

Abstract
Platform technologies based on plant virus nano-particles (VNPs) and virus-like particles (VLPs) are attracting the attention of researchers and clinicians because the particles are biocompatible, biodegradable, noninfectious in mammals, and can readily be chemically and genetically engineered to carry imaging agents and drugs. When the Physalis mottle virus (PhMV) coat protein is expressed in Escherichia coli, the resulting VLPs are nearly identical to the viruses formed in vivo. Here, we isolated PhMV-derived VLPs from ClearColi cells and carried out external and internal surface modification with fluorophores using reactive lysine-N-hydroxysuccinimide ester and cysteine-maleimide chemistries, respectively. The uptake of dye-labeled particles was tested in a range of cancer cells and monitored by confocal microscopy and flow cytometry. VLPs labeled internally on cysteine residues were taken up with high efficiency by several cancer cell lines and were colocalized with the endolysosomal marker LAMP-1 within 6 h, whereas VLPs labeled externally on lysine residues were taken up with lower efficiency, probably reflecting differences in surface charge and the propensity to bind to the cell surface. The infusion of dye and drug molecules into the cavity of the VLPs revealed that the photosensitizer (PS), Zn-EpPor, and the drugs crystal violet, mitoxantrone (MTX), and doxorubicin (DOX) associated stably with the carrier via noncovalent interactions. We confirmed the cytotoxicity of the PS-PhMV and DOX-PhMV particles against prostate cancer, ovarian and breast cancer cell lines, respectively. Our results show that PhMV-derived VLPs provide a new platform technology for the delivery of imaging agents and drugs, with preferential uptake into cancer cells. These particles could therefore be developed as multifunctional tools for cancer diagnosis and therapy.
Graphical abstract
INTRODUCTION
Nanocarrier platforms based on natural biological building blocks offer new opportunities in the biomedical and materials sciences.1 Virus nanoparticles (VNPs) are self-assembling supramolecular systems that can be used to develop bioinspired nanomaterials and nanocarriers due to their simple and inexpensive production, well-defined structural features, unique shapes and sizes, genetic programmability, and robust chemistries.1–6 VNPs based on plant viruses are particularly advantageous in medicine because they are biocompatible and biodegradable, but the plant viruses do not infect humans and other mammals. Such VNPs can carry drugs, imaging agents, and other nanoparticles in their internal cavity by assembly, infusion, or internal surface modification, and the external surface can be chemically or genetically engineered to attach targeting ligands for tissue-specific delivery.7–13 Plant VNPs have already overcome many of the challenges of nanoparticle delivery, such as low stability in biological fluids, and in some cases, targeting specificity.12–15 We and several others have established a broad range of plant VNPs such as those based on Cowpea mosaic virus (CPMV), Cowpea chlorotic mottle virus (CCMV), Brome mosaic virus (BMV), Potato virus X (PVX), and Tobacco mosaic virus (TMV), and further viruses are also being tested.6,12,13,16
Virus-like particles (VLPs) are a subset of VNPs that lack the viral genome and assemble spontaneously from virus structural proteins into noninfectious protein cage-like structures. Many different virus structural proteins form VLPs when expressed in standard heterologous expression systems such as Escherichia coli (E. coli),17 yeasts,18 plants,19 mammalian cells,20–22 and insect cells.23 Such VLPs tend to be structurally and morphologically similar to the wild-type virus particles and demonstrate similar cell tropism, uptake, and intracellular trafficking.3,13,24–27 Protein engineering can be used to introduce new functionalities at three distinct interfaces of VLPs: internal, external, and intersubunit. This allows the fine-tuning of surface charge, drug encapsulation, ligand display, and particle stability.28 VLPs have been developed as targeted delivery vehicles for drugs, siRNAs, RNA aptamers, proteins, and peptides, and as scaffolds to present epitopes in subunit vaccines.28–36 However, despite their advantages, few examples of plant VLPs have advanced through the development pipeline, and more work is required to make these structures amenable for specific biomedical applications.37–45
It is important to expand the library of VLPs under investigation from the current small collection of model viruses to facilitate the selection of useful properties for the delivery of drugs and imaging reagents. Although the list of VLPs under development is expanding, much less data are available compared to the myriad examples of synthetic nanoparticles including those made from lipids, polymers, and metals. Given the limited data available for VLPs, it is not yet possible to predict the in vivo fates of such particles using structural information and in silico approaches. For example, CPMV enables high-resolution vascular imaging when administered intravenously to mouse and chicken embryos,46 and this property was later attributed to interactions between the VLPs and vimentin.47 Whether other VLPs exhibit similar in vivo properties must be determined empirically until the volume of data is sufficient to make rational predictions. We decided to expand the VLP library by the investigation of VLPs based on Physalis mottle virus (PhMV) as a platform for imaging and drug delivery. PhMV was selected based on the availability of recombinant expression systems, its known stability in a range of aqueous environments, and its well-characterized structure, which is described below.
PhMV (Tymovirus, Tymoviridae) has a ~30 nm icosahedral capsid with T = 3 symmetry, containing a single-stranded, plus-sense RNA genome of 6.67 kb.48 The genome is encapsidated in a protein shell comprising 180 chemically identical 21 kDa coat protein subunits, with three distinct bonding patterns (A, B, and C). The A type subunits form 12 pentamers at the icosahedral five-fold axes (60 subunits), whereas the B and C type subunits form 20 hexamers at the icosahedral three-fold axes (120 subunits). The multiple copies of the asymmetric unit provide regularly spaced attachment sites on both the internal and external surfaces of the PhMV capsid. In earlier studies, the PhMV coat protein expressed in E. coli was shown to self-assemble into stable VLPs that were nearly identical to the viruses formed in vivo.49 These VLPs can be purified in large quantities (50–100 mg/L) and are exceptionally robust, maintaining their integrity within the pH range 4.2–9.0 and in the presence of up to 5 M urea. They are monodisperse, symmetrical, and polyvalent.49 Neither the deletion nor the addition of amino acids at the N-terminus of the PhMV coat protein hinders capsid assembly, making this an ideal site for modifications.50 The three-dimensional crystal structures of PhMV and its empty capsid have been determined to 3.8 and 3.2 Å resolution, respectively.51,52 The structures indicate that the empty shells correspond to the “swollen state” of the virus, with increased disorder in the N-terminal segments as well as some positively charged side chains lining the internal cavity.52
In earlier studies, PhMV-derived VLPs were genetically engineered to display diagnostic and immunogenic epito-pes.38,53,54 Here, we investigated the internal and external surface chemistries of PhMV and developed protocols to achieve the specific functionalization of these surfaces with different reagents. A library of functionalization protocols, including bioconjugation and noncovalent infusion was used to modify PhMV with dyes, drugs, and photosensitizers. These functionalized PhMV nanoparticles were then characterized by flow cytometry and confocal microscopy, and their cytotoxic efficacy was tested in a range of cell lines.
METHODS
Expression and Purification of PhMV VLPs
The 564-bp PhMV coat protein gene (GenBank S97776)55 was prepared as a synthetic construct by Invitrogen GeneArt, with XhoI and HindIII sites at the 5′ and 3′ ends, respectively, for insertion into pRSET-A (Invitrogen) at the same sites. The integrity of the recombinant vector (pR-PhMV-CP) was confirmed by automated DNA sequencing before the transformation of ClearColi BL21(DE3) cells. The expression and purification of PhMV VLPs were carried out as previously described49 with modifications. Briefly, a single colony carrying pR-PhMV-CP was inoculated into 50 mL of lysogeny broth (LB) containing 100 mg/mL ampicillin and was incubated for 20 h at 37 °C. We then used 5 mL of the preculture to inoculate 500 mL of LB with ampicillin as above. After 4–5 h of growth (OD600 nm = 0.6), expression was induced with 0.5 mM isopropyl-β-D-1-thiogalactopyranoside (IPTG), and the cells were incubated at 30 °C overnight. The culture was centrifuged (6000 rpm using a JLA 16.500 rotor, Beckman Coulter Inc., 10 min, 4 °C), and the pellet was suspended in 50 mM sodium citrate buffer, pH 5.5 (SCB). The suspension was then sonicated and centrifuged at 12 000 rpm using a JLA 16.200 rotor (Beckman Coulter Inc.) at 4 °C for 15 min. The supernatant was centrifuged at 35 000 rpm using a Ti 50.2 rotor (Beckman Coulter Inc.) at 4 °C for 3 h. The pellet was resuspended in SCB and layered onto a 10–40% linear sucrose gradient and centrifuged at 28 000 rpm in an SW 32 Ti rotor (Beckman Coulter Inc.) at 4 °C for 3 h. The light scattering zone was collected with a Pasteur pipet, diluted with SCB, and centrifuged at 42 000 rpm using 50.2 Ti rotor at 4 °C for 3 h. The final pellet was resuspended in SCB and stored at 4 °C. The protein concentration was measured using Bradford reagent (BioRad).
Bioconjugation Reactions
External lysine residues were conjugated to sulfo-Cy5 NHS ester (Lumiprobe), whereas internal cysteine residues were conjugated to Cy5.5-maleimide (Lumiprobe). The dyes were added to PhMV at a concentration of 1 mg/mL in KP buffer (0.01 M potassium phosphate buffer pH 7.0) at a molar excess of 900 Cy5 molecules per particle (five molecules per coat protein) and 360 Cy5.5 per particle (two molecules per coat protein). The final DMSO concentration was adjusted to 10% (v/v). The reaction was left for 2 h (Cy5) or overnight (Cy5.5) at room temperature with agitation in the dark. Both reaction mixtures were purified over a 30% (w/v) sucrose cushion by ultracentrifugation at 52 000 rpm using a TLA 55 rotor (Beckman Coulter Inc.) for 1 h. Pellets containing dye-labeled particles were resuspended in KP buffer overnight at 4 °C. For PhMV-KE-Cy5 particles, 10 kDa molecular weight cutoff (MWCO) centrifugal filters (Millipore) were also used to remove excess dye molecules. Any aggregates were removed by a clearing spin at 12 000 rpm for 10 min using a table-top centrifuge. For the biotin conjugation reactions, the VLPs were used at a final concentration of 1 mg/mL in KP buffer and were incubated with a 360-fold molar excess of biotin (biotin-NHS ester or biotin-maleimide) at room temperature overnight, with agitation. The final DMSO concentration was adjusted to 10% of the reaction volume. Particles were purified using 10 kDa MWCO centrifugal filter units (Millipore).
Infusion Protocol
The VLPs were loaded with rhodamine B, fluorescein, crystal violet, MTX dihydrochloride (all Sigma–Aldrich), PS (cationic zinc ethynylphenyl porphyrin, full name 5-(4-ethynylphenyl)-10,15,20-tris)4-methylpyridin-4-ium-1-yl)porphyrin zinc(II) triiodide), or DOX hydrochloride (Indofine Chemical Company). The VLPs (1 mg/mL in KP buffer) were incubated with a molar excess of 500, 2500, 5000, or 10 000 cargo molecules per particle overnight at room temperature in the dark, before purification over a 30% (w/v) sucrose cushion to remove excess reagents by ultracentrifugation at 52 000 rpm using a TLA 55 rotor (Beckman Coulter Inc.) at 4 °C for 1 h. PS-PhMV and DOX-PhMV were synthesized for further characterization using a 5000-fold molar excess.
UV–visible Spectroscopy
A NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) was used to characterize the UV/visible spectra of native and modified VLPs. The dye load was determined using the protein concentration (measured using the Bradford assay), the Beer–Lambert law and the following dye-specific extinction coefficients: rhodamine B, ε(553 nm) = 116 000 M−1 cm−1; DOX, ε(496 nm) = 11,500 M−1 cm−1; crystal violet, ε(590 nm) = 87 000 M−1 cm−1; PS, ε(450 nm) = 195 000 M−1 cm−1; MTX, ε(622 nm) = 25 000 M−1 cm−1; sulfo-Cy5 NHS ester, ε(646 nm) = 271 000 M−1 cm−1; Cy5.5-maleimide, ε(673 nm) = 209 000 M−1 cm−1. The following molecular weights were used: PhMV = 4.7 × 106 g mol−1; rhodamine B = 479.02 g mol−1; DOX = 579.98 g mol−1; crystal violet = 407.98 g mol−1; PS = 1130 g mol−1; mitoxantrone = 517.4 g mol−1; Cy5 = 777.95 g mol−1; Cy5.5 = 741.36 g mol−1; biotin NHS ester = 341.38 g mol−1; biotin maleimide = 451.54 g mol−1.
Native and Denaturing Gel Electrophoresis
Intact VLPs (10–20 μg per lane) were analyzed by 1% (w/v) agarose native gel electrophoresis in 0.1 M Tris-maleate running buffer (pH 6.5) as previously described.49 Denatured protein subunits (10 μg per lane) were analyzed by polyacrylamide gel electrophoresis using 4–12% NuPAGE gels and 1× MOPS buffer (Invitrogen). Samples were denatured by boiling in SDS loading dye for 10 min. Gels were photographed under UV or white light before staining with Coomassie Blue, and under white light after staining, using an AlphaImager system (Biosciences).
Size Exclusion Chromatography (SEC)
VLPs were analyzed by SEC using a Superose-6 column on the AkTA Explorer system (GE Healthcare). The column was loaded with 100 μL samples (1 mg/mL) at a flow rate of 0.5 mL min−1 in KP buffer.
Transmission Electron Microscopy (TEM)
VLPs suspended at 1 mg/mL in 20 μL of KP buffer were deposited onto Formvar carbon film coated copper TEM grids (Electron Microscopy Sciences) for 2 min at room temperature. The grids were then washed twice with deionized water for 45 s and stained twice with 2% (w/v) uranyl acetate (UAc) in deionized water for another 30 s. A Tecnai F30 transmission electron microscope was used to analyze the samples at 300 kV.
Zeta Potential Analysis
The zeta potential (ζ) of the VLPs was determined by placing 0.25 mg/mL solutions of each VLP in a 90Plus Zeta potential analyzer (Brookhaven Instruments) and conducting five measurements, each comprising six runs.
Avidin Agarose Affinity Binding Assay
Biotinylated VLPs and controls were tested for their ability to bind avidin agarose resin (Pierce). The batch method provided by the supplier was used, with some modifications: 100 μg samples in 100 μL of binding buffer (PBS with 0.1% SDS and 1% NP-40) were added to 100 μL of the resin, and the resulting 200 μL of slurry was mixed for 1 h at room temperature. The supernatant was then recovered and the resin washed six times in 100 μL of binding buffer. Bound VLPs were eluted in 100 μL of 0.1 M glycine-HCl buffer (pH 2.8), and the pH was immediately adjusted with 10 μL of 1 M Tris buffer (pH 7.5). Samples of the wash fractions and the eluate were analyzed by denaturing gel electrophoresis, 30 μL per lane.
Tissue Culture
All cell lines were obtained from the American Type Culture Collection (ATCC). HeLa (cervical cancer), RAW264.7 (leukemic macrophages), A2780 (ovarian cancer), MDA-MB-231 (breast cancer), and U87 (brain cancer) cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS, Atlanta Biologicals), 1% (w/v) penicillin/streptomycin (pen/strep, Thermo Fisher Scientific), and 1% (w/v) glutamine at 37 °C and 5% CO2. HT1080 (fibrosarcoma) cells were maintained in Minimum Essential Medium (MEM) supple mented with 10% (v/v) FBS, 1% (w/v) pen/strep, and 1% (w/v) glutamine as above. PC-3 cells (prostate cancer) were maintained in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% (v/v) FBS, 1% (w/v) pen/strep, and 1% (w/v) glutamine as above. NIH3T3 murine fibroblasts were maintained in DMEM/F12 medium containing 10% (v/v) newborn calf serum, 1% (w/v) pen/strep, and 1% (w/v) GlutaMax as above.
Confocal Microscopy
Cell lines were grown for 24 h on glass coverslips (25 000 cells per well) placed in an untreated 24-well plate in 200 μL of the appropriate medium. The cells were washed twice with Dulbecco’s PBS (DPBS) before addition of the PhMV-KE-Cy5 or PhMV-CI-Cy5.5 particles (2.5 × 106 particles per cell, corresponding to ~0.5 μg/well) and incubation for 6 h. The cells were washed twice in DPBS to remove unbound particles and fixed for 5 min at room temperature in DPBS containing 4% (v/v) paraformaldehyde and 0.3% (v/v) glutaraldehyde. Cell membranes were stained with 1 μg/mL wheat germ agglutinin conjugated to AlexaFluor-555 (Invitrogen) diluted 1:1000 in DPBS containing 5% (v/v) goat serum, and the cells were then incubated for 45 min at room temperature in the dark. Finally, the cells were washed three times with DPBS, and the coverslips were mounted on glass slides using Fluoroshield with 4′,6-diamidino-2-phenylindole (DAPI, Sigma–Aldrich) and sealed using nail polish. Confocal images were captured on a Leica TCS SPE confocal microscope, and the images were processed using ImageJ v1.44o.
For colocalization studies, A2780 and MDA-MB-231 cells were incubated for 6 h with PhMV-CI-Cy5.5 particles, then blocked in 10% (v/v) goat serum for 45 min to reduce nonspecific antibody binding. Endolysosomes were stained using a mouse antihuman LAMP-1 antibody (Biolegend) diluted 1:250 in 5% goat serum for 60 min, with DAPI staining and imaging as described above. VLPs were visualized by detecting the covalently attached Cy5.5 maleimide dye as described above.
Fluorescence-Activated Cell Sorting (FACS)
Cells were grown to confluency, collected in enzyme-free Hank’s-based cell dissociation buffer, and distributed in 200 μL aliquots at a concentration of 2 × 105 cell/mL in V-bottom 96-well plates. Dye-labeled VLPs (100 000 particles per cell, corresponding to ~1.6 μg/well) were added to the cells and incubated for 6 h. The cells were washed twice in FACS buffer (0.1 mL of 0.5 M EDTA, 0.5 mL of FBS, and 1.25 mL of 1 M HEPES, pH 7.0 in 50 mL of PBS without Ca2+ and Mg2+) and fixed in 2% (v/v) formaldehyde in FACS buffer for 10 min at room temperature. Cells were washed and resuspended in FACS buffer and analyzed using a BD LSR II flow cytometer. Triplicates of each sample were maintained, and at least 10 000 events (gated for live cells) were recorded. Data were analyzed using FlowJo v8.6.3.
LIVE/DEAD Assay
PC-3 cells were seeded (20 000 cells/500 μL RPMI/well) in a 24-well plate overnight. The cells were washed twice in PBS and incubated for 8 h in triplicates with 5.0 μM PS, 5.0 μM PS-PhMV, or the corresponding concentration of unloaded VLPs (236.70 μM or ~0.19 mg/mL). After washing twice in PBS, 500 μL of RPMI medium was added. Photodynamic therapy was then applied using a white light source (Phillips Silhouette High Output F39T5/841 HO, Alto collection, ~10 mW cm−2) for 30 min (18.1 J cm−2 at 430 nm), and cells were incubated for a further 48 h in the dark. Cell viability was determined using a LIVE/DEAD assay for mammalian cells (Thermo Fisher Scientific) following the manufacturer’s procedures for cell staining, and cells were observed under a Zeiss Axio Observer Z1 motorized FL inverted microscope.
MTT Cell Viability Assay
PC-3 cells were seeded (5000 cells/100 μL RPMI 1640/well) in a 96-well plate overnight. After two PBS washes to remove unbound and dead cells, free PS or PS-PhMV was added to the cells in triplicate at concentrations of 0.01, 0.025, 0.05, 0.1, 0.25, 0.5, 1, 2.5, and 5.0 μM PS and incubated for 8 h. Untreated cells and cells treated with unloaded VLPs at the equivalent protein concentration to PS-PhMV particles at the highest dose of PS were also used as controls. Free particles were washed with PBS, and 100 μL of fresh medium was added. The cells were then illuminated with white light for 30 min as above. In parallel, a duplicate plate was prepared and kept in the dark as a negative control. Following phototherapy, cells were incubated for another 48 h in the dark, and their viability was subsequently measured using an MTT cell proliferation assay kit (ATCC) based on the manufacturer’s instructions. A Tecan Infinite 200 PRO multimode plate reader was used to measure absorbance at 570 nm, and the percent cell viability was normalized to the untreated control. All assays were carried out at least three times. The efficacy of DOX-PhMV was tested by seeding A2780 and MDA-MB-231 cells as above (5000 cells/100 μL DMEM/well) and treating triplicate wells with free DOX or DOX-PhMV at concentrations of 0.01, 0.05, 0.1, 0.5, 1, 5.0, and 10.0 μM DOX for 24h. Untreated cells and cells treated with unloaded VLPs at a concentration equivalent to the highest dose of DOX-PhMV were used as controls. Washing steps and the MTT assay were then carried out as above.
RESULTS AND DISCUSSION
Purification and Characterization of PhMV-Derived VLPs Produced in ClearColi Cells
PhMV-derived VLPs were purified from ClearColi cells to avoid endotoxin contamination. The coat protein gene (564 bp) was inserted into the vector pRSET-A and expressed in ClearColi BL21(DE3). Optimum expression was achieved by inducing the culture with 0.5 mM IPTG and cultivating at 30 °C overnight. SDS-PAGE analysis confirmed the presence of the coat protein in total and soluble cell protein extracts (Figure S1A, lanes 1, 2), and VLPs were purified from the soluble fraction.49 A single light-scattering zone was observed in a 10–40% (w/v) linear sucrose density gradient (Figure S1B) and the VLP yield was 40–50 mg per liter of culture medium, as determined using the Bradford assay. Previous studies indicate that PhMV-derived VLPs expressed in E. coli contain nucleic acids and encapsidate mRNAs encoding the PhMV coat protein.49 To investigate whether PhMV VLPs used in this study contained RNA, we analyzed VLPs after electrophoretic separation on agarose gels stained with GelRed stain followed by visualization under UV light; indeed, the nucleic acid stain indicated that the VLPs contained nucleic acids (presence of the protein capsid was confirmed by Coomassie staining and visualization under white light, Figure S2).
The denatured VLP preparation revealed a single 26 kDa band corresponding to the coat protein (Figure S1C). Size exclusion chromatography (SEC) analysis confirmed that the particles eluted as a single peak at 7.5 mL, indicating they were intact and stable (Figure S1D). Transmission electron microscopy (TEM) revealed that the VLPs were approximately spherical and 29 ± 2 nm in diameter (Figure S1E), indicating that recombinant PhMV coat proteins are indeed capable of self-assembly. The zeta potential of the VLPs was +4.20 ± 0.46 mV (Table 1).
Table 1.
Zeta Potential Measurement of Functionalized PhMV Particles
| sample | zeta potential (standard error in parentheses) |
|---|---|
| native PhMV | +4.20 mV (0.46 mV) |
| PhMV-KE Cy5 | −7.92 mV (2.49 mV) |
| PhMV-CI Cy5.5 | +0.38 mV (3.32 mV) |
| DOX-PhMV | +9.38 mV (1.93 mV) |
| PS-PhMV | −0.81 mV (3.08 mV) |
Structure-Based Design of PhMV-Derived VLPs To Carry Dyes and Drugs
A reliable PhMV-based platform for chemical modification requires the identification of attachment sites on the capsid that do not compromise the structure of the asymmetric unit and its native biological functions but nevertheless allow for efficient bioconjugation reactions. The bioconjugation sites on the internal and external surfaces of the VLP were chosen by studying the structural contribution of each residue. Nine lysine residues are present on each PhMV coat protein subunit, four of which (K62, K143, K153, and K166) are exposed on the exterior, resulting in 720 addressable lysine residues per VLP that can be used for bioconjugation based on lysine/N-hydroxysuccinimide (NHS) ester chemistry (Figure 1A). The coat protein contains only a single cysteine residue (C75), and this is presented on the internal surface, resulting in 180 addressable cysteine residues per VLP potentially suitable for bioconjugation using thiol-maleimide chemistry (Figure 1A). As well as the development of bioconjugation protocols (Figure 1B, C), we also considered the encapsulation of cargo molecules in the cavity. The encapsulation of dyes and drugs via particle disassembly and assembly is not possible in the case of PhMV because the virus is stabilized predominantly by protein–protein interactions.56 We therefore developed an infusion protocol to load the VLPs with cargo (Figure 1D,E).
Figure 1.
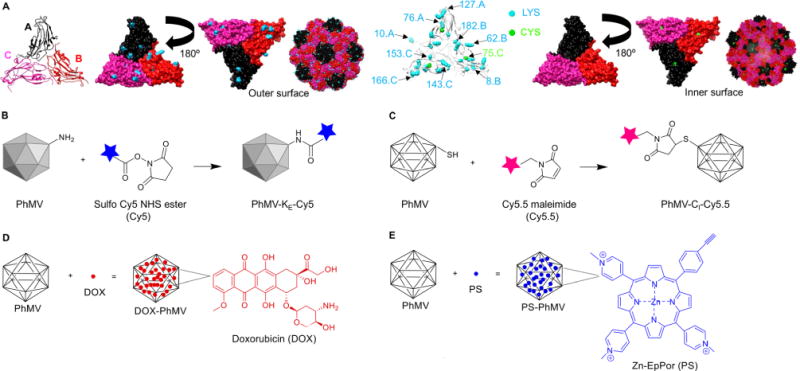
Structure of PhMV and the strategies used for the functionalization of PhMV-derived VLPs. (A) Ribbon diagram of the PhMV VLP icosahedral asymmetric unit consisting of A, B, and C subunits. Five A subunits make up pentameric capsomeres at the icosahedral five-fold axes, whereas B and C subunits form hexamers at the icosahedral three-fold axes. The A subunit is shown in black, the B subunit is shown in red, and the C subunit is shown in pink. Representation of internal and external surfaces (with 180° rotation) of the PhMV asymmetric unit, highlighting surface-exposed (K62, K143, K153, and K166) and buried (K8, K10, K76, K182, and K127) lysine residues (blue), and the single cysteine residue (C75, green). PhMV forms from 180 identical coat protein subunits arranged in a T = 3 icosahedral structure. Images created using UCSF Chimera (PDB: 1E57). The capsid is characterized by prominent protrusions of pentamers and hexamers. (B) Schematic of PhMV labeling with sulfo-Cy5 NHS ester (blue) using lysine-NHS ester chemistry. (C) Conjugation of Cy5.5-maleimide (pink) to internal cysteine residues using maleimide–thiol chemistry. (D) DOX (red) infusion into PhMV, leading to cargo-loaded particles. Washing and ultracentrifugation are used to remove excess DOX, yielding intact PhMV with infused DOX (DOX-PhMV). (E) Schematic of PS (blue) loading into PhMV via infusion, yielding PS-PhMV particles.
Bioconjugation of PhMV-Derived VLPs with Dyes
Surface-exposed lysine residues were conjugated to NHS-activated esters of sulfo-cyanine 5 succinimidyl ester (Cy5) by incubating for 2 h with a 900-fold molar excess of the dye, equivalent to five dye molecules per coat protein (Figure 1B). Similarly, the thiol groups on the internal cysteine residues were conjugated overnight using Cy5.5-maleimide (Cy5.5) at a 360-fold molar excess, equivalent to two dye molecules per coat protein (Figure 1C). The resulting VLP–dye conjugates (PhMV-KE-Cy5 and PhMV-CI-Cy5.5, where KE = external lysine and CI = internal cysteine) were purified by ultra-centrifugation, and PhMV-KE-Cy5 was purified further by ultrafiltration to remove free dye molecules. The Bradford protein assay was used to estimate the concentration of PhMV-KE-Cy5 and PhMV-CI-Cy5.5 particles. UV–visible spectroscopy was used to determine the number of dye molecules per particle based on the Beer–Lambert law and dye-specific extinction coefficients, revealing that the PhMV-KE-Cy5 particles contained 160–180 Cy5 molecules and the PhMV-CI-Cy5.5 particles contained 40–60 Cy5.5 molecules. The higher labeling capacity using lysine-NHS chemistry was expected due to the presence of 720 surface-exposed lysine residues compared to 180 internal cysteine residues.
Increasing the molar excess of Cy5.5-maleimide did not increase the internal labeling density, and maximum labeling efficiency was achieved with two dye molecules per coat protein. It may be possible to increase interior conjugation capabilities of the PhMV VLPs further by removing the nucleic acids (coat protein mRNA from the expression host, Figure S2),52,51 but this was not investigated in the present study. In contrast, we achieved a greater density of external labeling when the molar excess of sulfo-Cy5 was increased to 20 dye molecules per coat protein (Figure S3) but further increases were not tested because the dye aggregated at higher concentrations. For imaging applications, the spatial distribution of dye molecules is important, so we measured the distance between the surface-exposed lysine side chains using Chimera software and found the spacing lies between 1 and 5 nm (Table S1). However, if we assume random distribution of the Cy5 molecules conjugated to the external lysine side chains, the average distance between two fluorophores would be 2–4 nm assuming 1–2 dyes per coat protein. On the basis of the Foerster radius, which suggests quenching generally occurs when dyes are <10 nm apart, we would expect quenching to occur for the labeled VLPs due to cross-talk between the fluorophore centers.57–59 Nevertheless, we experienced no fluorescence detection issues, and in any case the dyes on these particles are likely to be cleaved from the VLP when taken up by cells, as previously shown.60 Further structure–function studies would be required for the development of fluorophore-labeled PhMV VLPs as optical probes.
We characterized the PhMV-KE-Cy5 and PhMV-CI-Cy5.5 particles using a combination of native and denaturing gel electrophoresis, zeta potential analysis, FPLC, and TEM. Native and denaturing gel electrophoresis followed by visualization under white light before Coomassie staining confirmed the covalent attachment of the dyes (Figure 2A,B; lanes 2, 3). The charge of the PhMV-KE-Cy5 particles was altered following bioconjugation, as evident from the mobility shift toward the anode during native electrophoresis (Figure 2B, lane 2). This was anticipated because the addition of the noncharged dye removes positive amines from the particle surface and thus reduces the overall positive surface charge. Accordingly, zeta potential measurements revealed that the PhMV-KE-Cy5 particles were negatively charged (−7.92 mV), whereas native PhMV particles were positively charged (+4.20 mV) (Table 1). In contrast, the mobility of the PhMV-CI-Cy5.5 particles was the same as wild-type PhMV because cysteine residues are uncharged (Figure 2B, lane 3). Zeta potential measurements nevertheless indicated a reduction in the net positive charge of the PhMV-CI-Cy5.5 particles (+0.38 mV), probably reflecting changes in surface charge distribution.
Figure 2.
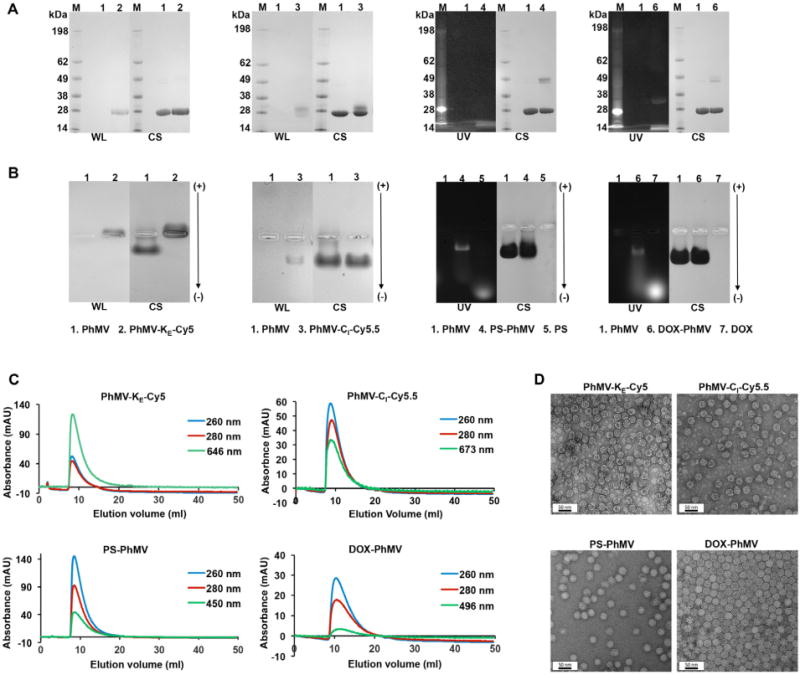
Characterization of fluorophore-labeled and drug-loaded VLPs. (A) SDS-PAGE analysis of PhMV-KE-Cy5, PhMV-CI-Cy5.5, PS-PhMV, and DOX-PhMV visualized under UV light (UV), white light (WL) before staining, and under white light after Coomassie blue staining (CS). M = SeeBlue Plus2 molecular weight (kDa) standard; 1 = Native PhMV; 2 = PhMV-KE-Cy5; 3 = PhMV-CI-Cy5.5; 4 = PS-PhMV; 5 = PS; 6 = DOX-PhMV; 7 = DOX. (B) Agarose gel electrophoresis of PhMV-KE-Cy5, PhMV-CI-Cy5.5, PS-PhMV, and DOX-PhMV visualized under UV light and white light before, and under white light after Coomassie blue staining (CS). Functionalized particles loaded in each lane was as described above for SDS-PAGE analysis (note: the white marks in the center of the gel are the pockets into which the samples were loaded prior to electrophoretic separation). (C) Size exclusion chromatograms of PhMV-KE-Cy5 [monitored at 260 nm (blue), 280 nm (red), and 646 nm (green), sulfo-Cy5 NHS ester absorbance], PhMV-CI-Cy5.5 [monitored at 260 nm (blue), 280 nm (red), and 673 nm (green), Cy5.5-maleimide absorbance], PS-PhMV [monitored at 260 nm (blue), 280 nm (red), and 450 nm (green), PS absorbance] and DOX-PhMV [monitored at 260 nm (blue), 280 nm (red), and 496 nm (green), DOX absorbance]. (D) Transmission electron micrographs of negatively stained (UAc) PhMV-KE-Cy5, PhMV-CI-Cy5.5, PS-PhMV, and DOX-PhMV.
FPLC and TEM analysis indicated that the dye-labeled VLPs were intact. Dye-labeled particles eluted as a single peak from the Superose-6 column at an elution volume of 7.5 mL, the same as wild-type particles, and the fluorophore coeluted with the PhMV-KE-Cy5 and PhMV-CI-Cy5.5 particles at 646 and 673 nm, respectively (Figure 2C; top panel). TEM analysis revealed that the labeled particles remained monodisperse, with a diameter of 28–29 nm based on ImageJ analysis (Figure 2D; top panel). Finally, the labeled particles remained stable when stored at 4 °C in KP buffer for several months (Figure S4A, B).
Spatial Distribution of Biotin in VLP–Biotin Conjugates
To confirm the position of the bioconjugation sites, we modified each site with biotin labels and mapped them using an avidin bead assay. This was necessary because although maleimide chemistry is thiol selective, some reports indicate cross-reactivity with lysine residues.41 External lysine residues were therefore labeled using a 360-fold molar excess of a NHS-reactive biotin probe to generate PhMV-KE-bio particles, and internal cysteine residues were labeled using a 360-fold molar excess of a maleimide-reactive biotin probe to generate PhMV-CI-bio particles. The resulting particles were analyzed by native and denaturing gel electrophoresis. As expected, we observed no difference in mobility between labeled and unlabeled particles in the denaturing gels (Figure 3A), whereas the mobility of PhMV-KE-bio but not PhMV-CI-bio differed from the unlabeled particles in the native gels due to the elimination of positive surface charges, consistent with the behavior of the dye-labeled particles above (Figure 3B).
Figure 3.
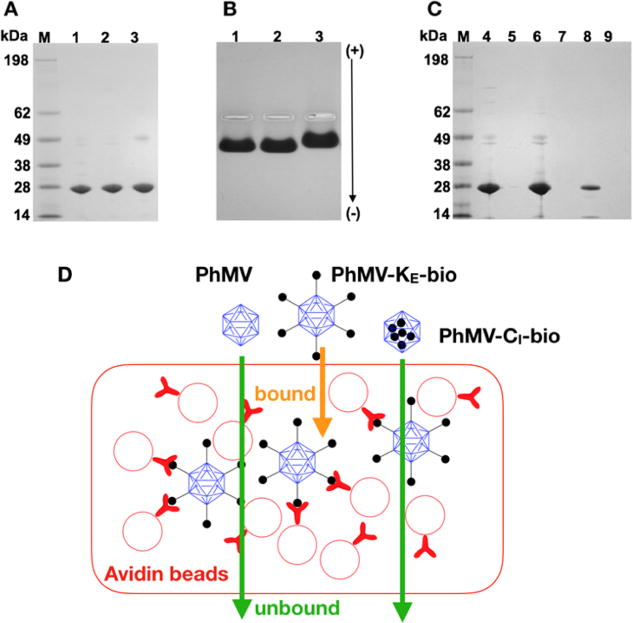
Characterization of PhMV-biotin conjugates. (A) Biotinylated PhMV particles separated by denaturing SDS-PAGE visualized after staining with Coomassie. M = SeeBlue Plus2 molecular weight marker. (1) Native PhMV; (2) PhMV-CI-bio; (3) PhMV-KE-bio. (B) Biotinylated PhMV particles separated by agarose gel electrophoresis visualized after Coomassie staining. (C) Flow through and eluted biotinylated particles from avidin bead binding assay separated by SDS-PAGE and stained with Coomassie. (4) Native PhMV flow through; (5) PhMV-KE-bio flow through; (6) PhMV-CI-bio flow through; (7) bound native PhMV; (8) bound PhMV-KE-bio; (9) bound PhMV-CI-bio. (D) Avidin bead assay: PhMV samples are exposed to avidin-coated beads; only particles with biotin on the external surface bind to the beads.
The avidin bead assay was used to selectively capture VLPs displaying biotin on the external surface. The avidin agarose beads were mixed with unlabeled VLPs, PhMV-KE-bio particles, or PhMV-CI-bio particles. The beads were then washed, and the particles were eluted in acidic buffer to disrupt avidin–biotin binding. The flow through from the washing steps and the eluent after acid treatment were collected and analyzed on a denaturing gel (Figure 3C). As expected, unlabeled VLPs and PhMV-CI-bio particles were detected in the flow through, indicating these formulations did not interact with the avidin agarose beads (Figure 3C, lanes 4, 6), whereas the externally labeled PhMV-KE-bio particles were not detected in the flow through (Figure 3C, lane 5) but were detected in the eluate (Figure 3C, lane 8). These data confirm that maleimide chemistry was selective in our experiments and that biotin labels in the PhMV-CI-bio particles were attached solely to the internal cysteine residues.
Cellular Internalization
The PhMV-KE-Cy5 particles were incubated with ovarian cancer (A2780), breast cancer (MDA-MB-231), and prostate cancer (PC-3) cell lines. Qualitative data were acquired by confocal microscopy (Figure 4A) and quantitative data by fluorescence activated cell sorting (FACS) (Figure 4B). We found that the PhMV-KE-Cy5 particles were internalized by all three cell lines with high efficiencies, given that the mean fluorescence intensity (MFI) of cells after incubation with the VLPs increased by more than three-fold (see inset in Figure 4B). The PhMV-CI-Cy5.5 particles were then incubated with a panel of cancer cell lines (as well as macrophages and fibroblast cells), and data were acquired as above. Again, we found that particles were taken efficiently up by most of the cancer cell lines, with the MFI increasing by 10–46-fold after VLP exposure. Although most of the cell lines efficiently internalized the VLPs, no internalization was observed for the HT1080 fibrosarcoma cells (Figure 4D) perhaps due to differences in cell surface proteins or metabolic rates compared to the other cells. The fibroblast cell line NIH/3T3 also showed inefficient particle uptake (Figure 4D). Particle uptake was also observed in RAW 264.7 macrophages as expected given their function as phagocytic cells.
Figure 4.
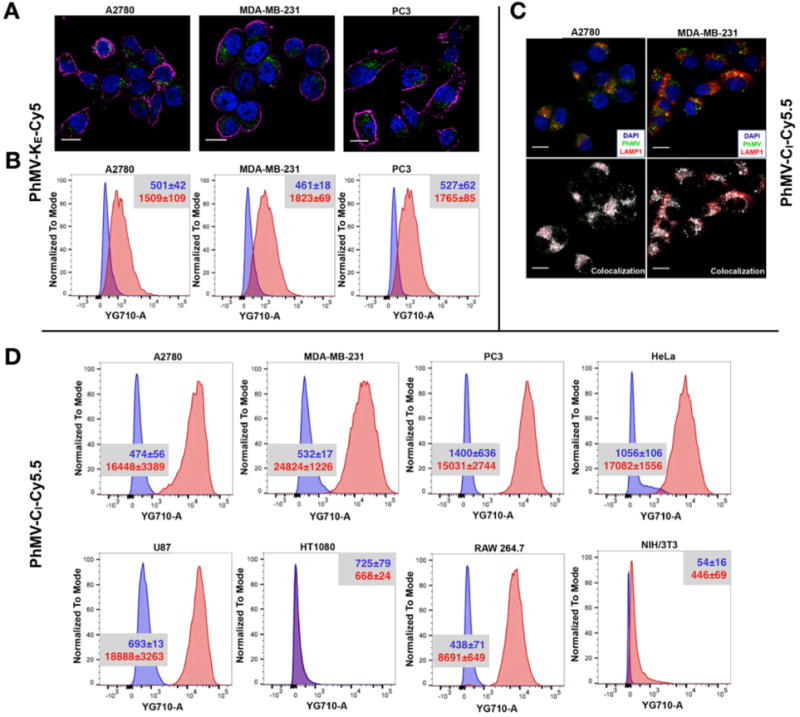
Cell uptake studies with fluorescence-labeled PhMV analyzed by confocal microscopy and FACS. (A) Confocal images representing the internalization of PhMV-KE-Cy5 by A2780, MDA-MB-231, and PC-3 cells. PhMV was tagged with sulfo-Cy5 NHS ester (pseudo green), the cell membrane was stained with wheat germ agglutinin (WGA)-Alexa Fluor 555 (pseudo pink), and the nucleus was stained with DAPI (blue). Scale bars = 25 μm. (B) Flow cytometry of A2780, MDA-MB-231, and PC-3 cells following 6 h incubation with PhMV-KE-Cy5 particles. Inset provides mean fluorescence intensities (MFIs) of cells for each sample quantified from three replicates with standard deviations (±) in the corresponding cell panels; blue = cells only, red = after incubation with VLPs. (C) Confocal imaging of A2780 and MDA-MB-231 cells showing colocalization of PhMV-CI-Cy5.5 particles with the endolysosomal marker LAMP-1 after 6 h. Nuclei are shown in blue, endolysosomes are stained with mouse antihuman LAMP-1 antibody (red) and PhMV-CI-Cy5.5 (pseudo green). Colocalization signals are shown in white (overlay, bottom panel). Scale bars = 25 μm. (D) FACS quantification of PhMV-CI-Cy5.5 uptake using A2780, MDA-MB-231, PC-3, HeLa, U87, HT1080, RAW 264.7, and NIH/3T3 cells. Inset provides mean fluorescence intensities (MFIs) of cells for each sample quantified from three replicates with standard deviations (±) in the corresponding cell panels; blue = cells only, red = after incubation with VLPs. All samples were measured in triplicates and analyzed using FlowJo software.
Cellular uptake was confirmed by confocal microscopy and cell surface staining using A555-labeled wheat germ agglutinin. The analysis of confocal images using ImageJ software confirmed the internalization of PhMV-CI-Cy5.5 particles (Figure S5). In A2780 and MDA-MB-231 cells, we confirmed that the internalization of PhMV-CI-Cy5.5 particles involved endocytosis, and the particles colocalized with the endolysosomal marker LAMP-1 (Figure 4C).
These results indicate that PhMV-CI-Cy5.5 particles are the most suitable candidates for future in vivo imaging and tumor homing studies among the particles we tested. The efficient internalization of these particles by cancer cells probably reflects the retained positive surface charge (+0.38 mV) as revealed by native gel electrophoresis (Figure 2B, lane 3). In contrast, PhMV-KE-Cy5 particles are taken up less efficiently due to the neutralization of positively charged lysine residues by NHS esterification (Figure 2B, lower panel, lane 2), resulting in a net negative charge of −7.92 mV lower panel, lane 2), resulting in a netthat would repel like charges on the cell membrane.
VLP Infusion and Characterization of Loaded Particles
The potential of the VLPs to encapsulate cargo molecules was tested using the tracer dye rhodamine B, fluorescein, the cancer drug doxorubicin (DOX), the anthelmintic drug crystal violet,61 the photodynamic therapeutic photosensitizer (PS) cationic zinc ethynylphenyl porphyr-in,62,63 and the cancer drug mitoxantrone (MTX), most of which are positively charged and fluorescent (exceptionally, fluorescein is neutral), allowing quantification by UV–visible spectroscopy based on absorbance.
Intact VLPs were incubated in a bathing solution containing the guest molecule at various molar excesses (500, 2500, 5000, and 10 000 molecules per VLP) in KP buffer with 10% (v/v) DMSO overnight at room temperature. After the reaction, excess guest molecules were removed by ultracentrifugation and the amounts of protein and cargo were quantified using the Bradford assay and by UV–visible spectroscopy, respectively. We applied the Beer–Lambert law and the specific extinction coefficient of each guest molecule to determine the number of cargo molecules per VLP (Figure 5). The VLPs showed the greatest affinity for crystal violet (2000 molecules per particle) and the lowest affinity for rhodamine B, fluorescein, and PS (~200 molecules per particle). The VLPs showed intermediate affinities for DOX and MTX, with 750 DOX and 450 MTX molecules per particle, respectively (Figure 5). The differences in loading probably reflect the density and distribution of charged and hydrophobic groups on the guest molecules. Furthermore, PhMV VLPs contain nucleic acids (coat protein mRNA packaged from the expression host, Figure S2) so differences in loading efficiency may also reflect the different affinities of the cargo molecules for nucleic acids.
Figure 5.
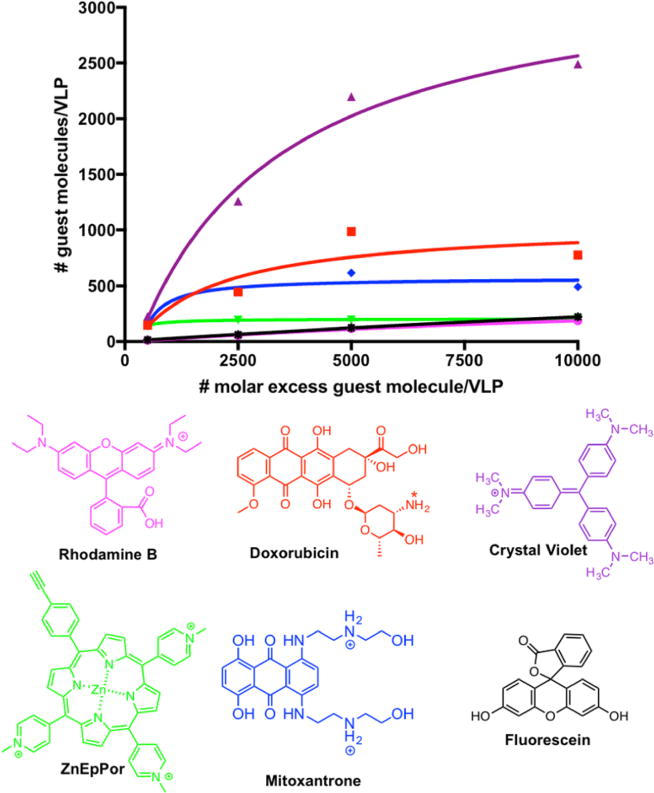
Number of dye/drug (guest) molecules loaded per VLP via infusion at different molar excesses (averaged data from two experiments are shown). The number of guest molecules per particle was determined by UV/vis absorbance and the Bradford assay was used to determine the protein concentration. The chemical structure of each guest molecule is depicted with their corresponding charge. The amine group of DOX is annotated with asterisks to indicate a site of protonation (positive charge) under physiological conditions.
Purification of the drug-loaded VLPs achieved 50–60% recovery. Two formulations (PS-PhMV and DOX-PhMV) were taken forward for complete characterization and testing for in vitro cargo delivery and cytotoxic efficacy. We used a 5000-fold molar excess of PS or DOX per particle for further experiments because this facilitated efficient loading without significant aggregation. Extending the incubation time did not increase the loading efficiency at this molar ratio (data not shown). We consistently observed the loading of 160–180 PS molecules and 600–800 DOX molecules per VLP (Figure 5).
A combination of denaturing and native gel electrophoresis and FPLC was used to confirm that PS and DOX were loaded into the cavity of the VLPs noncovalently. Gels were visualized under UV light before Coomassie staining and under white light afterward. In native gels, the PS-PhMV and DOX-PhMV coat proteins comigrated with the native PhMV coat protein. In contrast, denaturation released the encapsulated cargo (PS or DOX), which has a high mobility due to the low molecular weight of each molecule (PS = 1130 g mol−1, DOX = 579.98 g mol−1) resulting in a fluorescent buffer front (Figure 2A, lanes 4, 6). However, the DOX-PhMV coat proteins (~26 kDa) also showed evidence of fluorescence, indicating that some DOX remained associated with the protein even under denaturing conditions within the electrophoretic field (Figure 2A, lane 6). This may reflect hydrophobic interactions between DOX and aromatic amino acids on the VLP internal surface. Anthracycline drugs such as DOX bind to proteins such as human serum albumin and human α-1 acid glycoprotein via hydrophobic interactions that are stabilized by hydrogen bonds.64,65 We have previously proposed that DOX associates with PVX particles via hydrophobic interactions, specifically involving π–π stacking between the benzene rings of DOX and nonpolar aromatic amino acids such as phenylalanine, tyrosine, and tryptophan.66 Likewise, DOX can also interact via π–π stacking with other DOX molecules. After Coomassie staining, the PS-PhMV and DOX-PhMV coat proteins appeared as two bands representing the monomer and a dimer, indicating some degree of aggregation (Figure 2A, lanes 4, 6). However, interparticle aggregation was not apparent during FPLC and TEM analysis (see below).
Native agarose gel electrophoresis was used to confirm that PS and DOX were associated with the VLPs. After separating the loaded particles, the gels were visualized under UV light, stained with Coomassie, and imaged under white light (Figure 2B). Both particles showed fluorescence under UV light, indicating stable association with PS and DOX. Notably, trace amounts of free PS and DOX were apparently detected in the native gels whereas no free cargo was detected by FPLC (Figure 2B,C). We attribute this phenomenon to the electrophoretic separation of the cargo and carrier, followed by their independent migration through the electric field. The FPLC analysis of freshly prepared samples as well as samples that had been stored for weeks or months indicated that the compounds were indeed stably associated with the particles; therefore, no free PS or DOX was detected by this method (Figures 2C and S4).
Size exclusion chromatography (SEC) using FPLC and a Superose-6 column showed the typical elution profiles for intact drug-loaded VLPs, consistent with the elution profile of native PhMV (Figure 2C; bottom panel). The FPLC profiles also indicated the coelution of PS and DOX at 450 and 496 nm, respectively, confirming the successful loading of these molecules. Finally, TEM analysis confirmed the integrity of the particles after loading with PS or DOX. The TEM images clearly showed that the approximately spherical structure of the wild-type particles was unchanged after loading, and there was no significant change in size from the wild-type particle diameter of 28–29 nm (Figure 2D; bottom panel). The stability of the loaded VLPs was tested again by FPLC after storage for several weeks or months in KP buffer at 4 °C. The elution profiles did not change after storage, confirming that the cargo was stably encapsulated, and there was no evidence of particle aggregation (Figure S4C,D). Infusion therefore appears to be a suitable approach for the loading of cargo molecules into PhMV-based VLPs.
Efficacy of Photodynamic Therapy Using PS-PhMV Particles against PC-3 Cells
The delivery of PS-PhMV particles and their cytotoxic efficacy in vitro were evaluated in PC-3 cells because efficient internalization had already been demonstrated in these cells (Figures 4D and S5) and photodynamic therapy is a promising approach for the treatment of prostate cancer.67 To evaluate the efficacy of PS-PhMV compared to free PS, we incubated the cells with each reagent for 8 h, with concentrations of PS ranging from 0.01–5.0 μM. Treatment was induced by exposing the cells to white light, whereas controls were kept in the dark. Unloaded VLPs were used as additional controls, at the same protein concentration as the highest dose of drug-loaded particles. Cell viability was measured using an MTT assay to assess metabolic activity. We found that the efficacy of free PS (IC50 = 0.05 μM) was not significantly affected by encapsulation: the IC50 value of the PS-PhMV particles was 0.03 μM (Figure 6A). PS-PhMV controls maintained in the dark showed no evidence of cytotoxicity, nor did the unloaded VLPs, demonstrating the biocompatibility of the PhMV platform technology. The performance of the PS-PhMV particles was confirmed by setting up a LIVE/DEAD cell viability assay in 24-well plates. As above, confluent PC-3 cells were incubated for 8 h with 5.0 μM free PS or PS-PhMV (or corresponding controls), a concentration that achieved maximum cytotoxicity in the MTT assay. After any remaining extracellular PS/PS-PhMV was removed, white light was applied for 30 min. The cells were incubated overnight in the dark, then stained with a combination of calcein-AM and ethidium homodimer-1 to detect living and dead cells, respectively (Figure 6B). As above, PS and PS-PhMV controls maintained in the dark remained 100% viable, as did untreated cells and illuminated cells exposed to unloaded VLPs. In contrast, illuminated cells exposed to 5.0 μM PS or PS-PhMV showed 0% viability.
Figure 6.
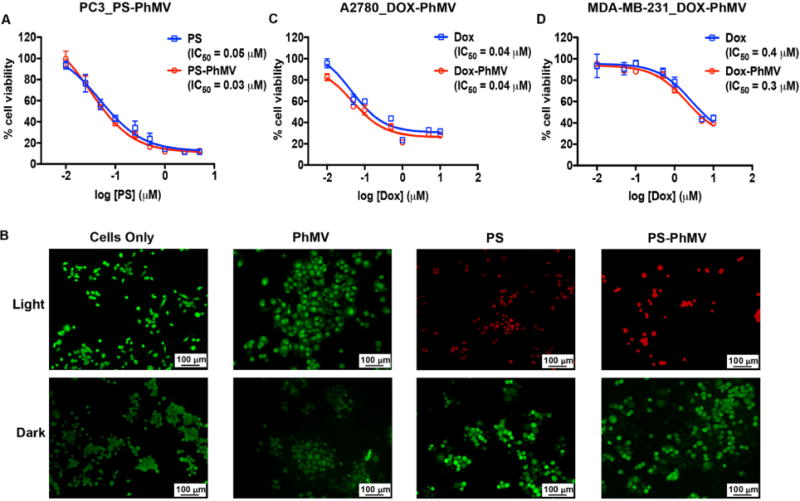
Evaluation of the cytotoxic efficacy of drug-loaded PhMV particles. (A) MTT cell viability assay of PC-3 cells using PS-PhMV. Cell viability was measured following 8 h incubation with varying concentrations of PS or PS-PhMV and 30 min illumination with white light (no cytotoxicity was observed when cells were incubated in the dark, not shown). (B) LIVE/DEAD assay of PC-3 cells showing representative images after photodynamic therapy of cells incubated with PS-PhMV or free PS and LIVE/DEAD cell staining. Calcein-AM staining of live cells is shown in green, and ethidium homodimer-1 staining of dead cells is shown in red. Scale bar = 100 μm. Illuminated cells incubated with PS-PhMV showed a slight increase in cytotoxic efficacy (IC50 = 0.03 μM) compared to free PS (IC50 = 0.05 μM). Dark controls show no cytotoxicity with PS-PhMV or PS. Scale bar = 100 μm. (C, D) Efficacy of DOX-PhMV (red line) versus DOX (blue line) using A2780 (human ovarian cancer) and MDA-MB-231 (human breast cancer) cells as determined by MTT assay. Cells were treated with DOX or DOX-PhMV corresponding to 0, 0.01, 0.05, 0.1, 0.5, 1, 5, and 10 μM for 24 h. IC50 values were determined using GraphPad Prism software.
These data are consistent with our previous reports in which CPMV or TMV were used to deliver PS.62,63 Other examples include dual-surface modified bacteriophage MS2 capsids encapsulating PS and carrying an external Jurkat-specific aptamer, resulting in the selective killing of Jurkat leukemia T cells.68 Similarly, the simultaneous modification of bacteriophage Qβ VLPs with a metalloporphyrin-based PS and a glycan ligand achieved the specific targeting of CD22+ cells.69
DOX-PhMV Delivery to Breast and Ovarian Cancer Cells
Finally, we investigated whether DOX retained its cytotoxic activity in the context of DOX-PhMV particles by exposing A2780 ovarian cancer cells and MDA-MB-231 breast cancer cells to a range of concentrations of free DOX (0.1 to 10 μM) and equivalent concentrations of DOX in the context of DOX-PhMV particles. We chose breast cancer and ovarian cancer cells because DOX chemotherapy is used for first-line and adjuvant treatment in women with breast or ovarian cancer.70 We found that the cytotoxicity of free DOX was not significantly affected by encapsulation. The IC50 value of the DOX-PhMV particles was 0.3 μM in the breast cancer cells and 0.04 μM in the ovarian cancer cells, equivalent to the corresponding values for the free drug (Figure 6C,D). In both cell lines, the unloaded VLPs showed no apparent cytotoxicity, as determined using an MTT assay, at the protein concentration corresponding to the highest dose of DOX-PhMV, confirming the biocompatible nature of the VLP delivery platform. On the basis of the cell uptake and imaging data, we propose that PhMV is taken up by endocytosis and is trafficked to the lysosome where the protein carrier is degraded,28 thus releasing the guest molecule (DOX in this case) that diffuses into the cytosol and kills the cell.
DOX is a highly potent drug used for cancer treatment, but delivery vehicles are required to overcome its dose-limiting toxicity toward healthy cells. Nonviral and viral delivery systems are undergoing preclinical and clinical testing.36,71 Although each carrier system has advantages and disadvantages, VLPs are robust, monodisperse, easy and inexpensive to produce, biocompatible, biodegradable, and noninfectious; they are also readily engineered for the site-specific introduction of new functionalities by genetic modification or bioconjugation, which can increase their solubility, reduce their immunogenicity, allow specific cell targeting, increase the efficiency of internalization, and increase their potency.36,72 Several plant viruses have been used to encapsulate DOX by infusion though gating or simple diffusion and caging mechanisms. For example, the depletion of divalent cations (Ca2+ and Mg2+) induces significant conformational changes in the Red clover necrotic mosaic virus (RCNMV) capsid, leading to the reversible formation of pores that allow the ingress of dyes and DOX molecules into the internal cavity, where they bind to the negatively charged virus genome.73 RCNMV was thus loaded with DOX and armed with targeting peptides as a multifunctional tool to target and deliver cargo to cancer cells.74 Another example is encapsulated DOX nanoconjugates targeted to folate-expressing cancer cells in vivo using the Cucumber mosaic virus (CMV) platform. The authors demonstrated reduced cardiotoxicity and increased antitumor responses compared to the free drug.75 We have demonstrated that CPMV, PVX, and TMV achieve the efficient delivery of DOX and targeted cell killing using bioconjugation and encapsulation protocols.66,76,77
Although the reports discussed above used replication-competent viruses that retain their genomic RNA, VLP platforms lack an infectious genome and thus offer a safer alternative. For example, the disassembly and reassembly of Hibiscus chlorotic ringspot virus (HCRSV) facilitated the encapsulation of anionic polyacids and polyacid-conjugated DOX in which loading was dependent on electrostatic interactions, generating HCRSV-derived VLPs suitable for drug delivery.37,78
Animal virus-based VLPs have also been studied and developed for DOX delivery. Self-assembled VLPs based on the rotavirus structural protein VP6 were chemically conjugated with DOX and lactobionic acid to specifically target the hepatoma cell line HepG2.79 VLPs based on Rous sarcoma virus displaying the single chain fragment variable (scFv) antibody hcc49 were produced in silkworm larvae and loaded with DOX and demonstrated the ability to kill colon carcinoma cells in vitro.80 Truncated hepatitis B virus core protein (tHBcAg) loaded with DOX and poly(acrylic acid) demonstrated a sustained drug release profile in vitro in colorectal cancer cells, leading to enhanced antitumor effects.81 Again, the studies described here and the growing body of data in the literature support the further development and testing of virus-based drug carriers.
CONCLUSIONS
In summary, we have developed bioconjugation chemistries and infusion protocols that enable the functionalization of PhMVbased VLPs, thus providing multiple approaches to modify the behavior and properties of the corresponding particles. The PhMV-derived VLPs are stable and inexpensive to produce, allowing their development as nanocarriers for in vitro drug delivery applications. The physical stability and batch-to-batch consistency are advantageous because the functionalized VLPs remain stable for long periods in storage. The production of PhMV-based VLPs could be scaled up by increasing the capacity of bacterial fermentation, but even greater scalability could be achieved by expression in plants. PhMV does not replicate in mammals, but is biocompatible and biodegradable, adding a layer of safety compared to mammalian virus-based systems. In future studies, the PhMV particle surface will be decorated with shielding molecules that prevent immune clearance or targeting ligands to direct the particles to specific cells and tissues. The PhMV platform is a welcome new addition to the current array of plant virus-based nanotechnologies.
Supplementary Material
Acknowledgments
H.M. acknowledges the United States–India Education Foundation (USIEF) for providing a Fulbright–Nehru Academic and Professional Excellence Fellowship (FBN-APE). H.M. also acknowledges DBT, New Delhi and Prof. H.S. Savithri, Prof. M.R.N. Murthy, IISc, Bangalore for their helpful discussions. This work was funded in part by NIH Grant No. R01-CA202814 (to N.F.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-mac.7b01196.
Characterization of native PhMV VLPs; confirmation of presence of nucleic acids in PhMV VLPs efficiency of PhMV labeling via external lysine and internal cysteine side chains; stability of functionalized PhMV particles determined by FPLC; interactions between cells and PhMV VLPs labeled on internal cysteine residues; distance between lysine side chains measured using Chimera software (PDF)
ORCID
Paul L. Chariou: 0000-0002-7115-3878
Sourabh Shukla: 0000-0001-9751-5833
Nicole F. Steinmetz: 0000-0002-0130-0481
Notes
The authors declare no competing financial interest.
References
- 1.Ma Y, Nolte RJ, Cornelissen JJ. Virus-Based Nanocarriers for Drug Delivery. Adv Drug Delivery Rev. 2012;64(9):811–825. doi: 10.1016/j.addr.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Yildiz I, Shukla S, Steinmetz NF. Applications of Viral Nanoparticles in Medicine. Curr Opin Biotechnol. 2011;22(6):901–908. doi: 10.1016/j.copbio.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, Qiao J, Niu Z, Wang Q. Natural Supramolecular Building Blocks: From Virus Coat Proteins to Viral Nanoparticles. Chem Soc Rev. 2012;41(18):6178–6194. doi: 10.1039/c2cs35108k. [DOI] [PubMed] [Google Scholar]
- 4.Koudelka KJ, Pitek AS, Manchester M, Steinmetz NF. Virus-Based Nanoparticles as Versatile Nanomachines. Annu Rev Virol. 2015;2(1):379–401. doi: 10.1146/annurev-virology-100114-055141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mateu MG. Assembly, Engineering and Applications of Virus-Based Protein Nanoparticles. Adv Exp Med Biol. 2016;940:83–120. doi: 10.1007/978-3-319-39196-0_5. [DOI] [PubMed] [Google Scholar]
- 6.Wen AM, Steinmetz NF. Design of Virus-Based Nanomaterials for Medicine, Biotechnology, and Energy. Chem Soc Rev. 2016;45(15):4074–4126. doi: 10.1039/c5cs00287g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lomonossoff GP, Evans DJ. Applications of Plant Viruses in Bionanotechnology. Curr Top Microbiol Immunol. 2011;375:61–87. doi: 10.1007/82_2011_184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen AM, Cho CF, Lewis JD, Steinmetz NF. The Application of Plant Viral Nanoparticles in Tissue-Specific Imaging. In: Mikhail YB, editor. Nanotechnology for Biomedical Imaging and Diagnostics: From Nano-particle Design to Clinical Applications. 1st. 2015. pp. 401–427. [Google Scholar]
- 9.Lebel ME, Chartrand K, Leclerc D, Lamarre A. Plant Viruses as Nanoparticle-Based Vaccines and Adjuvants. Vaccines (Basel, Switz) 2015;3(3):620–637. doi: 10.3390/vaccines3030620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shukla S, Steinmetz NF. Virus-Based Nanomaterials as Positron Emission Tomography and Magnetic Resonance Contrast Agents: From Technology Development to Translational Medicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(5):708–721. doi: 10.1002/wnan.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pokorski JK, Steinmetz NF. The Art of Engineering Viral Nanoparticles. Mol Pharmaceutics. 2011;8(1):29–43. doi: 10.1021/mp100225y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Czapar AE, Steinmetz NF. Plant Viruses and Bacteriophages for Drug Delivery in Medicine and Biotechnology. Curr Opin Chem Biol. 2017;38:108–116. doi: 10.1016/j.cbpa.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steele JFC, Peyret H, Saunders K, Castells-Graells R, Marsian J, Meshcheriakova Y, Lomonossoff GP. Synthetic Plant Virology for Nanobiotechnology and Nanomedicine. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(4):e1447. doi: 10.1002/wnan.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young M, Willits D, Uchida M, Douglas T. Plant Viruses as Biotemplates for Materials and Their Use in Nanotechnology. Annu Rev Phytopathol. 2008;46:361–384. doi: 10.1146/annurev.phyto.032508.131939. [DOI] [PubMed] [Google Scholar]
- 15.Ren Y, Wong SM, Lim LY. Application of Plant Viruses as Nano Drug Delivery Systems. Pharm Res. 2010;27(11):2509–2513. doi: 10.1007/s11095-010-0251-2. [DOI] [PubMed] [Google Scholar]
- 16.Singh P, Gonzalez MJ, Manchester M. Viruses and Their Uses in Nanotechnology. Drug Dev Res. 2006;67(1):23–41. [Google Scholar]
- 17.Huang X, Wang X, Zhang J, Xia N, Zhao Q. Escherichia Coli-Derived Virus-Like Particles in Vaccine Development. npj Vaccines. 2017;2(1):3. doi: 10.1038/s41541-017-0006-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Kim HJ. Yeast as an Expression System for Producing Virus-Like Particles: What Factors Do We Need to Consider? Lett Appl Microbiol. 2017;64(2):111–123. doi: 10.1111/lam.12695. [DOI] [PubMed] [Google Scholar]
- 19.Marsian J, Lomonossoff GP. Molecular Pharming - VLPs Made in Plants. Curr Opin Biotechnol. 2016;37:201–206. doi: 10.1016/j.copbio.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Wu CY, Yeh YC, Yang YC, Chou C, Liu MT, Wu HS, Chan JT, Hsiao PW. Mammalian Expression of Virus-Like Particles for Advanced Mimicry of Authentic Influenza Virus. PLoS One. 2010;5(3):e9784. doi: 10.1371/journal.pone.0009784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeltins A. Construction and Characterization of Virus-Like Particles: A Review. Mol Biotechnol. 2013;53(1):92–107. doi: 10.1007/s12033-012-9598-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pushko P, Pumpens P, Grens E. Development of Virus-Like Particle Technology from Small Highly Symmetric to Large Complex Virus-Like Particle Structures. Intervirology. 2013;56(3):141–165. doi: 10.1159/000346773. [DOI] [PubMed] [Google Scholar]
- 23.Yamaji H. Suitability and Perspectives on Using Recombinant Insect Cells for the Production of Virus-Like Particles. Appl Microbiol Biotechnol. 2014;98(5):1963–1970. doi: 10.1007/s00253-013-5474-9. [DOI] [PubMed] [Google Scholar]
- 24.Grgacic EV, Anderson DA. Virus-Like Particles: Passport to Immune Recognition. Methods. 2006;40(1):60–65. doi: 10.1016/j.ymeth.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ludwig C, Wagner R. Virus-Like Particles-Universal Molecular Toolboxes. Curr Opin Biotechnol. 2007;18(6):537–545. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noad R, Roy P. Virus-Like Particles as Immunogens. Trends Microbiol. 2003;11(9):438–444. doi: 10.1016/s0966-842x(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 27.Palucha A, Loniewska A, Satheshkumar S, Boguszewska-Chachulska AM, Umashankar M, Milner M, Haenni AL, Savithri HS. Virus-Like Particles: Models for Assembly Studies and Foreign Epitope Carriers. Prog Nucleic Acid Res Mol Biol. 2005;80:135–168. doi: 10.1016/S0079-6603(05)80004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Molino NM, Wang SW. Caged Protein Nanoparticles for Drug Delivery. Curr Opin Biotechnol. 2014;28:75–82. doi: 10.1016/j.copbio.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jennings GT, Bachmann MF. Immunodrugs: Therapeutic VLP-Based Vaccines for Chronic Diseases. Annu Rev Pharmacol Toxicol. 2009;49:303–326. doi: 10.1146/annurev-pharmtox-061008-103129. [DOI] [PubMed] [Google Scholar]
- 30.Lua LH, Connors NK, Sainsbury F, Chuan YP, Wibowo N, Middelberg AP. Bioengineering Virus-Like Particles as Vaccines. Biotechnol Bioeng. 2014;111(3):425–440. doi: 10.1002/bit.25159. [DOI] [PubMed] [Google Scholar]
- 31.Shirbaghaee Z, Bolhassani A. Different Applications of Virus-Like Particles in Biology and Medicine: Vaccination and Delivery Systems. Biopolymers. 2016;105(3):113–132. doi: 10.1002/bip.22759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frietze KM, Peabody DS, Chackerian B. Engineering Virus-Like Particles as Vaccine Platforms. Curr Opin Virol. 2016;18:44–49. doi: 10.1016/j.coviro.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zdanowicz M, Chroboczek J. Virus-Like Particles as Drug Delivery Vectors. Acta Biochim Pol. 2016;63(3):469–473. doi: 10.18388/abp.2016_1275. [DOI] [PubMed] [Google Scholar]
- 34.Lee KL, Twyman RM, Fiering S, Steinmetz NF. Virus-Based Nanoparticles as Platform Technologies for Modern Vaccines. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2016;8(4):554–578. doi: 10.1002/wnan.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naskalska A, Pyrc K. Virus Like Particles as Immunogens and Universal Nanocarriers. Polish J Microbiol. 2015;64(1):3–13. [PubMed] [Google Scholar]
- 36.Rohovie MJ, Nagasawa M, Swartz JR. Virus-Like Particles: Next-Generation Nanoparticles for Targeted Therapeutic Delivery. Bioengineering & Translational Medicine. 2017;2(1):43–57. doi: 10.1002/btm2.10049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ren Y, Wong SM, Lim LY. In Vitro-Reassembled Plant Virus-Like Particles for Loading of Polyacids. J Gen Virol. 2006;87(9):2749–2754. doi: 10.1099/vir.0.81944-0. [DOI] [PubMed] [Google Scholar]
- 38.Hema M, Nagendrakumar SB, Yamini R, Chandran D, Rajendra L, Thiagarajan D, Parida S, Paton DJ, Srinivasan VA. Chimeric Tymovirus-Like Particles Displaying Foot-and-Mouth Disease Virus Non-Structural Protein Epitopes and Its Use for Detection of Fmdv-Nsp Antibodies. Vaccine. 2007;25(25):4784–4794. doi: 10.1016/j.vaccine.2007.04.023. [DOI] [PubMed] [Google Scholar]
- 39.Natilla A, Hammond RW. Maize Rayado Fino Virus Virus-Like Particles Expressed in Tobacco Plants: A New Platform for Cysteine Selective Bioconjugation Peptide Display. J Virol Methods. 2011;178(1–2):209–215. doi: 10.1016/j.jviromet.2011.09.013. [DOI] [PubMed] [Google Scholar]
- 40.Lu X, Thompson JR, Perry KL. Encapsidation of DNA, a Protein and a Fluorophore into Virus-Like Particles by the Capsid Protein of Cucumber Mosaic Virus. J Gen Virol. 2012;93(Pt 5):1120–1126. doi: 10.1099/vir.0.040170-0. [DOI] [PubMed] [Google Scholar]
- 41.Wen AM, Shukla S, Saxena P, Aljabali AA, Yildiz I, Dey S, Mealy JE, Yang AC, Evans DJ, Lomonossoff GP, Steinmetz NF. Interior Engineering of a Viral Nanoparticle and Its Tumor Homing Properties. Biomacromolecules. 2012;13(12):3990–4001. doi: 10.1021/bm301278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hassani-Mehraban A, Creutzburg S, van Heereveld L, Kormelink R. Feasibility of Cowpea Chlorotic Mottle Virus-Like Particles as Scaffold for Epitope Presentations. BMC Biotechnol. 2015;15:80. doi: 10.1186/s12896-015-0180-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culver JN, Brown AD, Zang F, Gnerlich M, Gerasopoulos K, Ghodssi R. Plant Virus Directed Fabrication of Nanoscale Materials and Devices. Virology. 2015;479–480:200–212. doi: 10.1016/j.virol.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 44.Abraham A, Natraj U, Karande AA, Gulati A, Murthy MR, Murugesan S, Mukunda P, Savithri HS. Intracellular Delivery of Antibodies by Chimeric Sesbania Mosaic Virus (SeMV) Virus Like Particles. Sci Rep. 2016;6:21803. doi: 10.1038/srep21803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung B, Rao AL, Anvari B. Optical Nano-Constructs Composed of Genome-Depleted Brome Mosaic Virus Doped with a near Infrared Chromophore for Potential Biomedical Applications. ACS Nano. 2011;5(2):1243–1252. doi: 10.1021/nn1028696. [DOI] [PubMed] [Google Scholar]
- 46.Lewis JD, Destito G, Zijlstra A, Gonzalez MJ, Quigley JP, Manchester M, Stuhlmann H. Viral Nanoparticles as Tools for Intravital Vascular Imaging. Nat Med. 2006;12(3):354–360. doi: 10.1038/nm1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koudelka KJ, Destito G, Plummer EM, Trauger SA, Siuzdak G, Manchester M. Endothelial Targeting of Cowpea Mosaic Virus (CPMV) Via Surface Vimentin. PLoS Pathog. 2009;5(5):e1000417. doi: 10.1371/journal.ppat.1000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ranjith-Kumar CT, Gopinath K, Jacob AN, Srividhya V, Elango P, Savithri HS. Genomic Sequence of Physalis Mottle Virus and Its Evolutionary Relationship with Other Tymoviruses. Arch Virol. 1998;143(8):1489–1500. doi: 10.1007/s007050050392. [DOI] [PubMed] [Google Scholar]
- 49.Sastri M, Kekuda R, Gopinath K, Kumar CT, Jagath JR, Savithri HS. Assembly of Physalis Mottle Virus Capsid Protein in Escherichia Coli and the Role of Amino and Carboxy Termini in the Formation of the Icosahedral Particles. J Mol Biol. 1997;272(4):541–552. doi: 10.1006/jmbi.1997.1258. [DOI] [PubMed] [Google Scholar]
- 50.Sastri M, Reddy DS, Krishna SS, Murthy MR, Savithri HS. Identification of a Discrete Intermediate in the Assembly/Disassembly of Physalis Mottle Tymovirus through Mutational Analysis. J Mol Biol. 1999;289(4):905–918. doi: 10.1006/jmbi.1999.2786. [DOI] [PubMed] [Google Scholar]
- 51.Krishna SS, Hiremath CN, Munshi SK, Prahadeeswaran D, Sastri M, Savithri HS, Murthy MR. Three-Dimensional Structure of Physalis Mottle Virus: Implications for the Viral Assembly. J Mol Biol. 1999;289(4):919–934. doi: 10.1006/jmbi.1999.2787. [DOI] [PubMed] [Google Scholar]
- 52.Sri Krishna S, Sastri M, Savithri HS, Murthy MR. Structural Studies on the Empty Capsids of Physalis Mottle Virus. J Mol Biol. 2001;307(4):1035–1047. doi: 10.1006/jmbi.2001.4533. [DOI] [PubMed] [Google Scholar]
- 53.Chandran D, Shahana PV, Rani GS, Sugumar P, Shankar CR, Srinivasan VA. Display of Neutralizing Epitopes of Canine Parvovirus and a T-Cell Epitope of the Fusion Protein of Canine Distemper Virus on Chimeric Tymovirus-Like Particles and Its Use as a Vaccine Candidate Both against Canine Parvo and Canine Distemper. Vaccine. 2009;28(1):132–139. doi: 10.1016/j.vaccine.2009.09.093. [DOI] [PubMed] [Google Scholar]
- 54.Shahana PV, Das D, Gontu A, Chandran D, Maithal K. Efficient Production of Tymovirus Like Particles Displaying Immunodominant Epitopes of Japanese Encephalitis Virus Envelope Protein. Protein Expression Purif. 2015;113:35–43. doi: 10.1016/j.pep.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 55.Jacob AN, Murthy MR, Savithri HS. Nucleotide Sequence of the 3′ Terminal Region of Belladonna Mottle Virus-Iowa (Renamed Physalis Mottle Virus) RNA and an Analysis of the Relationships of Tymoviral Coat Proteins. Arch Virol. 1992;123(3–4):367–377. doi: 10.1007/BF01317270. [DOI] [PubMed] [Google Scholar]
- 56.Umashankar M, Murthy MR, Singh SA, Appu Rao AG, Savithri HS. The Role of Inter-Subunit Ionic Interactions in the Assembly of Physalis Mottle Tymovirus. Arch Virol. 2006;151(10):1917–1931. doi: 10.1007/s00705-006-0783-2. [DOI] [PubMed] [Google Scholar]
- 57.Clapp AR, Medintz IL, Mauro JM, Fisher BR, Bawendi MG, Mattoussi H. Fluorescence Resonance Energy Transfer between Quantum Dot Donors and Dye-Labeled Protein Acceptors. J Am Chem Soc. 2004;126(1):301–310. doi: 10.1021/ja037088b. [DOI] [PubMed] [Google Scholar]
- 58.Strat D, Dolp F, von Einem B, Steinmetz C, von Arnim CA, Rueck A. Spectrally Resolved Fluorescence Lifetime Imaging Microscopy: Forster Resonant Energy Transfer Global Analysis with a One- and Two-Exponential Donor Model. J Biomed Opt. 2011;16(2):026002. doi: 10.1117/1.3533318. [DOI] [PubMed] [Google Scholar]
- 59.Yefimova SL, Tkacheva TN, Kurilchenko IY, Sorokin AV, Malyukin YV. Spectroscopic Study of Interactions between Dye Molecules in Micelle and Liposome Nanovolumes. J Appl Spectrosc. 2013;79(6):914–921. [Google Scholar]
- 60.Wen AM, Infusino M, De Luca A, Kernan DL, Czapar AE, Strangi G, Steinmetz NF. Interface of Physics and Biology: Engineering Virus-Based Nanoparticles for Biophotonics. Bioconjugate Chem. 2015;26(1):51–62. doi: 10.1021/bc500524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chariou PL, Steinmetz NF. Delivery of Pesticides to Plant Parasitic Nematodes Using Tobacco Mild Green Mosaic Virus as a Nanocarrier. ACS Nano. 2017;11(5):4719–4730. doi: 10.1021/acsnano.7b00823. [DOI] [PubMed] [Google Scholar]
- 62.Lee KL, Carpenter BL, Wen AM, Ghiladi RA, Steinmetz NF. High Aspect Ratio Nanotubes Formed by Tobacco Mosaic Virus for Delivery of Photodynamic Agents Targeting Melanoma. ACS Biomater Sci Eng. 2016;2(5):838–844. doi: 10.1021/acsbiomaterials.6b00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wen AM, Lee KL, Cao P, Pangilinan K, Carpenter BL, Lam P, Veliz FA, Ghiladi RA, Advincula RC, Steinmetz NF. Utilizing Viral Nanoparticle/Dendron Hybrid Conjugates in Photo-dynamic Therapy for Dual Delivery to Macrophages and Cancer Cells. Bioconjugate Chem. 2016;27(5):1227–1235. doi: 10.1021/acs.bioconjchem.6b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Agudelo D, Bourassa P, Bruneau J, Berube G, Asselin E, Tajmir-Riahi HA. Probing the Binding Sites of Antibiotic Drugs Doxorubicin and N-(Trifluoroacetyl) Doxorubicin with Human and Bovine Serum Albumins. PLoS One. 2012;7(8):e43814. doi: 10.1371/journal.pone.0043814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Husain N, Agbaria RA, Warner IM. Spectroscopic Analysis of the Binding of Doxorubicin to Human.Alpha.−1 Acid Glycoprotein. J Phys Chem. 1993;97(41):10857–10861. [Google Scholar]
- 66.Le DH, Lee KL, Shukla S, Commandeur U, Steinmetz NF. Potato Virus X, a Filamentous Plant Viral Nanoparticle for Doxorubicin Delivery in Cancer Therapy. Nanoscale. 2017;9(6):2348–2357. doi: 10.1039/c6nr09099k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gheewala T, Skwor T, Munirathinam G. Photosensitizers in Prostate Cancer Therapy. Oncotarget. 2015;8(18):30524–30538. doi: 10.18632/oncotarget.15496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stephanopoulos N, Tong GJ, Hsiao SC, Francis MB. Dual-Surface Modified Virus Capsids for Targeted Delivery of Photodynamic Agents to Cancer Cells. ACS Nano. 2010;4(10):6014–6020. doi: 10.1021/nn1014769. [DOI] [PubMed] [Google Scholar]
- 69.Rhee JK, Baksh M, Nycholat C, Paulson JC, Kitagishi H, Finn MG. Glycan-Targeted Virus-Like Nanoparticles for Photodynamic Therapy. Biomacromolecules. 2012;13(8):2333–2338. doi: 10.1021/bm300578p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Breast Cancer; American Cancer Society. 2017 https://www.cancer.org/ (accessed October 17, 2017)
- 71.Hare JI, Lammers T, Ashford MB, Puri S, Storm G, Barry ST. Challenges and Strategies in Anti-Cancer Nanomedicine Development: An Industry Perspective. Adv Drug Delivery Rev. 2017;108:25–38. doi: 10.1016/j.addr.2016.04.025. [DOI] [PubMed] [Google Scholar]
- 72.Heddle JG, Chakraborti S, Iwasaki K. Natural and Artificial Protein Cages: Design, Structure and Therapeutic Applications. Curr Opin Struct Biol. 2017;43:148–155. doi: 10.1016/j.sbi.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 73.Loo L, Guenther RH, Lommel SA, Franzen S. Infusion of Dye Molecules into Red Clover Necrotic Mosaic Virus. Chem Commun (Cambridge, UK) 2008;1:88–90. doi: 10.1039/b714748a. [DOI] [PubMed] [Google Scholar]
- 74.Lockney DM, Guenther RN, Loo L, Overton W, Antonelli R, Clark J, Hu M, Luft C, Lommel SA, Franzen S. The Red Clover Necrotic Mosaic Virus Capsid as a Multifunctional Cell Targeting Plant Viral Nanoparticle. Bioconjugate Chem. 2011;22(1):67–73. doi: 10.1021/bc100361z. [DOI] [PubMed] [Google Scholar]
- 75.Zeng Q, Wen H, Wen Q, Chen X, Wang Y, Xuan W, Liang J, Wan S. Cucumber Mosaic Virus as Drug Delivery Vehicle for Doxorubicin. Biomaterials. 2013;34(19):4632–4642. doi: 10.1016/j.biomaterials.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 76.Aljabali AA, Shukla S, Lomonossoff GP, Steinmetz NF, Evans DJ. CPMV-Dox Delivers. Mol Pharmaceutics. 2013;10(1):3–10. doi: 10.1021/mp3002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bruckman MA, Czapar AE, VanMeter A, Randolph LN, Steinmetz NF. Tobacco Mosaic Virus-Based Protein Nanoparticles and Nanorods for Chemotherapy Delivery Targeting Breast Cancer. J Controlled Release. 2016;231:103–113. doi: 10.1016/j.jconrel.2016.02.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ren Y, Wong SM, Lim LY. Folic Acid-Conjugated Protein Cages of a Plant Virus: A Novel Delivery Platform for Doxorubicin. Bioconjugate Chem. 2007;18(3):836–843. doi: 10.1021/bc060361p. [DOI] [PubMed] [Google Scholar]
- 79.Zhao Q, Chen W, Chen Y, Zhang L, Zhang J, Zhang Z. Self-Assembled Virus-Like Particles from Rotavirus Structural Protein VP6 for Targeted Drug Delivery. Bioconjugate Chem. 2011;22(3):346–352. doi: 10.1021/bc1002532. [DOI] [PubMed] [Google Scholar]
- 80.Kato T, Yui M, Deo VK, Park EY. Development of Rous Sarcoma Virus-Like Particles Displaying hCC49 scFV for Specific Targeted Drug Delivery to Human Colon Carcinoma Cells. Pharm Res. 2015;32(11):3699–3707. doi: 10.1007/s11095-015-1730-2. [DOI] [PubMed] [Google Scholar]
- 81.Biabanikhankahdani R, Alitheen NB, Ho KL, Tan WS. Ph-Responsive Virus-Like Nanoparticles with Enhanced Tumour-Targeting Ligands for Cancer Drug Delivery. Sci Rep. 2016;6:37891. doi: 10.1038/srep37891. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.