM, an EC50 value (the half-maximal effective concentration of potentiator) was determined by fitting the following equation
| (1) |
to the mean composite concentration-response data normalized to the current in the absence of the potentiator (100%) where N equals the Hill slope and maximum is the maximal response predicted for saturating concentration of potentiator. The concentration that gave a 2-fold increase in the current response was determined by rearranging equation (1) to yield
| (2) |
Determination of pharmacokinetics and brain penetration was performed for compound 2 following IACUC-approved protocols and the animal welfare regulations outlined in the “Guide for the Care and Use of Laboratory Animals”. Briefly, 30–35 gm C57Bl/6 mice were injected intraperitoneal (i.p.) with 10 mg/kg of compound 2 at 1.25 mg/mL in 5% NMP/20% solutol/75% of a 20% solution of hydroxypropyl-β-cyclodextrin. No behavioral abnormalities were observed for all animals post-dosing. Animals were terminally anaesthetized by a raising the concentration of CO2, and following the cessation of breathing, the blood removed via cardiac punctureand collected in heparinized vials. The chest cavity was opened to expose the heart, an incision cut at the right auricle by surgical scissors and a syringe full of saline was pushed into the heart slowly via the left ventricle (saline volume ~20 mL for rat and 10 mL for mouse). After perfusion, the brain was removed, and samples were washed with saline, dried with clean surgical gauze, and then put into weighed tubes. The brain samples were stored at −75±15°C until analysis. Plasma samples for 0.25~0.5 h time points were prepared by sequentially adding 5 μL of blank solution, 2 μL of plasma sample, 8 μL of blank plasma, and fifteen volumes of acetonitrile (150 μL) for protein precipitation; the final compound concentration in plasma (ng/mL) was corrected by multiplying 5. Other plasma samples were prepared by adding 5 μL of blank solution, 10 μL of plasma samples, fifteen volumes of acetonitrile (150 μL) for protein precipitation. Brain samples were prepared by adding brain weight (g) to deionized water volume (mL) at a ratio of 1:4 for homogenization, and 30 μL of brain homogenate was added to 15 μL of blank solution, followed by addition of 150 μL of acetonitrile for protein precipitation. The mixtures were vortexed for 30 s, then centrifuged at 4000 rpm for 15 min, and the supernatant diluted 3 times with water. An aliquot of 3 μL for plasma or 1 μL for brain of the diluted supernatant was injected into the LC-MS/MS system that included a Shimadzu LC-30A coupled with Rack Changer Autosampler/AB API6500 and a Phenomenex Kinetex 2.6u F5 (30 × 2.1mm) column. A solvent gradient was obtained from 95% water/0.1% formic acid and 95% acetonitrile/0.1% formic acid; the flow rate was 0.6 ml/min at room temperature. Concentrations were determined from an 8 point standard curve generated in duplicate. Blood and brain samples were collected at 0, 15, 30, 60, 120, 240 min from three C57Bl/6 mice at each time point.
The metabolic stability of compound 138 was evaluated in human liver microsomes in the presence of NADPH, a cofactor required for cytochrome P450 enzymes (CYP) and flavin monooxygenases activity, plus UDPGA, a cofactor required for glucuronic acid conjugation (glucuronidation) by UGT enzymes (uridine-5′-diphospho-glucuronosyltransferases). A reversed phase gradient HPLC method with positive ion electrospray ionization (ESI) mass spectrometry (MS) detection employing an internal standard was utilized to quantify the concentrations of unchanged parent compound 138 and the potential amide metabolite 2, as well as positive controls that were run in parallel (verapamil and 7-hydroxycoumarin for Phase I and Phase II metabolism, respectively) to establish systems’ competency.
Compound 138 (5.1 μM or 2458 ng/mL) was incubated in triplicate in human liver microsomes (0.5 mg total protein), with and without the necessary cytochrome P450, flavin monooxygnease, and uridine-5′-diphospho-glucuronosyltransferase (UGT) cofactors (NADPH, UDPGA) to determine its metabolic stability. UGT Solution B (UGT-B; BD Biosciences) is a solution of 0.5 M (5X) Tris buffer and alamethicin (a pore-forming agent used to mitigate the latency of UGT enzymes). UGT Solution A (UGT-A) is a 25 mM UDPGA (uridine-5′-diphosphoglucuronic acid) solution, the cofactor necessary for glucuronidation. The NADPH regenerating system consists of a 2% (w/v) sodium bicarbonate with magnesium chloride, NADP, glucose-6-phosphate and glucose-6-phosphate dehydrogenase. Bicarbonate solution without the cofactors was used as a negative control. Incubations were performed in 13 × 100 mm glass culture tubes. Samples were placed in a water bath shaker set at 37°C and shaken at 150 rpm. Negative controls (without cofactors but containing drug and microsomes) and controls without microsomes were run in parallel to account for any chemical degradation. Incubations of the positive controls, verapamil (1 μM) and 7-hydroxycoumarin (261 μM), demonstrated that the incubation systems were competent with respect to oxidative activities and conjugation (glucuronidation). A control without drug was run and used for matrix standard preparation and to determine specificity. Aliquots of 100 μL were taken at 0, 15, 30, 60 and 120 min for the NADPH/UDPGA sample and at 0 and 120 minutes for the negative controls. These aliquots were mixed with 400 μL of an internal standard in a 1.5-mL conical polypropylene microcentrifuge tube that consisted of 1 μM the metabolite 2 in acetonitrile with 0.1% formic acid for the compound 138 assay and 5 μM of the metabolite D5–7-ethoxycoumarin in acetonitrile with 0.1 % formic acid for the verapamil and coumarin assays. Subsamples plus internal control from each time point were vortexed for 10 seconds and centrifuged for 2 minutes at 13,000 rpm. Supernatants were decanted into 2-mL LC vials and analyzed by LCMS/MS using a Zorbax column (Eclipse XDB-C18, 4.6 × 150 mm, 5μm) run at 35°C with a flow rate of 1000 μl/min and an injection volume of 5 μl. The mobile phase gradient was made from 50 mM ammonium acetate (aq) + 0.1% formic acid (v/v) and acetonitrile + 0.1% formic acid (v/v). Quantitation was performed using the mean response factor (analyte/internal standard ratios) of the time zero replicates. These samples were assigned a value of 100% and the subsequent time-point samples were calculated as a percentage relative to this time-zero average response. These values represented the % parent compound (138) remaining at the corresponding time-point. For the compound 2 quantitation, a 5-point calibration curve was built and the sample concentrations were calculated in ng/mL.
Chemistry Experimentals
Density Functional Theory Computational Methodology
The Schrodinger Small-Molecule Drug Discovery Suite 2016-2 (Small-Molecule Drug Discovery Suite 2016-2, Schrödinger, LLC, New York, NY, 2017) was used. Initial structures were generated using molecular mechanics modeling in combination with geometric constraints. These poses identify two conformations of the A ring either extended or adjacent to the C ring. These can be identified respectively as cis and trans conformations of the amide carbonyl or sulfonyl moiety with respect to the chiral carbon, as depicted in Supplemental Figure S1. These geometries were then optimized at the B3LYP/6-311-G(2d,2p) level using the TeraChem software package (Titov DOI: 10.1021/ct300321a) in the mixed precision mode. These calculations were conducted in vacuo and employing the polarizable continuum model (Liu DOI: 10.1021/acs.jctc.5b00370) of solvation with dielectric constants of 78.3 and 7 to represent the corresponding geometry in water (National Bureau of Standards - Research Paper 2641) and the peptide interior (Li DOI: 10.1021/ct400065j).
General Experimental
All reagents were purchased from commercial vendors and used without further purification. Thin layer chromatography (TLC) on precoated aluminum plates (silica gel 60 F254, 0.25 mm) or LCMS (Varian) were used to monitor reaction progress. Purification by flash column chromatography was done on a Teledyne ISCO Combiflash Companion using Teledyne Redisep normal phase columns. Proton and carbon NMR spectra were recorded on an INOVA-400 (400 MHz), VNMRS-400 (400 MHz), Mercury 300 Vx (300 MHz) or INOVA-600 (600 MHz). All chemical shifts were reported in parts per million and coupling constants were reported in Hertz (Hz). The spectra were referenced to the solvent peak. Mass spectra were performed by the Emory University Mass Spectroscopy Center on either a VG 70-S Nier Johnson or JEOL instrument. Purity was established by LC-MS (Varian) in two solvents systems (MeOH:water and ACN:water) unless indicated by combustion analysis. For each compound where purity was established with LC-MS, the retention time in both solvent conditions is given. The conditions were determined for each individual compound. Three compounds (98, 99, and 109) were not greater than 95% pure by LCMS, but all compounds were greater than 87% pure and the specific details for each compound are given below. Elemental analysis was performed by Atlantic Microlab, Inc (Norcross, GA) for C, H, and N, and agreed with proposed structures within 0.4 + of theoretical values. Optical rotation values were obtained using a Perkin-Elmer 314 instrument.
General preparation for phenethylcarbamate compounds (Procedure I)
Potassium carbonate (4.0 equiv) was added to a solution of tert-butyl 3-hydroxyphenethylcarbamate 3 (1.0 equiv) in dry DMF (44.4 ml) and the reaction was allowed to stir for 2 hours before the alkyl halogen (1.5 – 2.0 equiv) was added and the reaction was heated to 60°C. After stirring for 24 hours, the reaction was quenched with saturated NH4Cl and extracted into EtOAc. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was subjected to flash column chromatography to afford the title compound.
General preparation for phenethylcarbamate compounds (Procedure II)
Triphenylphosphine (1.0 equiv), the substituted alcohol (1.2 equiv), and triethylamine (1.0 equiv) were added to a solution of tert-butyl 3-hydroxyphenethylcarbamate (1.0 equiv) dissolved in dry THF. The reaction was brought to 0 °C with an ice bath and (Z)-diisopropyl diazene-1,2-dicarboxylate (1.0 equiv) was added dropwise. After stirring for 6 days, the reaction was quenched with 1M HCl and extracted into EtOAc. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was subjected to flash column chromatography to afford the title compound.
General preparation for phenylethanamine hydrochloride compounds (Procedure III)
The phenethylcarbamate compound was dissolved in a solution of ether and 12 N HCl (2:1 mixture) and allowed to stir at room temperature for 40 minutes. The volatiles were removed in vacuo to afford the title compound, which was carried forward without any further purification.
General preparation for 2-chloro-N-phenethylacetamide compounds (Procedure IV)
A solution of the phenethylamine (1 equiv.) in dry DCM was brought to 0°C using an ice bath. Triethylamine (2 equiv.) was added followed by the dropwise addition of 2-chloroacetyl chloride (1.2 equiv.) The reaction was allowed to cool to room temperature and stir for 3 hours. Upon completion by TLC, the reaction was quenched with 1M HCl, extracted into DCM, washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for N-(phenethyl)-2-(phenoxy)acetamide compounds (Procedure V)
To a solution of phenol (1.2 equiv.) dissolved in dry ACN was added Cs2CO3 (4 equiv.). The solution was allowed to stir for 2 hours before the 2-chloro-N-phenethylacetamide compound (1.0 equiv.) dissolved in dry ACN was added and the reaction was allowed to stir for 15 hours. Once TLC indicated complete conversion, the reaction was quenched with saturated NH4Cl and extracted into EtOAc. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for N-(3-iodophenethyl)-2-(phenoxy)acetamide compounds (Procedure VI)
A 10 mL sealed vial was charged with the N-(3-bromophenethyl)-2-phenoxyacetamide compound (1.0 equiv), copper(I) iodide (0.5 equiv), and sodium iodide (1.5 equiv). The vial was sealed, evacuated, and backfilled with argon. This was repeated twice more before N1,N2-dimethylethane-1,2-diamine (0.1 equiv) and dry dioxane were added and the reaction was stirred at 110 °C for 24 hours. The reaction was then allowed to cool to room temperature, quenched with a 30 % ammonium hydroxide solution, and diluted with water. The reaction was extracted into EtOAc, washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for N-(phenethyl)-2-(phenoxy)-acetamide compounds (Procedure VII)
A 10 mL sealed vial was charged with the N-(3-iodophenethyl)-2-(phenoxy)acetamide compounds (1.0 equiv), copper(I) iodide (0.1 equiv), 1,10-phenanthroline (0.2 equiv), and cesium carbonate (2.0 equiv). The vial was sealed, evacuated, and back-filled with argon. This was repeated twice more before the alcohol 3-pentanol (9.3 equiv) was added, and the vial was submerged in an oil bath heated to 110 °C. After stirring for 24 – 48 hours, the reaction was allowed to cool to room temperature, diluted with EtOAc, and filtered through a plug of celite rinsing with EtOAc. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 2-(4-methoxyphenoxy)-N-(3-(piperidin-1-yl)phenethyl)acetamide, N-(3-(cyclohexylamino)phenethyl)-2-(4-methoxyphenoxy)acetamide, and 2-(phenoxy)-N-(3-morpholinophenethyl)acetamide compounds (Procedure VIII)
A 10 mL sealed vial was charged with the N-(3-iodophenethyl)-2-(phenoxy)acetamide compounds (1.0 equiv), copper(I) iodide (0.2 equiv), potassium carbonate (2.0 equiv), and L-proline (0.2 equiv). The vial was sealed, evacuated, and back-filled with argon. This was repeated twice more before DMSO was added followed by cyclic amines or cyclohexanes (1.5 equiv) and the reaction was submerged in an oil bath heated to 80 °C. After stirring for 48 hours, the reaction appeared to be complete by LCMS and TLC. The reaction was allowed to cool to room temperature, diluted with EtOAc, and washed with water (3×), brine (6x), and 5.0 % LiCl solution (8x). The organic layer was dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for N-(3-(amino)phenethyl)-2-(4-methoxyphenoxy)acetamide compounds (Procedure IX)
The N-(3-(amino)phenethyl)-2-(4-methoxyphenoxy)acetamide compound (1.0 equiv) and paraformaldehyde (10 equiv) were dissolved in AcOH and sodium cyanoborohydride (5.0 equiv) was added in one portion at room temperature. After stirring for 4 – 20 hours the reaction was brought to 0°C was made basic using concentrated NH4OH. The reaction was extracted into EtOAc, washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 3,4-dihydroisoquinoline Compounds (Procedure X)
The amide (1.0 equiv) was suspended in dry toluene and the reaction mixture was brought to reflux. Phosphorous trichloride (3.0 equiv) was added and the reaction was allowed to stir until TLC indicated complete conversion. The reaction was allowed to cool to room temperature, the excess toluene was decanted, and the remaining residue was carried on without further purification.
General procedure for 3,4-dihydroisoquinoline Compounds (Procedure XI)
The amide (1.0 equiv) was suspended in dry toluene and the reaction mixture was brought to reflux. Phosphorous trichloride (3.0 equiv) was added and allowed to stir until TLC indicated complete conversion. The reaction was allowed to cool to room temperature, made basic (pH 13) with concentrated NH4OH, and the organic layer was extracted into DCM. The organic layer was then washed with water, brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was carried on without further purification.
General procedure for 1,2,3,4-tetrahydroisoquinoline Compounds (Procedure XII)
The dihydroisoquinoline (1.0 equiv) was dissolved in dry MeOH and brought to 0°C using an ice bath. Sodium borohydride (3.0 equiv) was added slowly and the reaction was allowed to stir overnight and warm to room temperature. The volatiles were concentrated in vacuo, and the resulting residue was extracted into DCM. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 3,4-dihydroisoquinolin-2(1H)-yl)methanone) Compounds (Procedure XIII)
The tetrahydroisoquinoline (1.0 equiv) was dissolved in dry DCM and brought to 0°C using an ice bath. Triethylamine (2.0 equiv) was added followed by the benzoyl chloride (1.2 equiv). The reaction mixture stirred for approximately 3 hours under an argon atmosphere, at which point TLC indicated complete conversion. The reaction was quenched with 1M HCl and extracted into DCM. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 3,4-dihydroisoquinolin-2(1H)-yl)methanone) Compounds (Procedure XIV)
Benzoic acid (1.0 equiv) was dissolved in dry DCM and brought to 0 °C using an ice bath. N1-((ethylimino)methylene)-N3,N3-dimethylpropane-1,3-diamine (1.2 equiv) and N,N-dimethylpyridin-4-amine (1.2 equiv) were added and the reaction was allowed to stir for 2 hours before tetrahydroisoquinoline (1.0 equiv) dissolved in dry DCM was added. The reaction was allowed to stir overnight and warm to room temperature. The reaction was quenched with DI water, extracted into DCM, washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 3,4-dihydroisoquinolin-2(1H)-yl)methanethione) Compounds (Procedure XV)
The dihydroisoquinoline (1 equiv) was dissolved in dry toluene and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.5 – 1 equiv) was added. The reaction was brought to reflux. After 2 – 8 hours, the solvent was removed under reduced pressure and the residue was dissolved in DCM. The solution was washed with a 10% solution of NaHCO3, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
General procedure for 3,4-dihydroisoquinolin-2(1H)-yl)methanethione) Compounds (Procedure XVI)
The dihydroisoquinoline (1 equiv) was dissolved in dry toluene and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.5 – 1 equiv) were dissolved in toluene in a sealed tube. The material was heated to 150°C for 35 minutes in the microwave. The reaction was diluted with DCM, quenched with saturated sodium bicarbonate. The organic layer was washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The resulting residue was then subjected to column chromatography to afford the title compound.
(3-Chlorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (2)
Tetrahydroisoquinoline 2 was prepared via Procedure XIII using tetrahydroisoquinoline 67 (0.15 g, 0.48 mmol) and 3-chlorobenzoyl chloride (0.061 mL, 0.45 mmol) in DCM (7.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.15 g, 70% mixture of two amide rotamers). 1HNMR (CDCl3, 400 MHz) δ: 8.04-7.93 (m, 0.5H), 7.56-7.52 (m, 0.5H),7.39-7.19 (m, 4H), 6.91-6.66 (m, 6H), 5.98 (t, J = 4.8 Hz, 0.5H), 5.14-5.10 (m, J = 3.6 Hz, J = 6.0 Hz, 0.5H), 4.87-4.82 (m, J = 4.8 Hz, J = 5.2 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.37-4.29 (m, 1H), 4.12 (t, J = 10 Hz, 0.5H), 3.93-3.90 (m, 0.5H), 3.74 (s, 3H), 3.72-3.64 (m, 1H), 3.29-3.01 (m, 1H), 2.92-2.71 (m, 1.5H), 1.32 (d, J = 6.0 Hz, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.6, 169.8, 157.6, 157.0, 154.4, 154.3, 153.1, 152.6, 138.3, 138.1, 136.6, 135.5, 134.9, 134.8, 134.6, 133.6, 130.4, 130.2, 129.9, 129.8, 128.7, 128.4, 128.3, 127.0, 125.9, 125.1, 124.9, 124.0, 116.2, 115.9, 115.5, 114.9, 114.8, 114.7, 114.5, 76.9, 71.2, 70.1, 57.3, 55.9, 52.0, 42.8, 35.5, 29.9, 28.5, 22.3, 22.2; HRMS calcd. for C27H29ClN1O4, 466.17784 [M + H]+; found 466.17796 [M + H]+; Anal. (C27H28N1O4Cl): C, H, N; mp: 72°C.
(6-(Benzylthio)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-chlorophenyl)methanone (87)
Dihydroisoquinoline 58 was prepared via procedure X using amide 36 (0.67 g, 2.0 mmol) and phosphorous trichloride (1.3 mL, 6.1 mmol) in dry toluene (11 mL). The crude residue (0.84 g) was carried on without further purification. HRMS calcd. for C19H22O3N1, 312.15942 [M + H]+; found 312.15912 [M + H]+. Tetrahydroisoquinoline 77 was prepared via procedure XII using dihydroisoquinoline 58 (0.84 g, 2.7 mmol) and sodium borohydride (0.31 g, 8.1 mmol) in dry MeOH (14 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 75 (0.21 g, 25 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. Tetrahydroisoquinoline 87 was prepared via procedure XIII using tetrahydroisoquinoline 77 (0.21 g, 0.68 mmol) and 3-chlorobenzoyl chloride (0.10 mL, 0.81 mmol) in DCM (11 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.059 g, 19 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.71; 1HNMR (CDCl3, 400 MHz) δ: 7.61-7.56 (m, 0.5H), 7.42-7.22 (m, 4H), 6.95-6.69 (m, 6.5H), 5.98-6.01 (m, 0.5H), 5.16-5.13 (m, J = 3.2 Hz, J = 9.2 Hz, 0.5H), 4.89-4.85 (m, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.37-4.32 (m, 1H), 4.15 (t, J = 10.4 Hz, 0.5H), 4.03 (q, J = 6.8 Hz, 2H), 3.96-3.92 (m, 0.5H), 3.77 (s, 3H), 3.73-3.67 (m, 1H), 3.31-3.12 (m, 1H), 2.95-2.74 (m, 1.5H), 1.42 (t, J = 6.8 Hz, 3H); 13CNMR (CDCl3, 100 MHz) δ: 170.5, 169.7, 158.6, 158.1, 154.3, 154.2, 153.1, 152.6, 141.4, 138.3, 138.2, 136.6, 135.6, 134.9, 134.6, 133.6, 131.7, 130.2, 129.9, 129.8, 128.7, 128.4, 128.3, 128.2, 127.0, 125.8, 125.2, 124.9, 124.2, 115.9, 115.5, 114.9, 114.4, 113.6, 113.4, 71.3, 70.2, 63.7, 57.3, 55.9, 51.9, 42.8, 35.5, 29.9, 28.5, 15.1, 14.1; HRMS calcd. for C26H27NO435Cl, 452.16231 [M + H]+; found 452.16203 [M + H]+. Anal. (C26H26N1O4Cl): C, H, N.
(3-Chlorophenyl)(6-isobutoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (88)
Dihydroisoquinoline 53 was prepared via Procedure X using 25 (1.1 g, 3.1 mmol) and phosphorous trichloride (1.9 mL, 3.1 mmol) in dry toluene (16 mL). The crude residue (0.82 g) was carried on without further purification. HRMS calcd. for C21H26O3N1, 340.19072 [M + H]+; found 340.19061 [M + H]+. Tetrahydroisoquinoline 72 was prepared via Procedure XII using dihydroisoquinoline 53 (0.82 g, 2.4 mmol) and sodium borohydride (0.27 g, 7.2 mmol) in dry MeOH (13 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 20 g column, 0–20% MeOH/DCM gradient) to afford the title compound as a yellow solid (0.45 g, 55 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. HRMS calcd. for C21H28O3N1, 342.20637 [M + H]+; found 342.20630 [M + H]+. Tetrahydroisoquinoline 88 was prepared via Procedure XIII using tetrahydroisoquinoline 72 (0.23 g, 0.66 mmol) and 3-chlorobenzoyl chloride (0.11 mL, 0.79 mmol) in DCM (10 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.087g, 27% yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.68; 1HNMR (CDCl3, 400 MHz) δ: 7.58 (s, 0.5H), 7.39-7.19 (m, 4H), 6.92-6.67 (m, 6.5H), 5.97-5.96 (m, 0.5H), 5.12-5.11 (m, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.37-4.30 (m, 1H), 4.13 (t, J =9.6 Hz, 0.5H), 3.93-3.89 (m, 1H), 3.72 (s, 3H), 3.69 (d, J = 6.4 Hz, 2H), 3.66-3.63 (m, 0.5H), 3.28-3.09 (m, 1H), 2.92-2.72 (m, 1.5H), 2.11-2.01 (m, 1H), 1.00 (d, J = 6.8 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.7, 158.9, 158.4, 154.4, 154.3, 153.0, 153.6, 138.4, 138.2, 136.6, 135.5, 134.9, 134.5, 130.2, 129.9, 129.8, 128.7, 128.3, 128.2, 127.0, 125.8, 125.1, 124.9, 124.1, 115.9, 115.4, 114.9, 114.8, 114.4, 113.7, 113.5, 74.7, 71.3, 70.2, 57.3, 55.9, 51.9, 42.9, 35.5, 29.9, 28.5, 19.5, 19.4; HRMS calcd. for C28H31NO435Cl, 480.19361 [M + H]+; found, 480.19346 [M + H]+; Anal. (C28H30N1O4Cl): C, H, N.
(3-Chlorophenyl)(1-((4-methoxyphenoxy)methyl)-6-(pentan-3-yloxy)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (89)
Tetrahydroisoquinoline 89 was prepared via Procedure XIII using tetrahydroisoquinoline 73 (0.16 g, 0.44 mmol) and 3-chlorobenzoyl chloride (0.069 mL, 0.52 mmol) in DCM (6.8 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.087 g, 41 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.69; 1HNMR (CDCl3, 400 MHz) δ: 7.59 (s, 1H), 7.37-7.18 (m, 4H), 6.91-6.67 (m, 7H), 5.97 (t, J = 4.8 Hz, 0.5 H), 5.13-5.10 (m, J = 3.6 Hz, J = 10 Hz, 0.5 H), 4.86-4.82 (m, J = 5.6 Hz, J = 13.2 Hz, 0.5 H), 4.37-4.29 (m, 1H), 4.14-4.06 (m, 1.5 H), 3.92-3.89 (m, J = 3.2 Hz, J = 13.2 Hz, 0.5 H), 3.75 (s, 3H), 3.69-3.62 (m, 1H), 3.27-3.08 (m, 1H), 2.93-2.71 (m, 1.5H), 1.69-1.63 (m, 4H), 0.95 (t, J = 7.2 Hz, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 158.4, 157.9, 154.4, 154.2, 153.1, 152.6, 138.4, 138.3, 136.6, 135.5, 134.8, 134.5, 130.2, 129.9, 129.8, 128.7, 128.3, 128.2, 127.0, 125.9, 125.1, 124.9, 123.9, 116.3, 115.9, 115.4, 114.9, 114.8, 114.5, 80.3, 71.2, 70.1, 57.3, 55.9, 51.9, 42.8, 35.4, 29.9, 28.6, 26.4, 9.8; HRMS calcd. for C29H33NO435Cl, 494.20962 [M + H]+; found 494.21000 [M + H]+; Anal. (C29H32N1O4Cl): C, H, N.
(3-Chlorophenyl)(6-(cyclohexyloxy)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (90)
Dihydroisoquinoline 55 was prepared via Procedure XI using amide 27 (0.52 g, 1.4 mmol) and phosphorus trichloride (0.84 mL, 4.1 mmol) in dry toluene (7.6 mL). The crude solid was carried on without further purification. HRMS calcd. for C23H28O3N, 366.20627 [M + H]+; found 366.20615 [M + H]+. Tetrahydroisoquinoline 74 was prepared via Procedure XII using 55 (0.58 g, 1.6 mmol) and sodium borohydride (0.18 g, 4.8 mmol) in dry MeOH (8.0 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 74 (0.32 g, 55 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. HRMS calcd. for C23H30NO3, 368.22202 [M + H]+; found, 368.22227 [M + H]+. Tetrahydroisoquinoline 90 was prepared via Procedure XIII using tetrahydroisoquinoline 74 (0.16 g, 0.44 mmol) and 3-chlorobenzoyl chloride (0.071 mL, 0.53 mmol) in DCM (6.9 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.038 g, 17% yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.72 ; 1HNMR (CDCl3, 400 MHz) δ: 7.57 (s, 0.5H), 7.39-7.17 (m, 3H), 7.25-7.23 (m, 1H), 7.19-7.17 (m, 0.5H), 6.90-6.66 (m, 6H), 5.96 (t, J = 4.8 Hz, 0.5H), 5.12-5.09 (m, 3.2 Hz, J = 9.2 Hz, 0.5H), 4.86-4.81 (m, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.33-4.29 (m, 1H), 4.23-4.20 (m, 1H), 4.14-4.09 (m, 0.5H), 3.92-3.89 (m, 0.5H), 3.75 (s, 3H), 3.72-3.62 (m, 1H), 3.27-3.08 (m, 1H), 2.90-2.70 (m, 1.5H), 1.98-1.95 (m, 2H), 1.79-1.77 (m, 2H), 1.57-1.23 (m, 8H); 13CNMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 167.2, 157.5, 156.9, 154.3, 153.1, 152.6, 138.4, 138.2, 136.6, 135.5, 134.8, 134.5, 132.0, 130.2, 129.8, 128.7, 128.3, 127.1, 125.8, 124.8, 124.1, 122.6, 117.7, 116.3, 116.0, 115.9, 115.4, 114.9, 114.8, 114.6, 71.3, 70.7, 70.2, 69.3, 66.6, 63.0, 59.0, 57.8, 57.3, 56.8, 55.9, 53.5, 51.9, 49.9, 48.0, 42.8, 37.5, 35.4, 31.9, 29.9, 28.5, 25.8, 23.9, 22.6; HRMS calcd. for C30H33NO435Cl, 506.20926 [M + H]+; found 506.20932 [M + H]+; Anal. (C30H32N1O4Cl): C, H, N.
(3-Chlorophenyl)(6-ethoxy-1-((4-ethoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (91)
Dihydroisoquinoline 56 was prepared via procedure X using amide 29 (0.58 g, 1.7 mmol) and phosphorus trichloride (0.84 mL, 4.1 mmol) in dry toluene (8.4 mL). The crude solid was carried on without further purification. HRMS calcd. for C20H24O3N, 326.17507 [M + H]+; found 326.17457 [M + H]+. Tetrahydroisoquinoline 75 was prepared via procedure XII using 56 (0.91 g, 2.8 mmol) and sodium borohydride (0.32 g, 8.4 mmol) in dry MeOH (14 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 77 (0.12 g, 14 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. Tetrahydroisoquinoline 91 was prepared via procedure XIII using tetrahydroisoquinoline 75 (0.061 g, 0.19 mmol) and 3-chlorobenzoyl chloride (0.029 mL, 0.22 mmol) in DCM (2.9 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.039 g, 45 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.70; 1HNMR (CDCl3, 400 MHz) δ: 7.6 (bs, 0.5H), 7.42-7.22 (m, 3.5H), 6.94-6.92 (m, 0.5H), 6.87-6.69 (m, 6.5H), 6.0-5.98 (m, 0.5H), 5.14-5.13 (m, 0.5H), 4.88-4.84 (m, 0.5H), 4.35-4.32 (m, 1H), 4.14 (t, J = 10 Hz, 0.5), 4.40 −3.92 (m, 4.5H), 3.79-3.66 (m, 1H), 3.31-3.11 (m, 1H), 2.98-2.74 (m 1.5H), 1.44-1.38 (m, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.4, 169.7, 158.7, 158.2, 153.8, 153.1, 152.6, 138.5, 138.3, 136.6, 135.6, 134.9, 134.6, 130.2, 129.9, 128.7, 128.3, 128.2, 127.1, 125.8, 125.4, 124.9, 124.4, 116.0, 115.8, 115.6, 114.9, 114.5, 113.7, 113.5, 71.4, 70.4, 64.3, 63.8, 57.4, 52.1, 42.9, 35.6, 30.0, 28.6, 15.2, 15.1; HRMS calcd. for C27H29NO4Cl, 446.17796 [M + H]+; found 446.17853 [M + H]+; Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.74 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 8 mins at 1 mL/min (retention time 5.55 minutes).
(3-Chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (92)
Tetrahydroisoquinoline 92 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.15 g, 0.44 mmol) and 3-chlorobenzoyl chloride (0.070 mL, 0.48 mmol) in dry DCM (7.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.11 g, 56 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.78 1HNMR (CDCl3, 400 MHz) δ: 7.37-7.18 (m, 4H), 6.91-6.74 (m, 5H), 6.74-6.66 (m, 2H) 5.97-5.85 (t, J = 5.2 Hz, 0.5H), 5.12-5.09 (dd, J = 3.6 Hz, J = 9.6 Hz, 0.5H), 4.86-4.81 (dd, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.35-4.32 (m, 1H), 4.14-4.09 (m, 0.5 H), 3.98-3.89 (m, 2.5 H), 3.75-3.66 (m, 1H), 3.25-3.09 (m, 1H), 2.88-2.70 (m, 1.5 H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.31 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 170.2, 169.6, 157.6, 157.1, 153.8, 152.5, 148.3, 138.4, 136.7, 135.6, 134.9, 134.6, 130.2, 129.9, 129.8, 128.8, 128.3, 128.2, 127.1, 125.9, 125.7, 124.9, 124.1, 116.2, 115.9, 115.7, 115.6, 115.5, 114.7, 114.5, 71.3, 70.3, 64.2, 57.3, 52.0, 42.9, 35.5, 29.9, 28.5, 22.3, 22.2, 15.2; HRMS calcd. for C28H31ClN1O4, 480.19361 [M + H]+; found 480.19337 [M + H]+. Anal. (C28H30ClN1O4): C, H, N.
(3-Chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isobutoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (93)
Tetrahydroisoquinoline 93 was prepared via Procedure XIII using tetrahydroisoquinoline 79 (0.18 g, 0.50 mmol) and 3-chlorobenzoyl chloride (0.076 mL, 0.60 mmol) in DCM (7.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.80 g, 33 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.67; 1HNMR (CDCl3, 400 MHz) δ: 7.59 (s, 0.5H), 7.42-7.21 (m, 4H), 6.94-6.92 (m, 0.5), 6.85-6.69 (m, 6H), 5.98 (t, J = 4.8 Hz, 0.5H), 5.15-5.12 (m, J = 3.6 Hz, J = 9.6 Hz, 0.5H), 4.89-4.84 (m, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.39-4.32 (m, 1H), 4.14 (t, J = 10.4 Hz, 0.5H), 3.98 (q, J = 6.8 Hz, 2H), 3.94-3.92 (m, 0.5H), 3.79-3.75 (m, 0.5H), 3.71 (d, J = 6.4 Hz, 2H), 3.68-3.65 (m, 0.5H), 3.31-3.11 (m, 1H), 2.94-2.74 (m, 1.5H), 2.11-2.05 (m, 1H), 1.40 (t, J = 6.8 Hz, 3H), 1.03 (d, J = 6.8 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.5, 169.7, 158.9, 158.5, 153.7, 153.6, 153.0, 152.5, 138.4, 138.2, 136.6, 138.2, 136.6, 135.5, 134.9, 134.6, 130.3, 129.9, 128.7, 128.3, 128.2, 127.1, 125.9, 125.2, 124.9, 124.1, 115.9, 115.7, 115.6, 115.5, 114.9, 114.4, 113.7, 113.5, 74.7, 71.3, 70.2, 64.2, 57.3, 52.0, 42.9, 35.5, 29.9, 28.5, 19.5, 15.2; HRMS calcd. for C29H33NO435Cl, 494.20926 [M + H]+; found, 494.20919 [M + H]+; mp: 75°C. Anal. (C29H32N1O4Cl): C, H, N
(3-Chlorophenyl)(6-(cyclopentyloxy)-1-((4-ethoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (94)
Tetrahydroisoquinoline 94 was prepared via procedure XIII using tetrahydroisoquinoline 78 (0.18 g, 0.50 mmol) and 3-chlorobenzoyl chloride (0.080 mL, 0.60 mmol) in DCM (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.17 g, 68% yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.69; 1HNMR (CDCl3, 400 MHz) δ: 8.04-7.94 (m, 0.5H), 7.55-7.53 (m, 0.5H), 7.40-7.25 (m, 3H), 7.20-7.17 (m, 0.5H), 6.90-6.75 (m, 5H), 6.70-6.64 (m, 1.5H), 5.98 (t, J = 5.2 Hz, 0.5H), 5.13-5.10 (m, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.87-4.82 (m, J = 5.2 Hz, 12.8 Hz, 0.5H), 4.37-4.29 (m, 1H), 4.13 (t, J = 10.0 Hz, 0.5H), 3.96 (q, J = 7.2 Hz, 2H), 3.92-3.89 (m, 0.5H), 3.76-3.63 (m, 1H), 3.29-3.09 (m, 1H), 2.29-2.70 (m, 1.5H), 1.88-1.74 (m, 6H), 1.68-1.60 (m, 2H), 1.38 (t, J = 6.8 Hz); 13C NMR (CDCl3, 100 MHz) δ: 170.5, 169.8, 169.2, 157.8, 157.2, 153.7, 153.6, 152.9, 152.5, 138.3, 136.5, 135.4, 134.9, 134.8, 134.5, 133.7, 130.4, 130.2, 129.9, 129.8, 128.7, 128.2, 127.0, 125.9, 124.9, 123.8, 115.9, 115.7, 115.6, 115.5, 115.4, 114.5, 114.2, 79.5, 71.3, 70.1, 64.2, 57.3, 52.0, 42.8, 35.5, 33.1, 29.9, 28.6, 24.2, 15.2; HRMS calcd. for C30H33NO435Cl, 506.20925 [M + H]+; found, 506.20991 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 2.30 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.42 minutes).
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(phenyl)methanone (95)
Tetrahydroisoquinoline 95 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.20 g, 0.59 mmol) and benzoyl chloride (0.080 mL, 0.70 mmol) in DCM (9.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.06 g, 24 % mixture of two rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.54 1HNMR (CDCl3, 400 MHz) δ: 7.51-7.19 (m, 5H), 6.89-6.65 (m, 6H), 5.96-5.99 (m, 0.5 H), 5.18-5.16 (d, J = 5.6 Hz, 0.5H), 4.88-4.84 (dd, J = 4.4 Hz, J = 12.4 Hz, 0.5H), 4.55-4.47 (m, 1H), 4.35-4.33 (m, 1H), 4.12-4.07 (m, 0.5 H), 3.98-3.89 (m, 2.5H), 3.80-3.62 (m, 1H), 3.27-3.09 (m, 1H), 2.93-2.68 (m,1.5 H), 1.39-1.35 (t, J = 6.8 Hz, 3H), 1.33-2.31 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 171.9, 171.2, 157.5, 156.9, 153.7, 153.5, 153.1, 152.7, 136.7, 135.7, 129.8, 129.6, 128.8, 128.5, 128.3, 127.7, 126.8, 125.6, 124.5, 116.1, 115.9, 115.6, 114.6, 114.3, 71.4, 70.3, 70.1, 64.2, 57.2, 51.8, 42.8, 35.4, 30.0, 28.7, 22.3, 22.2, 15.2; ; HRMS calcd. for C28H32N1O4, 446.23259 [M + H]+; found 446.23242 [M + H]+. Anal. (C29H32N1O4): C, H, N.
(3-Bromophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (96)
Tetrahydroisoquinoline 96 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.15 g, 0.44 mmol) and 3-bromobenzoyl chloride (0.070 mL, 0.48 mmol). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.12 g, 51 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.80 1HNMR (CDCl3, 400 MHz) δ: 7.55-7.41 (m, 2H), 7.29-7.18 (m, 2H), 6.91-6.66 (m, 7H), 5.94-5.93 (m, 0.5 H), 5.12-5.09 (dd, J = 3.2 Hz, J = 9.2 Hz, 0.5H), 4.85-4.81 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.33-4.30 (m, 1H), 4.14-4.09 (m, 0.5 H), 3.98-3.89 (m, 2.5 H), 3.74-3.66 (m, 1H), 3.27-3.09 (m, 1H), 2.9-2.71 (m, 1.5H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.31 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 170.3, 169.5, 157.6, 157.1, 153.7, 153.6, 153.0, 152.5, 138.6, 136.6, 132.9, 132.8, 132.7, 131.0, 130.5, 130.1, 129.9, 128.8, 128.3, 126.3, 125.3, 125.2, 124.1, 122.9, 122.7, 116.2, 115.9, 115.6, 115.5, 115.3, 114.8, 114.5, 71.3, 70.2, 70.1, 64.2, 57.3, 51.9, 42.8, 35.8, 29.9, 28.5, 22.3, 22.2, 15.2; HRMS calcd. for C28H31BrN1O4, 524.14310 [M + H]+; found 524.14327 [M + H]+. Anal. (C28H30BrN1O4): C, H, N.
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanone (97)
Tetrahydroisoquinoline 97 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.15 g, 0.44 mmol) and 3-fluorobenzoyl chloride (0.060 mL, 0.48 mmol) in DCM (7.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.090 g, 46 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.80 1HNMR (CDCl3, 400 MHz) δ: 7.42-7.29 (m, 2H), 7.21-7.06 (m, 2H), 6.90-6.66 (m, 7H), 5.97-5.95 (t, J = 4.4 Hz, 0.5H), 5.14-5.11 (dd, J = 4.0 Hz, J = 10.0 Hz, 0.5H), 4.86-4.81 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.55-4.48 (m, 1H), 4.37-4.29 (m, 1H), 4.13-4.08 (m, 0.5 H), 3.98-3.89 (2.5 H), 3.76-3.66 (m, 1H), 3.28-3.08 (m, 1H), 2.90-2.69 (m, 1.5 H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.31 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 170.5, 169.8, 163.9, 161.4, 157.6, 157.0, 153.7, 153.6, 153.0, 152.5, 138.6, 136.6, 135.6, 130.7, 130.6, 130.2, 128.7, 128.3, 125.2, 124.2, 123.5, 122.5, 116.9, 116.8, 116.7, 116.6, 116.2, 115.9, 115.7, 115.6, 115.5, 114.7, 114.4, 114.0, 71.3, 70.2, 64.2, 57.3, 51.9, 42.9, 35.5, 29.9, 28.5, 22.3, 22.2, 15.1; HRMS calcd. for C28H31FN1O4, 464.22316 [M + H]+; found 464.22293 [M + H]+; mp: 75°C.Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 1.56 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 1.25 minutes).
(6-Isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (98)
Tetrahydroisoquinoline 98 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.13 g, 0.40 mmol) and 3-(trifluoromethyl)benzoyl chloride (0.070 mL, 0.43 mmol) in DCM (7.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.19 g, 55 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.66 1HNMR (CDCl3, 400 MHz) δ: 7.70-7.48 (m, 3.5 H), 7.22-7.19 (m, 0.5 H), 6.89-6.66 (m, 6.5 H), 6.00-5.98 (t, J = 5.2 Hz, 0.5H), 5.09-5.07 (dd, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.89-4.84 (dd, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.36-4.34 (m, 0.5 H), 4.15-4.09 (m, 1H), 3.93-3.89 (m, 0.5 H), 3.74 (s, 3H), 3.69-3.68 (m, 1H), 3.29-3.12 (m, 1H), 2.91-2.72 (m, 1.5 H), 1.33-1.12 (dd, J = 1.6 Hz, J = 6.4 Hz, 6H); 13CNMR (100 MHz, CDCl3) δ: 170.5, 169.7, 168.2, 157.6, 157.1, 154.4, 154.3, 153.0, 152.5, 137.4, 137.2, 136.6, 135.4, 133.5, 131.1, 130.9, 130.2, 129.4, 129.3, 129.1, 128.7, 128.3, 126.5, 123.9, 116.2, 115.9, 115.4, 115.3, 114.9, 114.8, 114.5, 71.3, 70.1, 57.4, 55.9, 52.1, 42.9, 35.5, 29.9, 28.5, 22.3, 22.2; HRMS calcd. for C29H29F3N1O4, 500.20432 [M + H]+; found 500.20415 [M + H]+; mp: 85°C. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 1.77 minutes). Compound 98 was 87% pure (impurity retention time =1.82 minutes)
Biphenyl-3-yl(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (99)
Tetrahydroisoquinoline 99 was prepared via procedure XIV using biphenyl-3-carboxylic acid (0.29 g, 1.5 mmol), N1-((ethylimino)methylene)-N3,N3-dimethylpropane-1,3-diamine (.25 g, 1.6 mmol), N,N-dimethylpyridin-4-amine (0.19 g, 1.6 mmol), and tetrahydroisoquinoline 76 (0.46 g, 1.4 mmol). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.10 g, 15 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.65 1HNMR (CDCl3, 400 MHz) δ: 7.79-7.56 (m, 4H), 7.52-7.30 (m, 5H), 7.24-7.21 (m, 0.5 H), 6.91-6.66 (m, 6.5 H), 6.03-6.00 (t, J = 4.4 Hz, 0.5H), 5.25-5.21 (dd, J = 3.6 Hz, J = 9.2 Hz, 0.5H), 4.91-4.87 (dd, J = 5.6 Hz, J = 13.2 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.41-4.34 (m, 1H), 4.17-4.12 (m, 0.5 H), 3.95-3.92 (m, 1.5 H), 3.89-3.65 (m, 1H), 3.28-3.14 (m, 1H), 2.95-2.69 (m, 2.5 H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.32 (m, 6H);13CNMR (CDCl3,100 MHz) δ: 171.9, 171.1, 157.5, 156.9, 153.7, 153.45, 153.1, 152.6, 141.8, 141.5, 140.6, 140.5, 137.3, 137.1, 136.7, 135.7, 129.3, 129.1, 129.0, 128.9, 128.8, 128.5, 128.4, 127.9, 127.8, 127.4, 127.3, 126., 125.6, 125.5, 5124.6, 116.2, 115.9, 115.8, 115.6, 115.4, 114.3, 71.4, 70.4, 70.1, 64.2, 57.3, 51.9, 42.9, 35.5, 30.1, 28.6, 22.5, 22.3, 15.2; ; HRMS calcd. for C45H36N1O4, 522.26389 [M + H]+; found 522.26390 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 2.07 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.20 minutes). Compound 230 was only 94% pure (retention time of impurity = 2.07 minutes)
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(m-tolyl)methanone (100)
Tetrahydroisoquinoline 100 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.21 g, 0.62 mmol) and 3-methylbenzoyl chloride (0.11 mL, 0.74 mmol) in DCM (9.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.22 g, 78 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.88; 1HNMR (CDCl3, 400 MHz) δ: 7.47-7.45 (m, 1H), 7.35-7.33 (m, 1H), 7.26-7.21 (m, 2H), 6.95-6.71 (m, 1H), 6.02-6.01 (m, 0.5H), 5.27-5.25 (m, 0.5H), 4.90 (m, J = 4.4 Hz, J = 12.0 Hz, 0.5H), 4.61-5.54 (m, 1H), 4.39 (d, J = 5.2 Hz, 1H), 4.18-4.10 (m, 0.5H), 4.00 (q, J = 7.2 Hz, 2H), 3.89-3.67 (m, 1H), 3.31-3.13 (m, 1H), 2.98-2.72 (m, 2H), 2.41 (s, 3H), 1.42 (t, J = 6.4 Hz, 3H), 1.37 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 172.2, 171.4, 157.4, 156.9, 153.5, 153.1, 153.7, 138.7, 138.4, 136.7, 136.5, 135.8, 130.4, 128.8, 128.6, 128.3, 127.4, 125.6, 124.6, 123.6, 116.1, 115.9, 115.8, 115.6, 115.5, 114.5, 114.3, 71.4, 70.3, 70.0, 64.2, 57.1, 51.7, 42.8, 35.4, 30.1, 28.7, 22.3, 22.2, 21.6, 15.2; HRMS calcd. for C29H34NO4, 460.24824 [M + H]+; found, 460.24844 [M + H]+; Anal. (C29H33N1O4): C, H, N.
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-methoxyphenyl)methanone (101)
Tetrahydroisoquinoline 101 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.21 g, 0.62 mmol) and 3-methoxybenzoyl chloride (0.10 mL, 0.74 mmol) in DCM (9.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.20 g, 68 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.69; 1HNMR (CDCl3, 400 MHz) δ: 7.35-7.22 (m, 1H), 7.09-7.07 (m, 1H), 6.97-6.68 (m, 9H), 5.99 (t, J = 4.8 Hz, 0.5H), 5.23-5.20 (m, J = 3.6 Hz, J = 8.8 Hz, 0.5H), 4.89-4.85 (m, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.58-4.49 (m, 1H), 4.37-4.33 (m, 1H), 4.16-4.09 (m, 0.5H), 4.00-3.95 (q, J = 6.8 Hz, 2H), 3.82-3.79 (d, J = 11.2 Hz, 3H), 3.70-3.61 (m, 0.5H), 3.30-3.11 (m, 1H), 2.95-2.71 (m, 1.5H), 1.41-1.38 (t, J = 7.2 Hz, 3H), 1.35-1.34 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 171.7, 170.9, 159.9, 159.7, 156.9, 153.7, 153.5, 153.1, 152.7, 138.0, 137.8, 136.7, 135.8, 129.9, 129.6, 128.8, 128.3, 125.5, 124.6, 119.9, 118.8, 116.1, 115.9, 115.7, 115.6, 114.6, 114.3, 113.1, 112.0, 71.4, 70.4, 70.0, 64.2, 57.1, 55.6, 51.8, 42.8, 35.5, 30.0, 28.6, 22.7, 15.2; HRMS calcd. for C29H34NO5, 476.24315 [M + H]+; found, 476.24324 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.19 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.96 minutes).
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(2-fluorophenyl)methanone (102)
Tetrahydroisoquinoline 102 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.14 g, 0.41 mmol) and 2-fluorobenzoyl chloride (0.049 mL, 0.41 mmol) in DCM (6.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an yellow foam (0.10 g, 53 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.86; 1HNMR (CDCl3, 400 MHz) δ: 7.44-7.36 (m, 1H), 7.27-7.11 (m, 3H), 6.97-6.68 (m, 7H), 6.02 (t, J = 4.8 Hz, 0.5H), 5.02-4.92 (m, 0.5H), 4.57-4.51 (m, 1H), 4.39-4.31 (m, 1H), 4.14-4.06 (m, 0.5H), 4.00-3.90 (m, 2.5H), 3.75-3.67 (m, 1H), 3.31-3.09 (m, 1H), 2.93-2.73 (m, 1.5H), 1.39 (t, J = 7.2 Hz, 3H), 1.36-1.33 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ: 166.7, 166.4, 157.4, 157.0, 153.5, 153.1, 152.6, 136.5, 132.9, 131.4, 128.8, 128.3, 125.3, 124.9, 124.2, 115.9, 115.7, 115.6, 115.4, 114.6, 114.4, 71.4, 70.3, 70.1, 64.2, 60.6, 51.9, 44.6, 42.5, 35.6, 29.9, 28.8, 22.3, 15.1; HRMS calcd. for C28H31N1O4F, 464.22316 [M + H]+; found, 464.22228 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.14 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.94 minutes).
(2-Chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (103)
Tetrahydroisoquinoline 103 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.14 g, 0.41 mmol) and 2-chlorobenzoyl chloride (0.062 mL, 0.49 mmol) in DCM (6.3 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an yellow foam (0.090 g, 56 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.86; 1HNMR (CDCl3, 400 MHz) δ: 7.59-7.21 (m, 4H), 6.96-6.67 (m, 7H), 6.06-6.00 (m, 0.5H), 4.99-4.95 (m, J = 4.8 Hz, J = 12.0 Hz, 0.5H), 4.88-4.84 (m, J = 4.0 Hz, J = 9.2 Hz, 0.5H), 4.59-4.50 (m, 1H), 4.41-4.31 (m, 1H), 4.09 (t, J = 9.6 Hz, 0.5H), 4.01-3.90 (m, 2H),3.91-3.75 (m, 0.5H), 3.64-3.49 (m, 1H), 3.30-3.04 (m, 1H), 2.89-2.72 (m, 1.5H), 1.41-1.34 (m, 9H); 13C NMR (CDCl3, 100 MHz) δ: 168.2, 167.8, 157.3, 157.0, 153.6, 153.5, 152.9, 152.6, 136.5, 136.1, 135.9, 135.7, 135.0, 130.8, 130.5, 130.3, 130.1, 129.9, 129.8, 129.3, 128.8, 128.7, 128.3, 128.1, 127.7, 127.5, 127.4, 127.1, 126.7, 125.4, 125.2, 124.5, 115.9, 115.8, 115.7, 115.6, 115.5, 115.4, 114.6, 114.3, 71.3, 70.4, 70.0, 64.2, 56.8, 51.7, 42.7, 42.3, 35.7, 29.9, 29.8, 28.8, 22.3, 15.2; HRMS calcd. for C28H31N1O4Cl, 480.19361 [M + H]+; found, 480.19269 [M + H]+. Anal. (C28H30N1O4Cl): C, H, N.
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(4-fluorophenyl)methanone (104)
Tetrahydroisoquinoline 104 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.21 g, 0.62 mmol) and 4-fluorobenzoyl chloride (0.089 mL, 0.74 mmol) in DCM (9.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.19 g, 65 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.81; 1HNMR (CDCl3, 400 MHz) δ: 7.55-7.54 (m, 1H), 7.40-7.42 (m, 1H), 7.24-7.21 (m, 0.5H), 7.13-7.06 (m, 2H), 6.94-6.68 (m, 6.5H), 5.98-5.97 (m, 0.5H), 5.17-5.16 (m, 0.5H), 4.88-4.83 (m, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.59-4.50 (m, 1H), 4.36 (s, 1H), 4.15-4.10 (m, 0.5H), 3.97 (q, J = 6.8 Hz, 2H), 3.79-3.67 (m, 1H), 3.29-3.11 (m, 1H), 2.91-2.73 (m, 2H), 1.39 (t, J = 7.2 Hz, 3H), 1.34 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 171.1, 170.3, 164.8, 162.3, 157.5, 156.9, 153.7, 153.0, 152.6, 136.7, 135.6, 132.7, 130.1, 129.1, 128.8, 128.3, 125.4, 124.3, 116.2, 115.9, 115.6, 114.7, 114.4, 71.3, 70.3, 70.0, 64.2, 57.4, 51.9, 42.9, 35.5, 29.9, 28.5, 22.3, 22.2, 15.1; HRMS calcd. for C28H31NO4F, 464.22316 [M + H]+; found, 464.22329 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.17 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.94 minutes).
(4-Chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (105)
Tetrahydroisoquinoline 105 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.21 g, 0.62 mmol) and 4-chlorobenzoyl chloride (0.095 mL, 0.74 mmol) in DCM (9.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.12 g, 40 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.87; 1HNMR (CDCl3, 400 MHz) δ: 7.50-7.48 (m, 1H), 7.42-7.34 (m, 3H), 7.23-7.19 (m, 0.5H), 6.93-6.68 (m, 6.H), 5.99-5.97 (m, 0.5H), 5.16-5.14 (m, 0.5H), 4.89-4.83 (m, J = 4.8 Hz, J = 12.4 Hz, 0.5H), 4.57-4.51 (m, 1H), 4.36-4.34 (m, 1H), 4.14-4.08 (m, 0.5H), 3.97 (q, J = 6.8 Hz, 2H), 3.76-3.69 (m, 1H), 3.29-3.12 (m, 1H), 2.92-2.72 (m, 2H), 1.40 (t, J = 7.2 Hz, 3H), 1.34 (d, J = 5.6 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.9, 170.2, 157.5, 157.0, 153.8, 153.6, 153.0, 152.5, 136.6, 135.8, 135.7, 135.6, 135.1, 129.4, 129.1, 128.8, 128.4, 128.3, 125.3, 124.2, 116.2, 115.7, 115.6, 115.5, 114.7, 114.4, 71.3, 70.2, 70.1, 64.2, 57.3, 51.9, 42.9, 35.4, 29.9, 28.7, 22.3, 22.2, 15.2; HRMS calcd. for C28H31NO4Cl, 480.19361 [M + H]+; found, 480.19379 [M + H]+; Anal. (C28H30N1O4Cl): C, H, N.
(3,4-Difluorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (106)
Tetrahydroisoquinoline 106 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.21 g, 0.62 mmol) and 3,4-difluorobenzoyl chloride (0.094 mL, 0.74 mmol) in DCM (9.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.18 g, 62 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.86; 1HNMR (CDCl3, 400 MHz) δ: 7.50-7.46 (m, 0.5H), 7.29-7.16 (m, 3H), 6.94-6.92 (m, 0.5H), 6.83-6.69 (m, 6H), 5.97 (bs, 0.5H), 5.16-5.15 (m, 0.5H), 4.84-4.82 (m, 0.5H), 4.59-4.51 (m, 1H), 4.35 (s, 1H), 4.15 (t, J = 10.4 Hz, 0.5H), 3.98 (q, J = 6.8 Hz, 2H), 3.76-3.71 (m, 0.5H), 3.29-3.14 (m, 1H), 2.90-2.75 (m, 2H), 1.40 (t, J = 6.8 Hz, 3H), 1.34 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 169.7, 168.9, 157.6, 157.1, 153.8, 153.6, 152.9, 152.4, 150.1, 149.9, 136.6, 135.4, 133.3, 128.7, 125.1, 124.5, 123.9, 117.9, 117.6, 117.4, 116.8, 116.2, 115.8, 115.5, 114.8, 114.5, 71.2, 70.1, 64.2, 57.4, 53.1, 42.9, 35.5, 29.9, 28.4, 22.3, 22.2, 15.1; HRMS calcd. for C28H30NO4F2, 482.21374 [M + H]+; found, 482.21387 [M + H]+; Anal. (C28H29N1O4F2): C, H, N.
(3,4-Dichlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (107)
Tetrahydroisoquinoline 107 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.25 g, 0.73 mmol) and 3,4-dichlorobenzoyl chloride (0.18 g, 0.89 mmol) in DCM (12 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.16 g, 42 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.59 1HNMR (CDCl3, 400 MHz) δ: 7.71-7.42 (m, 2H), 7.36-7.34 (m, 0.5 H), 7.22-7.15 (m, 1H), 6.92-6.89 (m, 0.5 H), 6.84-6.66 (m, 6H), 5.96-5.92 (m, 0.5 H), 5.11-5.08 (dd, J = 3.2 Hz, J = 9.2 Hz, 0.5H), 4.83-4.79 (dd, J = 4.8 Hz, J = 12.8 Hz, 0.5H), 4.56-4.48 (m, 1H), 4.36-4.29 (m, 1H), 4.15-4.09 (m, 0.5 H), 3.98-3.91 (m, 2.5 H), 3.71-3.67 (m, 1H), 3.27-3.08 (m, 1H), 2.91-2.72 (m, 1.5 H), 1.39-1.36 (t, J = 6.8 Hz, 3H), 1.33-1.32 (d, J = 1.6 Hz, 3H), 1.31-1.30 (d, J = 1.6 Hz, 3H); 13CNMR (CDCl3, 100 MHz) δ: 169.6, 168.8, 157.6, 157.1, 153.8, 153.6, 152.9, 152.3, 136.5, 136.4, 136.3, 135.4, 134.1, 133.9, 133.3, 132.8, 130.9, 130.6, 130.3, 129.1, 128.7, 128.3, 127.2, 126.2, 125.0, 123.9, 116.2, 115.8, 115.7, 115.6, 115.4, 115.2, 114.8, 114.5, 71.2, 70.1, 64.2, 57.4, 52.1, 42.8, 35.5, 29.9, 28.5, 22.3, 22.2, 15.2; HRMS calcd. for C28H30Cl2N1O4, 514.15464 [M + H]+; found 514.15456 [M + H]+. Anal. (C29H32N1O4): C, H, N.
(3-Chloro-4-fluorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (108)
Tetrahydroisoquinoline 108 was prepared via procedure XIV using 3-chloro-4-fluorobenzoic acid (0.085 g, 0.49 mmol), N1-((ethylimino)methylene)-N3,N3-dimethylpropane-1,3-diamine (0.082 g, 0.53 mmol), N,N-dimethylpyridin-4-amine (0.055 g, 0.45 mmol), and tetrahydroisoquinoline 76 (0.14 g, 0.41 mmol). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.14 g, 69 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.32; 1HNMR (CDCl3, 400 MHz) δ: 7.74-7.72 (m, 0.5H), 7.45 (s, 1H), 7.29-7.13 (m, 2H), 6.97-6.93 (m, 0.5H), 6.82-6.69 (m, 5H), 5.98 (s, 0.5H), 5.15 (d, J = 6.4 Hz, 0.5H), 4.86-4.81 (m, J = 4.4 Hz, J = 12.0 Hz, 0.5H), 4.59-4.50 (m, 1H), 4.36 (m, 1H), 4.16 (t, J = 10.0 Hz, 0.5H), 4.01-3.96 (m, 2.5H), 3.75-3.68 (m, 1H), 3.30-3.15 (m, 1H), 2.93-2.75 (m, 1.5H), 1.40 (t, J = 6.8 Hz, 3H), 1.36-1.33 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ: 169.8, 168.9, 160.2, 157.6, 157.1, 153.8, 153.6, 152.9, 152.4, 126.6, 135.4, 133.5, 130.9, 129.7, 128.7, 128.3, 128.1, 127.1, 125.1, 123.9, 121.5, 117.2, 116.8, 116.2, 115.8, 115.7, 115.6, 115.4, 114.8, 114.5, 71.2, 70.1, 64.2, 57.5, 52.2, 42.9, 35.6, 29.9, 28.4, 22.3, 22.2, 15.2; HRMS calcd. for C28H29N1O4ClF, 498.18419 [M + H]+; found, 498.18419 [M + H]+. Anal. (C28H29N1O4ClF): C, H, N.
3,5-Dichlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanone (109)
Tetrahydroisoquinoline 109 was prepared via procedure XIII using tetrahydroisoquinoline 76 (0.20 g, 0.59 mmol) and 3,5-dischlorobenzoyl chloride (0.15 g, 0.70 mmol) in DCM (9.0 mL). The crude residue was purified by silica gel title compound as an off-white foam (0.070 g, 23 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.71 1HNMR (CDCl3, 400 MHz) δ: 7.46-7.38 (m, 2H), 7.21-7.17 (m, 1H), 6.93-6.66 (m, 7H), 5.94-5.92 (m, 0.5 H), 5.08-5.50 (dd, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.83-4.78 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.56-4.48 (m, 1H), 4.33-4.28 (m, 1H), 4.17-4.14 (m, 0.5 H), 4.00-3.89 (m, 2.5 H), 3.68-3.67 (m, 1H), 3.28-3.07 (m, 1H), 2.93-2.72 (m, 1.5 H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.32 (d, 6H); 13CNMR (CDCl3, 100 MHz) δ: 168.9, 168.2, 157.6, 157.2, 153.8, 152.9, 152.4, 139.4, 136.5, 135.7, 135.4, 129.8, 129.7, 128.7, 128.4, 126.5, 125.3, 124.9, 123.7, 116.2, 115.9, 115.7, 115.6, 115.3, 114.8, 114.5, 71.1, 70.1, 64.1, 57.4, 52.2, 42.8, 35.5, 29.9, 28.3, 22.3, 22.2, 15.1; HRMS calcd. for C28H30Cl2N1O4, 514.15464 [M + H]+; found 514.15456 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 2.24 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.49 minutes). Compound 109 was only 93% pure (impurity retention time = 2.30 minutes
(3-Chlorophenyl)(6-isopropoxy-1-((3-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone) (110)
Dihydroisoquinoline 50 was prepared via Procedure X using amide 21 (1.4 g, 4.3 mmol) and phosphorus trichloride (1.2 mL, 13 mmol) in dry toluene (24 mL). The crude solid was carried on without further purification. HRMS calcd. for C20H23O3N1, 326.17507 [M + H]+; found 326.17588 [M + H]+. Tetrahydroisoquinoline 69 was prepared via Procedure XII using 50 (4.9 g, 15 mmol) and sodium borohydride (1.7 g, 45 mmol) in dry MeOH (75 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 38b (0.26 g, 5.0 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. HRMS calcd. for C19H23NO3, 314.17507 [M + H]+; found, 314.17484 [M + H]+. Tetrahydroisoquinoline 110 was prepared via Procedure XIII using tetrahydroisoquinoline 69 (0.26 g, 0.81 mmol) and 3-chlorobenzoyl chloride (0.12 mL, 0.89 mmol) in DCM (13 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.094 g, 31%, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.88 ; 1HNMR (CDCl3, 400 MHz) δ: 7.60-7.13 (m, 5H), 6.92-6.75 (m, 1H), 6.71-6.66 (m, 2H), 6.52-6.42 (m, 3H), 6.00-5.98 (t, J = 5.2 Hz, 0.5H), 5.15-5.12 (dd, J = 2.8 Hz, J = 9.2 Hz, 0.5H), 4.87-4.82 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.40-4.33 (m, 1H), 4.19-4.09 (m, 0.5 H), 3.95-3.91 (dd, J = 3.6 Hz, J = 10.4 Hz, 0.5H), 3.78-3.62 (m, 4H), 3.28-3.09 (m, 1H), 2.92-2.71 (m, 1.5 H), 1.33-1.32 (d, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 168.6, 161.1, 160.9, 160.1, 159.6, 157.9, 157.6, 157.1, 138.4, 138.2, 136.6, 135.6, 134.9, 134.6, 131.9, 130.2, 130.1, 129.9, 129.4, 128.8, 128.4, 127.1, 125.7, 125.1, 124.9, 123.9, 123.5, 122.7, 116.2, 115.9, 115.1, 114.5, 109.3, 108.2, 107.3, 107.1, 106.4, 102.3, 101.3, 100.9, 70.5, 70.1, 69.5, 57.2, 55.6, 51.8, 42.8, 42.3, 35.4, 29.9, 29.6, 28.5, 22.3; HRMS calcd. for C27H29ClN1O4, 466.17796 [M + H]+; found 466.17733 [M + H]+. Anal. (C27H28ClN1O4): C, H, N.
(3-Chlorophenyl)(6-isopropoxy-1-((2-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (111)
Tetrahydroisoquinoline 111 was prepared via Procedure XIII using tetrahydroisoquinoline 68 (0.20 g, 0.61 mmol) and 3-chlorobenzoyl chloride (0.090 mL, 0.67 mmol) in DCM (9.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.15 g, 53 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.72 1HNMR (CDCl3, 400 MHz) δ: 7.75-7.52 (m, 1H), 7.42-7.19 (m, 3.5 H), 6.96-6.66 (m, 6.5 H), 6.05-6.02 (t, J = 5.6 Hz, 0.5H), 5.19-5.15 (dd, J = 4.0 Hz, J = 10.4 Hz, 0.5H), 4.87-4.83 (dd, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.54-4.48 (m, 1H), 4.45-4.35 (m, 1H), 4.24-4.19 (m, 0.5 H), 4.02-3.98 (dd, J = 4.0 Hz, J = 10.4 Hz, 0.5H), 3.90-3.74 (m, 4H), 3.29-3.09 (m, 1H), 2.88-2.73 (m, 1.5 H), 1.34-1.31 (d, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.6, 169.9, 157.6, 157.1, 149.8, 148.1, 138.2, 136.6, 135.7, 134.5, 130.2, 129.8, 129.7, 128.7, 128.4, 128.3, 127.2, 126.2, 125.3, 124.9, 124.2, 122.1, 122.0, 121.2, 120.8, 116.3, 115.9, 114.7, 114.4, 113.4, 112.7, 111.9, 71.6, 70.2, 70.1, 57.3, 56.2, 55.8, 51.8, 42.7, 35.3, 29.9, 28.4, 22.3, 22.2; HRMS calcd. for C27H29ClN1O4, 466.17796 [M + H]+; found 466.17790 [M + H]+. Anal. (C27H28ClN1O4): C, H, N.
(3-Chlorophenyl)(6-isopropoxy-1-((4-(trifluoromethoxy)phenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (112)
Dihydroisoquinoline 52 was prepared via procedure X using amide 24 (1.58 g, 4.0 mmol) and phosphorous trichloride (1.0 mL, 11.9 mmol) in dry toluene (20 mL). The crude residue (1.33 g) was carried on without further purification. HRMS calcd. for C20H21O3N1F3, 380.14680 [M + H]+; found 380.14716 [M + H]+. Tetrahydroisoquinoline 71 was prepared via procedure XII using dihydroisoquinoline 52 (1.33 g, 3.5 mmol) and sodium borohydride (0.40 g, 11 mmol) in dry MeOH (19 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 71 (0.30 g, 19 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. Tetrahydroisoquinoline 112 was prepared via procedure XIII using tetrahydroisoquinoline 71 (0.15 g, 0.38 mmol) and 3-chlorobenzoyl chloride (0.058 mL, 0.46 mmol) in DCM (5.9 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.089 g, 45 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.65; 1HNMR (CDCl3, 300 MHz) δ: 8.12 – 7.52 (m, 0.5H), 7.44 – 7.28 (m, 3H), 7.21 – 7.03 (m, 2.5H), 6.97 – 6.48 (m, 5H), 5.99 (t, J = 5.2 Hz, 0.5H), 5.16 (dd, J = 9.9, 3.1 Hz, 0.5H), 4.86 (dd, J = 11.5, 4.8 Hz, 0.5H), 4.61 – 4.47 (m, 1H), 4.38 (d, J = 5.1 Hz, 1H), 4.18 (t, J = 10.0 Hz, 0.5H), 3.95 (dd, J = 10.0, 3.6 Hz, 0.5H), 3.79 – 3.62 (m, 1H), 3.36 – 3.05 (m, 1H), 2.97 – 2.64 (m, 1.5H), 1.34 (m, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.16, 169.70, 169.52, 157.48, 157.13, 156.97, 138.01, 137.85, 136.42, 135.38, 134.74, 134.40, 130.03, 129.82, 129.70, 129.68, 128.49, 128.06, 127.92, 126.81, 125.51, 124.63, 124.61, 123.45, 122.60, 122.49, 122.37, 121.54, 119.51, 116.04, 115.69, 115.49, 115.12, 115.11, 114.57, 114.33, 56.86, 51.58, 42.74, 35.27, 29.71, 28.30, 22.07; HRMS calcd. for C27H26NO4Cl1F3, 520.14970 [M + H]+; found 520.14947 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 2.34 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 8 mins at 1 mL/min (retention time 5.89 minutes).
(3-Chlorophenyl)(6-isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (113)
Tetrahydroisoquinoline 113 was prepared via procedure XIII using tetrahydroisoquinoline 70 (0.13 g, 0.36 mmol) and 3-chlorobenzoyl chloride (0.056 mL, 0.44 mmol) in dry DCM (5.8 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.048 g, 26 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:3, v/v) Rf = 0.55; 1HNMR (CDCl3, 400 MHz) δ: 7.39-7.21 (m, 4H), 6.87-6.85 (m, 6H), 6.0-5.98 (m, 0.5H), 5.14-5.13 (m, 0.5H), 4.88-4.84 (m, J = 5.2 Hz, J = 12.0 Hz, 0.5), 4.60-4.53 (m, 1H), 4.46-4.40 (m, 1H), 4.36-4.32 (m, 1H), 4.17-4.06 (m, 0.5H), 3.95-3.92 (m, 0.5H), 3.74-3.66 (m, 1H), 3.30-2.73 (m, 2.5H), 1.35 (d, J = 6.4 Hz, 6H), 1.31 (d, J = 6.0 Hz, 6H); 13CNMR (100 MHz, CDCl3) δ: 157.3, 156.8, 152.9, 152.4, 152.3, 152.1, 138.2, 137.9, 136.4, 135.3, 134.6, 134.6, 129.9, 129.6, 129.5, 129.2, 128.5, 128.1, 127.9, 126.8, 125.6, 124.9, 124.6, 123.9, 117.5, 117.3, 117.2, 115.9, 115.6, 115.2, 115.1, 114.5, 114.2, 71.0, 70.8, 69.8, 57.1, 51.7, 42.6, 35.2, 29.7, 28.3, 22.1; HRMS calcd. for C29H33ClN1O4,494.20926 [M + H]+; found 494.20919 [M + H]+; mp: 103°C. Anal. (C29H32FN1O4): C, H, N.
(3-Fluorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (114)
Tetrahydroisoquinoline 114 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.10 g, 0.32 mmol) and 3-fluorobenzoyl chloride (0.044 mL, 0.38 mmol) in DCM (5.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.17 g, 53 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.53; 1HNMR (CDCl3, 400 MHz) δ: 7.41-7.08 (m, 5H), 6.91-6.66 (m, 6H), 5.98-5.96 (m, 0.5H), 5.14-5.13 (m, 0.5H), 4.86-4.82 (m, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.56-4.49 (m, 1H), 4.34-4.30 (m, 1H), 4.12 (t, J = 9.6 Hz, 0.5H), 3.94-3.91 (m, 0.5H), 3.74 (s, 3H), 3.69-3.63 (m, 1H), 3.27-3.08 (m, 1H), 2.91-2.70 (m, 1.5H), 1.32 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.6, 169.7, 164.0, 161.4, 157.6, 157.5, 154.4, 154.3, 153.1, 152.6, 138.5, 136.6, 135.6, 130.7, 130.6, 130.3, 130.2, 128.7, 128.3, 125.2, 124.1, 123.4, 122.5, 116.9, 116.8, 116.7, 116.6, 116.1, 115.9, 115.5, 115.2, 114.9, 114.8, 114.7, 114.4, 114.2, 114.0, 71.3, 70.2, 70.1, 57.3, 55.9, 51.9, 42.7, 35.5, 29.9, 28.6, 22.3, 22.2; HRMS calcd. for C28H29NO4F, 450.20751 [M + H]+; found, 450.20722 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.03 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.77 minutes).
(3-Fluorophenyl)(6-isopropoxy-1-((4-(trifluoromethoxy)phenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (115)
Dihydroisoquinoline 52 was prepared via procedure X using amide 24 (1.58 g, 4.0 mmol) and phosphorous trichloride (1.0 mL, 11.9 mmol) in dry toluene (20 mL). The crude residue (1.33 g) was carried on without further purification. HRMS calcd. for C20H21O3N1F3, 380.14680 [M + H]+; found 380.14716 [M + H]+. Tetrahydroisoquinoline 71 was prepared via procedure XII using dihydroisoquinoline 52 (1.33 g, 3.5 mmol) and sodium borohydride (0.40 g, 11 mmol) in dry MeOH (19 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 71 (0.30 g, 19 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. Tetrahydroisoquinoline 115 was prepared via procedure XIII using tetrahydroisoquinoline 71 (0.15 g, 0.38 mmol) and 3-fluorobenzoyl chloride (0.055 mL, 0.46 mmol) in DCM (5.9 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.10 g, 53 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.63; 1HNMR (CDCl3, 300 MHz) δ: 7.88 – 7.28 (m, 1H), 7.23 – 7.03 (m, 5H), 7.03 – 6.65 (m, 5H), 5.99 (t, J = 4.9 Hz, 0.5H), 5.18 (dd, J = 9.7, 3.2 Hz, 0.5H), 4.87 (dd, 0.5H), 4.61 – 4.47 (m, 1H), 4.38 (d, J = 4.9 Hz, 1H), 4.18 (m, 0.5H), 3.96 (dd, J = 10.2, 3.4 Hz, 0.5H), 3.84 – 3.56 (m, 1H), 3.37 – 3.04 (m, 1H), 3.00 – 2.69 (m, 1.5H), 1.45 – 1.25 (m, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.25, 169.62, 163.58, 161.62, 161.52, 157.49, 157.14, 156.97, 156.61, 143.15, 143.12, 142.98, 142.97, 142.95, 138.27, 138.17, 138.11, 136.42, 136.40, 135.41, 130.52, 130.46, 130.15, 130.08, 130.05, 129.99, 128.49, 128.03, 125.76, 124.65, 123.49, 123.13, 123.10, 122.58, 122.49, 122.37, 122.19, 121.55, 120.42, 120.25, 119.50, 116.98, 116.80, 116.67, 116.64, 116.51, 116.06, 115.70, 115.48, 115.12, 115.05, 114.87, 114.56, 114.55, 114.32, 114.31, 114.01, 113.82, 70.74, 69.92, 69.71, 56.80, 51.57, 42.69, 35.28, 29.72, 28.31, 22.03. HRMS calcd. for C27H26NO4F4, 504.17925 [M + H]+; found 504.17909 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.89 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 5.54 minutes).
(3-Fluorophenyl)(6-isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (116)
Tetrahydroisoquinoline 116 was prepared via procedure XIII using tetrahydroisoquinoline 70 (0.80 g, 0.23 mmol) and 3-fluorobenzoyl chloride (0.031 mL, 0.27 mmol) in dry DCM (3.5 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.074 g, 70 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:3, v/v) Rf = 0.76; 1HNMR (CDCl3, 400 MHz) δ: 7.44-7.10 (m, 5H), 6.94-6.68 (m, 6H), 5.99 (t, J = 6.4 Hz, 0.5H), 5.18-5.13 (m, J = 5.6 Hz, J = 4.8 Hz, 0.5H), 4.89-4.48 (m, J = 5.6 Hz, J = 6.8 Hz, 0.5H), 4.58-4.48 (m, 1H), 4.47-4.36 (m, 1H), 4.39-4.31 (m, 1H), 4.17-4.10 (m, 0.5H), 3.97-3.92 (m, 0.5H), 3.76-3.64 (m, 1H), 3.32-3.09 (m, 1H), 2.96-2.72 (m, 1.5H), 1.36-1.29 (m, 12H); 13CNMR (100 MHz, CDCl3) δ: 170.5, 169.7, 164.0, 163.8, 161.6, 161.4, 157.6, 157.1, 153.1, 152.6, 152.5, 152.4, 138.7, 138.5, 136.6, 135.6, 130.7, 130.6, 130.3, 130.2, 130.1, 128.7, 128.3, 125.9, 125.2, 124.2, 123.4, 122.5, 120.5, 117.7, 117.6. 117.2. 116.9, 116.8, 116.7, 116.6, 116.2, 115.8, 115.4, 115.2, 114.7, 114.4, 114.3, 114.0, 71.3, 71.1, 71.0, 70.6, 70.2, 70.1, 60.6, 57.3, 53.7, 51.9, 42.8, 35.4, 31.8, 31.2, 29.9, 28.6, 22.9, 22.3, 22.2, 21.3, 14.4; HRMS calcd. for C29H33FN1O4, 478.23881 [M + H]+; found 478.23858 [M + H]+; Anal. (C29H32FN1O4): C, H, N
(3-chlorophenyl)(6-(dimethylamino)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (117)
Tetrahydroisoquinoline 117 was prepared via Procedure XIII using tetrahydroisoquinoline 83 (0.26 g, 0.85 mmol) and 3-chlorobenzoyl chloride (0.14 mL, 1.0 mmol) in DCM (13 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.31 g, 81 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.43; 1HNMR (CDCl3, 400 MHz) δ: 7.59-7.16 (m, 4.5H), 6.89-6.76 (m, 4.5 H), 6.68-6.57 (m, 1H), 6.52-6.48 (m, 1H), 5.97-5.95 (t, J = 5.2 Hz, 0.5H), 5.12-5.09 (dd, J = 3.2 Hz, J = 9.2 Hz, 0.5H), 4.86-4.82 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.33-4.31 (m, 1H), 4.14-4.09 (m, 0.5H), 3.92-3.88 (dd, J = 3.2 Hz, J = 6.8 Hz), 3.73 (s, 3H), 3.70-3.65 (m, 1H), 3.27-3.09 (m, 1H), 2.94 (s, 6H), 2.87-2.71 (m, 1.5 H); 13CNMR (100 MHz, CDCl3) δ: 170.4, 169.6, 154.3, 153.2, 152.7, 150.2, 138.6, 138.4, 136.1, 134.8, 134.5, 130.3, 130.2, 129.9, 129.8, 129.7, 128.3, 128.2, 127.9, 127.1, 125.8, 124.9, 115.9, 115.4, 114.9, 114.8, 112.7, 111.2, 71.2, 70.2, 57.3, 55.9, 51.8, 42.9, 40.7, 35.6, 30.2, 28.8; HRMS calcd. for C26H27ClN2O3, 451.17830 [M + H]+; found 451.17929 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 3.67 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 2.43 minutes).
(3-bromophenyl)(6-(dimethylamino)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (118)
Tetrahydroisoquinoline 118 was prepared via Procedure XIII using tetrahydroisoquinoline 83 (0.26 g, 0.85 mmol) and 3-bromobenzoyl chloride (0.14 mL, 1.0 mmol) in DCM (13 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.30 g, 76 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.43; 1HNMR (CDCl3, 400 MHz) δ: 7.75-7.41 (m, 2H), 7.31-7.15 (m, 2H), 6.89-6.85 (m, 2H), 6.82-6.77 (m, 3H), 6.67-6.57 (m, 1H), 6.52-6.47 (m, 1H), 5.97-5.94 (t, J = 5.2 Hz, 0.5H), 5.12-5.09 (dd, J = 3.6 Hz, J = 9.6 Hz, 0.5H), 4.86-4.82 (dd, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.33-4.28 (m, 1H), 4.14-4.05 (m, 0.5 H), 3.92-3.88 (dd, J = 3.6 Hz, J = 10.0 Hz, 0.5H), 3.75 (s, 3H), 3.71-3.61 (m, 1H), 3.29-3.08 (m, 1H), 2.94 (s, 6H), 2.89-2.71 (m, 1.5H); 13CNMR (100 MHz, CDCl3) δ: 170.3, 169.5, 154.3, 154.2, 153.2, 152.6, 150.2, 149.8, 138.8, 138.6, 135.9, 134.9, 132.8, 132.7, 131.0, 130.4, 130.1, 129.8, 128.3, 127.9, 126.3, 125.3, 122.9, 122.6, 119.9, 115.9, 115.4, 114.8, 112.7, 112.4, 11.9, 111.2, 78.6, 71.2, 70.2, 57.3, 55.9, 51.9, 42.9, 40.8, 40.8, 35,7, 30.2, 28.8; HRMS calcd. for C26H27BrN2O3, 495.12778 [M + H]+; found 495.12898 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 5.5 mins at 1 mL/min (retention time = 2.75 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 3 mins at 1 mL/min (retention time 1.40 minutes).
(3-chlorophenyl)(6-(isopropyl(methyl)amino)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (119)
Tetrahydroisoquinoline 119 was prepared via procedure XIII using tetrahydroisoquinoline 85 (0.13 g, 0.39 mmol) and 3-chlorobenzoyl chloride (0.050 mL, 0.39 mmol) in DCM (6.2 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.098 g, 52 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.72; 1HNMR (CDCl3, 400 MHz) δ: 7.42-7.17 (m, 4H), 6.91-6.80 (m, 5H), 6.75-6.54 (m, 2H), 5.99 (t, J = 5.2 Hz, 0.5H), 5.14-5.11 (m, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.89-4.85 (m, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.36-4.29 (m, 0.5H), 4.18-4.07 (m, 1.5H), 3.96-3.91 (m, 0.5H), 3.78-3.60 (m, 4H), 3.31-3.12 (m, 1H), 2.94-2.74 (m, 5H), 1.20-1.18 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 154.3, 153.2, 152.7, 149.7, 149.4, 138.6, 138.4, 136.1, 134.9, 134.5, 130.2, 129.8, 129.7, 128.4, 128.2, 128.0, 127.0, 125.9, 124.9, 120.8, 119.5, 115.9, 115.4, 114.8, 114.6, 114.4, 112.9, 112.7, 112.3, 111.4, 71.2, 70.2, 57.2, 55.9, 51.8, 48.9, 48.8, 42.9, 35.7, 30.3, 30.0, 28.9, 19.7; HRMS calcd. for C28H32N2O3Cl, 479.20960 [M + H]+; found, 479.20975 [M + H]+; Anal. (C28H31N2O3Cl): C, H, N.
(3-chlorophenyl)(6-(isopropylamino)-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (120)
Tetrahydroisoquinoline 120 was prepared via procedure XIII using tetrahydroisoquinoline 84 (0.039 g, 0.12 mmol) and 3-chlorobenzoyl chloride (0.015 mL, 0.12 mmol) in DCM (1.8 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.024 g, 42 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.59; 1HNMR (CDCl3, 400 MHz) δ: 7.60-7.25 (m, 4H), 6.89-6.78 (m, 5H), 6.52-6.36 (m, 2H), 5.95 (t, J = 5.2 Hz, 0.5H), 5.10-5.07 (m, J = 2.8 Hz, J = 9.2 Hz, 0.5H), 4.86-4.81 (m, J = 5.6 Hz, J = 13.2 Hz, 0.5H), 4.33-4.29 (m, 1H), 4.15-4.06 (m, 0.5H), 3.92-3.88 (dd, J = 3.6 Hz, J = 10.0 Hz, 0.5H), 3.77 (s, 3H), 3.71-3.67 (m, 1H), 3.65-3.57 (m, 1H), 3.28-3.06 (m, 1.5H), 2.89-2.67 (m, 2H), 1.23-1.21 (m, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 154.3, 153.1, 152.6, 138.5, 138.4, 136.4, 135.4, 134.9, 134.5, 130.2, 129.8, 129.7, 128.6, 128.2, 127.0, 125.9, 124.9, 115.9, 115.4, 114.9, 114.8, 114.1, 113.3, 71.2, 70.2, 57.3, 55.9, 51.9, 42.8, 35.5, 29.9, 28.6, 22.9, 22.7; HRMS calcd. for C27H30N2O3Cl, 465.19395 [M + H]+; found, 465.19225 [M + H]+; mp: 108°C. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 0.89 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 3 mins at 1 mL/min (retention time 1.84 minutes).
(3-chlorophenyl)(1-((4-methoxyphenoxy)methyl)-6-(piperidin-1-yl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (121)
Tetrahydroisoquinoline 121 was prepared via Procedure XIII using tetrahydroisoquinoline 80 (0.11 g, 0.32 mmol) and 3-chlorobenzoyl chloride (0.050 mL, 0.39 mmol) in DCM (5.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.11 g, 67 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.48; 1HNMR (CDCl3, 400 MHz) δ: 7.58 (s, 0.5H), 7.37-7.23 (m, 3H), 7.18-7.16 (m, 0.5H), 6.89-6.7 (m, 7H), 5.96 (t, J = 4.8 Hz, 0.5H), 5.12-5.08 (m, J = 3.6 Hz, J = 3.6 Hz, 0.5H), 4.86-4.82 (m, J = 5.6 Hz, J = 13.2 Hz, 0.5H), 4.36-4.29 (m, 1H), 4.12 (t, J = 10.4 Hz, 0.5H), 3.92-3.89 (m, 0.5H), 3.75 (s, 3H), 3.72-3.62 (m, 0.5H), 3.28-3.32 (m, 1.5H), 3.15-3.08 (m, 4H), 2.91-2.69 (m, 1.5H), 1.76-1.66 (m, 4H), 1.58-1.57 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 167.9, 153.3, 154.2, 153.1, 152.6, 151.7, 138.5, 138.3, 135.9, 134.9, 134.5, 133.1, 130.2, 129.8, 129.7, 128.8, 128.2, 127.9, 127.0, 125.9, 124.8, 122.4, 116.6, 115.9, 115.7, 115.4, 114.9, 114.8, 71.2, 70.1, 57.3, 55.9, 51.9, 50.6, 42.9, 35.6, 30.1, 29.9, 25.9, 24.4; HRMS calcd. for C29H32N2O335Cl, 491.20960 [M + H]+; found, 491.20961 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 0.85 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 3 mins at 1 mL/min (retention time 1.30 minutes).
(3-chlorophenyl)(1-((4-methoxyphenoxy)methyl)-6-morpholino-3,4-dihydroisoquinolin-2(1H)-yl)methanone (122)
Tetrahydroisoquinoline 122 was prepared via Procedure XIII using tetrahydroisoquinoline 81 (0.23 g, 0.67 mmol) and 3-chlorobenzoyl chloride (0.10 mL, 0.80 mmol) in DCM (11 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.18 g, 55 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.74; 1HNMR (CDCl3, 400 MHz) δ: 7.57 (s, 0.5H), 7.39-7.19 (m, 4H), 6.93-6.65 (m, 6.5H), 5.96 (t, J = 5.2 Hz, 0.5H), 5.12-5.09 (m, J = 3.6 Hz, J = 9.2 Hz, 0.5H), 4.86-4.81 (m, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.36-4.29 (m, 1H), 4.14-4.09 (m, 0.5H), 3.92-3.89 (m, 0.5H), 3.83 (t, J = 4.8 Hz, 4H), 3.78 (s, 3H), 3.69-3.62 (m, 1.5H), 3.28-3.20 (m, 0.5H), 3.17-3.13 (m, 4H), 2.92-2.70 (m, 1.5H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.7, 154.4, 154.2, 153.1, 152.6, 150.9, 150.4, 138.9, 138.2, 136.1, 135.11, 134.8, 134.5, 130.3, 129.9, 129.8, 128.4, 128.2, 128.1, 127.0, 125.8, 124.9, 124.6, 123.4, 115.9, 115.4, 114.9, 114.8, 114.2, 71.2, 70.1, 67.0, 57.3, 55.9, 51.9, 49.4, 49.2, 42.9, 35.6, 30.1, 28.7, 21.3; HRMS calcd. for C28H30N2O435Cl, 493.18886 [M + H]+; found, 493.18900 [M + H]+; mp: 108°C. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 1.33 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 0.85 minutes).
(3-bromophenyl)(1-((4-methoxyphenoxy)methyl)-6-morpholino-3,4-dihydroisoquinolin-2(1H)-yl)methanone (123)
Tetrahydroisoquinoline 123 was prepared via Procedure XIII using tetrahydroisoquinoline 81 (0.23 g, 0.67 mmol) and 3-bromobenzoyl chloride (0.11 mL, 0.80 mmol) in DCM (11 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.16 g, 46 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.72; 1HNMR (CDCl3, 400 MHz) δ: 7.72 (s, 0.5H), 7.55-7.49 (m, 1.5H), 7.41-7.19 (m, 2H), 6.93-6.91 (m, 0.5H), 6.86-6.65 (m, 6.5H), 5.95 (t, J = 4.8 Hz, 0.5H), 5.12-5.09 (m, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.86-4.82 (m, J = 5.2 Hz, J = 12.0 Hz, 0.5H), 4.39-4.29 (m, 1H), 4.12 (t, J = 10.4 Hz, 0.5H), 3.92-3.89 (m, 0.5H), 3.84 (t, J = 4.8 Hz, 4H), 3.74 (s, 3H), 3.69-3.56 (m, 1.5H), 3.27-3.20 (m, 0.5H), 3.13 (s, 4H), 2.91-2.70 (m, 1.5H); 13C NMR (CDCl3, 100 MHz) δ: 170.3, 169.5, 154.4, 154.2, 153.1, 152.6, 150.9, 150.5, 138.6, 138.5, 137.2, 135.1, 132.8, 130.9, 130.5, 130.2, 129.9, 128.4, 128.1, 126.3, 125.3, 124.6, 123.4, 122.9, 122.6, 116.1, 115.9, 115.5, 114.9, 114.8, 114.2, 71.2, 70.1, 67.0, 62.2, 57.3, 55.9, 51.9, 49.4, 49.2, 42.9, 35.6, 30.1, 28.7; HRMS calcd. for C28H30N2O479Br, 537.13835 [M + H]+; found, 537.13859 [M + H]+; Anal. (C28H29N2O4Br): C, H, N.
(3-chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-morpholino-3,4-dihydroisoquinolin-2(1H)-yl)methanone (124)
Tetrahydroisoquinoline 124 was prepared via Procedure XIII using tetrahydroisoquinoline 82 (0.21 g, 0.57 mmol) and 3-chlorobenzoyl chloride (0.086 mL, 0.67 mmol) in DCM (8.8 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.11 g, 38 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.32; 1HNMR (CDCl3, 400 MHz) δ: 7.57 (s, 0.5H), 7.37-7.19 (m, 4H), 6.93-6.91 (m, 0.5H),6.82-6.77 (m, 5H), 6.71-6.65 (m, 1H), 5.97-5.95 (m, 0.5H), 5.12-5.09 (m, J = 5.2 Hz, J = 9.2 Hz, 0.5H), 4.33-4.29 (m, 1H), 4.14-4.09 (m, 0.5H), 3.95 (q, J = 7.2 Hz, 2H), 3.84 (t, J = 4.4 Hz, 4H), 3.73-3.64 (m, 1H), 3.28-3.14 (m, 5H), 2.90-2.70 (m, 2H), 1.37 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 170.4, 169.6, 154.2, 153.7, 153.6, 153.0, 152.5, 150.9, 150.5, 138.4, 138.2, 136.2, 135.1, 134.9, 134.5, 133.4, 130.2, 129.9, 128.5, 128.1, 127.0, 125.8, 124.7, 123.5, 115.8, 115.7, 115.6, 115.4, 114.8, 114.2, 71.2, 70.1, 67.1, 64.2, 57.3, 55.9, 51.9, 49.4, 49.2, 42.9, 40.0, 35.6, 30.1, 28.7, 26.3, 15.2; HRMS calcd. for C29H32N2O435Cl, 507.20451 [M + H]+; found, 507.20466 [M + H]+. Anal. (C21H26NO3Br): C, H, N.
(3-bromophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (125)
Tetrahydroisoquinoline 125 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.15 g, 0.46 mmol) and 3-bromobenzoyl chloride (0.070 mL, 0.51 mmol) in DCM (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.13 g, 56 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.73; 1HNMR (CDCl3, 400 MHz) δ: 7.74-7.50 (m, 2H), 7.42-7.18 (m, 2H), 6.91-6.66 (m, 6H), 5.97-5.95 (t, J = 5.2 Hz, 0.5H), 5.13-5.10 (dd, J = 3.2 Hz, J = 9.6 Hz, 0.5H), 4.86-4.81 (dd, 5.2 Hz, J = 12.4 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.37-4.29 (m, 1H), 4.15-4.08 (m, 0.5 H), 3.92-3.89 (dd, J = 3.6 Hz, J = 9.6, 0.5H), 3.75 (s, 3H), 3.73-3.62 (m, 1H), 3.27-3.08 (m, 1H), 2.29-2.70 (m, 1.5 H), 1.33-1.31 (d, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.3, 169.6, 157.7, 157.1, 154.4, 154.3, 153.1, 152.8, 138.6, 138.4, 136.6, 135.6, 133.2, 132.8, 131.1, 130.4, 130.1, 129.9, 128.8, 128.3, 126.3, 125.3, 124.1, 122.9, 122.6, 116.2, 115.9, 115.5, 114.9, 114.8, 114.7, 114.5, 71.3, 70.1, 57.3, 55.9, 51.9, 42.8, 35.5, 29.9, 28.5, 22.3, 22.2; HRMS calcd. for C21H27BrO3N1, 510.12745 [M + H]+; found 510.12750 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/15% water to 95%ACN/5% water over 6 mins at 1 mL/min (retention time = 1.62 minutes). Method 2: 85% MeOH/15% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 1.34 minutes).
(3-iodophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (126)
Tetrahydroisoquinoline 126 was prepared via procedure XIV using 3-iodobenzoic acid (0.18 g, 0.71 mmol) dissolved in dry DCM (2.3 ml) and THF (1.7 ml), N1-((ethylimino)methylene)-N3, N3-dimethylpropane-1,3-diamine hydrochloride (0.16 g, 0.85 mmol), N, N-dimethylpyridin-4-amine (0.096 g, 0.78 mmol), and tetrahydroisoquinoline 67 (0.23 g, 0.71 mmol). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.15 g, 38 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.64; 1HNMR (CDCl3, 400 MHz) δ: 7.96-7.73 (m, 1.5H), 7.48 (d, J = 7.2 Hz, 0.5H), 7.36 (d, J = 7.6 Hz, 0.5H), 7.23-7.12 (m, 1.5H), 6.94-6.69 (m, 7H), 5.98 (t, J = 5.2 Hz, 0.5H), 5.16-5.13 (m, J = 3.6 Hz, J = 9.2 Hz, 0.5H), 4.88-4.84 (m, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.59-4.52 (m, 1H), 4.39-4.32 (m, 1H), 4.17-4.12 (m, 0.5H), 3.95-3.92 (dd, J = 3.2 Hz, J = 9.6Hz, 0.5H), 3.77 (s, 3H), 3.75-3.64 (m, 1H), 3.30-3.11 (m, 1H), 2.94-2.73 (m, 1.5H), 1.35 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.2, 169.4, 157.6, 157.0, 154.4, 154.3, 153.1, 152.6, 142.2, 139.1, 138.8, 138.6, 138.5, 136.7, 136.6, 135.6, 130.5, 130.2, 129.4, 128.7, 128.4, 126.8, 125.8, 125.2, 124.1, 116.2, 115.9, 115.5, 114.9, 114.8, 114.7, 115.5, 71.2, 70.1, 57.3, 55.9, 51.9, 42.8, 35.4, 29.9, 28.5, 22.3, 22.2; HRMS calcd. for C27H28N1O4I, 558.11358 [M + H]+; found, 558.11362 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.34 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 4.29 minutes).
(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (127)
Tetrahydroisoquinoline 127 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.17 g, 0.49 mmol) and 3-(trifluoromethyl)benzoyl chloride (0.080 mL, 0.54 mmol) in DCM (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.13 g, 54 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.66 1HNMR (CDCl3, 400 MHz) δ: 7.88-7.48 (m, 4H), 7.22-7.20 (m, 0.5 H), 6.89-6.66 (m, 6.5 H), 5.99-5.96 (m, 0.5 H), 5.09-5.06 (dd, J = 3.2 Hz, J = 6.4 Hz, 0.5H), 4.89-4.84 (dd, J = 5.6 Hz, J = 12.8 Hz, 0.5H), 4.55-4.49 (m, 1H), 4.37-4.34 (m, 1H), 4.15-4.10 (m, 0.5 H), 3.98-3.89 (m, 2.5H), 3.70-3.69 (m, 1H), 3.27-3.12 (m, 1H), 2.91-2.72 (m, 1.5H), 1.39-1.35 (t, J = 7.2 Hz, 3H), 1.33-1.32 (d, J = 1.6 Hz, 3H), 1.31-1.30 (d, J = 1.6 Hz, 3H); 13CNMR (100 MHz, CDCl3) δ: 170.5, 169.7, 168.4, 157.6, 157.1, 153.8, 153.6, 152.9, 152.4, 137.4, 137.3, 136.5, 135.4, 133.5, 131.5, 131.1, 130.8, 130.2, 129.4, 129.3, 129.1, 128.7, 127.2, 126.5, 125.2, 123.9, 116.2, 115.9, 115.6, 115.5, 115.4, 115.3, 114.8, 114.5, 76.9, 71.2, 70.1, 64.2, 57.4, 52.1, 42.9, 35.5, 29.8, 28.5, 22.3, 22.2, 15.1; HRMS calcd. for C29H31F3N1O4, 514.21914 [M + H]+; found 514.21949 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.34 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 4.29 minutes).
(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-nitrophenyl)methanone (128)
Tetrahydroisoquinoline 128 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.33 g, 0.99 mmol) and 3-nitrobenzoyl chloride (0.22 mL, 1.2 mmol) in DCM (16 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an yellow foam (0.26 g, 53 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.45; 1HNMR (CDCl3, 400 MHz) δ: 8.53-8.27 (m, 1.5H), 7.85-7.22 (m, 2.5H), 6.92-6.70 (m, 7H), 5.99-6.01 (m, 0.5H), 5.09-5.07 (m, 0.5H), 4.91-4.86 (m, J = 5.2 Hz, J = 12.4 Hz, 0.5H), 4.59-4.52 (m, 1H), 4.40-4.37 (m, 1H), 4.18 (t, J = 10.4 Hz, 0.5H), 3.96-3.93 (m, J = 3.6 Hz, J = 9.2 Hz, 0.5H), 3.77 (s, 3H), 3.75-3.73 (m, 1H), 3.34-3.14 (m, 1H), 2.98-2.77 (m, 1.5H), 1.35 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 169.4, 168.6, 157.7, 157.2, 154.5, 153.0, 152.4, 148.4, 148.2, 138.2, 136.5, 135.3, 133.8, 132.9, 130.2, 129.7, 128.7, 128.3, 124.9, 124.6, 124.5, 123.6, 123.4, 122.1, 116.3, 115.9, 115.4, 114.9, 114.6, 71.3, 70.1, 69.9, 57.5, 55.9, 52.3, 42.9, 35.5, 29.8, 28.5, 22.3; HRMS calcd. for C27H29N2O6, 477.20201 [M + H]+; found, 477.20169 [M + H]+. Anal. (C27H28N2O6): C, H, N.
(3-hydroxyphenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (131)
Tetrahydroisoquinoline 130 (.15 g, 0.27 mmol) was dissolved in THF (1.59 ml) and brought to 0 °C using an ice bath. Tetrabutylammonium fluoride (0.14 g, 0.53 mmol) was added and the reaction was allowed to stir for 5 minutes before the reaction was warmed to room temperature. The reaction stirred for 1 hour, at which point it was complete by TLC. The reaction was quenched with 1M HCl, extracted into EtOAc, washed with water and brine, dried with MgSO4, filtered, and concentrated in vacuo. The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.11 g, 66 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.42; 1HNMR (CDCl3, 400 MHz) δ: 7.19-7.03 (m, 2H), 6.93-6.64 (m, 9H), 5.97-5.94 (t, J = 5.2 Hz, 0.5H), 5.23-5.19 (dd, J = 4.4 Hz, J = 9.2 Hz, 0.5H), 4.83-4.78 (dd, J = 5.6 Hz, J = 13.2 Hz, 0.5H), 4.54-4.48 (m, 1H), 4.33-4.26 (m, 1H), 4.14-4.04 (m, 0.5 H), 3.92-3.88 (dd, J = 4.0 Hz, J = 10.0 Hz, 0.5H), 3.83-3.79 (m, 0.5 H), 3.71 (s, 3H), 3.63-3.55 (m, 0.5 H), 3.51-3.05 (m, 1H), 2.89-2.64 (m, 1.5 H), 1.32-1.30 (d, 3H, J = 2.4 Hz), 1.31-1.30 (d, 3H, J = 2.4 Hz); 13CNMR (CDCl3, 100 MHz) δ: 172.7, 171.9, 157.5, 157.2, 157.0, 154.3, 154.2, 153.1, 152.6, 136.8, 136.7, 136.4, 135.7, 129.9, 129.6, 128.8, 128.4, 125.2, 124.4, 118.6, 117.6, 116.2, 116.1, 115.9, 115.8, 115.6, 115.5, 114.9, 114.7, 114.4, 114.3, 71.3, 70.3, 70.1, 60.7, 57.2, 55.9, 52.0, 42.9, 35.9, 29.9, 28.6, 22.3, 22.3; HRMS calcd. for C27H30N1O5, 448.21185[M + H]+; found 448.21131 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 0.87 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 0.84 minutes).
(3-aminophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (132)
To a suspension of tetrahydroisoquinoline 128 (0.24 g, 0.49 mmol) in THF (0.82 ml) and 2-propanol (0.82 ml) was added palladium on carbon (0.024 g, 0.015 mmol). The reaction was hydrogenated at room temperature using a balloon. After stirring overnight the reaction was filtered through a pad of celite washing with MeOH and DCM and the volatiles were removed in vacuo. The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.20 g, 90 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.20; 1HNMR (CDCl3, 400 MHz) δ: 7.23-7.14 (m, 2H), 6.93-6.78 (m, 5.5H), 6.74-6.63 (m, 3.5H), 5.98 (t, J = 5.2 Hz, 0.5H), 5.26-5.23 (m, J = 4.4 Hz, J = 8.8 Hz, 0.5H), 4.87-4.83 (m, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.57-4.51 (m, 1H), 4.37 (d, J = 5.6 Hz, 1H), 4.16-3.94 (m, 0.5H), 3.97-3.83 (m, 2H), 3.76 (s, 3H), 3.67-3.60 (m, 0.5), 3.26-3.11 (m, 0.5H), 2.92-2.68 (m, 1H), 1.34 (d, J = 1.6 Hz, 3H), 1.33 (d, J = 1.6 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 172.1, 171.4, 157.4, 156.9, 154.2, 153.2, 152.9, 147.0, 146.8, 137.8, 137.6, 136.6, 135.9, 129.7, 129.5, 128.8, 128.4, 125.6, 124.8, 117.5, 116.4, 116.2, 115.9, 115.8, 115.7, 114.9, 114.6, 114.3, 114.2, 113.2, 71.4, 70.6, 70.1, 57.0, 55.9, 51.7, 42.7, 35.5, 30.1, 28.7, 22.3, 22.2; HRMS calcd. for C27H31N2O4, 447.22783 [M + H]+; found, 447.22712 [M + H]+. Anal. (C27H30N2O4): C, H, N.
(4-chlorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (133)
Tetrahydroisoquinoline 133 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.15 g, 0.46 mmol) and 4-chlorobenzoyl chloride (0.06 mL, 0.55 mmol) in DCM (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.07 g, 31 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.79 1HNMR (CDCl3, 400 MHz) δ: 7.47-7.30 (m, 4H), 7.23-7.16 (m, 0.5 H), 6.91-6.65 (m, 6.5 H), 5.61-5.95 (m, 0.5 H), 5.13-5.11 (dd, 0.5 H, J = 8.8 Hz, J = 2.8 Hz), 4.85-4.81 (dd, 0.5 H, J = 5.2 Hz, J = 12.4 Hz), 4.54-4.48 (m, 1H), 4.33-4.32 (m, 1H), 4.12-4.07 (m, 0.5 H), 3.93-3.89 (dd, 0.5 H, J = 4.0 Hz, J = 9.6 Hz), 3.74 (s, 3H), 3.69-3.66 (m, 1H), 3.26-3.07 (m, 1H), 2.89-2.69 (m, 1.5 H), 1.33-1.31 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 171.0, 170.2, 157.6, 157.0, 154.4, 154.2, 153.1, 152.6, 136.6, 135.9, 135.7, 135.6, 134.9, 134.8, 131.7, 129.3, 129.1, 128.9, 129.0, 128.9, 128.8, 128.4, 128.3, 128.1, 125.2, 124.1, 116.2, 115.9, 115.6, 115.4, 114.9, 114.8, 114.7, 114.4, 71.3, 70.2, 70.1, 57.3, 55.9, 51.9, 42.8, 35.4, 29.9, 28.6, 22.3, 22.2; HRMS calcd. for C27H29ClN1O4, 466.17796 [M + H]+; found 466.17812 [M + H]+. Anal. (C27H28ClN1O4): C, H, N.
(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(4-methoxyphenyl)methanone (134)
Tetrahydroisoquinoline 134 was prepared via procedure XIII using tetrahydroisoquinoline 67 (0.10 g, 0.31 mmol) and 4-methoxybenzoyl chloride (0.06 mL, 0.33 mmol) in DCM (10. mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.08 g, 56 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.88 1HNMR (CDCl3, 400 MHz) δ: 7.50-7.48 (m, 1H), 7.37-7.35 (m, 1H), 7.21-7.19 (m, 0.5 H), 6.89-6.66 (m, 8.5 H), 5.95 (m, 0.5 H), 5.28-5.23 (m, 0.5 H), 4.82-4.79 (d, J = 7.6 Hz, 0.5H), 4.55-4.47 (m, 1H), 4.35-4.33 (m, 1H), 4.13-4.08 (m, 0.5 H), 3.94-3.84 (m, 1H), 3.82 (s, 3H), 3.74 (m, 3H), 3.69-3.63 (m, 0.5 H), 3.22-3.12 (m, 1H), 2.89-2.69 (m, 1.5 H), 1.32-1.30 (d, 6H); 13CNMR (100 MHz, CDCl3) δ: 171.9, 171.2, 160.8, 157.5, 156.9, 154.2, 153.2, 152.8, 136.8, 135.8, 129.7, 128.9, 128.3, 125.9, 124.8, 116.1, 115.9, 115.6, 114.9, 114.6, 114.3, 114.0, 113.8, 71.5, 70.5, 70.1, 66.1, 57.4, 56.0, 55.9, 55.6, 51.9, 43.0, 35.6, 30.1, 29.9, 28.6, 22.3, 22.2; HRMS calcd. for C28H32N1O5, 462.22750 [M + H]+; found 462.22672 [M + H]+. Anal. (C28H32N1O5): C, H, N.
(6-ethoxy-1-((4-ethoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanone (135)
Dihydroisoquinoline 56 was prepared via procedure X using amide 29 (0.58 g, 1.7 mmol) and phosphorus trichloride (0.84 mL, 4.1 mmol) in dry toluene (8.4 mL). The crude solid was carried on without further purification. HRMS calcd. for C20H24O3N, 326.17507 [M + H]+; found 326.17457 [M + H]+. Tetrahydroisoquinoline 75 was prepared via procedure XII using 56 (0.91 g, 2.8 mmol) and sodium borohydride (0.32 g, 8.4 mmol) in dry MeOH (14 mL). The crude residue was subjected to flash column chromatography (ISCO, Redisep 24 g column, 0–10% MeOH/DCM gradient) to afford tetrahydroisoquinoline 75 (0.12 g, 14 %) in an impure form. The impurities were inseparable by chromatography. The product was visible by LCMS and was carried on without further purification. Tetrahydroisoquinoline 135 was prepared via procedure XIII using tetrahydroisoquinoline 75 (0.061 g, 0.19 mmol) and 3-fluorobenzoyl chloride (0.026 mL, 0.22 mmol) in DCM (2.9 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10–80% EtOAc/hexanes gradient) to afford the title compound as a white foam (0.025 g, 30 % yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.70; 1HNMR (CDCl3, 400 MHz) δ: 7.43-7.09 (m, 4H), 6.94-6.91 (m, 0.5H), 6.86-6.73 (m, 6H), 6.68 (s, 0.5H), 6.0-5.98 (m, 0.5H), 5.16-5.14 (m, 0.5H), 4.88-4.84 (m, J = 5.6 Hz, J = 5.2 Hz, 0.5H), 4.36-4.32 (m, 1H), 4.12 (t, J = 10 Hz, 0.5H), 4.03-3.92 (m, 4.5H), 3.78-3.65 (m, 1H), 3.30-3.11 (m, 1H), 2.94-2.73 (m, 1.5H), 1.44-138 (m, 6H); 13CNMR (CDCl3, 100 MHz) δ: 170.6., 169.8, 162.8 (J = 247 Hz), 162.7 (J = 247 Hz), 158.6, 158.1, 153.8, 153.6, 153.0, 152.5, 138.8, 138.7, 138.6, 138.5, 136.6, 135.6, 130.7, 130.6, 130.4, 130.3, 128.7, 128.3, 125.3, 124.3, 123.4, 122.5, 116.9, 116.8, 116.7, 116.6, 115.9, 115.7, 115.6, 115.5, 115.4, 115.2, 114.9, 115.2, 114.9, 114.4, 114.3, 114.0, 113.6, 113.4, 71.4, 70.2, 64.2, 63.8, 63.7, 57.3, 51.9, 42.8, 35.4, 30.0, 28.6, 15.1, 15.0; HRMS calcd. for C27H29NO4F, 450.20751 [M + H]+; found 450.20805 [M + H]+. Anal. (C28H31N1O5): C, H, N.
(1-((4-ethoxyphenoxy)methyl)-6-isobutoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanone (136)
Tetrahydroisoquinoline 136 was prepared via Procedure XIII using tetrahydroisoquinoline 79 (0.18 g, 0.50 mmol) and 3-fluorobenzoyl chloride (0.073 mL, 0.60 mmol) in DCM (7.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.61 g, 27 % yield, mixture of two amide rotamers). TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.83; 1HNMR (CDCl3, 400 MHz) δ: 7.47-7.09 (m, 5H), 6.94-6.92 (m, 0.5H), 6.87-6.74 (m, 5H),6.68-6.69 (m, 0.5H), 5.98 (t, J = 4.8 Hz, 0.5H), 5.16-5.14 (m, J = 6.4 Hz, J = 9.6 Hz, 0.5 H), 4.89-4.84 (m, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 4.36-4.31 (m, 1H), 4.15-4.10 (m, 0.5H), 4.00-3.95 (q, J = 6.4 Hz, 2H), 3.93-3.91 (m, 0.5H), 3.79-3.74 (m, 0.5H), 3.71 (d, J = 6.4 Hz, 2H), 3.68-3.65 (m, 0.5H), 2.11-2.03 (m, 1H), 1.40 (t, J = 6.8 Hz, 3H), 1.03 (d, J = 6.8 Hz, 6H); 13C NMR (CDCl3, 100 MHz) δ: 170.6, 169.8, 164.0, 158.9, 158.4, 153.7, 153.6, 153.0, 152.5, 138.6, 136.6, 135.5, 130.7, 130.3, 128.7, 126.1, 125.2, 124.1, 123.4, 122.5, 120.7, 117.2, 116.8, 116.6, 115.8, 115.7, 115.6, 115.4, 115.2, 114.8, 114.4, 114.3, 114.1, 113.7, 113.4, 111.2, 74.6, 71.3, 70.2, 64.2, 57.3, 51.9, 42.8, 35.5, 29.9, 28.4, 19.5, 15.2; HRMS calcd. for C29H33NO4F, 478.23881 [M + H]+; found, 478.23887 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.33 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 6 mins at 1 mL/min (retention time 5.50 minutes).
(6-(cyclopentyloxy)-1-((4-ethoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanone (137)
Tetrahydroisoquinoline 137 was prepared via Procedure XIII using tetrahydroisoquinoline 78 (0.18 g, 0.50 mmol) and 3-fluorobenzoyl chloride (0.073 mL, 0.60 mmol) in DCM (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.18 g, 64% yield, mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:1, v/v) Rf = 0.69; 1HNMR (CDCl3, 400 MHz) δ: 7.85-7.71 (m, 0.5H), 7.43-7.09 (m, 4H), 6.90-6.74 (m, 5H), 6.69 (s, 1H), 6.64 (s, 0.5H), 5.97 (t, J = 4.4 Hz, J = 9.2 Hz, 0.5H), 5.15-5.11 (m, J = 2.8 Hz, J = 9.2 Hz, 0.5H), 4.87-4.82 (m, J = 5.6 Hz, J = 13.2 Hz, 0.5H), 4.74-4.71 (m, 1H), 4.37-4.29 (m, 1H), 4.11 (t, J = 10.4 Hz, 0.5H), 3.95 (q, J = 7.2 Hz, 2H), 3.91-3.89 (m, 0.5H), 3.76-3.63 (m, 1H), 3.28-3.08 (m, 1H), 2.93-2.70 (m, 1.5H), 1.88-1.76 (m, 6H), 1.68-1.60 (m, 2H), 1.37 (t, J = 6.8 Hz, 3H); 13C NMR (CDCl3, 100 MHz) δ: 170.6, 169.8, 164.0, 163.8, 161.6, 161.4, 157.8, 157.3, 153.7, 153.6, 153.0, 152.5, 138.7, 138.6, 138.5, 138.4, 136.5, 135.5, 130.7, 130.6, 130.3, 128.6, 128.2, 126.0, 124.9, 123.9, 123.5, 122.4, 120.4, 117.2, 116.9, 116.8, 116.7, 116.6, 115.9, 115.6, 115.5, 115.4, 115.2, 114.5, 114.3, 114.2, 114.0, 113.8, 106.2, 103.0, 94.5, 82.1, 79.5, 71.3, 70.2, 64.2, 57.3, 51.9, 42.8, 35.5, 33.1, 29.9, 28.6, 24.2, 15.2; HRMS calcd. for C30H33NO4F, 490.23881 [M + H]+; found, 490.23958 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 1.99 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 1.83 minutes).
(3-Chlorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (138)
Tetrahydroisoquinoline 138 was prepared via procedure XV using tetrahydroisoquinoline 2 (0.045 g, 0.97 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.039 g, 0.97 mmol) in toluene (5.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.032 g, 68%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.80; 1H NMR (CDCl3, 400 MHz) δ: 7.32-7.25 (m, 3.5H), 7.14-7.13 (m, 0.5H), 6.87-6.70 (m, 7H), 5.75-5.70 (m, J = 5.2 Hz, J = 5.2 Hz, 0.5H), 5.14-5.38 (m, J = 4.0 Hz, J = 3.6 Hz, 0.5H), 4.70-4.69 (doublet of doublet, J = 4.0 Hz, J = 4.0 Hz, 0.5H), 4.58-4.52 (m, 1H), 4.50-4.46 (doublet of doublet, J = 4.4 Hz, J = 4.0 Hz, 0.5H), 4.20-4.15 (m, 0.5H), 4.05-3.87 (m, 2H), 3.77 (s, 3H), 3.72-3.64 (m, 0.5H), 3.36-3.28 (m, 0.5H), 2.98-2.90 (m, 1H), 2.83-2.79 (m, 0.5H), 1.36-1.34 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200.4, 198.9, 157.9, 158.1, 154.5, 154.4, 152.9, 152.4, 145.0, 144.7, 134.9, 134.7, 130.2, 128.8, 128.6, 128.5, 128.1, 124.9, 123.5, 116.0, 115.9, 115.7, 115.1, 114.9, 114.8, 114.7, 70.8, 70.2, 61.5, 58.4, 55.9, 48.1, 30.1, 28.1, 22.3, 22.2; HRMS calcd. for C27H28ClNO3S, 482.15512 [M + H]+; 488.15514 [M + H]+ found; mp: 80°C. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 6 mins at 1 mL/min (retention time = 1.94 minutes).
(3-Fluorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (139)
Tetrahydroisoquinoline 139 was prepared via procedure XV using tetrahydroisoquinoline 114 (0.028 g, 0.61 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.025 g, 0.61 mmol) in toluene (4.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.19 g, 65%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.83; 1H NMR (CDCl3, 400 MHz) δ: 7.34-7.25 (m, 1H), 7.08-6.97 (m, 2H), 6.88-6.69 (m, 8H), 5.75-5.70 (m, J = 4.4 Hz, J = 4.8 Hz, 0.5H), 5.42-5.39 (m, J = 4.0 Hz, J = 4.4 Hz, 0.5H), 4.71-4.67 (doublet of doublet, J = 4.4 Hz, J = 4.4 Hz, 0.5H), 4.56-4.52 (m, 1H), 4.50-4.46 (m, J = 4.4 Hz, J = 4.4 Hz, 0.5H), 4.21-4.16 (t, J = 9.6 Hz, 0.5H), 4.05-3.87 (m, 1.5H), 3.77-3.59 (m, 3H), 3.36-3.27 (m, 0.5H), 2.99-2.91 (m, 1H), 2.83-2.78 (m, 0.5H), 1.36-1.34 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200.5, 199.1, 164.4, 161.1, 157.9, 157.1, 154.6, 154.4, 152.9, 152.4, 154.4, 145.3, 136.2, 134.9, 130.7, 130.6, 130.1, 128.5, 128.1, 124.9, 123.5, 116.0, 115.9, 115.8, 115.7, 115.6, 115.4, 115.1, 114.9, 114.8, 114.6, 70.9, 70.3, 70.2, 70.1, 61.4, 58.4, 55.9, 40.1, 42.7, 30.1, 28.1, 22.3; HRMS calcd. for C27H38FNO3SNa, 488.16661 [M + H]+; 488.16668 [M + H]+ found. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.64 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 6 mins at 1 mL/min (retention time 4.58 minutes).
(3-Chlorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (140)
Tetrahydroisoquinoline 140 was prepared via procedure XV using tetrahydroisoquinoline 92 (0.067 g, 0.14 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.029 g, 0.071 mmol) in toluene (5.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.024 g, 33%, mixture of two thioamide rotamers) TLC (EtOAc:hexanes, 1:5, v/v) Rf = 0.58; 1H NMR (CDCl3, 400 MHz) δ: 7.32-7.22 (m, 3H), 7.14-7.13 (m, 1H), 6.86-6.70 (m, 7H), 5.75-5.70 (m, J = 5.2 Hz, J = 12.8 Hz, 0.5H), 5.41-5.38 (m, J = 4.0 Hz, J = 9.6 Hz, 0.5H), 4.70-4.67 (m, J = 4.0 Hz, J = 10.0 Hz, 0.5H), 4.60-4.52 (m, 1H), 4.49-4.46 (m, J = 4.8 Hz, J = 10.4 Hz, 0.5H), 4.23-4.13 (m, 1H), 4.08-3.87 (m, 3.5H), 3.71-3.63 (m, 0.5H), 3.36-3.27 (m, 0.5H), 2.98-2.79 (m, 1.5H), 1.42-1.33 (m, 9H); 13C NMR (75 MHz, CDCl3) δ: 200.4, 198.9, 157.8, 157.1, 153.9, 152.7, 152.9, 152.3, 145.0, 144.8, 136.2, 134.9, 134.8, 130.2, 129.7, 128.8, 128.6, 128.5, 128.2, 124.9, 123.5, 116.0, 115.9, 115.7, 115.6, 115.5, 115.1, 114.8, 70.8, 70.2, 70.1, 64.2, 61.5, 58.4, 48.1, 42.7, 30.1, 28.1, 22.3, 15.1; HRMS calcd. for C28H31NO3ClS, 496.17077 [M + H]+; 496.17044 [M + H]+ found. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.39 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 6 mins at 1 mL/min (retention time 4.17 minutes).
(3-Bromophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (141)
Tetrahydroisoquinoline 141 was prepared via procedure XV using tetrahydroisoquinoline 96 (0.033 g, 0.081 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.043 g, 0.081 mmol) in toluene (3.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.030 g, 68%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.80; 1H NMR (CDCl3, 400 MHz) δ: 7.50-7.40 (m, 1.5H), 7.25-7.17 (m, 1.5H), 6.88-6.69 (m, 8H), 5.74-5.70 (m, J = 4.8 Hz, J = 12.0 Hz, 0.5H), 5.41-5.38 (m, J = 4.0 Hz, J = 9.6 Hz, 0.5H), 4.70-4.66 (m, J = 4.0 Hz, J = 5.6 Hz, 0.5H), 4.59-4.52 (m, 1H), 4.49-4.45 (m, J = 4.8 Hz, J = 10.4 Hz, 0.5H), 4.20-4.12 (m, 0.5H), 4.04-3.87 (m, 4H), 3.71-3.63 (m, 0.5H), 3.36-3.27 (m, 0.5H), 2.98-2.90 (m, 1H), 2.83-2.79 (m, 0.5H), 1.42-1.33 (m, 9H); 13C NMR (100 MHz, CDCl3) δ: 200.2, 198.8, 157.9, 157.1, 153.8, 153.7, 152.8, 152.3, 145.3, 144.9, 136.2, 131.7, 131.5, 130.4, 128.5, 128.1, 125.9, 123.5, 122.8, 116.0, 115.9, 115.7, 115.6, 115.5, 115.1, 114.6, 70.8, 70.1, 64.2, 61.5, 58.4, 48.1, 42.7, 30.1, 29.9, 28.1, 22.3, 22.2, 15.1; HRMS calcd. for C28H30BrNO3S, 540.12025 [M + H]+; 540.12060 [M + H]+ found. Anal. (C28H30N1O3SBr): C, H, N.
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanethione (142)
Tetrahydroisoquinoline 142 was prepared via procedure XV using tetrahydroisoquinoline 97 (0.10 g, 0.22 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.045 g, 0.11 mmol) in toluene (8.0 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.035 g, 35%, mixture of two thioamide rotamers) TLC (EtOAc:hexanes, 1:1, v/v) Rf =0.69; 1HNMR (CDCl3, 400 MHz) δ: 7.34-7.33 (m, 1H), 7.07-6.97 (m, 2.5H), 6.88-6.69 (m, 7.5H), 5.75-5.70 (m, 5.2 Hz, 4.8 Hz, 0.5H), 5.42-5.38 (m, 3.6 Hz, 4.0 Hz, 0.5H), 4.70-4.67 (doublet of doublet, J = 4.4 Hz, J = 10.0 Hz, 0.5H), 4.59-4.52 (m, 1H), 4.49-4.46 (doublet of doublet, J = 4.8 Hz, J = 10.0 Hz, 0.5H), 4.21-4.13 (m, 0.5H), 4.05-3.86 (m, 2H), 3.77 (s, 3H), 3.71-3.64 (m, 0.5H), 3.36-3.27 (m, 0.5H), 2.99-2.90 (m, 1H), 2.83-2.77 (m, 0.5H), 1.42-1.33 (m, 9H); 13C NMR (100 MHz, CDCl3) δ: 199.2, 161.5, 157.9, 157.1, 153.9, 153.8, 152.9, 145.4, 136.2, 134.9, 130.7, 130.6, 128.5, 128.2, 124.9, 123.6, 116.0, 115.9, 115.8, 115.7, 115.6, 115.5, 115.4, 115.1, 114.7, 70.9, 64.3, 61.5, 58.4, 48.1, 42.8, 30.2, 29.9, 28.1, 22.4, 22.3, 15.2; HRMS calcd. for C28H31NO3FS, 480.20032 [M + H]+; 480.19774 [M + H]+ found; mp: 75°C; Anal. (C28H30N1O3SF): C, H, N.
(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanethione (143)
Tetrahydroisoquinoline 143 was prepared via procedure XV using tetrahydroisoquinoline 98 (0.084 g, 0.16 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.066 g, 0.16 mmol) in toluene (8.2 mL). After stirring for 8 hours, the reaction was worked up. The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.021 g, 56%, mixture of two thioamide rotamers) TLC (EtOAc:hexanes, 1:1, v/v) Rf =0.85; 1HNMR (CDCl3, 300 MHz) δ: 7.63-7.43 (m, 3H), 7.28-7.25 (m, 1H), 6.89-6.70 (m, 7H), 5.75-5.73 (m, 0.5H), 5.37-5.33 (m, J = 4.2 Hz, J = 3.6 Hz, 0.5H), 4.73-4.68 (m, 0.5H), 4.59-4.47 (m, 1.5H), 4.23-4.16 (m, 0.5H), 4.01-3.87 (m, 4H), 3.75-3.65 (m, 0.5H), 3.40-3.29 (m, 0.5H), 2.98-2.76 (m, 1.5H), 1.42-1.34 (9H); 13C NMR (100 MHz, CDCl3) δ: 200.3, 198.9, 157.9, 157.2, 153.9, 153.7, 152.8, 152.2, 144.2, 143.9, 136.2, 134.8, 129.5, 128.9, 128.5, 128.1, 125.4, 124.9, 123.3, 116.0, 115.9, 115.8, 115.6, 115.2, 114.7, 70.9, 70.2, 64.2, 61.6, 58.6, 48.2, 42.7, 30.0, 28.1, 22.2, 15.1; HRMS calcd. for C29H31NO3F3S, 530.19822 [M + H]+; 530.19888 [M + H]+ found; mp: 93°C. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 1.70 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 6 mins at 1 mL/min (retention time 4.69 minutes).
(3-Chloro-4-fluorophenyl)(1-((4-ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (144)
Tetrahydroisoquinoline 144 was prepared via procedure XV using tetrahydroisoquinoline 108 (0.072 g, 0.14 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.058 g, 0.14 mmol) in toluene (7.2 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.026 g, 35%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.95; 1H NMR (CDCl3, 400 MHz) δ: 7.32-7.24 (m, 1H), 7.15-7.14 (m, 1H), 6.89-6.70 (m, 8H), 5.73-5.68 (m, J = 6.0 Hz, J = 5.6 Hz, 0.5H), 5.14-5.38 (m, J = 4.0 Hz, J = 3.6 Hz, 0.5H), 4.70-4.66 (m, J = 4.0 Hz, J = 4.0 Hz, 0.5H), 4.58-4.51 (m, 1H), 4.49-4.45 (m, J = 4.4 Hz, J = 4.4 Hz, 0.5H), 4.23-4.14 (m, 0.5H), 4.01-3.89 (m, 4H), 3.70-3.63 (m, 0.5H), 3.37-3.29 (m, 0.5H), 2.98-2.81 (m, 1.5H), 1.42-1.34 (m, 9H); 13C NMR (100 MHz, CDCl3) δ: 199.6, 198.1, 159.4, 158.6, 157.9, 157.2, 157.1, 156.9, 153.9, 153.8, 152.8, 152.2, 140.5, 140.2, 136.2, 134.8, 128.5, 128.1, 124.8, 123.4, 116.9, 116.1, 115.9, 115.7, 115.6, 115.5, 115.4, 114.7, 70.8, 70.2, 70.1, 64.2, 64.1, 61.6, 58.6, 48.2, 42.7, 30.1, 27.9, 22.3, 22.2, 15.2; HRMS calcd. for C28H30ClFNO3S, 514.16135 [M + H]+; 514.16130 [M + H]+ found. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 2.12 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 8 mins at 1 mL/min (retention time 4.64 minutes).
(3-Chlorophenyl)(6-isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (145)
Tetrahydroisoquinoline 145 was prepared via procedure XVI using tetrahydroisoquinoline 113 (0.026 g, 0.053 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.032 g, 0.080 mmol) in toluene (0.53 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.014 g, 51%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:3, v/v) Rf = 0.76; 1H NMR (CDCl3, 400 MHz) δ: 7.33-7.25 (m, 3.5 H), 7.14-7.13 (m, 0.5H), 6.88-6.69 (m, 7H), 5.75-5.71 (m, J = 5.6 Hz, J = 4.0 Hz, 0.5H), 5.41-5.37 (m, J = 4.0 Hz, J = 4.4 Hz, 0.5H), 4.69-4.68 (m, 0.5H), 4.58-4.52 (m, 1H),4.49-4.40 (m, 1.5), 4.22-4.12 (m, 0.5H), 4.04-3.88 (m, 2H), 3.71-3.64 (m, 0.5), 3.36-3.28 (m, 0.5H), 2.98-2.90 (m, 1H), 2.83-2.79 (m, 0.5H), 1.36-1.34 (m, 6H), 1.32-1.29 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200.1, 198.7, 157.6, 156.9, 152.7, 152.4, 152.3, 152.2, 144.8, 144.5, 135.9, 134.7, 134.6, 129.9, 128.6, 128.4, 128.3, 127.9, 124.7, 123.3, 117.5, 117.3, 115.8, 115.6, 115.4, 115.3, 115.1, 114.9, 70.9, 70.6, 69.9, 61.3, 58.2, 47.9, 42.5, 29.8, 29.7, 27.8, 22.1, 22.0; HRMS calcd. for C29H33ClNO3S, 510.18642 [M + H]+; 510.18687 [M + H]+ found. Anal. (C29H32N1O3SCl): C, H, N.
(3-Fluorophenyl)(6-isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanone (146)
Tetrahydroisoquinoline 146 was prepared via procedure XVI using tetrahydroisoquinoline 116 (0.043 g, 0.090 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.073 g, 0.18 mmol) in toluene (0.902 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.030 g, 67 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:3, v/v) Rf = 0.89; 1HNMR (CDCl3, 400 MHz) δ: 7.38-7.32 (m, 0.8H), 7.25 (s, 0.2H), 7.07-6.97 (m, 2H), 6.88-6.80 (m, 5H), 6.78-6.72 (m, 2.5H), 6.70-6.69 (m, 0.5H), 5.75-5.70 (m, J = 4.8 Hz, J = 5.2 Hz, 0.5H), 5.42-5.38 (m, J = 4.0 Hz, J = 4.4 Hz, 0.5H), 4.71-4.67 (m, 0.5H), 4.58-4.52 (m, 1H), 4.49-4.39 (m, 2H), 4.19 (t, J = 10 Hz, 0.5H), 5.06-3.87 (m, 1.5H), 3.72-3.64 (m, 0.5H), 3.36-3.27 (m, 0.5H), 2.99-2.91 (m, 1H), 2.83-2.78 (m, 0.5H), 1.36-134 (m, 6H), 1.32-1.30 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200.4, 199.0, 162.6 (J = 247 Hz), 157.8, 157.0, 152.9, 152.6, 152.5, 152.3, 145.3, 145.2, 136.1, 134.9, 130.6, 130.5, 128.4, 128.0, 124.9, 123.5, 117.6, 117.5, 115.9, 115.8, 115.7, 115.6, 115.5, 115.4, 115.3, 115.0, 114.6, 71.1, 71.0, 70.7, 70.2, 70.1, 70.0, 61.4, 58.3, 48.0, 42.7, 30.0, 28.0, 22.3, 22.2; HRMS calcd. for C29H33FNSO3, 429.21597 [M + H]+; found 429.21623 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 85% ACN/25% water over 3 mins at 1 mL/min (retention time = 2.73 minutes). Method 2: 75% MeOH/25% Water to 95% MeOH/5% water over 6 mins at 1 mL/min (retention time 4.74 minutes).
(6-Isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanone (147)
Tetrahydroisoquinoline 147 was prepared via procedure XIII using tetrahydroisoquinoline 70 (0.80 g, 0.23 mmol) and 3-(trifluoromethyl)benzoyl chloride chloride (0.041 mL, 0.27 mmol) in dry DCM (3.5 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.090 g, 77 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:3, v/v) Rf = 0.83; 1HNMR (CDCl3, 400 MHz) δ: 7.91 (s, 0.5H), 7.69-7.49 (m, 3H), 7.25-7.22 (m, 0.5H), 6.92-6.69 (m, 8H), 5.99-6.00 (m, 0.5H), 5.10-5.08 (m, 0.5H), 4.90-4.86 (m, 0.5H), 4.59-4.48 (m, 1H), 4.47-4.38 (m, 2H), 4.18-4.12 (m, 0.5H), 3.95-3.92 (m, 0.5H), 3.73-3.71 (m, 1H), 3.33-3.12 (m, 1H), 2.94-2.74 (m, 1.5H), 1.36-1.29 (m, 12H); 13CNMR (100 MHz, CDCl3) δ: 170.5, 169.7, 157.6, 157.1, 153.1, 152.6, 152.5, 137.5, 137.3, 136.6, 135.5, 131.2, 130.2, 129.5, 129.3, 129.1, 128.7, 128.3, 126.5, 125.2, 123.9, 117.7, 117.6, 116.2, 115.9, 115.4, 114.8, 114.5, 71.3, 71.1, 70.1, 57.5, 53.7, 52., 42.9, 35.5, 29.9, 28.5, 22.3, 22.2; HRMS calcd. for C30H33F3N1O4, 528.23562 [M + H]+; found 528.23530 [M + H]+. Anal. (C30H32N1O4F3): C, H, N.
(6-Isopropoxy-1-((4-isopropoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)(3-(trifluoromethyl)phenyl)methanethione (148)
Tetrahydroisoquinoline 148 was prepared via procedure XVI using tetrahydroisoquinoline 147 (0.043 g, 0.093 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.075 g, 0.18 mmol in toluene (0.93 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 10 – 80% EtOAc/hexanes gradient) to afford the title compound as an off-white foam (0.035 g, 69 % mixture of two amide rotamers) TLC (EtOAc: hexanes, 1:3, v/v) Rf = 0.62; 1HNMR (CDCl3, 400 MHz) δ: 7.63-7.44 (m, 3H), 6.88-6.80 (m, 5H), 6.78-6.70 (m, 3H), 5.73 (bs, 0.5H), 5.36-5.33 (m, J = 4.0 Hz, J = 4.4 Hz, 0.5H), 4.73-4.58 (m, 0.5H), 5.57-4.40 (m, 3H), 4.19 (t, J = 10 Hz, 0.5H), 3.99-3.89 (m, 1.5H), 3.73-3.66 (m, 0.5H), 3.38-3.30 (m, 0.5H), 2.97-2.91 (m, 1H), 2.85-2.79 (m, 0.5H), 1.36-1.34 (m, 6H), 1.32-1.30 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200.1, 198.7, 157.7, 156.9, 152.7, 152.5, 152.3, 152.1, 143.9, 143.6, 134.6, 129.2, 128.8, 128.3, 127.8, 125.2, 125.1, 125.1, 124.6, 123.1, 117.4, 117.3, 115.9, 115.8, 115.6, 115.4, 114.1, 114., 115.6, 114.4, 70.8, 70.6, 70.0, 69.9, 69.8, 61.3, 58.3, 47.9, 29.8, 27.8, 22.0, 21.9; HRMS calcd. for C30H33F3N1O3S, 544.21278 [M + H]+; found 544.21250 [M + H]+. Purity was established using an Agilent pump on a Zorbax XBD-C18 column (46 mm × 50 mm, 3.5 μm). Method 1: 75% MeOH/25% Water to 95% MeOH/5% water over 5 mins at 1 mL/min (retention time 4.04 minutes).
Separation of 2 enantiomers
Semi-preparative separation of 2 enantiomers from racemic 2 (0.020 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 8 mL injection volume (2 mg / 1 mL), 65% hexanes / 35% IPA over 60 minutes to afford S-(−)-2: tR: 26.8; R-(+)-2: tR: 40.3 minutes. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 65% hexanes / 35% IPA; S-(−)-2: tR: 14.5 minutes, 100% ee, [α]D20 = −70 (c 0.10, dry CHCl3), R-(+)-2: tR: 21.4 minutes, 100% ee, [α]D20 = +70 (c 0.10, dry CHCl3). The proton spectrum for each enantiomer was identical to that of the racemic mixture.
Separation of 97 enantiomers
Semi-preparative separation of 97 enantiomers from racemic 97 (0.14 g) was done using a ChiralPak AD-H (30 mm × 250 mm) with the following conditions: 20 mL/min flow rate, 8 mL injection volume (2 mg / 1 mL), 70% hexanes / 30% IPA over 45 minutes to afford S-(−)-97 (0.055 g): tR: 25.2; R-(+)-97 (0.056 g): tR: 46.1 minutes. The enantiomeric excess (ee) was determined using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 1 mL/min flow rate, 10 μL injection volume, 80% hexanes / 20% IPA; S-(−)-97: tR: 18.1 minutes, 100% ee, [α]D20 = −102 (c 0.1, dry CHCl3), R-(+)-97: tR: 33.4 minutes, 100% ee, [α]D20 = +103 (c 1.0, dry CHCl3). The proton spectrum for each enantiomer was identical to that of the racemic mixture.
(S)-(3-Chlorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanethione (S-138)
Tetrahydroisoquinoline S-138 was prepared via procedure XV using tetrahydroisoquinoline S-2 (0.033 g, 0.071 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.029 g, 0.071 mmol) in toluene (3.5 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 4 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.022 g, 65%, a mixture of rotamers) TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.80; 1H NMR (CDCl3, 300 MHz): 7.33-7.25 (m, 3.5H), 7.14-7.13 (m, 0.5H), 6.86-6.70 (m, 7H), 5.75-5.70 (m, J = 5.2 Hz, J = 13.2 Hz, 0.5H), 5.41-5.38 (m, J = 4.0 Hz, J = 9.6 Hz, 0.5H), 4.70-4.67 (double of doublets, J = 4.4 Hz, J = 10.4 Hz, 0.5H), 4.58-4.52 (m, 1H), 4.50-4.46 (doublet of doublet, J = 4.8 Hz, J = 10.0 Hz, 0.5H), 4.20-4.17 (m, 0.5H), 4.04-3.87 (m, 2H), 3.77 (s, 3H), 3.71-3.63 (m, 0.5H), 3.36-3.28 (m, 0.5H), 2.99-2.92 (m, 1H), 2.83-2.79 (m, 0.5H), 1.36-1.34 (m, 6H); 13C NMR (100 MHz, CDCl3) δ: 200. 4, 199.0, 157.8, 157.1, 154.5, 154.4, 152.9, 152.4, 145.0, 144.8, 136.2, 134.9, 130.2, 129.7, 128.7, 128.6, 128.5, 128.1, 124.9, 123.5, 115.9, 115.7, 115.5, 115.1, 114.9, 114.8, 114.7, 70.9, 70.1, 70.1, 61.5, 58.4, 55.9, 48.1, 42.7, 30.1, 28.1, 22.3; HRMS calcd. for C27H28ClNO3SNa, 482.15512 [M + H]+; 482.15515 [M + H]+ found; [α]D20 = −142.0 (c 0.10, dry CHCl3); The enantiomeric excess was determined to be 96% ee using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 10% IPA/90% hexanes over 40 min, 1.0 mL per min flow rate, read at 254 nm, tR1 = 24.18 min, tR2 = 32.19.
(R)-(3-Chlorophenyl)(6-isopropoxy-1-((4-methoxyphenoxy)methyl)-3,4-dihydroisoquinolin-2(1H)-yl)methanethione ((R)-138)
Tetrahydroisoquinoline R-138 was prepared via procedure XV using tetrahydroisoquinoline R-2 (0.022 g, 0.048 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.019 g, 0.048 mmol) in toluene (2.7 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 4 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.011 g, 49%, a mixture of rotamers) TLC (EtOAc:hexanes, 1:1, v/v) Rf = 0.79 ; 1H NMR (CDCl3, 300 MHz): 7.33-7.25 (m, 3.5H), 7.14-7.13 (m, 0.5H), 6.86-6.70 (m, 7H), 5.75-5.70 (m, J = 5.2 Hz, J = 13.2 Hz, 0.5H), 5.41-5.38 (m, J = 4.0 Hz, J = 9.6 Hz, 0.5H), 4.70-4.67 (dd, J = 4.4 Hz, J = 10.4 Hz, 0.5H), 4.58-4.52 (m, 1H), 4.50-4.46 (dd, J = 4.8 Hz, J = 10.0 Hz, 0.5H), 4.20-4.17 (m, 0.5H), 4.04-3.87 (m, 2H), 3.77 (s, 3H), 3.71-3.63 (m, 0.5H), 3.36-3.28 (m, 0.5H), 2.99-2.92 (m, 1H), 2.83-2.79 (m, 0.5H), 1.36-1.34 (m, 6H) HRMS calcd. for C27H28ClNO3SNa, 482.15512 [M + H]+; 482.15516 [M + H]+ found; [α]D20 = +114.0 (c 0.10, dry CHCl3); The enantiomeric excess (ee) was determined to be 100% ee using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 10% IPA/90% hexanes over 40 min, 1.0 mL per min flow rate, read at 254 nm, tR1 = 24.18 min, tR2 = 32.19.
S-(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanethione (S-(−)-142)
Tetrahydroisoquinoline S-(−)-142 was prepared via procedure XVI using tetrahydroisoquinoline S-(−)-97 (0.047 g, 0.096 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.039 g, 0.096 mmol) in toluene (3.6 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–80% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.026 g, 56%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:3, v/v) Rf = 0.70; [α]D20 = −121.0 (c 0.10, dry CHCl3); Following recrystallization with DCM/hexanes, the enantiomeric excess was determined to be 100% ee using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 10% IPA/90% hexanes over 100 min, 0.025 mL per min flow rate, read at 254 nm, tR1 = 73.9 min, tR2 = 78.7 min. The proton spectrum was identical to that of racemic 142.
(R)-(1-((4-Ethoxyphenoxy)methyl)-6-isopropoxy-3,4-dihydroisoquinolin-2(1H)-yl)(3-fluorophenyl)methanethione (R-(+)-142)
Tetrahydroisoquinoline R-(+)-142 was prepared via procedure XVI using tetrahydroisoquinoline R-(+)-97 (0.046 g, 0.096 mmol) and 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide (0.040 g, 0.096 mmol) in toluene (3.6 mL). The crude residue was purified by silica gel chromatography (ISCO, Redisep 12 g column, 0–50% EtOAc/hexanes gradient) to afford the title compound as a yellow foam (0.031 g, 65%, mixture of two thioamide rotamers); TLC (EtOAc:hexanes, 1:3, v/v) Rf = 0.70; [α]D20 = +129.0 (c 0.10, dry CHCl3); Following recrystallization with DCM/hexanes, the enantiomeric excess was determined to be 100% ee using an Agilent pump on a ChiralPak AD-H column (4.6 mm × 150 mm, 5 μm) with the following conditions: 10% IPA/90% hexanes over 100 min, 0.025 mL per min flow rate, read at 254 nm, tR1 = 73.9 min, tR2 = 78.7 min. The proton spectrum was identical to that of racemic 142.
Supplementary Material
Acknowledgments
This project was partially supported by the ELSI Origins Network (EON), which is supported by a grant from the John Templeton Foundation, and NIH-NINDS (NS036654, NS065371 SFT). The authors thank the Emory Institute for Drug Development and George Painter, Jose Marengo, and Richard Arrendale for performing the studies of metabolic stability in liver microsomes. The authors also thank Janssen Research & Development, LLC, Brian Scott, Tatiana Koudriakova, and Tim Lovenberg for their support and help with pharmacokinetic analysis. The authors also thank Phuong Le and Jing Zhang for excellent technical assistance. The opinions expressed in this publication are those of the author(s) and do not necessarily reflect the views of the NIH or John Templeton Foundation.
Abbreviations
- ABD
agonist binding domain
- ATD
amino terminal domain
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- CTD
carboxy terminal domain
- CNS
central nervous system
- FDA
Food and Drug Administration
- HEK
human embryonic kidney
- NMDA
N-methyl-D-aspartic acid
- NMDAR
NMDA receptor
- OCD
obsessive compulsive disorder
- PK
pharmacokinetics
- PTSD
post-traumatic stress disorder
- SEM
standard error of the mean
- SAR
structure activity relationship
- TMD
transmembrane domain
Footnotes
Supporting Information Available: Chemistry experimental procedures are available along with crystal structure experimental information and computational data.
References
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate Receptor Ion Channels: Structure, Regulation, and Function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic Enhancement of Learning and Memory in Mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- 3.Collingridge GL, Volianskis A, Bannister N, France G, Hanna L, Mercier M, Tidball P, Fang G, Irvine MW, Costa BM, Monaghan DT, Bortolotto ZA, Molnár E, Lodge D, Jane DE. The NMDA Receptor as a Target for Cognitive Enhancement. Neuropharmacology. 2013;64:13–26. doi: 10.1016/j.neuropharm.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okabe S, Collin C, Auerbach JM, Meiri N, Bengzon J, Kennedy MB, Segal M, McKay RDG. Hippocampal Synaptic Plasticity in Mice Overexpressing an Embryonic Subunit of the NMDA Receptor. J Neurosci. 1998;18:4177–4188. doi: 10.1523/JNEUROSCI.18-11-04177.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bliss TVP, Collingridge GL. A Synaptic Model of Memory: Long-Term Potentiation in the Hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 6.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium Gates Glutamate-Activated Channels in Mouse Central Neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 7.Johnson JW, Ascher P. Glycine Potentiates the NMDA Response in Cultured Mouse Brain Neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 8.Mayer ML, Westbrook GL, Guthrie PB. Voltage-Dependent Block by Mg2+ of NMDA Responses in Spinal Cord Neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 9.MacDermott AB, Mayer ML, Westbrook GL, Smith SJ, Barker JL. NMDA-Receptor Activation Increases Cytoplasmic Calcium Concentration in Cultured Spinal Cord Neurones. Nature. 1986;321:519–522. doi: 10.1038/321519a0. [DOI] [PubMed] [Google Scholar]
- 10.Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, Yuan H, Le P, Lyuboslavsky PN, Micale N, Jørgensen L, Clausen RP, Wyllie DJA, Snyder JP, Traynelis SF. Subunit-Specific Agonist Activity at NR2A-, NR2B-, NR2C-, and NR2D-Containing N-Methyl-D-Aspartate Glutamate Receptors. Mol Pharmacol. 2007;72:907–920. doi: 10.1124/mol.107.037333. [DOI] [PubMed] [Google Scholar]
- 11.Erreger K, Dravid SM, Banke TG, Wyllie DJA, Traynelis SF. Subunit-Specific Gating Controls Rat NR1/NR2A and NR1/NR2B NMDA Channel Kinetics and Synaptic Signalling Profiles. J Physiol. 2005;563:345–358. doi: 10.1113/jphysiol.2004.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dravid SM, Prakash A, Traynelis SF. Activation of Recombinant NR1/NR2C NMDA Receptors. J Physiol. 2008;586:4425–4439. doi: 10.1113/jphysiol.2008.158634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wyllie DJA, Béhé P, Colquhoun D. Single-Channel Activations and Concentration Jumps: Comparison of Recombinant NR1a/NR2A and NR1a/NR2D NMDA Receptors. J Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vance KM, Hansen KB, Traynelis SF. GluN1 Splice Variant Control of GluN1/GluN2D NMDA Receptors. J Physiol. 2012;590:3857–3875. doi: 10.1113/jphysiol.2012.234062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and Pharmacological Differences Between RecombinantN-Methyl-D-Aspartate Receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 16.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and Regional Expression in the Rat Brain and Functional Properties of Four NMDA Receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 17.Akazawa C, Shigemoto R, Bessho Y, Nakanishi S, Mizuno N. Differential Expression of Five N-Methyl-D-Aspartate Receptor Subunit mRNAs in the Cerebellum of Developing and Adult Rats. J Comp Neurol. 1994;347:150–160. doi: 10.1002/cne.903470112. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe M, Inoue Y, Sakimura K, Mishina M. Developmental Changes in Distribution of NMDA Receptor Channel Subunit mRNAs. NeuroReport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- 19.Karakas E, Furukawa H. Crystal Structure of a Heterotetrameric NMDA Receptor Ion Channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee CH, Lü W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA Receptor Structures Reveal Subunit Arrangement and Pore Architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ. Memantine in Moderate-to-Severe Alzheimer’s Disease. N Engl J Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 22.Choi DW, Rothman SM. The Role of Glutamate Neurotoxicity in Hypoxic-Ischemic Neuronal Death. Annu Rev Neurosci. 1990;13:171–182. doi: 10.1146/annurev.ne.13.030190.001131. [DOI] [PubMed] [Google Scholar]
- 23.Yuan H, Myers SJ, Wells G, Nicholson KL, Swanger SA, Lyuboslavsky P, Tahirovic YA, Menaldino DS, Ganesh T, Wilson LJ, Liotta DC, Snyder JP, Traynelis SF. Context-Dependent GluN2B-Selective Inhibitors of NMDA Receptor Function Are Neuroprotective with Minimal Side Effects. Neuron. 2015;85:1305–1318. doi: 10.1016/j.neuron.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant Effects of Ketamine in Depressed Patients. Biol Psychiatry. 2000;47(4):351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 25.Hallett PJ, Standaert DG. Rationale for and Use of NMDA Receptor Antagonists in Parkinson’s Disease. Pharmacol Ther. 2004;102:155–174. doi: 10.1016/j.pharmthera.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 26.Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Mol Pharmacol. 2015;88:203–217. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coyle JT. NMDA Receptor and Schizophrenia: A Brief History. Schizophr Bull. 2012;38(5):920–926. doi: 10.1093/schbul/sbs076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olney JW, Newcomer JW, Farber NB. NMDA Receptor Hypofunction Model of Schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- 29.Kemp JA, McKernan RM. NMDA Receptor Pathways as Drug Targets. Nat Neurosci. 2002;5:1039–1042. doi: 10.1038/nn936. [DOI] [PubMed] [Google Scholar]
- 30.Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC. Novel NMDA Receptor Modulators: An Update. Expert Opin Ther Pat. 2012;22:1337–1352. doi: 10.1517/13543776.2012.728587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strong KL, Jing Y, Prosser AR, Traynelis SF, Liotta DC. NMDA Receptor Modulators: An Updated Patent Review (2013–2014) Expert Opin Ther Pat. 2014;24:1349–1366. doi: 10.1517/13543776.2014.972938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huettner JE, Bean BP. Block of N-Methyl-D-Aspartate-Activated Current by the Anticonvulsant MK-801: Selective Binding to Open Channels. Proc Natl Acad Sci. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sobolevsky AI, Koshelev SG, Khodorov BI. Interaction of Memantine and Amantadine with Agonist-Unbound NMDA-Receptor Channels in Acutely Isolated Rat Hippocampal Neurons. J Physiol. 1998;512:47–60. doi: 10.1111/j.1469-7793.1998.047bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen HSV, Lipton SA. The Chemical Biology of Clinically Tolerated NMDA Receptor Antagonists. J Neurochem. 2006;97:1611–1626. doi: 10.1111/j.1471-4159.2006.03991.x. [DOI] [PubMed] [Google Scholar]
- 35.Taylor CP, Traynelis SF, Siffert J, Pope LE, Matsumoto RR. Pharmacology of Dextromethorphan: Relevance to Dextromethorphan/Quinidine (Nuedexta®) Clinical Use. Pharmacol Ther. 2016;164:170–182. doi: 10.1016/j.pharmthera.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 36.Hasselmann H. Ketamine as Antidepressant? Current State and Future Perspectives. Curr Neuropharmacol. 2014;12:57–70. doi: 10.2174/1570159X113119990043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez CI, Kegeles LS, Levinson A, Feng T, Marcus SM, Vermes D, Flood P, Simpson HB. Randomized Controlled Crossover Trial of Ketamine in Obsessive-Compulsive Disorder: Proof-of-Concept. Neuropsychopharmacology. 2013;38:2475–2483. doi: 10.1038/npp.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feder A, Parides MK, Murrough JW, Perez AM, Morgan JE, Saxena S, Kirkwood K, Aan Het Rot M, Lapidus KA, Wan LB, Iosifescu D, Charney DS. Efficacy of Intravenous Ketamine for Treatment of Chronic Posttraumatic Stress Disorder: A Randomized Clinical Trial. JAMA Psychiatry. 2014;71:681–688. doi: 10.1001/jamapsychiatry.2014.62. [DOI] [PubMed] [Google Scholar]
- 39.Lipton SA. Failures and Successes of NMDA Receptor Antagonists: Molecular Basis for the Use of Open-Channel Blockers like Memantine in the Treatment of Acute and Chronic Neurologic Insults. NeuroRx. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams K. Ifenprodil Discriminates Subtypes of the N-Methyl-D-Aspartate Receptor: Selectivity and Mechanisms at Recombinant Heteromeric Receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 41.Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De Costa DL, Ducat MF, Dumont ML, Fox CB. (1S, 2S)-1-(4-Hydroxyphenyl)-2-(4-Hydroxy-4-Phenylpiperidino)-1-Propanol: A Potent New Neuroprotectant Which Blocks N-Methyl-D-Aspartate Responses. J Med Chem. 1995;38:3138–3145. doi: 10.1021/jm00016a017. [DOI] [PubMed] [Google Scholar]
- 42.Gotti B, Duverger D, Bertin J, Carter C, Dupont R, Frost J, Gaudilliere B, MacKenzie ET, Rousseau J, Scatton B. Ifenprodil and SL 82.0715 as Cerebral Anti-Ischemic Agents. I. Evidence for Efficacy in Models of Focal Cerebral Ischemia. J Pharmacol Exp Ther. 1988;247:1211–1221. [PubMed] [Google Scholar]
- 43.Taniguchi K, Shinjo K, Mizutani M, Shimada K, Ishikawa T, Menniti FS, Nagahisa A. Antinociceptive Activity of CP-101,606, an NMDA Receptor NR2B Subunit Antagonist. Br J Pharmacol. 1997;122:809–812. doi: 10.1038/sj.bjp.0701445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chizh BA, Headley PM, Tzschentke TM. NMDA Receptor Antagonists as Analgesics: Focus on the NR2B Subtype. Trends Pharmacol Sci. 2001;22:636–642. doi: 10.1016/s0165-6147(00)01863-0. [DOI] [PubMed] [Google Scholar]
- 45.Boyce S, Wyatt A, Webb JK, O’Donnell R, Mason G, Rigby M, Sirinathsinghji D, Hill RG, Rupniak NMJ. Selective NMDA NR2B Antagonists Induce Antinociception without Motor Dysfunction: Correlation with Restricted Localisation of NR2B Subunit in Dorsal Horn. Neuropharmacology. 1999;38:611–623. doi: 10.1016/s0028-3908(98)00218-4. [DOI] [PubMed] [Google Scholar]
- 46.Bettini E, Sava A, Griffante C, Carignani C, Buson A, Capelli AM, Negri M, Andreetta F, Senar-Sancho SA, Guiral L, Cardullo F. Identification and Characterization of Novel NMDA Receptor Antagonists Selective for NR2A- over NR2B-Containing Receptors. J Pharmacol Exp Ther. 2010;335:636–644. doi: 10.1124/jpet.110.172544. [DOI] [PubMed] [Google Scholar]
- 47.Edman S, McKay S, MacDonald LJ, Samadi M, Livesey MR, Hardingham GE, Wyllie DJA. TCN 201 Selectively Blocks GluN2A-Containing NMDARs in a GluN1 Co-Agonist Dependent but Non-Competitive Manner. Neuropharmacology. 2012;63:441–449. doi: 10.1016/j.neuropharm.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hansen KB, Ogden KK, Traynelis SF. Subunit-Selective Allosteric Inhibition of Glycine Binding to NMDA Receptors. J Neurosci Off J Soc Neurosci. 2012;32:6197–6208. doi: 10.1523/JNEUROSCI.5757-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Volkmann RA, Fanger CM, Anderson DR, Sirivolu VR, Paschetto K, Gordon E, Virginio C, Gleyzes M, Buisson B, Steidl E, Mierau SB, Fagiolini M, Menniti FS. MPX-004 and MPX-007: New Pharmacological Tools to Study the Physiology of NMDA Receptors Containing the GluN2A Subunit. PLOS ONE. 2016;11:e0148129. doi: 10.1371/journal.pone.0148129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, Traynelis SF. Mechanism for Noncompetitive Inhibition by Novel GluN2C/D N-Methyl-D-Aspartate Receptor Subunit-Selective Modulators. Mol Pharmacol. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Bräuner-Osborne H, Liotta DC, Traynelis SF. Quinazolin-4-One Derivatives: A Novel Class of Noncompetitive NR2C/D Subunit-Selective N-Methyl-D-Aspartate Receptor Antagonists. J Med Chem. 2010;53:5476–5490. doi: 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hansen KB, Traynelis SF. Structural and Mechanistic Determinants of a Novel Site for Noncompetitive Inhibition of GluN2D-Containing NMDA Receptors. J Neurosci. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Acker TM, Khatri A, Vance KM, Slabber C, Bacsa J, Snyder JP, Traynelis SF, Liotta DC. Structure Activity Relationships and Pharmacophore Model of a Noncompetitive Pyrazoline Containing Class of GluN2C/GluN2D Selective Antagonists. J Med Chem. 2013;56:6434–6456. doi: 10.1021/jm400652r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Skifter DA, Jane DE, Monaghan DT. A Novel Family of Negative and Positive Allosteric Modulators of NMDA Receptors. J Pharmacol Exp Ther. 2010;335:614–621. doi: 10.1124/jpet.110.174144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monaghan DT, Irvine MW, Costa BM, Fang G, Jane DE. Pharmacological Modulation of NMDA Receptor Activity and the Advent of Negative and Positive Allosteric Modulators. Neurochem Int. 2012;61:581–592. doi: 10.1016/j.neuint.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costa BM, Irvine MW, Fang G, Eaves RJ, Mayo-Martin MB, Laube B, Jane DE, Monaghan DT. Structure-Activity Relationships for Allosteric NMDA Receptor Inhibitors Based on 2-Naphthoic Acid. Neuropharmacology. 2012;62:1730–1736. doi: 10.1016/j.neuropharm.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Irvine MW, Costa BM, Volianskis A, Fang G, Ceolin L, Collingridge GL, Monaghan DT, Jane DE. Coumarin-3-Carboxylic Acid Derivatives as Potentiators and Inhibitors of Recombinant and Native N-Methyl-D-Aspartate Receptors. Neurochem Int. 2012;61:593–600. doi: 10.1016/j.neuint.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swanger SA, Vance KM, Pare JF, Sotty F, Fog K, Smith Y, Traynelis SF. NMDA Receptors Containing the GluN2D Subunit Control Neuronal Function in the Subthalamic Nucleus. J Neurosci. 2015;35:15971–15983. doi: 10.1523/JNEUROSCI.1702-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perszyk RE, DiRaddo JO, Strong KL, Low CM, Ogden KK, Khatri A, Vargish GA, Pelkey KA, Tricoire L, Liotta DC, Smith Y, McBain CJ, Traynelis SF. GluN2D-Containing NMDA Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Mol Pharmacol. 2016;90:689–702. doi: 10.1124/mol.116.105130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF. A Subunit-Selective Potentiator of NR2C- and NR2D-Containing NMDA Receptors. Nat Commun. 2010;1:90. doi: 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santangelo Freel RM, Ogden KK, Strong KL, Khatri A, Chepiga KM, Jensen HS, Traynelis SF, Liotta DC. Synthesis and Structure Activity Relationship of Tetrahydroisoquinoline-Based Potentiators of GluN2C and GluN2D Containing N-Methyl-D-Aspartate Receptors. J Med Chem. 2013;56:5351–5381. doi: 10.1021/jm400177t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zimmerman SS, Khatri A, Garnier-Amblard EC, Mullasseril P, Kurtkaya NL, Gyoneva S, Hansen KB, Traynelis SF, Liotta DC. Design, Synthesis, and Structure–Activity Relationship of a Novel Series of GluN2C-Selective Potentiators. J Med Chem. 2014;57:2334–2356. doi: 10.1021/jm401695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Khatri A, Burger PB, Swanger SA, Hansen KB, Zimmerman S, Karakas E, Liotta DC, Furukawa H, Snyder JP, Traynelis SF. Structural Determinants and Mechanism of Action of a GluN2C-Selective NMDA Receptor Positive Allosteric Modulator. Mol Pharmacol. 2014;86:548–560. doi: 10.1124/mol.114.094516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hackos DH, Lupardus PJ, Grand T, Chen Y, Wang TM, Reynen P, Gustafson A, Wallweber HJA, Volgraf M, Sellers BD, Schwarz JB, Paoletti P, Sheng M, Zhou Q, Hanson JE. Positive Allosteric Modulators of GluN2A-Containing NMDARs with Distinct Modes of Action and Impacts on Circuit Function. Neuron. 2016;89:983–999. doi: 10.1016/j.neuron.2016.01.016. [DOI] [PubMed] [Google Scholar]
- 65.Volgraf M, Sellers BD, Jiang Y, Wu G, Ly CQ, Villemure E, Pastor RM, Yuen P, Lu A, Luo X, Liu M, Zhang S, Sun L, Fu Y, Lupardus PJ, Wallweber HJA, Liederer BM, Deshmukh G, Plise E, Tay S, Reynen P, Herrington J, Gustafson A, Liu Y, Dirksen A, Dietz MGA, Liu Y, Wang TM, Hanson JE, Hackos D, Scearce-Levie K, Schwarz JB. Discovery of GluN2A-Selective NMDA Receptor Positive Allosteric Modulators (PAMs): Tuning Deactivation Kinetics via Structure-Based Design. J Med Chem. 2016;59:2760–2779. doi: 10.1021/acs.jmedchem.5b02010. [DOI] [PubMed] [Google Scholar]
- 66.Williams K, Zappia AM, Pritchett DB, Shen YM, Molinoff PB. Sensitivity of the N-Methyl-D-Aspartate Receptor to Polyamines Is Controlled by NR2 Subunits. Mol Pharmacol. 1994;45:803–809. [PubMed] [Google Scholar]
- 67.Traynelis SF, Hartley M, Heinemann SF. Control of Proton Sensitivity of the NMDA Receptor by RNA Splicing and Polyamines. Science. 1995;268:873–876. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- 68.Masuko T, Kuno T, Kashiwagi K, Kusama T, Williams K, Igarashi K. Stimulatory and Inhibitory Properties of Aminoglycoside Antibiotics at N-Methyl-D-Aspartate Receptors. J Pharmacol Exp Ther. 1999;290:1026–1033. [PubMed] [Google Scholar]
- 69.Malayev A, Gibbs TT, Farb DH. Inhibition of the NMDA Response by Pregnenolone Sulphate Reveals Subtype Selective Modulation of NMDA Receptors by Sulphated Steroids. Br J Pharmacol. 2002;135:901–909. doi: 10.1038/sj.bjp.0704543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao X, Cui Z, Feng R, Tang YP, Qin Z, Mei B, Tsien JZ. Maintenance of Superior Learning and Memory Function in NR2B Transgenic Mice during Ageing. Eur J Neurosci. 2007;25:1815–1822. doi: 10.1111/j.1460-9568.2007.05431.x. [DOI] [PubMed] [Google Scholar]
- 71.Jacobs SA, Tsien JZ. Genetic Overexpression of NR2B Subunit Enhances Social Recognition Memory for Different Strains and Species. PLoS ONE. 2012;7:e36387. doi: 10.1371/journal.pone.0036387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cui Yihui, Jin Jing, Zhang Xuliang, Xu Hao, Yang Liguo, Du Dan, Zeng Qingwen, Tsien Joe Z, Yu Huiting, Cao Xiaohua. Forebrain NR2B Overexpression Facilitating the Prefrontal Cortex Long-Term Potentiation and Enhancing Working Memory Function in Mice. PLoS ONE. 2011;6:1–10. doi: 10.1371/journal.pone.0020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.White TL, Youngentob SL. The Effect of NMDA-NR2B Receptor Subunit over-Expression on Olfactory Memory Task Performance in the Mouse. Brain Res. 2004;1021:1–7. doi: 10.1016/j.brainres.2004.05.114. [DOI] [PubMed] [Google Scholar]
- 74.Wolter M, Nordmann G, Job GE, Buchwald SL. Copper-Catalyzed Coupling of Aryl Iodides with Aliphatic Alcohols. Org Lett. 2002;4:973–976. doi: 10.1021/ol025548k. [DOI] [PubMed] [Google Scholar]
- 75.Klapars A, Buchwald SL. Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J Am Chem Soc. 2002;124:14844–14845. doi: 10.1021/ja028865v. [DOI] [PubMed] [Google Scholar]
- 76.Ma D, Cai Q, Zhang H. Mild Method for Ullmann Coupling Reaction of Amines and Aryl Halides. Org Lett. 2003;5:2453–2455. doi: 10.1021/ol0346584. [DOI] [PubMed] [Google Scholar]
- 77.Xu H, Wolf C. Efficient Copper-Catalyzed Coupling of Aryl Chlorides, Bromides and Iodides with Aqueous Ammonia. Chem Commun. 2009;21:3035–3037. doi: 10.1039/b904188e. [DOI] [PubMed] [Google Scholar]
- 78.Ono M, Watanabe R, Kawashima H, Kawai T, Watanabe H, Haratake M, Saji H, Nakayama M. 18F- Labeled Flavones for in Vivo Imaging of β-Amyloid Plaques in Alzheimer’s Brains. Bioorg Med Chem. 2009;17:2069–2076. doi: 10.1016/j.bmc.2009.01.025. [DOI] [PubMed] [Google Scholar]
- 79.Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The Majority of N-Methyl-D-Aspartate Receptor Complexes in Adult Rat Cerebral Cortex Contain at Least Three Different Subunits (NR1/NR2A/NR2B) Mol Pharmacol. 1997;51:79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- 80.Rauner C, Köhr G. Triheteromeric NR1/NR2A/NR2B Receptors Constitute the Major N-Methyl-D-Aspartate Receptor Population in Adult Hippocampal Synapses. J Biol Chem. 2011;286:7558–7566. doi: 10.1074/jbc.M110.182600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing Subunit Composition of Heteromeric NMDA Receptors during Development of Rat Cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 82.Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA Receptors at Hippocampal Synapses. J Neurosci. 2013;33(21):9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogden KK, Khatri A, Traynelis SF, Heldt SA. Potentiation of GluN2C/D NMDA Receptor Subtypes in the Amygdala Facilitates the Retention of Fear and Extinction Learning in Mice. Neuropsychopharmacology. 2014;39:625–637. doi: 10.1038/npp.2013.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Santangelo Freel RM, Ogden KK, Strong KL, Khatri A, Chepiga KM, Jensen HS, Traynelis SF, Liotta DC. Correction to Synthesis and Structure Activity Relationship of Tetrahydroisoquinoline-Based Potentiators of GluN2C and GluN2D Containing N-Methyl-D-Aspartate Receptors. J Med Chem. 2014;57:4975–4975. doi: 10.1021/jm500710w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dravid SM, Erreger K, Yuan H, Nicholson K, Le P, Lyuboslavsky P, Almonte A, Murray E, Mosley C, Barber J, French A, Balster R, Murray TF, Traynelis SF. Subunit-Specific Mechanisms and Proton Sensitivity of NMDA Receptor Channel Block. J Physiol. 2007;581:107–128. doi: 10.1113/jphysiol.2006.124958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct Functional and Pharmacological Properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA Receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.