

Abstract
Disclosed is a mild, scalable, visible-light-promoted cross-coupling reaction between thiols and aryl halides for the construction of C–S bonds in the absence of both transition metal and photoredox catalysts. The scope of aryl halides and thiol partners includes over 60 examples and therefore provides an entry point into various aryl thioether building blocks of pharmaceutical interest. Furthermore, to demonstrate its utility, this C–S coupling protocol was applied in drug synthesis and late-stage modifications of active pharmaceutical ingredients. UV–vis spectroscopy and time-dependent density functional theory calculations suggest that visible-light-promoted intermolecular charge transfer within the thiolate–aryl halide electron donor–acceptor complex permits the reactivity in the absence of catalyst.
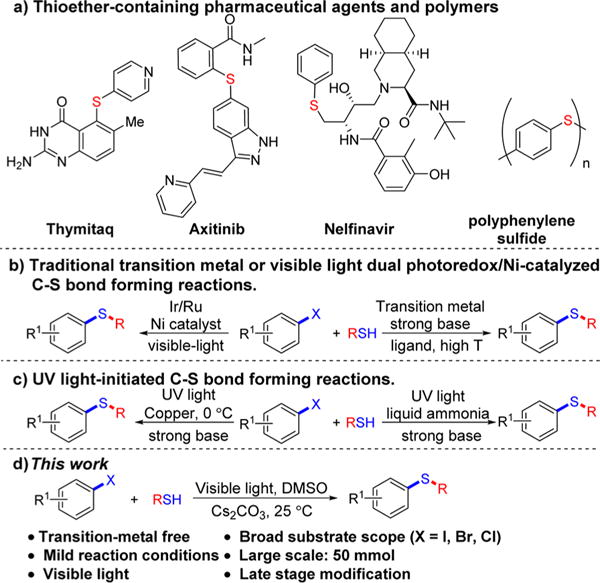
Aromatic thioethers are prevalent in a wide range of bioactive natural products and pharmaceuticals, including thymitaq, axitinib, and nelfinavir (Scheme 1a).1a Furthermore, aromatic thioethers are also valuable architectures in drug development,1b,c organic materials,1d and polymers.1e Therefore, the development of environmentally friendly and atom-economical methods for constructing C–S bonds is of significant importance with broad impact across the areas of small-molecule synthesis and materials.2
Scheme 1.
Importance of C–S Bonds and Approaches for Their Formation
Traditionally, transition metal catalysts are employed to catalyze cross-coupling reactions of thiols with aryl halides, providing a useful approach for C–S bond construction (Scheme 1b).3 However, most of the reported methods require a strong base (e.g., t-BuONa), specific or air-sensitive ligands, and high temperature. Such concerns motivate efforts to develop alternative approaches for C–S bond formation.
Recently, photoredox catalysis has become a powerful strategy for the development of a wide range of reactions under mild conditions, including C–S bond formation (Scheme 1b).4 In 2013, Noël and co-workers reported a one-pot Stadler–Ziegler process to form C–S bonds by employing Ru(bpy)3Cl2·6H2O as a photoredox catalyst.5 Dual photoredox/Ni-catalyzed cross-coupling of thiols with (hetero)aryl halides was then developed in 2016.6 More recently, [fac-Ir(ppy)3] was implemented to catalyze the arylation of thiols with aryl halides without the need for a nickel catalyst.7 Nevertheless, ruthenium and iridium photoredox catalysts may introduce limitations in terms of the scalability and sustainability of these processes. In addition, UV-photoinduced8 coupling of thiophenoxide with aryl iodides in liquid ammonia was also reported (Scheme 1c).8a However, the required high-energy UV irradiation can lead to side reactions.8a These side reactions could potentially be minimized by the use of lower-energy visible light, leading to increased functional group tolerance in producing aryl thioethers.
Herein we report the visible-light-induced C–S cross-coupling reactions between (hetero)aryl halides (ArX, where X = I, Br, or Cl) and (hetero)aryl thiols across a broad substrate scope (>60 examples). Notably, these C–S bond formations were carried out at room temperature in the absence of both photoredox catalysts and transition metals typically required to effect cross-coupling reactions (Scheme 1d).
In our early attempts to achieve visible-light-promoted and transition-metal-free conditions for C–S cross-coupling, we applied strongly reducing N,N-diaryldihydrophenazines9a or N-arylphenoxazines9b as organic photoredox catalysts, which we previously developed for use in organocatalyzed atom transfer radical polymerization as well as in small-molecule transformations.9c,d We hypothesized that these organic photoredox catalysts, in the photoexcited state, could directly reduce an aryl halide and generate a radical anion capable of partaking in C–S bond formation.10 Encouragingly, we observed the C–S cross-coupled product in high yield with white light-emitting diode (LED) irradiation of a solution containing 4′-bromoacetophe-none (1a), 4-methylbenzenethiol (2a), Cs2CO3, and the organic photoredox catalyst 5,10-di-1-naphthyl-5,10-dihydrophenazine in dimethyl sulfoxide (DMSO).
However, control experiments revealed that desired product was also isolated in high yield (97%) in the absence of the organic photoredox catalyst after 1 h of white LED irradiation at room temperature (Table S1, entry 1).11 Further studies revealed that white LED irradiation, the presence of base, and the absence of oxygen were each essential for this transformation (entries 2, 5, and 6). These control experiments suggest a radical mechanism involving thiyl and aryl radicals formed as a result of visible-light-promoted intermolecular charge transfer12 that are subsequently quenched to yield the C–S cross-coupled product (vide infra).
The effect of varying the base, base loading, and solvent was also investigated (Table S1). We obtained increasingly higher yields as the Cs2CO3 loading was increased from 0.0 equiv (0%; entry 5) to 1.5 equiv (97%; entry 1). It is noteworthy that K2CO3, which is much less expensive than Cs2CO3, is also an excellent base for this transformation (91%; entry 7), although Na2CO3 gave a much lower yield (24%; entry 8). To eliminate the possibility of trace impurities being responsible for this transformation, we employed other sources of base, including Cs2CO3 (99.995%) and K2CO3 (99.997%), and observed similarly high yields.11 DMSO was determined to be the best solvent compared with other polar aprotic solvents (entries 1 and 9–11).
With the optimized conditions in hand, the scope of this mild, visible-light-promoted, and procedurally simple C–S cross-coupling method was further explored. With respect to aryl thiols, diverse thiophenols (2) served as effective cross-coupling partners with 1a to form C–S bonds (Scheme 2). C–S-coupled products were obtained in good to excellent yields (50–97%) with thiols containing hindered (3c), electron-rich (3c–e, 3h, 3i, 3l), and electron-poor (3f, 3g) functional groups. Alkyl thiols were also successfully coupled (3l). It is worth highlighting that aryl thiol substrates containing free hydroxyl, amine, or carboxyl groups were also tolerated under our C–S coupling conditions (products 3h–j), eliminating the need for protecting groups. To illustrate the robustness and preparative-scale utility of this C–S cross-coupling method, we scaled up the production of 3b to 50 mmol and suffered only a small loss in yield (9.71 g, 80%). In addition, 3b was produced in high yield (86%) under sunlight irradiation, demonstrating the potential for sustainable preparation of aromatic thiothers using solar energy.11
Scheme 2. Scope of Thiolsa.
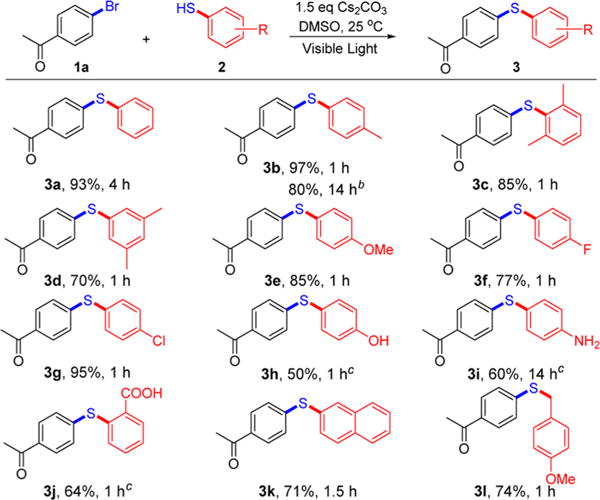
aGeneral conditions: 1a (0.2 mmol), 2 (1.5 equiv), Cs2CO3 (1.5 equiv), DMSO (1.5 mL). Isolated yields are provided. b50 mmol scale. c2.0 equiv of Cs2CO3.
Various aryl iodides, bromides, and chlorides were successfully coupled to thiol nucleophiles (Scheme 3). The electronic effects of the aryl iodides resulted in significant differences in reactivity. For example, both longer irradiation times (e.g., 20–24 h) and the use of electron-rich thiophenols were required to obtain reasonable yields with electron-neutral or -rich aryl iodides (4a– f). Conversely, shorter reaction times and higher yields were obtained with electron-poor aryl iodides (3b, 4g–o). Aryl bromides also resulted in good reactivity (4j, 4l, 4m, 4o–v, 4x). Overall, this C–S cross-coupling method is compatible with aryl halides containing a wide range of functional groups, including carbonyl (3b, 4g, 4h, 4l), formyl (4i), ester (4j), nitryl (4k), cyano (4m, 4n), trifluoromethyl (4o), extended aromatic (4p– u), amide (4v, 4w), and amino groups (4x, 4y).
Scheme 3. Scope of Aryl Chlorides/Bromides/Iodidesa.
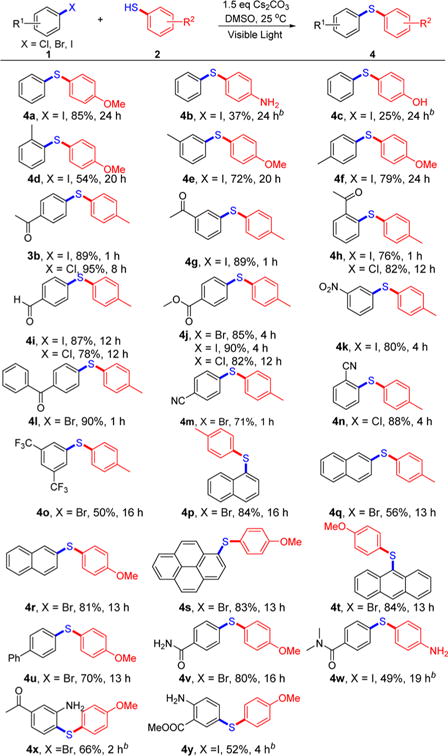
aGeneral conditions: 1 (0.2 mmol), 2 (1.5 equiv), Cs2CO3 (1.5 equiv), DMSO (1.5 mL). Isolated yields are provided. b2.0 equiv of Cs2CO3.
In comparison with aryl iodides and bromides, aromatic chlorides are more attractive for synthetic applications because they are inexpensive and available in great structural diversity.13 However, aromatic chlorides as cross-coupling partners in photoredox-catalyzed thioether formation are less common.7 Here we extended the visible-light-promoted arylation of thiols to aryl chloride substrates. A number of aryl chlorides containing electron-withdrawing groups were successfully coupled to 2a, and the corresponding products (3b, 4h–j, 4n, 5i) were isolated in good to excellent yields.
We next investigated C–S cross-couplings involving various heterocycles, which are ubiquitous among pharmaceutical products (Scheme 4). (Hetero)aryl halides (5i–l), mercaptopyridines (5a, 5b), mercaptopyrimidines (5c–e), 2-mercaptobenzimidazole (5g), 2-mercaptobenzothiazole (5f), and 7-mercapto-4-methylcoumarin (5h) were all effectively coupled in this protocol (70–92% yield).
Scheme 4. Scope of (Hetero)arene Coupling Partnersa.
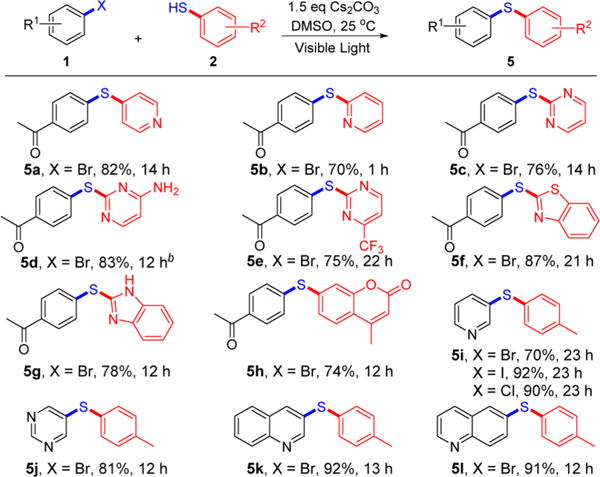
aGeneral conditions: 1 (0.2 mmol), 2 (1.5 equiv), Cs2CO3 (1.5 equiv), DMSO (1.5 mL). Isolated yields are provided. b2.0 equiv of Cs2CO3.
To illustrate potential pharmaceutical applications (Scheme 5), we first applied our visible-light-promoted C–S cross-coupling methodology in the synthesis of the key structure of 11β-HSD1 inhibitors (6, 84%; 7, 93%). Compared with reported methods,14 our transformation involves milder conditions and requires less time. Moreover, we evaluated our method for late-stage functionalization applications. In particular, we subjected indometacin, fenofibrate, moclobemide, and hydrochlorothiazide pharmaceutical ingredients (containing an aryl chloride) to thiophenols under our reaction conditions and obtained the thiolated compounds 8–13 in 50%–79% yield.
Scheme 5.
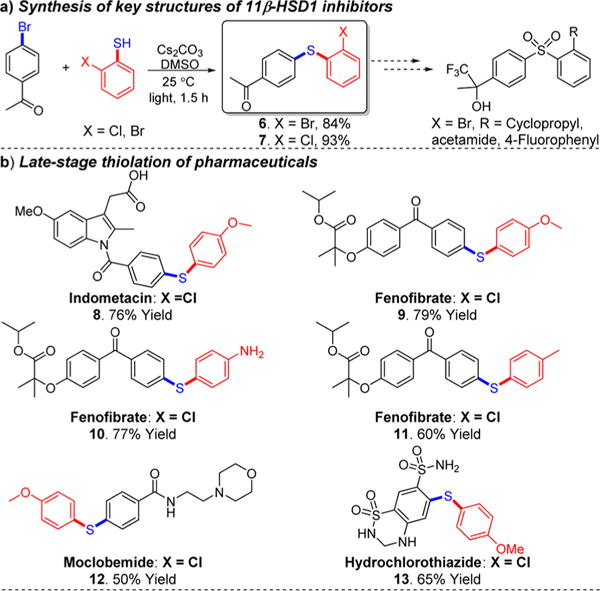
Synthetic Applications of the Visible-Light-Promoted C–S Bond Formation
To gain insight into the C–S cross-coupling mechanism, we performed UV–vis spectroscopic measurements on various combinations of 1a, 2a, and Cs2CO3 in DMSO at 6 × 10−4 M concentration for each species (Figure 1a). A red shift in 2a’s absorption upon Cs2CO3 addition was observed. This shift was attributed to the thiolate anion’s absorption (deprotonated 2a) and is supported by the upfield shift of the NMR signal when 2a and Cs2CO3 were mixed.11 Furthermore, we observed the formation of a new peak (λmax = 306 nm) when 1a, 2a, and Cs2CO3 were combined; this peak is proposed to result from the absorption of an electron donor–acceptor (EDA) complex12 formed by the association of the thiolate anion and aryl bromide 1a. At the higher concentration of 0.1 M, a solution containing this EDA complex is visibly yellow and has visible-light absorption tailing to the 400–515 nm region (Figure 1b).15
Figure 1.
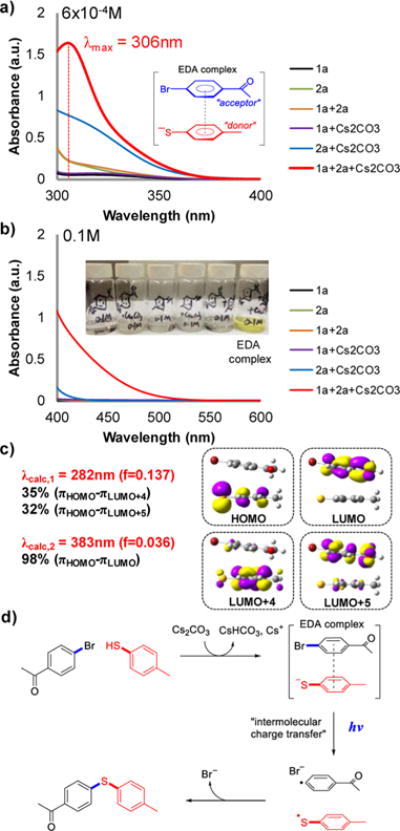
(a, b) UV–vis absorption spectra of mixtures of 1a, 2a, and Cs2CO3 in DMSO (path length = 1 cm) at concentrations of (a) 6 × 10−4 M and (b) 0.1 M for each species. The inset of (b) shows the formation of a yellow compound (proposed EDA complex) upon mixing of 1a, 2a, and Cs2CO3. (c) Time-dependent DFT calculations to predict UV–vis absorptions of the EDA complex. (d) Proposed mechanism for visible-light induced C–S cross-coupling.
Density functional theory (DFT) calculations support the proposed EDA complex formation. The electron-rich thiolate anion (deprotonated 2a) and the electron-poor aryl bromide 1a interact via a π–π interaction with a shortest π-stacking distance of approximately 3.4 Å (Figure 1c). Additionally, time-dependent DFT (TD-DFT) calculations computed at the CAM-B3LYP/6-31+G(d,p) level assigned the observed λmax = 306 nm to have both local and charge-transfer excitation characteristics; this peak was predicted to be λcalc,1 = 282 nm with an oscillator strength (f) of 0.137. Specifically, 35% of the 306 nm absorption is contributed by a local excitation involving the thiolate π orbitals (πHOMO → πLUMO+4) while 32% is contributed by a charge-transfer excitation from the thiolate to 1a (πHOMO → πLUMO+5). Moreover, TD-DFT calculations also predicted a significantly red-shifted peak at 383 nm, albeit with weaker absorption (f = 0.036). This peak has almost exclusively charge-transfer character (98%) involving π orbitals of the thiolate (πHOMO) and 1a (πLUMO) and is proposed to be responsible for the observed visible-light absorption.
On the basis of these analyses, we propose the following visible-light-induced C–S cross-coupling mechanism (Figure 1d). A thiolate anion and an aryl halide first associate to form an EDA complex. As a result of the charge-transfer absorption of this EDA complex, visible-light-induced electron transfer from the thiolate anion to the aryl halide generates the intermediate halide anion, thiyl radical, and aryl radical; these radicals subsequently couple to yield the desired C–S cross-coupled product.
In summary, we have developed a mild, efficient, visible-light-promoted protocol for the C–S cross-coupling of thiols and aryl halides. A wide range of C–S bonds were constructed (>60 examples) under visible-light irradiation without the use of either transition metal or photoredox catalysts. UV–vis spectroscopy and TD-DFT calculations suggest the formation of an EDA complex between the electron-rich thiolate anion and the electron-poor aryl halide. The EDA complex absorbs visible light to effect intermolecular charge transfer; the subsequently formed thiyl and aryl radicals then couple to form the desired C– S cross-coupled product. Furthermore, the potential utility of this transformation has been demonstrated by late-stage functionalization of active pharmaceutical reagents and by the synthesis of key structures of 11β-HSD1 inhibitors. The extension of this work to polymer synthesis is currently in progress.
Supplementary Material
Acknowledgments
This work was supported by Colorado State University, University of Colorado Boulder, the Advanced Research Projects Agency-Energy (DE-AR0000683), and NIH NIGMS (R35GM119702). We acknowledge the use of XSEDE supercomputing resources (NSF ACI-1053575).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07390.
Experimental procedures and additional data (PDF)
ORCID
Garret M. Miyake: 0000-0003-2451-7090
Notes
The authors declare no competing financial interest.
References
- 1.(a) Feng M, Tang B, Liang S, Jiang X. Curr Top Med Chem. 2016;16:1200. doi: 10.2174/1568026615666150915111741. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Patani GA, LaVoie EJ. Chem Rev. 1996;96:3147. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]; (c) Ilardi EA, Vitaku E, Njardarson JT. J Med Chem. 2014;57:2832. doi: 10.1021/jm401375q. [DOI] [PubMed] [Google Scholar]; (d) Boyd DA. Angew Chem, Int Ed. 2016;55:15486. doi: 10.1002/anie.201604615. [DOI] [PubMed] [Google Scholar]; (e) Rahate AS, Nemade KR, Waghuley SA. Rev Chem Eng. 2013;29:471. [Google Scholar]
- 2.(a) Hartwig JF. Acc Chem Res. 2008;41:1534. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beletskaya IP, Ananikov VP. Chem Rev. 2011;111:1596. doi: 10.1021/cr100347k. [DOI] [PubMed] [Google Scholar]; (c) Song S, Zhang Y, Yeerlan A, Zhu B, Liu J, Jiao N. Angew Chem, Int Ed. 2017;56:2487–2491. doi: 10.1002/anie.201612190. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kwong FY, Buchwald SL. Org Lett. 2002;4:3517. doi: 10.1021/ol0266673. [DOI] [PubMed] [Google Scholar]; (b) Murata M, Buchwald SL. Tetrahedron. 2004;60:7397. [Google Scholar]; (c) Fernańdez-Rodríguez MA, Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:2180. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]; (d) Alvaro E, Hartwig JF. J Am Chem Soc. 2009;131:7858. doi: 10.1021/ja901793w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sayah M, Organ MG. Chem – Eur J. 2011;17:11719. doi: 10.1002/chem.201102158. [DOI] [PubMed] [Google Scholar]; (f) Gogoi P, Hazarika S, Sarma MJ, Sarma K, Barman P. Tetrahedron. 2014;70:7484. [Google Scholar]
- 4.(a) Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; (c) Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582. [DOI] [PubMed] [Google Scholar]
- 5.Wang X, Cuny GD, Noël T. Angew Chem, Int Ed. 2013;52:7860. doi: 10.1002/anie.201303483. [DOI] [PubMed] [Google Scholar]
- 6.(a) Oderinde MS, Frenette M, Robbins DW, Aquila B, Johannes JW. J Am Chem Soc. 2016;138:1760. doi: 10.1021/jacs.5b11244. [DOI] [PubMed] [Google Scholar]; (b) Jouffroy M, Kelly CB, Molander GA. Org Lett. 2016;18:876. doi: 10.1021/acs.orglett.6b00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang M, Li H, Yang H, Fu H. Angew Chem, Int Ed. 2017;56:874. doi: 10.1002/anie.201610414. [DOI] [PubMed] [Google Scholar]
- 8.(a) Bunnett JF, Creary X. J Org Chem. 1974;39:3173. [Google Scholar]; (b) Uyeda C, Tan Y, Fu GC, Peters JC. J Am Chem Soc. 2013;135:9548. doi: 10.1021/ja404050f. [DOI] [PubMed] [Google Scholar]; (c) Johnson MW, Hannoun KI, Tan Y, Fu GC, Peters JC. Chem Sci. 2016;7:4091. doi: 10.1039/c5sc04709a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Theriot JC, Lim CH, Yang H, Ryan MD, Musgrave CB, Miyake GM. Science. 2016;352:1082. doi: 10.1126/science.aaf3935. [DOI] [PubMed] [Google Scholar]; (b) Pearson RM, Lim CH, McCarthy BG, Musgrave CB, Miyake GM. J Am Chem Soc. 2016;138:11399. doi: 10.1021/jacs.6b08068. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Du Y, Pearson RM, Lim CH, Sartor SM, Ryan MD, Yang H, Damrauer NH, Miyake GM. Chem – Eur J. 2017;23:10962. doi: 10.1002/chem.201702926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Theriot JC, McCarthy BG, Lim CH, Miyake GM. Macromol Rapid Commun. 2017;38:1700040. doi: 10.1002/marc.201700040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rossi RA, Pierini AB, Peñeń̃ory AB. Chem Rev. 2003;103:71. doi: 10.1021/cr960134o. [DOI] [PubMed] [Google Scholar]
- 11.See the Supporting Information for further information.
- 12.For reviews, see:; (a) Rosokha SV, Kochi JK. Acc Chem Res. 2008;41:641. doi: 10.1021/ar700256a. [DOI] [PubMed] [Google Scholar]; (b) Lima CGS, Lima TdM, Duarte M, Jurberg ID, Paixão MW. ACS Catal. 2016;6:1389. [Google Scholar]
- 13.Grushin V, Alper H. Chem Rev. 1994;94:1047. [Google Scholar]
- 14.Yan X, Wang Z, Sudom A, Cardozo M, DeGraffenreid M, Di Y, Fan P, He X, Jaen JC, Labelle M, Liu J, Ma J, McMinn D, Miao S, Sun D, Tang L, Tu H, Ursu S, Walker N, Ye Q, Powers JP. Bioorg Med Chem Lett. 2010;20:7071. doi: 10.1016/j.bmcl.2010.09.097. [DOI] [PubMed] [Google Scholar]
- 15.For examples of visible-light-induced EDA chemistry, see:; (a) Arceo E, Jurberg ID, Álvarez-Fernańdez A, Melchiorre P. Nat Chem. 2013;5:750. doi: 10.1038/nchem.1727. [DOI] [PubMed] [Google Scholar]; (b) Beatty JW, Douglas JJ, Miller R, McAtee RC, Cole KP, Stephenson CR. J Chem. 2016;1:456. doi: 10.1016/j.chempr.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sun X, Wang W, Li Y, Ma J, Yu S. Org Lett. 2016;18:4638. doi: 10.1021/acs.orglett.6b02271. [DOI] [PubMed] [Google Scholar]; (d) Deng Y, Wei X-J, Wang H, Sun Y, Noël T, Wang X. Angew Chem, Int Ed. 2017;56:832. doi: 10.1002/anie.201607948. [DOI] [PubMed] [Google Scholar]; (e) Candish L, Teders M, Glorius F. J Am Chem Soc. 2017;139:7440. doi: 10.1021/jacs.7b03127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.