

Abstract
Objective
To measure the role of platelets and red blood cells (RBC) on thrombus propagation in an in vitro model of venous valvular stasis.
Approach and Results
A microfluidic model with dimensional similarity to human venous valves consists of a sinus distal to a sudden expansion, where for sufficiently high Reynolds numbers, two countercurrent vortices arise due to flow separation. The primary vortex is defined by the points of flow separation and reattachment. A secondary vortex forms in the deepest recess of the valve pocket characterized by low shear rates. An initial fibrin gel formed within the secondary vortex of a tissue factor (TF) coated valve sinus. Platelet accumulated at the interface of the fibrin gel and the primary vortex. RBC at physiologic hematocrits were necessary to provide an adequate flux of platelets to support thrombus growth out of the valve sinus. A subpopulation of platelets that adhered to fibrin expose phosphatidylserine (PS). Platelet-dependent thrombus growth was attenuated by inhibition of glycoprotein VI (GPVI) with a blocking Fab fragment or D-dimer.
Conclusions
A three-step process regulated by hemodynamics was necessary for robust thrombus propagation: First, immobilized TF initiates coagulation and fibrin deposition within a low flow niche defined by a secondary vortex in the pocket of a model venous valve. Second, a primary vortex delivers platelets to the fibrin interface in a RBC-dependent manner. Third, platelets adhere to fibrin, activate through GPVI, express PS, and subsequently promote thrombus growth beyond the valve sinus and into the bulk flow.
Keywords: coagulation, hemorheology, biotransport
Subject Codes: Hemodynamics, Pathophysiology, Platelets, Thrombosis
INTRODUCTION
The initiation and propagation of venous thrombosis (VT) is poorly characterized compared to arterial thrombosis.1 This is due, in part, to the lack of animal and in vitro models that replicate the hemodynamics and microenvironment of venous valves where most VT in humans originates. The most common animal models of VT are initiated via partial or total ligation of vessels like the inferior vena cava that do not have valves.2,3 In vitro flow chambers are often used to simulate the flows and forces that regulate thrombus formation in straight or stenotic channels, but few recreate the geometry of venous valves.4–6 In this study, we describe a scaled model of human venous valves that recreates their essential hemodynamics in order to determine the influence of blood flow, red blood cells (RBC), and platelets on thrombus propagation initiated by immobilized tissue factor (TF).
VT is thought to result from a combination of flow stasis, hypoxia-induced activation of the endothelium, and subsequent accumulation of procoagulant factors in the valve sinus.7,8 Flow stasis broadly refers to a complete lack of blood flow, but also disturbed flows that result in low flow niches. Venous valve stasis caused by aging and immobility reduces blood flow into and within the valve sinus.9 These unusual hemodynamics conditions in the valve sinus yield a hypoxic environment that results in the presentation of P- and E-selectin and von Willebrand factor (VWF) on endothelial cells,10 which in turn supports adhesion of blood cells and microparticles.11 Accumulation of TF positive microparticles, monocytes, and platelets initiates coagulation through the extrinsic pathway.1 The intrinsic pathway may also play a role in thrombus propagation via exposure of neutrophil extracellular traps.11 Computational models of VT suggest that a threshold quantity of TF must accumulate prior to the initiation of coagulation.12 However, following this initiation, the biophysical mechanisms that regulate the propagation of a thrombus into the lumen of the vein are yet to be quantified.
The histology of venous thrombi implies a role for blood flow in thrombus propagation. The alternating layered structure of red, fibrin-rich regions that begin at the vessel wall in the valve sinus followed by white, platelet-rich regions suggests a mechanism of platelet accumulation to the initial fibrin-rich regions.13 The geometry of the valve sinus defined by a large cavity distal to the expansion created by the valve leaflets yields unique flow patterns. Flow through fixed venous valves of dogs shows a large primary vortex adjacent to the valve cusps, and a secondary vortex in the deepest recess of the valve pocket.14 The fluid velocity in this secondary vortex is extremely slow and corresponds to the most hypoxic area of the valve sinus.19 Vortical flows in the valve sinus have also been observed by ultrasound in human venous valves.15
Vortical flows caused by flow separation downstream of a stenosis or sudden expansion support thrombus formation.16 Blood cells that enter these flows have a long residence time that promotes platelet-platelet collisions, and ultimately aggregation in sudden annular expansions.17 RBC enhance the accumulation of platelets in vortical flows in a hematocrit (HCT) dependent manner.17–19 The low wall shear rates in vortical flows support platelet adhesion to collagen and neutrophil adhesion to P-selectin, with the most accumulation occurring near reattachment points in sudden annular expansions and backwards facing steps.17,20,21 These geometries are relevant to arterial thrombosis or vascular injuries where collagen is exposed to flowing blood, however they do not incorporate TF-dependent coagulation or fibrin deposition, which are central to the pathophysiology of VT.
In this study, we present TF-initiated thrombus propagation in a model venous valve. We use scaling arguments to fabricate valve geometries that have geometric similarity to human valves. A sudden expansion geometry with undercuts of different angles result in primary and secondary vortices at sufficiently high flow rates. An initial fibrin-rich thrombus forms in the secondary vortex, however RBC and platelets within the primary vortex are necessary to propagate the thrombus.
MATERIALS AND METHODS
Materials
Materials are available in the online-only Data Supplement.
Computational Fluid Mechanics
Flow through the model venous valve was simulated at steady state for the 90°, 120°, 135° and 150° angles using the same dimensions as the device using computational fluid dynamics software (COMSOL Multiphysics, COMSOL Inc, Burlington, MA). The entrance condition was set as a constant flow rate to match Re for a Newtonian fluid with a viscosity of 3.5 cP. The outlet condition was set as a zero pressure. The other boundary conditions were set to no-slip. The solution to the Navier-Stokes equation was calculated at a finer mesh size (1,379,416 elements). The simulations were repeated at a fine (422,701 elements), normal (159,625 elements) and coarse (76,256 elements) mesh to insure grid independence (Supplemental Fig. I).
Reynolds Number Matching
The Reynolds number, Re, is a non-dimensional number that compares viscous to inertial forces in flow. It is given by
where ρ is the fluid density, μ the fluid viscosity, v the average velocity and D is the characteristic dimension (the channel height in our case). When we increased the HCT, we increased the viscosity of the suspension. At the same flow rate, this would lead to a smaller Re. In order to achieve dynamic similarity and match Re across different HCT we estimate the viscosity using
where μrel is the relative viscosity of the suspension versus the bulk, and HCT the hematocrit.22 These flow rates are in Supplemental Tables I and II for plasma and buffer, respectively.
Microfluidic Devices
The flow chamber consists of 150 μm wide PMDS channels expanding into a 450 μm wide channels at angles 90°, 120°, 135° or 150°, where larger angles represent a more severe undercut. A vacuum chamber surrounds the channels to aid in the removal of air bubbles. Fabrication details are found in the online-only Data Supplement.
Visualization of streaklines
To measure streaklines, 3 μm FITC-labeled polystyrene beads at a number density of 200,000/μL in HBS were mixed with washed RBC at HCT of 0, 0.2, 0.4 and 0.6. A 500 μL Hamilton Gastight syringe was filled with the suspension and connected to a microfludic device via 0.01″ ID tubing. Flows rates were set to match desired Re of 0.1, 10, and 25. Images were acquired with an inverted microscope (Olympus IX81, 20X NA = 0.45, ex/em 475/505nm) at 100 ms exposure.
Blood collection
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (University of Colorado, Boulder) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all subjects for being included in the study. Blood was collected from healthy donors by venipuncture into 4.5 mL vacutainer tubes containing 3.2% sodium citrate. The first tube of blood collected was treated as waste to eliminate any activated platelets due to venipuncture. Donors had not consumed alcohol within 48 h prior to the draw, nor had they taken any prescription or over-the-counter drugs within the previous 10 days excluding oral contraception.
Procedures for platelet rich plasma, reconstituted blood, and plasma with RBC suspensions
Platelet rich plasma (PRP) and packed red blood cells (RBC) were obtained by centrifugation of citrated whole blood at 200g for 20 min. The top fraction above the buffy coat was collected for PRP and platelet counts were measured by flow cytometry (EMD Millipore, guava esayCyte 6 2L, Hayward, CA) gated by size. The bottom fraction contained packed RBC. Hematocrit of the packed RBC was measured with a CritSpin (Beckman Coulter, Brea, CA) following the manufacturer’s instructions. For experiments where RBC were added into PRP to give reconstituted blood, packed RBC and PRP were combined to achieve the desired hematocrit without wash steps (e.g., if the assay was to be run at HCT 0.4 we prepared the blood at HCT 0.44 to account for the additional volume of the recalcification buffer). For experiments where RBC were added into NPP, the RBC were first washed three times with RBC buffer for 5 min at 2000g. NPP was added back to the RBCs to achieve the desired hematocrit (HCT) concentrations. DiOC6 (1μM) and Alexa 555 labeled fibrinogen was added to each preparation (NPP, PRP, NPP + RBC, reconstituted blood, final concentration 28 μg/mL) for 15 min at 37°C prior to introduction into the device.
Reconstituted blood rheology
Packed RBC (see above for centrifugation procedure) were added to autologous donor plasma or NPP to achieve a hematocrit of 0.4. The RBC suspension (17 mL) was introduced into the cup (diameter = 30.02 mm, depth = 78.01 mm) of the Peltier Concentric Cylinder system of the DH-3 rheometer, after which the bob (diameter = 27.96 mm, depth = 41.91 mm) was lowered into position with an operating gap of 5.91 mm and temperature was maintained at 37 °C. FC-40 oil (1 mL, viscosity = 1.5 cSt) was carefully pipetted on top of the suspension to prevent aggregation at the air-liquid interface. The shear rate sweep from 0.1 to 1000 s−1 was performed for 10 seconds at each shear rate to measure the shear-dependent viscosity of the RBC suspension blood. Measurements were taken every 5 minutes for 45 minutes in total. The viscosity of the autologous donor plasma and NPP were measured at 100 s−1 to calculate the relative viscosity for each suspension.
Microfluidic Device Operation
Stock Innovin tissue factor (TF) was diluted 1:9 in HBS and used to pattern the post expansion half of the channel including the pockets by carefully backfilling via pipette aspiration. The device was connected to vacuum to ensure that there were no air bubbles in the valve pocket. After a one hour incubation of TF at room temperature, the small tubing, connector, and large tubing were connected as shown in Supplemental Fig. II. A 60 mL syringe was filled with 2% BSA in HBS to the 25 mL mark, and a 3 mL syringe is filled with recalcification solution to the 3 mL mark. The 3 mL syringe is connected first to the calcium inlet and recalcification buffer is perfused until it comes out of the blood inlet. Then the 60 mL syringe is connected. This order insures that the dead volume between the calcium inlet and the T-junction is filled with calcium buffer and not BSA solution. The syringes were placed onto a syringe pump (Harvard Apparatus PhD 2000), the flow rate set to match the desired Re (Supplemental Table I), and the solutions were perfused through the tubing for 5 minutes to insure a tight connection with the pump as well as to block the tubing along the path of blood flow. The tubing was then connected to the inlet of the microfluidic device and an additional tubing (60 cm, 0.01° tubing) containing HBS was connected to an outlet of the device. The whole system was perfused for 5 minutes and allowed to block with 2% BSA in HBS for an additional hour.
Reconstituted blood studies
When the plasma, RBC suspension, platelet rich plasma, reconstituted blood, or whole blood was ready for perfusion, the 60 mL syringe was emptied of the blocking solution, and filled with one of these solutions or suspensions to the same volume it held prior to removal insure it will fit on the syringe pump without adjustments. Flow was started immediately. Images of the valve pocket were captured every 20 seconds in a confocal microscope (Olympus FV10i, 60X objective NA = 0.95) at three z-locations to make sure the cover glass surface was in focus in at least one for at least 30 minutes. Alternatively, for thrombus area assays, images were captured with an inverted microscope (Olympus IX81, 20X NA = 0.45, ex/em 475/505 nm, 545/580 nm) every 10 seconds.
Whole blood and platelet inhibitor studies
Citrated whole blood from one blood draw was separated into two 13 mL samples. The first sample was supplemented with vehicle control (HBS for the abxicimab, ACT017, and D-dimer, DMSO for atopaxar) or the inhibitor (20 μg/mL abxicimab,23 500 nM atopaxar,24 100 μg/mL ACT017,25 30 μg/mL D-Dimer26). DiOC6 (1 μM) and Alexa 555 labeled fibrinogen (final concentration 28 μg/mL) was added to the preparation for 15 min at 37°C. The sample was then perfused as in the reconstituted blood case (HCT 0.4) at 372 μL/min through the device for 30 minutes. The second sample was then loaded with the drug or vehicle control and labeled for 15 min at 37°C and perfused through a second device under the same conditions. Images were captured with an inverted microscope (Olympus IX81, 10X NA = 0.3, ex/em 475/505 nm, 545/580 nm) every 10 seconds.
Phophatidylserine labeling
Assays were run with reconstituted whole blood (HCT 0.4) at Re = 10. After 25 min of perfusion, flow was stopped and the channel was rinsed three times with Annexin V binding buffer in the same direction of flow (blood inlet to blood outlet) using a pipette. The clot was then fixed by filling the channel with 2% glutaraldehyde in HBS for 10 minutes. The channel was then rinsed with the Annexin V binding buffer, and then filled with the Annexin V label mixed 1:1 with the binding buffer. The sample was incubated in the dark at room temperaturefor 45 min and then rinsed with Annexin V binding buffer and imaged by confocal microscopy.
Image Quantification
Thrombus areas were measured by thresholding the overlay of platelet and fibrin(ogen) images using ImageJ (NIH, Bethesda, MD). Integrated fluorescence of platelet accumulation was measured using ImageJ, the average greyscale pixel brightness normalized by the maximum (4096 for a 16-bit TIFF).
Statistical Analysis
Data was determined normally distributed by the Anderson-Darling test. Statistical differences were measured using a Student t-test or Mann-Whitney U-test (for data sets that failed and Anderson-Darling test) for pairs and one-way ANOVA for groups followed by a post hoc Tukey’s honestly significant difference procedure to compare pairs in MATLAB (R2016b, MathWorks, Natick, MA).
RESULTS
Design and characterization of model venous valves
The flow through human venous valves depends on vessel size and body position. For example in the common femoral vein, mean peak velocities ranged from 37.3 cm/s in supine leg up position to 1.3 cm/s in a sitting position.27 At this scale, a prohibitive volume of blood would be required for observing thrombus formation in a single pass device. Therefore, we used a scaling approach to develop a model vein and venous valve with dimensional similarity to human venous valves.28 The vessel stenosis ratio—vessel diameter:distance between valve leaflets—is the primary geometric ratio that dictates flow in an expansion. This ratio is approximately 3:1 in the human greater saphenous vein and superficial femoral vein.15,29 In the scaled model, we use a 450 μm wide channel as the vessel and a 150 μm wide channel as the stenosis to achieve a 3:1 stenosis ratio. Previous studies have examined hemodynamics in a sudden expansions or stenosis to model arteriosclerotic geometries,30,31 however the existence of a valve sinus distal to the expansion created by the valve leaflets likely influences the flow field in large veins. In order to model the valve sinus, we fabricated devices with undercut angles of 90°, 120°, 135° or 150° (Fig. 1A). We refer to the area near the opening of the valve sinus the valve cusp and the deeper regions as the valve pocket. The Reynolds numbers (Re) in veins can vary from 1–600, although in large human veins is typically Re>100.27,32,33 The Re is a dimensionless quantity that characterizes the relative importance of inertial forces to viscous forces. To avoid shear-induced platelet activation34 we limited ourselves to Re=1–25, which is comparable to reports in fixed canine valves.14 In flows with Re > 1 inertial forces dominate. For sufficiently high Re, flow separation arises due to a counter acting pressure gradient against the direction of flow immediately downstream of the sudden expansion (Fig. 1B). Fluid far from the wall has more inertia and is able to overcome this pressure gradient and continue downstream, but viscous forces acting on fluid in the near wall region reduce the inertia so that it cannot overcome the pressure gradient, causing recirculation. This flow separation is characterized by detachment and reattachment points (Fig. 1C) as well as regions of low wall shear rates that are favorable for fibrin deposition. The area of the valve pocket with a wall shear rate below 50 s−1, which can support fibrin deposition in the absence of blood cells,35 depends on the Re and, more strongly, the undercut angle as predicted by simulations (Fig. 1D). Above Re of 10, which is characteristic of large veins,29 the low shear rate area was independent of Re. This suggests that valve geometry, more than blood flow, dictates flow in the valve pocket.
Figure 1.

(A) Geometry of the model venous valve. The device consists of an expansion from 150 μm wide channel to 450 μm wide channel with a constant height of 141 μm. The angle of expansion was either 90°, 120°, 135° or 150°. The zone behind the expansion is designated the valve cusp. The deeper part of the sinus is designated as the valve pocket. (B) Visualization of the gauge pressure after a 150° expansion for a Reynolds number (Re) of 10. The pressure in the valve sinus is lower than the downstream pressure, resulting in a reversed flow. The arrows roughly depict average flow directions. (C) Streamlines for the 150°expansion for Re of 10. The flow reattachment point distance is measured from the corner to the end of the primary vortex.(D) The area with a wall shear rate of less than 50 s as calculated by the simulation for Re of 1, 10, 25 and 100 and the expansion angles of 90°, 120°, 135° or 150°.
To visualize the flow field in the absence of blood cells, we perfused suspensions of 3 μm fluorescent particles through the scaled model and measured their streaklines (Fig. 2A). At Re = 1 and all angles, we observe a particle free region in the valve pocket as indicated by the dark zones where few fluorescent particles were observed over a one hour experiment. Those particles that entered the corners of these zones had little apparent convective velocity, suggesting no or a small circulating flow. At Re = 10, we observed flow separation and the entrainment of particles in a primary vortex at undercut angles of 135° and 150° as indicated by particle streaklines that move countercurrent to the bulk flow. At Re = 25, we observed particle accumulation in the primary vortex at all angles except for 90°. The reattachment length as defined in Fig. 1C increased with increasing Re (Supplemental Fig. III). For a given Re, increasing the angle also increases the reattachment length (Supplemental Fig. III). At angles of 135° and 150°, particles were also observed in a secondary vortex for Re of 10 and 25, which was designated by slow moving particles in the valve pocket (Supplemental Video 1).
Figure 2.

Visualization of the flow field in model venous valves. (A) Perfusion of 3 μm fluorescent beads in buffer through the device for the 90°, 120°, 135° and 150° expansion at Re of 1, 10, and 25. (B) Perfusion of 3 μm fluorescent beads at HCT 0.2 in buffer through the device for the 90°, 120°, 135° and 150° expansion at Re of 1, 10, and 25 The primary vortex grows as a function of Re at both angles. The low flow, cell poor region (valve pocket) is visible at higher undercut angles, and more beads enter the vortices in the presence of RBC. Scale bar = 200 μm.
RBC enrich the valve pocket with platelets and platelet sized particles
In the presence of RBC, we observed enhanced entrainment of fluorescent particles in the valve pocket for HCT of 0.2, 0.4, and 0.6 compared to experiments with no RBC (Fig. 2B, Supplemental Fig. IV, Supplemental Video 1). To provide a faithful comparison of the flow field independent of HCT, the Re rather than the flow rate were matched. A HCT dependent function for the viscosity was used to calculate Re (see Methods).22 The reattachment length remained constant across matched Re for all HCT values, confirming that this is the appropriate scaling parameter (Supplemental Fig. III). In experiments where TF was immobilized in the valve pocket, the rate of platelet accumulation in the valve pocket was enhanced by roughly three-fold in reconstituted blood with a HCT of 0.4 compared to platelet rich plasma (PRP) (p= 0.0213, Supplemental Fig. V). In the 135° and 150° expansion angles, RBC also entered and became trapped in the valve pocket and the initial fibrin gel (Supplemental Video 2). These data show that RBC enhance the transport of platelet and platelet sized particles into the valve pocket.
Low flow regions in model valve pockets support initial fibrin formation and thrombus growth
To determine the influence of undercut angle on the initiation of thrombus formation, PRP and reconstituted blood (PRP with RBC) were perfused at a Re of 10 through devices with undercut angles of 90° and 150°, where TF was immobilized on the surface of the model valve sinus (Fig. 3A, Supplemental Videos 3 and 4). Fibrin deposition occurred in the lowest flow regions within ten minutes for the 90° undercut angle, and within five minutes for the 150° undercut angle. Initial fibrin deposition coincided with areas with a wall shear rate of less than 50 s−1 (Fig. 3B). For a 90° undercut angle, simulations predict a single vortex (Fig. 3C) and only moderate fibrin formation was observed followed by platelet accumulation (Fig. 3A). For a 150° undercut angle, a fibrin gel first fills the area coinciding with the predicted secondary vortex (Fig. 3C), followed by platelet adhesion and aggregation which then supports spatial propagation of the thrombus that ultimately grows out of the valve pocket (Fig. 3A). These data suggest that the valve pocket geometry, and in particular regions of very low flow, protect coagulation and fibrin polymerization reactions and support initial platelet accumulation.
Figure 3.

The effect of undercut angle on thrombus formation and the flow field. (A) Thrombus formation in the 150° (left) and 90° (right) undercut angle for reconstituted blood (HCT=0.4) after 15 min in TF-coated device at Re = 10. Platelets are shown in green, fibrin(ogen) is shown in red. Scale bar = 150 μm. Wall shear rate contours (B) and streamlines (C) from simulations for undercut angles of 150° (left) and 90° (right) at Re = 10.
Platelets and RBC are necessary for thrombus propagation out of the valve sinus
To determine the roles of platelets and RBC on thrombus growth in our model valve sinus, normal pooled plasma (NPP), platelet rich plasma (PRP), NPP containing RBC, and reconstituted blood (PRP with RBC) was perfused at a Re of 10 through devices with a 150° undercut angle (Fig. 4A).
Figure 4.

The role of platelets and RBC on thrombus propagation. (A) Normal pooled plasma (NPP) platelet rich plasma (PRP), washed RBC in NPP (NPP + RBC, HCT=0.4) and reconstituted whole blood (HCT=0.4) were perfused at Re = 10 through the device. NPP, PRP, and RBC+NPP show a slow moving fibrin front that corresponds to the edge of the primary vortex and the transition to the valve pocket. For the reconstituted whole blood, RBC fill the valve pocket, diminishing the fluorescence signal of the fibrin(ogen). Focal plane 10 μm from the bottom surface. Platelets are shown in green (DiOC6) and fibrin(ogen) is shown in red. Scale bar = 100 μm. (B)The area of the thrombus was measured at 30 minutes for RBC in NPP at (black bars) and reconstituted blood (grey bars) for HCT of 0, 0.4, and 0.6 (n=5). The means and standard error are plotted. Statistical differences were measured by ANOVA followed by Tukey’s test for multiple comparisons.
For NPP, fibrin formed within 3–5 minutes in the valve pocket region with the lowest shear rate as described above, but did not extend beyond the area defined by the initial secondary vortex (Fig. 4A, Supplemental Video 4). Note the concave interface of the fibrin gel mirrors the interface between the primary and secondary vortex from particle streaklines (Fig. 2) and simulations (Fig. 3). Fibrin fibers were densest at the interface suggesting an accumulation of coagulation proteins and fibrin(ogen) transported by flow into the primary vortex. The interface moved slowly; it extended just 50 μm over 30 min. Control experiments in the absence of immobilized TF showed fibrin deposition only in the deepest part of the valve pocket and only after 30 min of perfusion (Supplemental Video 5).
PRP yielded similar sized thrombi as NPP (Fig. 4A, Supplemental Video 4). Platelets tend to accumulate near the detachment point at the valve cusp, and to a lesser extent at the fibrin gel interface with the primary vortex. In regions near adhered platelets, the fibrin gel grows denser with time relative to gels formed with NPP, possibly due to platelet retraction or additional coagulation. However, thrombus size is similar to that of NPP after 30 minutes (Fig. 4B). Thus, platelets in the absence of RBC do not support the propagation of thrombi beyond the low flow region defined by the secondary vortex.
NPP with suspended RBC at HCT of 0.4 and 0.6 yielded fibrin deposition that was also limited to the valve pocket. Here, the morphology of the fibrin gel is different because RBC were incorporated in the fibrin gel (Fig. 4A). However, these RBC did not support additional thrombus growth beyond what was observed for NPP after 30 min (Fig. 4B, Supplemental Video 4). In control experiments without TF, RBC became trapped in the valve pocket, but no fibrin fibers were observed (Supplemental Video 5). It does not appear that RBC alone support additional coagulation that results in observable enhancement of thrombus growth compared to plasma alone. The shear-dependent viscosity of RBC suspended in NPP or autologous plasma was statistically similar (Supplemental Fig. VI), suggesting that RBC agglutination was not enhanced in NPP.
Reconstituted blood containing both platelets and RBC supports robust thrombus growth out of the valve pocket and into the bulk flow. These thrombi were roughly double the size of those formed with NPP, PRP, and NPP with RBC at 30 minutes (Fig. 4B, Supplemental Video 4). Compared to PRP, experiments with reconstituted blood show that platelets in the primary vortex adhere in a dense layer to the initial fibrin gel formed in the valve pocket, followed by the rapid formation of a thrombus that extends past the primary vortex (Fig. 4A, Supplemental Video 4). The thrombi grew beyond the field of view during some experiments, so exact thrombus sizes were not available for all assays. HCT of 0.4 and 0.6 yielded similar thrombus growth rates and sizes (Fig. 4B). Alternating platelet- and RBC-rich zones are apparent as the thrombus penetrated into the main channel (Supplemental Videos 2, 4).
Reconstituted blood (RBC, platelets, and plasma) and whole blood yield similar results in terms of thrombus area and growth rate (Supplemental Fig. VII). These data suggest that leukocytes do not play a significant role in this model of thrombus propagation.
Platelets adhered to fibrin in the valve sinus can become procoagulant
In both PRP and reconstituted blood experiments, we observed regions in the valve pocket where the DiOC6 dye, which labels the mitochondrial membrane, began to fade at around ten minutes after initial adhesion (Fig. 5A–B, Supplemental Video 4). We hypothesized that this was indicative of mitochondrial depolarization due to platelet activation and would lead to subsequent exposure to phosphatidylserine (PS).33 To test this hypothesis, we performed experiments with reconstituted blood (HCT 0.4) at Re of 10 for 25 minutes, fixed the thrombi, and then used annexin V to label PS (Fig. 5C). We found annexin V positive platelets were localized in dense fibrin networks near the fluid-thrombus interface and in large platelet aggregates. Platelets that lost their DiOC6 signal during the perfusion experiment were found to be annexin V positive following fixation.
Figure 5.
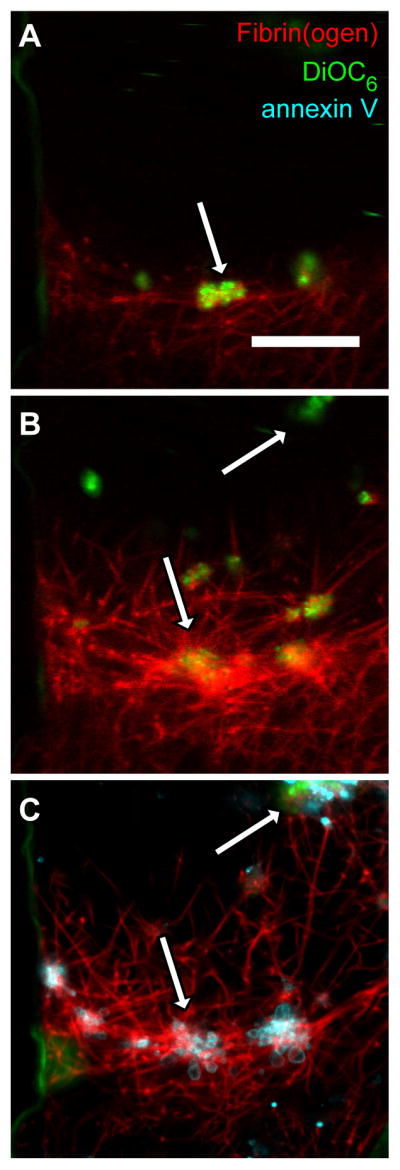
Fibrin adhered platelets become procoagulant. (A) Initial platelet (green) deposition on fibrin fibers (red) in the valve pocket after reconstitute blood (HCT=0.4) perfused at a Re of 10 for 5 min in a device with an undercut angle of 150°. (B) The same field-of-view as (A) at 20 minutes. The platelet aggregate denoted by the arrow in (A) became a site for fibrin deposition. (C) The same field-of-view as (A) after a 25 min perfusion and staining with annexin V (cyan). Annexin V localized on the edges of platelet aggregates and in the fibrin network as denoted by arrows. Scale bar = 20 μm.
Platelets aggregation in the valve sinus is supported by GPVI signaling
Finally, to determine what interactions mediate activation of platelets adhered to fibrin we conducted experiments with a series of platelet receptor inhibitors. Whole blood was treated with the αIIbβ3 inhibitor abciximab, the PAR1 inhibitor atopaxar, an anti-GPVI humanized Fab fragment ACT017,36 or D-dimer which binds to GPVI.26,37 Blood was perfused at Re = 10 through TF-coated devices with a 150° undercut angle. In all cases, an initial fibrin gel was formed as in reconstituted blood and plasma conditions. Abciximab abrogates platelet buildup beyond the first layer of platelets on the fibrin gel (Fig. 6). Atopaxar, a PAR1 inhibitor showed modest, but not significant differences in thrombus area compared to vehicle controls. Treatment with ACT017 and D-dimer did reduce total thrombus are in a spatially dependent manner; platelets still aggregate around the corner proximal to the expansion, but their accumulation adjacent to fibrin in the valve pocket or on the distal wall was diminished.
Figure 6.

Thrombus growth is driven by platelet activation through GPVI. Representative images of thrombus formation with whole blood for vehicle control (A) and treatments with atopaxar (B), D-dimer (C), anti-GPVI antibody ACT017 (D), and abciximab (E) after 30 minute perfusion. Scale bar = 100 μm. (E) Thrombus areas for vehicle controls and each treatment. The means (n = 6) and standard error are plotted. Statistical differences were measured by ANOVA followed by Tukey’s test for multiple comparisons.
DISCUSSION
In this study, we designed a model of TF-initiated venous thrombosis that captures some of the important hemodynamic features of human venous valves, specifically, primary and secondary vortices and flow separation in an expansion geometry with an undercut. An initial fibrin gel formed in the low flow region of the secondary vortex in the deepest part of the valve pocket independent of blood cells. Both RBC and platelets were necessary for thrombus growth beyond the valve pocket. Platelets support thrombus growth beyond the initial fibrin gel by adhering, activating, and for at least a subpopulation, becoming procoagulant. These results suggest a biophysical mechanism of thrombus propagation that depends on both flow-enhanced platelet-RBC interactions upstream of a valve and low flow niches that protect coagulation reactions and support platelet adhesion to fibrin (Supplemental Fig. VIII).
The initiation of coagulation and fibrin polymerization require protection from dilution by blood flow.38 Computational models of venous thrombosis suggest that a threshold concentration of TF must first accumulate on the valve wall before coagulation can initiate.39 Here, we have skipped the steps leading up the accumulation of TF, and started with a surface TF concentration capable of initiating coagulation. Previous studies found that even at high thrombin or TF concentrations, fibrin can only form at wall shear rates below 100 s−1 in the absence of adhered blood cells.6,35 That is consistent with observations in this study, where the low flow region defined by shear rates of less than 50 s−1 in the valve pocket was the nidus for fibrin deposition. The size of this initial fibrin gel was dictated by the degree of undercut, more so than the Re.
RBC promote thrombus propagation by enhancing the transport of platelets into the valve pocket at a sufficient rate to support coagulation beyond the TF-coated valve. Based on studies in non-biological dense suspensions in sudden expansions where margination occurs due to size exclusion effects,40 we would expect this is due to platelet margination upstream from the expansion point, rather than increased collision frequency within the vortices. Elevated hematocrit is correlated with increased risk for VT.41 In our model, we did not observe a significant difference in thrombus growth between hematocrits of 0.4 and 0.6. However, these experiments were conducted at a constant Re to provide faithful comparison of the hemodynamics, while the cardiovascular system is likely regulated by other parameters like oxygen demand.
The procoagulant activity of RBC reported by others42 was not sufficient in these studies to initiate fibrin deposition in the absence of TF, nor to enhance it in absence of platelets. This observation is consistent with in vivo, in vitro, and in silico models of arterial thrombosis where the primary influence of an elevated hematocrit is to promote platelet dependent thrombus growth rather than coagulation.43 RBC do contribute architecture and mechanics of venous thrombi; they are retained in fibrin-rich thrombi via FXIII-dependent fibrin α-chain cross-linking 44,45 and organize to form densely packed structures upon platelet-mediated contraction46 that could limit transport of coagulation products into and out of the growing thrombus. RBC can also adhere to activated platelets and fibrin at low shear rates such as those found in vortices.47
Platelet adhesion to fibrin and subsequent activation through GPVI is essential in this model for thrombus propagation. Platelets adhered to fibrin along the entire periphery of the primary vortex and formed a platelet-rich layer reminiscent of the laminate structure of thrombi formed in the venous valves of humans.13 Some platelets within this layer become procoagulant, supporting the formation of an adjacent fibrin-rich thrombus that penetrates from the valve sinus into the bulk flow. In agreement with previous studies,48,49 some platelets adhered to fibrin became exposed PS. Treatments with the GPVI blocking Fab fragment (ACT017) or D-dimer reduced thrombus growth. This is consistent with reports that GPVI can bind to the D-domain on fibrin(ogen), although whether the monomer or dimer of GPVI governs this interaction is a subject of debate.26,37 Importantly, D-dimer may also limit thrombus growth by inhibiting fibrin polymerization and αIIbβ3 interactions. Even when inhibiting GPVI there was platelet accumulation around the corner that defines the valve cusp. Platelet accumulation in this region is possibly driven by activation-independent aggregation as previously shown downstream of stenotic geometries.4
There is not an established relationship between GPVI and VT risk in humans. However, reduced platelet responsiveness to GPVI agonists, which is presumed to be a consequence of continuous activation, is associated with a higher risk of venous thromboembolism. 50 Our finding that platelets are necessary for thrombus propagation in an in vitro model using human blood are consistent with similar murine IVC flow restriction model.11 In our model, platelets do not play a significant role in the initiation of a thrombus, which is consistent with a lack of platelets at the thrombus nidus in vivo (the valve pocket in our model), but presence in successive layers (the valve cusp and beyond).51 In some animal models of VT, platelets play an essential role in early events thrombus growth,11,52 although it is difficult to delineate initiation from propagation in these models.
There are several limitations of this study. First, because the model valve leaflet is fixed, we do not capture the hemodynamics associated with valve opening and closing. This is an approach similar to other in vitro and computational studies of VT12,39 and we argue it is a relevant model for valvular stasis. Second, in order to use a non-prohibitive volume of human blood and avoid shear-induced platelet activation we used scaling arguments to create a small version of a human venous valve. This model captures the essential hemodynamics, but the time scale for thrombus propagation is naturally shortened due to the smaller length scales. Third, leukocytes and specifically neutrophils and neutrophil extracellular traps (NETs) may play a role in the initiation of venous thrombosis.11,52,53 These mechanisms were not explicitly studied here, however the presence of leukocytes in whole blood experiments did not appear to influence thrombus growth or size compared to reconstituted blood without leukocytes. Fourth, the surface-bound anticoagulants thrombomodulin and heparin sulfate are not present in this model. Computational studies suggest that coagulation can be initiated in the presence of these anticoagulants with a sufficient quantity of TF.12,54
Supplementary Material
HIGHLIGHTS.
A miniaturized model of a venous valve was created with dynamic and dimensional similarity to human valves.
Flow patterns in model valves include primary and secondary vortices in the valve sinus.
Red blood cells promote transport of platelets into the primary vortex and subsequent adhesion to fibrin formed in the valve pocket
Platelet adhesion to fibrin and activation through GPVI is essential for thrombus propagation out of the valve sinus
Acknowledgments
The authors would like to thank Anticor Biotech for the kind gift of the anti-GPVI Fab ACT017.
SOURCES OF FUNDING
This work was supported by the NSF CAREER (CBET-1351672), American Heart Association (14GRNT20410094), and the National Institutes of Health (R01HL120728).
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- GPVI
Glycoprotein VI
- HCT
Hematocrit
- NPP
Normal pooled plasma
- RBC
Red blood cells
- Re
Reynolds number
- PRP
Platelet rich plasma
- PS
Phosphatidylserine
- TF
Tissue factor
- VT
Venous thrombosis
Footnotes
DISCLOSURES
None.
References
- 1.Versteeg HH, Heemskerk JWM, Levi M, Reitsma PH. New Fundamentals in Hemostasis. Physiological Reviews. 2013;93:327–358. doi: 10.1152/physrev.00016.2011. [DOI] [PubMed] [Google Scholar]
- 2.Mackman N. Mouse Models, Risk Factors, and Treatments of Venous Thrombosis. Arterioscl Throm Vas. 2012;32:554–555. doi: 10.1161/ATVBAHA.112.246173. [DOI] [PubMed] [Google Scholar]
- 3.Singh I, Smith A, Vanzieleghem B, Collen D, Burnand K, Saint-Remy J-M, Jacquemin M. Antithrombotic effects of controlled inhibition of factor VIII with a partially inhibitory human monoclonal antibody in a murine vena cava thrombosis model. 2002;99:3235–3240. doi: 10.1182/blood.v99.9.3235. [DOI] [PubMed] [Google Scholar]
- 4.Nesbitt W, Westein E, Tovar-Lopez F, Tolouei E, Mitchell A, Fu J, Carberry J, Fouras A, Jackson S. A shear gradient–dependent platelet aggregation mechanism drives thrombus formation. Nat Med. 2009;15:665–673. doi: 10.1038/nm.1955. [DOI] [PubMed] [Google Scholar]
- 5.Sakariassen KS, Aarts PA, de Groot PG, Houdijk WP, Sixma JJ. A perfusion chamber developed to investigate platelet interaction in flowing blood with human vessel wall cells, their extracellular matrix, and purified components. J Lab Clin Med. 1983;102:522–535. [PubMed] [Google Scholar]
- 6.Onasoga-Jarvis AA, Puls TJ, O’Brien SK, Kuang L, Liang HJ, Neeves KB. Thrombin generation and fibrin formation under flow on biomimetic tissue factor-rich surfaces. J Thromb Haemost. 2014;12:373–382. doi: 10.1111/jth.12491. [DOI] [PubMed] [Google Scholar]
- 7.Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–2336. doi: 10.1172/JCI60229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malone PC, Agutter PS. Deep venous thrombosis: The valve cusp hypoxia thesis and its incompatibility with modern orthodoxy. Medical Hypotheses. 2016;86:60–66. doi: 10.1016/j.mehy.2015.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Chiu J-J, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiological Reviews. 2011;91:327–387. doi: 10.1152/physrev.00047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bovill EG, van der Vliet A. Venous Valvular Stasis–Associated Hypoxia and Thrombosis: What Is the Link? Annu Rev Physiol. 2011;73:527–545. doi: 10.1146/annurev-physiol-012110-142305. [DOI] [PubMed] [Google Scholar]
- 11.Bruhl von ML, Stark K, Steinhart A, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. Journal of Experimental Medicine. 2012;209:819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elizondo P, Fogelson AL. A Mathematical Model of Venous Thrombosis Initiation. Biophysical Journal. 2016;111:2722–2734. doi: 10.1016/j.bpj.2016.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sevitt S. The structure and growth of valve-pocket thrombi in femoral veins. J Clin Pathol. 1974;27:517–528. doi: 10.1136/jcp.27.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karino T, Motomiya M. Flow through a venous valve and its implication for thrombus formation. Thromb Res. 1984;36:245–257. doi: 10.1016/0049-3848(84)90224-x. [DOI] [PubMed] [Google Scholar]
- 15.Lurie F, Kistner RL, Eklof B, Kessler D. Mechanism of venous valve closure and role of the valve in circulation: a new concept. Journal of Vascular Surgery. 2003;38:955–961. doi: 10.1016/s0741-5214(03)00711-0. [DOI] [PubMed] [Google Scholar]
- 16.Karino T, Goldsmith HL, Motomiya M, Mabuchi S, Sohara Y. Flow Patterns in Vessels of Simple and Complex Geometries. Ann NY Acad Sci. 1987;516:422–441. doi: 10.1111/j.1749-6632.1987.tb33063.x. [DOI] [PubMed] [Google Scholar]
- 17.Karino T, Goldsmith HL. Aggregation of human platelets in an annular vortex distal to a tubular expansion. Microvascular Research. 1979;17:217–237. doi: 10.1016/s0026-2862(79)80001-1. [DOI] [PubMed] [Google Scholar]
- 18.Zhao R, Marhefka JN, Shu F, Hund SJ, Kameneva MV, Antaki JF. Micro-flow visualization of red blood cell-enhanced platelet concentration at sudden expansion. Ann Biomed Eng. 2008;36:1130–1141. doi: 10.1007/s10439-008-9494-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan A, David T, Homer-Vanniasinkam S, Graham A, Walker P. The effects of margination and red cell augmented platelet diffusivity on platelet adhesion in complex flow. Biorheology. 2004;41:641–653. [PubMed] [Google Scholar]
- 20.Skilbeck C, Westwood SM, Walker PG, David T, Nash GB. Population of the Vessel Wall by Leukocytes Binding to P-Selectin in a Model of Disturbed Arterial Flow. Arterioscl Throm Vas. 2001;21:1294–1300. doi: 10.1161/hq0801.093506. [DOI] [PubMed] [Google Scholar]
- 21.Skilbeck C, Westwood SM, Walker PG, David T, Nash GB. Dependence of adhesive behavior of neutrophils on local fluid dynamics in a region with recirculating flow. Biorheology. 2001;38:213–227. [PubMed] [Google Scholar]
- 22.Pries AR, Neuhaus D, Gaehtgens P. Blood viscosity in tube flow: dependence on diameter and hematocrit. Am J Physiol. 1992;263:H1770–H1778. doi: 10.1152/ajpheart.1992.263.6.H1770. [DOI] [PubMed] [Google Scholar]
- 23.Maxwell MJ, Westein E, Nesbitt WS, Giuliano S, Dopheide SM, Jackson SP. Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. 2007;109:566–576. doi: 10.1182/blood-2006-07-028282. [DOI] [PubMed] [Google Scholar]
- 24.French SL, Arthur JF, Lee H, Nesbitt WS, Andrews RK, Gardiner EE, Hamilton JR. Inhibition of protease-activated receptor 4 impairs platelet procoagulant activity during thrombus formation in human blood. J Thromb Haemost. 2016;14:1642–1654. doi: 10.1111/jth.13293. [DOI] [PubMed] [Google Scholar]
- 25.Mangin PH, Tang C, Bourdon C, Loyau S, Freund M, Hechler B, Gachet C, Jandrot-Perrus M. A Humanized Glycoprotein VI (GPVI) Mouse Model to Assess the Antithrombotic Efficacies of Anti-GPVI Agents. J Pharmacol Exp Ther. 2012;341:156–163. doi: 10.1124/jpet.111.189050. [DOI] [PubMed] [Google Scholar]
- 26.Onselaer M-B, Hardy AT, Wilson C, Sanchez X, Babar AK, Miller JLC, Watson CN, Watson SK, Bonna A, Philippou H, Herr AB, Mezzano D, Ariëns RAS, Watson SP. Fibrin and D-dimer bind to monomeric GPVI. Blood Advances. 2017;1:1495–1504. doi: 10.1182/bloodadvances.2017007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashby EC, Ashford NS, Campbell MJ. Posture, blood velocity in common femoral vein, and prophylaxis of venous thromboembolism. Lancet. 1995;345:419–421. doi: 10.1016/s0140-6736(95)90402-6. [DOI] [PubMed] [Google Scholar]
- 28.Mccarty OJT, Ku D, Sugimoto M, King MR, Cosemans JMEM, Neeves KB the Subcommittee on Biorheology. Dimensional analysis and scaling relevant to flow models of thrombus formation: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14:619–622. doi: 10.1111/jth.13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nam K-H, Yeom E, Ha H, Lee S-J. Velocity field measurements of valvular blood flow in a human superficial vein using high-frequency ultrasound speckle image velocimetry. Int J Cardiovasc Imaging. 2010;28:69–77. doi: 10.1007/s10554-010-9778-x. [DOI] [PubMed] [Google Scholar]
- 30.Karino T, Goldsmith HL. Flow Behaviour of Blood Cells and Rigid Spheres in an Annular Vortex. Philosophical Transactions of the Royal Society B: Biological Sciences. 1977;279:413–445. doi: 10.1098/rstb.1977.0095. [DOI] [PubMed] [Google Scholar]
- 31.Wootton DM, Ku DN. Fluid mechanics of vascular systems, diseases, and thrombosis. Annu Rev Biomed Eng. 1999;1:299–329. doi: 10.1146/annurev.bioeng.1.1.299. [DOI] [PubMed] [Google Scholar]
- 32.Ku DN, Klafta JM, Gewertz BL, Zarins CK. The contribution of valves to saphenous vein graft resistance. J Vasc Surg. 1987;6:274–279. [PubMed] [Google Scholar]
- 33.Helps EPW, Mcdonald DA. Observations on laminar flow in veins. The Journal of Physiology. 1954;124(3):631–639. doi: 10.1113/jphysiol.1954.sp005135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. 1996;88:1525–1541. [PubMed] [Google Scholar]
- 35.Neeves KB, Illing DAR, Diamond SL. Thrombin flux and wall shear rate regulate fibrin fiber deposition state during polymerization under flow. Biophysical Journal. 2010;98:1344–1352. doi: 10.1016/j.bpj.2009.12.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lebozec K, Jandrot-Perrus M, Avenard G, Favre-Bulle O, Billiald P. Design, development and characterization of ACT017, a humanized Fab that blocks platelet’s glycoprotein VI function without causing bleeding risks. mAbs. 2017;9:945–958. doi: 10.1080/19420862.2017.1336592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Induruwa I, Moroi M, Bonna A, Malcor JD, Howes JM, Warburton EA, Farndale RW, Jung SM. Platelet collagen receptor GPVI-dimer recognizes fibrinogen and fibrin through their D-domains, contributing to platelet adhesion and activation during thrombus formation. J Thromb Haemost. 2018 doi: 10.1111/jth.13919. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rana K, Neeves KB. Blood flow and mass transfer regulation of coagulation. Blood Reviews. 2016;30:357–368. doi: 10.1016/j.blre.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dydek EV, Chaikof EL. Simulated thrombin responses in venous valves. Journal of Vascular Surgery: Venous and Lymphatic Disorders. 2016;4:329–335. doi: 10.1016/j.jvsv.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moraczewski T, Tang H, Shapley NC. Flow of a concentrated suspension through an abrupt axisymmetric expansion measured by nuclear magnetic resonance imaging. Journal of Rheology. 2005;49:1409–1428. [Google Scholar]
- 41.Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Hansen JB. Hematocrit and risk of venous thromboembolism in a general population. The Tromso study. Haematologica. 2010;95:270–275. doi: 10.3324/haematol.2009.008417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. 2012;120:3837–3845. doi: 10.1182/blood-2012-05-427856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walton BL, Lehmann M, Skorczewski T, Holle LA, Beckman JD, Cribb JA, Mooberry MJ, Wufsus AR, Cooley BC, Homeister JW, Pawlinski R, Falvo MR, Key NS, Fogelson AL, Neeves KB, Wolberg AS. Elevated hematocrit enhances platelet accumulation following vascular injury. Blood. 2017;129:2537–2546. doi: 10.1182/blood-2016-10-746479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aleman MM, Byrnes JR, Wang J-G, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. 2014;124:3590–3600. doi: 10.1172/JCI75386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam WA, Meijers JCM, Ni H, Ariëns RAS, Wolberg AS. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin α-chain crosslinking. 2015;126:1940–1948. doi: 10.1182/blood-2015-06-652263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, Rauova L, Lowery TJ, Weisel JW. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2014;123:1596–1603. doi: 10.1182/blood-2013-08-523860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goel MS, Diamond SL. Adhesion of normal erythrocytes at depressed venous shear rates to activated neutrophils, activated platelets, and fibrin polymerized from plasma. 2002;100:3797–3803. doi: 10.1182/blood-2002-03-0712. [DOI] [PubMed] [Google Scholar]
- 48.Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. 2015;126:1601–1608. doi: 10.1182/blood-2015-04-641654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, Mangin PH, Jandrot-Perrus M. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. 2015;126:683–691. doi: 10.1182/blood-2015-02-629717. [DOI] [PubMed] [Google Scholar]
- 50.Riedl J, Kaider A, Marosi C, Prager GW, Eichelberger B, Assinger A, Pabinger I, Panzer S, Ay C. Decreased platelet reactivity in patients with cancer is associated with high risk of venous thromboembolism and poor prognosis. Thromb Haemost. 2017;117:90–98. doi: 10.1160/TH16-02-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reitsma PH, Versteeg HH, Middeldorp S. Mechanistic view of risk factors for venous thromboembolism. Arterioscl Throm Vas. 2012;32:563–568. doi: 10.1161/ATVBAHA.111.242818. [DOI] [PubMed] [Google Scholar]
- 52.Heestermans M, Salloum-Asfar S, Salvatori D, Laghmani EH, Luken BM, Zeerleder SS, Spronk HMH, Korporaal SJ, Wagenaar GTM, Reitsma PH, van Vlijmen BJM. Role of platelets, neutrophils, and factor XII in spontaneous venous thrombosis in mice. Blood. 2016;127:2630–2637. doi: 10.1182/blood-2018-05-854596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brill A, Fuchs TA, Savchenko A, Thomas GM, Martinod K, De Meyer SF, Bhandari AA, Wagner DD. Neutrophil Extracellular Traps Promote Deep Vein Thrombosis in Mice. J Thromb Haemost. 2012;10:136–144. doi: 10.1111/j.1538-7836.2011.04544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan SW, Chaikof EL. Simulated Surface-Induced Thrombin Generation in a Flow Field. Biophysical Journal. 2011;101:276–286. doi: 10.1016/j.bpj.2011.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.