

Abstract
Objective
HIV is a cardiovascular disease (CVD) risk factor. However, CVD risk is often underestimated in HIV-infected women. C-reactive protein (CRP) may improve CVD prediction in this population. We examined the association of baseline plasma CRP with subclinical CVD in women with and without HIV.
Design
Retrospective cohort study
Methods
572 HIV-infected and 211 HIV-uninfected women enrolled in the Women’s Interagency HIV Study underwent serial high-resolution B-mode carotid artery ultrasonography between 2004-2013 to assess carotid intima-media thickness (CIMT) and focal carotid artery plaques. We used multivariable linear and logistic regression models to assess the association of baseline high (≥3 mg/l) high-sensitivity (hs) CRP with baseline CIMT and focal plaques, and used multivariable linear and Poisson regression models for the associations of high hsCRP with CIMT change and focal plaque progression. We stratified our analyses by HIV status.
Results
Median (IQR) hsCRP was 2.2 mg/l(0.8-5.3) in HIV-infected, and 3.2 mg/l(0.9-7.7) in HIV-uninfected, women (p=0.005). There was no statistically significant association of hsCRP with baseline CIMT [adjusted mean difference -3.5 μm (95%CI:-19.0-12.1)] or focal plaques [aOR:1.31(0.67-2.67)], and no statistically significant association of hsCRP with CIMT change [adjusted mean difference 11.4 μm(-2.3-25.1). However, hsCRP ≥3 mg/l was positively associated with focal plaque progression in HIV-uninfected [aRR:5.97(1.46-24.43)], but not in HIV-infected [aRR:0.81(0.47-1.42)] women (p=0.042 for interaction).
Conclusion
In our cohort of women with similar CVD risk factors, higher baseline hsCRP is positively associated with carotid plaque progression in HIV-uninfected, but not HIV-infected, women, suggesting that subclinical CVD pathogenesis may be different HIV-infected women.
Keywords: HIV, subclinical CVD, C-reactive protein, atherosclerosis, women
Introduction
Persons living with human immunodeficiency virus (HIV) who are treated with antiretroviral therapy (ART) can expect to attain a near-normal life expectancy [1];however, they have an increased risk of age-related comorbidities, including cardiovascular disease (CVD) [2]. Compared with the general population, HIV-infected individuals have greater rates of myocardial infarctions (MIs) and ischemic strokes, especially at younger ages [3-7]. Additionally, although the incidence of CVD is greater among men than women, the relative contribution of HIV to CVD risk may be greater among HIV-infected women than HIV-infected men: in observational studies, HIV-infected women had a 2-3-fold increased risk of MI and stroke compared with their HIV-uninfected counterparts, while HIV-infected men had an approximately 1.5-fold increased risk [6-9]. However, CVD risk prediction tools, including the 2013 American College of Cardiology/American Heart Association (ACC/AHA) Pooled Cohort equations, do not include HIV as a CVD risk factor, and underestimate CVD risk in HIV-infected individuals [10, 11], particularly among women and minority populations [12].
There is a clear need to improve CVD risk assessment in these populations. C-reactive protein (CRP) is an acute phase reactant associated with subclinical CVD progression and clinical CVD in the general population [13, 14], and is included in the ACC/AHA guidelines as an option to improve risk-stratification among intermediate-risk persons [10]. However, its association with both subclinical and hard clinical CVD outcomes in the HIV population is less clear [15, 16], and data in HIV-infected women are particularly lacking. To determine the association of CRP with subclinical CVD prevalence and progression among HIV-infected women, we retrospectively analyzed data from the Women’s Interagency HIV Study (WIHS) carotid ultrasound substudy, in which measures of subclinical CVD, including carotid intima-media thickness (CIMT) and carotid plaque formation, which are associated with subsequent clinical CVD events [17], were obtained. We tested for differences in association by HIV serostatus.
Materials and Methods
Study population
WIHS is a multi-site, longitudinal cohort of HIV-infected and at-risk HIV-uninfected women in the United States [18]. Study design has been described previously [18, 19]. Briefly, participants undergo semiannual detailed interviews, physical examinations, and specimen collections. In 2004, WIHS initiated a longitudinal carotid artery ultrasound substudy; all WIHS participants at all six clinical sites were eligible for enrollment in the substudy [20]. For this retrospective analysis, we included all substudy participants with at least two carotid artery ultrasound substudy visits who had high sensitivity (hs) CRP measured at the baseline substudy visit. WIHS participants who HIV seroconverted during the follow-up period were excluded from the analysis (figure 1). All participants provided informed consent, and the study was approved by each participating institution’s Institutional Review Board.
Figure 1.
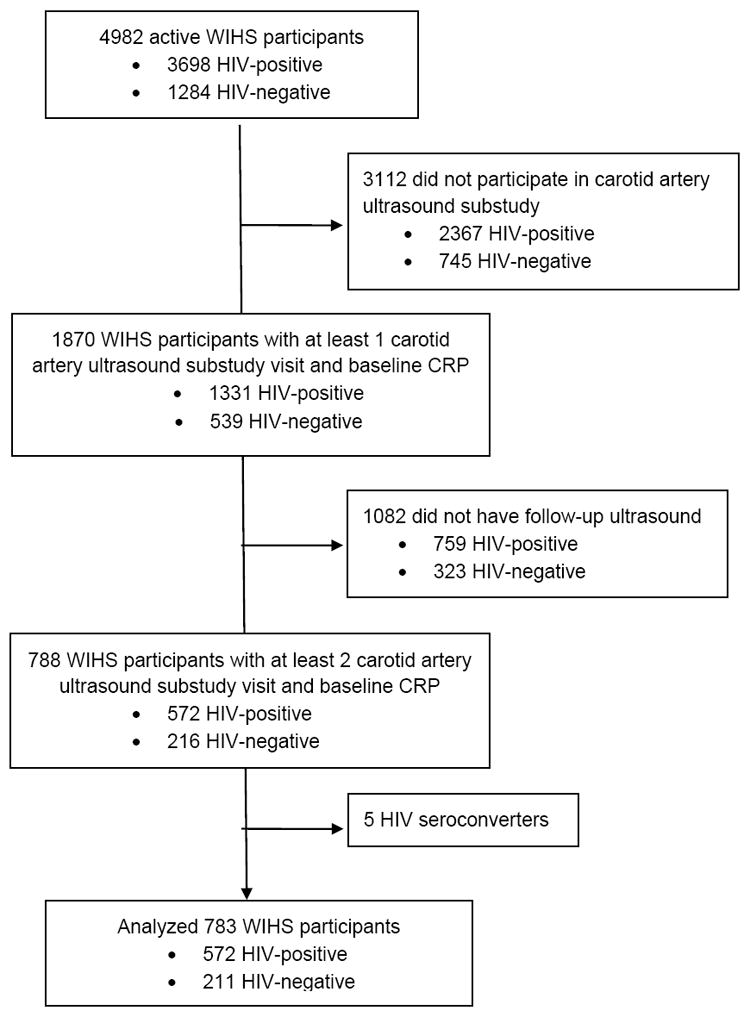
Flow diagram for eligibility for study analysis.
Data collection
CIMT and focal carotid plaques were measured by high-resolution B-mode ultrasonography according to previously published procedures [21] at a baseline substudy visit between 2004 and 2006, and at a final substudy visit between 2010 and 2013. All sites used a standardized protocol, and outcome measures were obtained at a centralized reading center (University of Southern California) [20, 22]. The coefficient of variation for repeated measures of CIMT in the WIHS cohort has been published previously [20]. Citrate plasma hsCRP was obtained at the baseline substudy visit on fasting samples obtained in duplicate and measured by a nephelometric immunoassay (Dade-Behring BN II) [23]. Demographic, clinical, and laboratory data were collected by standardized protocols at the WIHS core visit closest to the baseline substudy visit.
Outcome measures
Primary outcomes measures were baseline CIMT, presence of any baseline focal plaques, change in CIMT, and focal plaque progression. CIMT was measured at a 1-cm segment of the right common carotid artery far wall just distal to the carotid bifurcation. Focal plaque was defined as an area of focal IMT>1.5 mm in any of the 6 imaged segments: near and far walls of the common carotid artery, the carotid bifurcation, and the internal carotid artery [22]. Change in CIMT was defined as the difference between the final and baseline CIMT measurements. Focal plaque progression was defined as the number of new plaques formed in each participant between the baseline and final visits.
Measurement of covariates
HIV infection was determined by serologic testing using ELISA and confirmed with Western blot assays. Plasma HIV RNA levels (viral load) were quantified using nucleic-acid-sequence-based amplification assays with a lower limit of quantification of 80 copies/ml (bioMérieux, Boxtel, NC), total peripheral CD4 T cell counts were measured with standard flow cytometric methods. CD4 T cell nadir was defined as the lowest CD4 T cell count prior to ART initiation. ART use was defined as the use of three or more antiretroviral medications. History of clinical CVD was defined as a history of MI, angina, revascularization, transient ischemic attack or stroke. Diabetes was defined as fasting glucose ≥126 mg/dl, glycated hemoglobin (HbA1c) ≥6.5% or self-reported anti-diabetic medication use. Chronic hepatitis C virus (HCV) infection was defined as a positive enzyme immunoanalysis (EIA) and detectable HCV RNA. Lipid levels were obtained following an 8-hour fast. Estimated glomerular filtration rate (eGFR) was determined by the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) equation [24]. Age, race/ethnicity, crack/cocaine use, current smoking status, and antihypertensive and lipid-lowering medication use were self-reported.
Statistical analysis
Baseline characteristics of HIV-infected and HIV-uninfected participants were compared by chi-square for categorical variables, and Student’s t-tests or Wilcoxon rank-sum tests for continuous variables, as appropriate. For each outcome, hsCRP was analyzed as a categorical variable with a cutpoint of ≥3 mg/l [25], and as a continuous (natural log-transformed) variable. Because chronic HCV is associated with lower hsCRP levels [26], we also assessed the difference in hsCRP by HIV serostatus after excluding all participants with chronic HCV.
Multivariable linear and logistic regression models were used to assess the association of baseline hsCRP with baseline CIMT, and with the presence of baseline focal plaques, respectively. Multivariable linear regression and Poisson regression models were used to assess the association of baseline hsCRP with change in CIMT, and with focal plaque progression, respectively. After checking for collinearity, models were adjusted for age, race/ethnicity, systolic blood pressure, antihypertensive and lipid-lowering medication use, low-density lipoprotein (LDL) cholesterol, diabetes, history of CVD, eGFR, current smoking status, crack/cocaine use, and HCV. For each model, we assessed the interaction between dichotomized hsCRP (above or below 3 mg/l) and HIV status; non-significant interaction terms were removed from the model. Separate analyses by HIV status were conducted, and models for HIV-infected individuals were also adjusted for CD4 count, CD4 nadir, HIV viral load, and ART use. Finally, we examined the association of baseline hsCRP with subclinical CVD progression outcomes among HIV-infected women with persistent virologic suppression defined as an HIV viral load <80 copies/ml at all substudy visits while on continuous ART. Consistent with prior studies [22], we allowed for one virologic “blip” if the viral load remained <500 copies/ml. We also performed the same analyses among HIV-infected women who did not have persistent virologic suppression.
Analyses were conducted using SAS v9.4 (SAS Institute, Cary, NC). Statistical significance was determined using a p-value of <0.05.
Results
Baseline characteristics
1,870 WIHS participants participated in the carotid ultrasound study. Of these, 788 had more than one carotid ultrasound and baseline hsCRP. Five women were excluded for HIV seroconversion, so 783 (572 HIV-infected and 211 HIV-uninfected) women were included in this analysis (figure 1). The median [interquartile range (IQR)] time between baseline and final carotid artery ultrasound was 6.6 (6.4-7.0) years. Baseline characteristics stratified by HIV status are shown in Table 1. The median age at the first substudy visit was 41 years. 62% of the women were black and 29% were Hispanic. HIV-infected women had lower baseline body mass index (BMI), systolic blood pressure, high-density lipoprotein (HDL) cholesterol, and prevalence of diabetes, higher HCV prevalence, and similar LDL levels as HIV-uninfected women. Three percent of women in each group had a history of clinical CVD, and the average 10-year Framingham CVD risk [27] in both groups was 1%. More HIV-uninfected than HIV-infected women were current smokers and used crack/cocaine. Among HIV-infected participants, the median CD4 count was 452 cells/mm3; 46% had HIV viral load <80 copies/ml, and 62% were on ART. Median (IQR) baseline hsCRP was higher among HIV-uninfected women than HIV-infected women [3.2(0.9-7.7) vs. 2.2(0.8-5.3) mg/l, p=0.005], but baseline focal plaques and median CIMT were not different between groups. In women without chronic HCV, there was a trend toward higher median baseline hsCRP among HIV-uninfected women compared with HIV-infected women that was not statistically significant [3.4(1.0-7.9) vs. 2.6(1.2-5.6) mg/l, p=0.08)].
Table 1.
Baseline demographic and clinical data for all women and by HIV serostatus
| Characteristic, median (IQR) or n (%) | Entire cohort (n=783) | HIV-positive (n=572) | HIV-negative (n=211) | p-value |
|---|---|---|---|---|
| Age, years | 40.6 (35.2-46.7) | 40.8 (35.3-46.8) | 40.6 (34.3-46.5) | 0.51 |
| Follow-up time (years) | 6.6 (6.4-7.0) | 6.6 (6.4-7.0) | 6.6 (6.4-7.0) | 0.39 |
| Race | 0.36 | |||
| White, non-Hispanic | 57 (7) | 45 (8) | 12 (6) | |
| Black, non-Hispanic | 484 (62) | 344 (60) | 140 (66) | |
| Hispanic | 225 (29) | 169 (30) | 56 (27) | |
| Other, non-Hispanic | 17 (2) | 14 (2) | 3 (1) | |
| Systolic blood pressure, mm Hg | 117 (109-127) | 116 (108-127) | 120 (111-132) | 0.007 |
| Diastolic blood pressure, mm Hg | 71 (64-79) | 71 (64-78) | 71 (65-80) | 0.55 |
| Antihypertensive medication use | 142 (18) | 109 (19) | 33 (16) | 0.27 |
| Total cholesterol, mg/dL | 175 (150-201) | 174 (149-201) | 177 (154-204) | 0.26 |
| HDL cholesterol, mg/dl | 47 (39-58) | 46 (37-56) | 52 (43-63) | <0.001 |
| LDL cholesterol, mg/dl | 102 (79-124) | 102 (78-123) | 103 (79-125) | 0.40 |
| Triglycerides, mg/dl | 104 (73-152) | 112 (77-158) | 90 (66-127) | <0.001 |
| Lipid-lowering medication use | 42 (5) | 36 (6) | 6 (3) | 0.06 |
| Diabetes | 106 (14) | 69 (12) | 37 (18) | 0.047 |
| Body mass index, kg/m2 | 28.3 (24.3-33.5) | 27.6 (24.1-32.1) | 30.3 (25.3-37.8) | <0.001 |
| eGFR, ml/min/1.73 m2 (CKD-epi) | 100.9 (87.2-114.2) | 101.5 (86.1-115.0) | 99.8 (87.7-111.3) | 0.81 |
| History of clinical CVD* | 25 (3) | 18 (3) | 7 (3) | 0.90 |
| Framingham risk score | 8 (3-12) | 8 (3-12) | 9 (3-13) | 0.43 |
| 10-year CVD risk by FRS | 1% (1%-1%) | 1% (1%-1%) | 1% (1%-2%) | 0.13 |
| Current smoker | 374 (48) | 257 (45) | 117 (55) | 0.011 |
| Crack/cocaine use | <0.001 | |||
| Never | 456 (58) | 355 (62) | 101 (48) | |
| Former | 259 (33) | 180 (32) | 79 (37) | |
| Current | 65 (8) | 34 (6) | 31 (15) | |
| Current alcohol use | <0.001 | |||
| None | 399 (51) | 314 (55) | 85 (40) | |
| 1-7 drinks/week | 311 (40) | 214 (38) | 97 (46) | |
| > 7 drinks/week | 70 (9) | 41 (7) | 29 (14) | |
| Chronic HCV | 154 (20) | 122 (21) | 32 (15) | 0.054 |
| HIV-associated characteristics | ||||
| CD4 count, cells/mm3 | -- | 452 (288-658) | -- | |
| CD4 nadir, cells/mm3 | -- | 281 (161-409) | -- | |
| HIV viral load, cop/ml | -- | 180 (80-6900) | -- | |
| Undetectable (<80 cop/ml) | -- | 260 (46) | -- | |
| < 1000 cop/ml | -- | 84 (15) | -- | |
| ≥ 1000 cop/ml | -- | 227 (40) | -- | |
| History of clinical AIDS | -- | 270 (47) | -- | |
| ART use | -- | 356 (62) | -- | |
| NRTI use | -- | 355 (62) | -- | |
| NNRTI use | -- | 136 (24) | -- | |
| PI use | -- | 209 (37) | -- | |
| Outcome variables | ||||
| hsCRP | 2.4 (0.9-5.7) | 2.2 (0.8-5.3) | 3.2 (0.9-7.7) | 0.005 |
| hsCRP ≥ 3.0 mg/l | 344 (46) | 234 (43) | 110 (53) | 0.019 |
| Baseline carotid plaques | 54 (7) | 39 (7) | 15 (8) | 0.78 |
| Focal plaque progression** | 80 (11) | 65 (12) | 15 (8) | 0.11 |
| Baseline CIMT, μm | 709 (652-779) | 707 (649-776) | 714 (657-794) | 0.20 |
| Final CIMT, μm | 727 (674-804) | 724 (670-803) | 740 (684-814) | 0.25 |
| CIMT change***, μm | 25.4 (-1.0-49.0) | 19 (-2.0-49.0) | 23.0 (1.0-48.0) | 0.83 |
Abbreviations: HDL, high density lipoprotein; LDL, low density lipoprotein; eGFR, estimated glomerular filtration rate; CVD, cardiovascular disease; FRS, Framingham risk score; HCV, hepatitis C virus; ART, antiretroviral therapy; NRTI, nucleoside reverse transcriptase inhibitor; NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor, hsCRP, high-sensitivity C-reactive protein; CIMT, carotid artery intima-media thickness
Clinical CVD defined as a history of MI, stroke/TIA, angina, peripheral arterial disease, or revascularization procedure
Plaque progression defined as increase in number of total plaques from baseline visit to final visit
CIMT change defined as difference in CIMT from baseline visit to final visit
Association of hsCRP with baseline subclinical CVD
The interaction between HIV status and dichotomized hsCRP was not statistically significantly associated with either baseline CIMT or baseline focal plaques (p=0.20 and 0.72, respectively). After multivariable adjustment, there was no association between dichotomized hsCRP and baseline CIMT among all women [estimated mean difference high vs. low hsCRP -3.5 μm(95% confidence interval (CI):-19.0-12.1)] (figure 2a). High baseline hsCRP was positively associated with focal plaque prevalence among all women [adjusted odds ratio (aOR) 1.31(95%CI:0.67-2.67)], but the association was not statistically significant (figure 2b). Similar associations were observed when hsCRP was analyzed as a continuous variable (data not shown).
Figure 2.
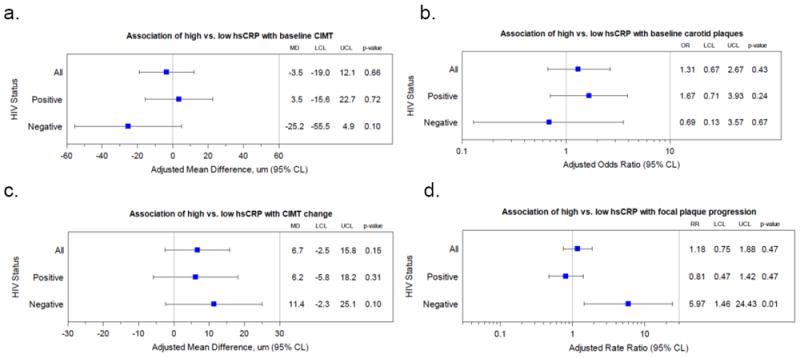
Association of high (≥3 mg/l) vs. low (<3 mg/l) baseline hsCRP with (a) baseline focal plaques, (b) baseline CIMT, (c) focal plaque progression, and (d) CIMT change. Models are adjusted for age, race/ethnicity, systolic blood pressure, antihypertensive and lipid-lowering medication use, low-density lipoprotein (LDL) cholesterol, diabetes, history of CVD, eGFR, current smoking status, crack/cocaine use, and HCV. Models for HIV-infected also adjusted for CD4 count, CD4 nadir, HIV viral load, and ART use.
Abbreviations: hsCRP, high-sensitivity C-reactive protein; MD, mean difference; OR, odds ratio; LCL, lower confidence limit; UCL, upper confidence limit; CIMT, carotid intima-media thickness; RR, rate ratio
Association of hsCRP with progression of subclinical CVD
There was no statistically significant association of an interaction of HIV status and dichotomized hsCRP with CIMT progression (p=0.67). After multivariable adjustment, dichotomized hsCRP was positively associated with CIMT progression among all women (estimated mean difference high vs. low hsCRP 11.4 μm(95%CI:-2.3-25.1), although this association was not statistically significant (figure 2c). However, high baseline hsCRP was positively associated with focal plaque progression among HIV-uninfected women [adjusted rate ratio (aRR) 5.97(95%CI:1.46-24.43)], but not among HIV-infected women [aRR 0.81(95%CI:0.47-1.42)], p=0.042 for interaction (figure 2d). Among 143 persistently virologically suppressed women, there was no statistically significant association between high baseline hsCRP and carotid plaque progression [aRR 0.55(95%CI:0.14-2.20)], or CIMT change [adjusted mean difference 4.5 μm(95%CI:-16.1-25.2)]. Similar associations were observed when hsCRP was analyzed as a continuous variable (data not shown). Finally, among the 379 women without persistent virologic suppression, there was no statistically significant association between high baseline CRP and carotid plaque progression [aRR 0.92(95%CI:0.51-1.65)] or CIMT change [adjusted mean difference 9.4 μm(95%CI:-4.9-23.7)]. Similar associations were observed when hsCRP was analyzed as a continuous variable (data not shown).
Discussion
In a well-characterized cohort of HIV-infected and at-risk HIV-uninfected women with similar 10-year Framingham risk scores, we report that baseline plasma hsCRP levels ≥3 mg/l are positively associated with progression of focal carotid plaques among HIV-uninfected, but not HIV-infected, women. We did not find any statistically significant association between hsCRP levels and baseline prevalence of subclinical CVD, nor did we find any statistically significant association between baseline hsCRP and CIMT progression, although there was a trend towards greater CIMT progression with high baseline hsCRP in both groups. Both groups had low Framingham CVD risk and similar subclinical CVD outcomes at baseline. Although HIV infected participants had lower systolic blood pressure, BMI, and prevalence of diabetes, and higher HDL cholesterol than HIV-uninfected participants, these differences were small and unlikely to be clinically significant. Rates of smoking in both groups was higher than the national average [28], and were even higher among HIV-uninfected participants. Similarly, crack and/or cocaine use was higher among HIV-uninfected participants, although current crack/cocaine use was low overall.
We also report higher baseline hsCRP levels in the HIV-uninfected group. This finding was somewhat unexpected because HIV infection has been shown to be positively associated with hsCRP elevation [29]. However, this association seems to be stronger in men than women [30, 31], which may account for differences between our study and others that have included men. Furthermore, hsCRP levels increase with ART administration in men and women [23]. In our analysis, only 62% of HIV-infected women were on ART, which may contribute to the lower hsCRP in that group. This discrepancy also may be due in part to the higher rate of chronic HCV in the HIV-infected group. CRP is synthesized by hepatocytes, and prior data have shown that HCV infection is associated with lower plasma hsCRP [26, 32]. Indeed, when all HCV-infected participants were excluded from the analysis, the difference in hsCRP was no longer statistically significant. However, since HCV status was included in the multivariable models, this difference should not affect our overall conclusions. It is also possible that other causes of inflammation, including smoking status and crack/cocaine use, which were higher in the HIV-uninfected group, contributed to the higher baseline hsCRP in this population. Finally, HIV-infected participants had a higher rate of lipid-lowering medication use, although the overall use was low (6% vs. 3%). It is possible that more statin use among HIV-infected women contributed to the lower hsCRP in this group, but it is unlikely to be a major factor since the overall prevalence of statin use in both groups was very low.
This study adds to a growing body of literature concerning CVD biomarkers in the HIV population. HsCRP is positively associated with CVD events in HIV-infected persons in most observational studies [16, 33, 34]. Its association with subclinical CVD measures in this population is more variable [15], possibly due to different mechanisms of atherogenesis, including the greater role of innate and adaptive immune activation in the setting of HIV infection [30, 35]. In our study, the lack of association between baseline hsCRP and subclinical CVD progression outcomes among HIV-infected women both with and without persistent virologic suppression, unlike in HIV-uninfected women, suggests there are factors beyond HIV viremia that contribute to subclinical CVD progression in HIV-infected women. Therefore, it is possible that hsCRP, which is positively associated with atherosclerosis and MI in the general population, may not be associated as strongly with these outcomes in persons living with HIV.
Our study has limitations. Its retrospective design limits our ability to control for all potential confounders. We used subclinical rather than clinical endpoints. Although carotid artery plaques and CIMT are correlated with CVD events in the general population [36], their association with CVD in the HIV-infected population is unknown. Also, we examined only two measures of atherosclerosis at the carotid artery. Examination of other vessels and other measures of atherosclerosis, such as echogenicity and arterial stiffness, may yield different results. In addition, our cohort was composed of relatively young women with few CVD risk factors and limited clinical CVD. It is possible that the association of hsCRP with subclinical CVD progression would have become apparent with longer follow-up. In addition, as with any multivariable modeling, we were limited in the number of predictors that we could use without over-fitting our models, and were unable to account for all possible covariates that might contribute to CVD risk including duration of HIV infection, duration of protease inhibitor therapy, and abacavir use [37]. Finally, our findings may not be generalizable to men. However, an important scientific strength of this study is the inclusion of women, especially those from racial and ethnic minorities, who have been historically underrepresented in both HIV and CVD clinical trials [38, 39].
In conclusion, we report that hsCRP predicts focal carotid artery plaque progression in HIV-uninfected, but not HIV-infected, women. These findings suggest that hsCRP is not a reliable biomarker for CVD in women in the setting of HIV infection, which could be because the pathogenesis of subclinical CVD may be different in HIV-infected women. Further investigation of alternative CVD biomarkers and the pathogenesis of CVD in the setting of HIV infection is warranted.
Acknowledgments
Data in this manuscript were collected by the Women’s Interagency HIV Study (WIHS). The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH). WIHS (Principal Investigators): Atlanta WIHS (Ighovwerha Ofotokun and Gina Wingood), U01-AI-103408, Bronx WIHS (Kathryn Anastos), U01-AI-035004; Brooklyn WIHS (Howard Minkoff and Deborah Gustafson), U01-AI-031834; Chicago WIHS (Mardge Cohen and Audrey French), U01-AI-034993; Metropolitan Washington WIHS (Seble Kassaye), U01-AI-034994; Connie Wofsy Women’s HIV Study, Northern California (Ruth Greenblatt, Bradley Aouizerat, and Phyllis Tien), U01-AI-034989; WIHS Data Management and Analysis Center (Stephen Gange and Elizabeth Golub), U01-AI-042590; Southern California WIHS (Joel Milam), U01-HD-032632 (WIHS I – WIHS IV). The WIHS is funded primarily by the National Institute of Allergy and Infectious Diseases (NIAID), with additional co-funding from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), the National Cancer Institute (NCI), the National Institute on Drug Abuse (NIDA), and the National Institute on Mental Health (NIMH). Targeted supplemental funding for specific projects is also provided by the National Institute of Dental and Craniofacial Research (NIDCR), the National Institute on Alcohol Abuse and Alcoholism (NIAAA), the National Institute on Deafness and other Communication Disorders (NIDCD), and the NIH Office of Research on Women’s Health. WIHS data collection is also supported by UL1-TR000004 (UCSF CTSA), and UL1-TR000454 (Atlanta CTSA). Other funding sources include R01HL126543 to R. C. K., R01HL132794 to R. C. K., R01HL083760 to R. C. K., R01HL095140 to R. C. K., K01-HL-137557 to D. B. H. and 1K23AI114407 to A. N. S. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author contribution: C.A.M., A.Q., R.C.K., and I.O. were involved in the study conception, design, implementation, and writing of the manuscript. C. C. M. was involved in study design, conducted the statistical analyses, and significant editing of the manuscript. A.N.S., D.B.H., D.R.G., M.W.P., W.J.M., P.C.T., A.L.F., and E.T.G. were involved in study conception, design, and significant editing of the manuscript. All authors approve of the final version of the manuscript.
Conflicts of Interest: The authors declare no conflicts of interest.
References
- 1.Marcus JL, Chao CR, Leyden WA, Xu L, Quesenberry CP, Jr, Klein DB, et al. Narrowing the Gap in Life Expectancy Between HIV-Infected and HIV-Uninfected Individuals With Access to Care. Journal of acquired immune deficiency syndromes (1999) 2016;73(1):39–46. doi: 10.1097/QAI.0000000000001014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aberg JA. Aging, inflammation, and HIV infection. Topics in antiviral medicine. 2012;20(3):101–105. [PMC free article] [PubMed] [Google Scholar]
- 3.Hanna DB, Ramaswamy C, Kaplan RC, Kizer JR, Anastos K, Daskalakis D, et al. Trends in Cardiovascular Disease Mortality Among Persons With HIV in New York City, 2001-2012. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2016;63(8):1122–1129. doi: 10.1093/cid/ciw470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Losina E, Hyle EP, Borre ED, Linas BP, Sax PE, Weinstein MC, et al. Projecting 10-year, 20-year, and Lifetime Risks of Cardiovascular Disease in Persons Living With Human Immunodeficiency Virus in the United States. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2017;65(8):1266–1271. doi: 10.1093/cid/cix547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gutierrez J, Albuquerque ALA, Falzon L. HIV infection as vascular risk: A systematic review of the literature and meta-analysis. PloS one. 2017;12(5):e0176686. doi: 10.1371/journal.pone.0176686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang S, Mary-Krause M, Cotte L, Gilquin J, Partisani M, Simon A, et al. Increased risk of myocardial infarction in HIV-infected patients in France, relative to the general population. AIDS (London, England) 2010;24(8):1228–1230. doi: 10.1097/QAD.0b013e328339192f. [DOI] [PubMed] [Google Scholar]
- 7.Chow FC, Regan S, Feske S, Meigs JB, Grinspoon SK, Triant VA. Comparison of ischemic stroke incidence in HIV-infected and non-HIV-infected patients in a US health care system. Journal of acquired immune deficiency syndromes (1999) 2012;60(4):351–358. doi: 10.1097/QAI.0b013e31825c7f24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, et al. HIV infection and the risk of acute myocardial infarction. JAMA internal medicine. 2013;173(8):614–622. doi: 10.1001/jamainternmed.2013.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Womack JA, Chang CC, So-Armah KA, Alcorn C, Baker JV, Brown ST, et al. HIV infection and cardiovascular disease in women. Journal of the American Heart Association. 2014;3(5):e001035. doi: 10.1161/JAHA.114.001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goff DC, Jr, Lloyd-Jones DM, Bennett G, Coady S, D’Agostino RB, Gibbons R, et al. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129(25 Suppl 2):S49–73. doi: 10.1161/01.cir.0000437741.48606.98. [DOI] [PubMed] [Google Scholar]
- 11.Thompson-Paul AM, Lichtenstein KA, Armon C, Palella FJ, Jr, Skarbinski J, Chmiel JS, et al. Cardiovascular Disease Risk Prediction in the HIV Outpatient Study. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2016;63(11):1508–1516. doi: 10.1093/cid/ciw615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feinstein MJ, Nance RM, Drozd DR, Ning H, Delaney JA, Heckbert SR, et al. Assessing and Refining Myocardial Infarction Risk Estimation Among Patients With Human Immunodeficiency Virus: A Study by the Centers for AIDS Research Network of Integrated Clinical Systems. JAMA cardiology. 2017;2(2):155–162. doi: 10.1001/jamacardio.2016.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mincu RI, Janosi RA, Vinereanu D, Rassaf T, Totzeck M. Preprocedural C-Reactive Protein Predicts Outcomes after Primary Percutaneous Coronary Intervention in Patients with ST-elevation Myocardial Infarction a systematic meta-analysis. Scientific reports. 2017;7:41530. doi: 10.1038/srep41530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldassarre D, De Jong A, Amato M, Werba JP, Castelnuovo S, Frigerio B, et al. Carotid intima-media thickness and markers of inflammation, endothelial damage and hemostasis. Annals of medicine. 2008;40(1):21–44. doi: 10.1080/07853890701645399. [DOI] [PubMed] [Google Scholar]
- 15.Vos AG, Idris NS, Barth RE, Klipstein-Grobusch K, Grobbee DE. Pro-Inflammatory Markers in Relation to Cardiovascular Disease in HIV Infection. A Systematic Review. PloS one. 2016;11(1):e0147484. doi: 10.1371/journal.pone.0147484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Triant VA, Meigs JB, Grinspoon SK. Association of C-reactive protein and HIV infection with acute myocardial infarction. Journal of acquired immune deficiency syndromes (1999) 2009;51(3):268–273. doi: 10.1097/QAI.0b013e3181a9992c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peters SAE, den Ruijter HM, Bots ML, Moons KGM. Improvements in risk stratification for the occurrence of cardiovascular disease by imaging subclinical atherosclerosis: a systematic review. Heart. 2012;98(3):177–184. doi: 10.1136/heartjnl-2011-300747. [DOI] [PubMed] [Google Scholar]
- 18.Barkan SE, Melnick SL, Preston-Martin S, Weber K, Kalish LA, Miotti P, et al. The Women’s Interagency HIV Study. WIHS Collaborative Study Group. Epidemiology (Cambridge, Mass) 1998;9(2):117–125. [PubMed] [Google Scholar]
- 19.Bacon MC, von Wyl V, Alden C, Sharp G, Robison E, Hessol N, et al. The Women’s Interagency HIV Study: an observational cohort brings clinical sciences to the bench. Clin Diagn Lab Immunol. 2005;12(9):1013–1019. doi: 10.1128/CDLI.12.9.1013-1019.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan RC, Kingsley LA, Gange SJ, Benning L, Jacobson LP, Lazar J, et al. Low CD4+ T-cell count as a major atherosclerosis risk factor in HIV-infected women and men. AIDS (London, England) 2008;22(13):1615–1624. doi: 10.1097/QAD.0b013e328300581d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodis HN, Mack WJ, Lobo RA, Shoupe D, Sevanian A, Mahrer PR, et al. Estrogen in the prevention of atherosclerosis. A randomized, double-blind, placebo-controlled trial. Annals of internal medicine. 2001;135(11):939–953. doi: 10.7326/0003-4819-135-11-200112040-00005. [DOI] [PubMed] [Google Scholar]
- 22.Hanna DB, Post WS, Deal JA, Hodis HN, Jacobson LP, Mack WJ, et al. HIV Infection Is Associated With Progression of Subclinical Carotid Atherosclerosis. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2015;61(4):640–650. doi: 10.1093/cid/civ325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palella FJ, Jr, Gange SJ, Benning L, Jacobson L, Kaplan RC, Landay AL, et al. Inflammatory biomarkers and abacavir use in the Women’s Interagency HIV Study and the Multicenter AIDS Cohort Study. AIDS (London, England) 2010;24(11):1657–1665. doi: 10.1097/QAD.0b013e3283389dfa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, 3rd, Feldman HI, et al. A new equation to estimate glomerular filtration rate. Annals of internal medicine. 2009;150(9):604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ridker PM. Clinical application of C-reactive protein for cardiovascular disease detection and prevention. Circulation. 2003;107(3):363–369. doi: 10.1161/01.cir.0000053730.47739.3c. [DOI] [PubMed] [Google Scholar]
- 26.Shah S, Ma Y, Scherzer R, Huhn G, French AL, Plankey M, et al. Association of HIV, hepatitis C virus and liver fibrosis severity with interleukin-6 and C-reactive protein levels. AIDS (London, England) 2015;29(11):1325–1333. doi: 10.1097/QAD.0000000000000654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Estimate of 10-Year Risk for Coronary Heart Disease Framingham Point Scores. National Institutes of Health: National Heart, Lung, and Blood Institute [Google Scholar]
- 28.Trends in current cigarette smoking among high school students and adults, United States, 1965-2014. Atlanta, GA: Centers for Disease Control and Prevention; 2017. [Google Scholar]
- 29.Dolan SE, Hadigan C, Killilea KM, Sullivan MP, Hemphill L, Lees RS, et al. Increased cardiovascular disease risk indices in HIV-infected women. Journal of acquired immune deficiency syndromes (1999) 2005;39(1):44–54. doi: 10.1097/01.qai.0000159323.59250.83. [DOI] [PubMed] [Google Scholar]
- 30.Hanna DB, Lin J, Post WS, Hodis HN, Xue X, Anastos K, et al. Association of Macrophage Inflammation Biomarkers With Progression of Subclinical Carotid Artery Atherosclerosis in HIV-Infected Women and Men. The Journal of infectious diseases. 2017;215(9):1352–1361. doi: 10.1093/infdis/jix082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reingold J, Wanke C, Kotler D, Lewis C, Tracy R, Heymsfield S, et al. Association of HIV infection and HIV/HCV coinfection with C-reactive protein levels: the fat redistribution and metabolic change in HIV infection (FRAM) study. Journal of acquired immune deficiency syndromes (1999) 2008;48(2):142–148. doi: 10.1097/QAI.0b013e3181685727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Floris-Moore M, Howard AA, Lo Y, Schoenbaum EE, Arnsten JH, Klein RS. Hepatitis C infection is associated with lower lipids and high-sensitivity C-reactive protein in HIV-infected men. AIDS Patient Care STDS. 2007;21(7):479–491. doi: 10.1089/apc.2006.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duprez DA, Neuhaus J, Kuller LH, Tracy R, Belloso W, De Wit S, et al. Inflammation, coagulation and cardiovascular disease in HIV-infected individuals. PloS one. 2012;7(9):e44454. doi: 10.1371/journal.pone.0044454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ford ES, Greenwald JH, Richterman AG, Rupert A, Dutcher L, Badralmaa Y, et al. Traditional risk factors and D-dimer predict incident cardiovascular disease events in chronic HIV infection. AIDS (London, England) 2010;24(10):1509–1517. doi: 10.1097/QAD.0b013e32833ad914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Longenecker CT, Sullivan C, Baker JV. Immune activation and cardiovascular disease in chronic HIV infection. Current opinion in HIV and AIDS. 2016;11(2):216–225. doi: 10.1097/COH.0000000000000227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inaba Y, Chen JA, Bergmann SR. Carotid plaque, compared with carotid intima-media thickness, more accurately predicts coronary artery disease events: a meta-analysis. Atherosclerosis. 2012;220(1):128–133. doi: 10.1016/j.atherosclerosis.2011.06.044. [DOI] [PubMed] [Google Scholar]
- 37.Friis-Moller N, Ryom L, Smith C, Weber R, Reiss P, Dabis F, et al. An updated prediction model of the global risk of cardiovascular disease in HIV-positive persons: The Data-collection on Adverse Effects of Anti-HIV Drugs (D:A:D) study. European journal of preventive cardiology. 2016;23(2):214–223. doi: 10.1177/2047487315579291. [DOI] [PubMed] [Google Scholar]
- 38.Downing NS, Shah ND, Neiman JH, Aminawung JA, Krumholz HM, Ross JS. Participation of the elderly, women, and minorities in pivotal trials supporting 2011-2013 U.S. Food and Drug Administration approvals. Trials. 2016;17:199. doi: 10.1186/s13063-016-1322-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kragholm K, Halim SA, Yang Q, Schulte PJ, Hochman JS, Melloni C, et al. Sex-Stratified Trends in Enrollment, Patient Characteristics, Treatment, and Outcomes Among Non-ST-Segment Elevation Acute Coronary Syndrome Patients: Insights From Clinical Trials Over 17 Years. Circulation Cardiovascular quality and outcomes. 2015;8(4):357–367. doi: 10.1161/CIRCOUTCOMES.114.001615. [DOI] [PMC free article] [PubMed] [Google Scholar]