

SUMMARY
Defects in mitochondrial metabolism have been increasingly linked with age-onset protein-misfolding dieases such as Alzheimer’s, Parkinson’s, and Huntington’s. In response to protein-folding stress, compartment-specific unfolded protein responses (UPRs) within the ER, mitochondria, and cytosol work in parallel to ensure cellular protein homeostasis. While perturbation of individual compartments can make other compartments more susceptible to protein stress, the cellular conditions that trigger cross-communication between the individual UPRs remain poorly understood. We have uncovered a conserved, robust mechanism linking mitochondrial protein homeostasis and the cytosolic folding environment through changes in lipid homeostasis. Metabolic restructuring caused by mitochondrial stress or small-molecule activators trigger changes in gene expression coordinated uniquely by both the mitochondrial and cytosolic UPRs, protecting the cell from disease-associated proteins. Our data suggest an intricate and unique system of communication between UPRs in response to metabolic changes that could unveil new targets for diseases of protein misfolding.
Graphical abstract
In Brief: How is mitochondrial stress signaled to other cellular compartments?
INTRODUCTION
One of the most complex tasks the cell faces is the need for constant surveillance of the imbalances among the independently acting subcellular regions amid volatile changes in external conditions. The evolution of membrane-enclosed, subcellular structures has given the eukaryotic cell the capacity to maintain distinct microenvironments, facilitating the efficient compartmentalization of processes and regulating the concentration, transport, and diffusion of the thousands of molecules required for proper cellular function. Compartmentalization also allows the cell to physically contain its damage, sequestering misfolded proteins and aberrant molecules away from other parts of the still-functioning cell. Accordingly, each subcellular location has evolved a large, unique, defensive fingerprint of genes and proteins that become differentially regulated upon the application of stress—a network of beneficial genes highly specialized for its own environment. These collections of typically hundreds of genes, referred to as an unfolded protein response (UPR), are often regulated by the activation of as little as a single transcription factor and play well-described roles in the stress-responsive regulation of the ER, cytoplasm, and mitochondria (Haynes and Ron, 2010; Kirstein-Miles and Morimoto, 2010; Liu and Chang, 2008).
As a loss in homeostasis in one organelle has deleterious consequences on the function of all of the sub-compartments across the cell, each organelle remains reliant upon the appropriate function of every other organelle for its survival (Hughes and Gottschling, 2012; Veatch et al., 2009). In such a setting, cross-communication between compartments is essential for ensuring cellular homeostasis. Coordinated reactions to cellular stress have been well described in relationship to the ER and mitochondria, for example, and cytosolic health necessarily affects the function of multiple organelles (Hu and Liu, 2011; Ron and Walter, 2007; Senft and Ronai, 2015; Vannuvel et al., 2013). Aberrant communication between organelles has been associated with the advent and severity of protein-folding diseases, including neurodegenerative diseases, cardiovascular disease, and diabetes (Indiveri et al., 2011; Jovaisaite et al., 2014; Kirstein-Miles and Morimoto, 2010; Senft and Ronai, 2015). Cross-communication between compartments is also critical in the regulation and distribution of lipid stores. Lipid synthesis in the ER and mitochondria is required for the appropriate function across cellular compartments. Importantly, imbalances in lipid stores within individual organelles cause changes in cellular functions across compartments and have been associated with a wide number of disease states.
Responsibility for the execution of stress responses in individual organelles belongs in part to the Hsp70 family of chaperones, which play a large and central role in the maintenance of cellular protein homeostasis (Mayer and Bukau, 2005). Hsp70 chaperones are among the most highly conserved proteins known and are, remarkably, present in every organism described to date. Their roles encompass a wide range of functions, including the folding of newly synthesized proteins, the appropriate translocation and folding of proteins within organelles, and the refolding of aggregating or misfolded proteins (Bukau and Horwich, 1998). Their members are sometimes redundant but often unique, and they can be either induced or constitutively expressed. Each of the Hsp70 family members is targeted primarily to a single specific subcellular compartment, such as the mitochondria, cytoplasm, ER, or chloroplast, and has highly optimized its functions for that specific subcellular location (Daugaard et al., 2007). In the cytosol, for example, Hsp70 chaperones are required for diverse processes such as transport and nascent chain folding. Within the ER, the Hsp70 chaperones BiP and Hyou1 work in concert to ensure the correct folding of proteins targeted for extracellular secretion. Within the mitochondria, the Hsp70 family member mortalin ensures both the translocation and folding of cytosol-synthesized mitochondrial proteins (Tavaria et al., 1996). A loss of Hsp70 function in a specific subcellular compartment will elicit an upregulation of the compartment-specific UPR and will negatively alter the protein-folding landscape for that compartment (Kim et al., 1995; Wang et al., 2009).
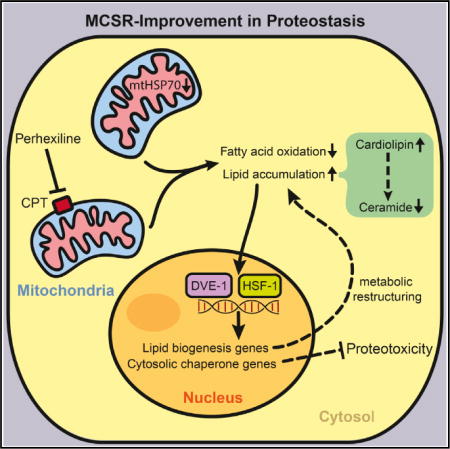
We hypothesized that Hsp70 family members facilitate communication between compartments, thus predicting that a loss in compartment-specific Hsp70 function may elicit the crosscompartment upregulation of additional UPRs. To test this, we systematically knocked down each of the 12 compartment-specific variants of Hsp70 family proteins in Caenorhabditis elegans and analyzed their effects on protein homeostasis across the cell. We found that reduced expression of the mitochondrial chaperone hsp-6 (mortalin/Grp75/mtHSP70) is sufficient to induce a previously unidentified mitochondrial-to-cytosolic stress response. We have termed this response the mitochondrial-to-cytosolic stress response (MCSR). The induction of the MCSR after hsp-6 RNAi depends upon dve-1 and hsf-1, transcription factors involved in the mitochondrial UPR (UPRmt) and cytosolic heat shock response (HSR), respectively. Induction of the MCSR requires the global alteration of fat metabolism: fatty acid synthesis is required for the mitochondrial mediated induction of the MCSR, while the increased synthesis of fatty acids, in contrast, is sufficient to induce the MCSR. The MCSR accompanies a specific increase in the inhibitors of ceramide synthesis, cardiolipins, indicating that a metabolic shift that involves ceramide levels plays an integral role in MCSR induction, and genetic or pharmacological manipulation of these species is sufficient to alter MCSR induction. Finally, genetic and pharmacologically mediated mitochondrial-dependent induction of the MCSR protects against the proteotoxicity caused by polyglutamine (polyQ) protein expression in C. elegans and human cells. Collectively, these data support a model in which the cytosol senses mitochondrial stress by recognizing the aberrant intracellular accumulation of lipids or a shift in metabolism, resulting in the upregulation of cross-compartmental defense mechanisms and a reshaping of the protein-folding landscape within the cell. These results suggest a potential therapeutic effect of fatty acid metabolism in the prevention of protein-misfolding diseases originating from disparate compartments in the cell.
RESULTS
Cross-Communication between the UPRmt and HSR
To test whether an UPR induction in one compartment could communicate to unaffected compartment-specific stress responses, we reduced the function of each Hsp70 family member using RNAi in C. elegans and analyzed the induction of the compartment-specific UPRs. Using this approach, we discovered that RNAi against the nematode mitochondrial chaperone Hsp70, hsp-6 (mortalin/Grp75/mtHSP70), was sufficient to upregulate hsp-16.2p::GFP, a marker for the cytosolic HSR, in otherwise unstressed conditions (Figure 1A). While reduction of hsp-6 also induced the UPRmt, it had no effect on the ER UPR (UPRER) (Figures 1A and 1B; Table S1). This effect also appeared unique: RNAi targeting any of the other 11 Hsp70 family members failed to elicit a cross-compartmental stress response. RNAi against ER resident Hsp70 family members BiP (hsp-3 and hsp-4), Hyou-1 (T14G8.3 and T24H7.2), or Stch (stc-1) had no effect on the cytosolic HSR or UPRmt (Figure 1A; Table S1). RNAi against the cytosolic hsp70 family members C12C8.1, F44E5.5, F44E5.4, hsp-110, hsp-1, or F11F1.1 likewise failed to induce the UPRmt or UPRER (Table S1). These data suggested that mitochondrial Hsp70 (mtHSP70) plays a distinctive and unidirectional role in the communication of mitochondrial perturbations and induction of protective responses in the cytosol. hsp-6 RNAi was sufficient to upregulate not only hsp-16.2 but also multiple cytosolic chaperones, suggesting a widespread response to the loss in mitochondrial homeostasis (Figure S1A). For convenience, we named this response the mitochondrial-to-cytosolic stress response, or MCSR.
Figure 1. Knockdown of Mitochondrial HSP70, hsp-6, Induces Cytosolic Heat Shock Response via UPRmt.
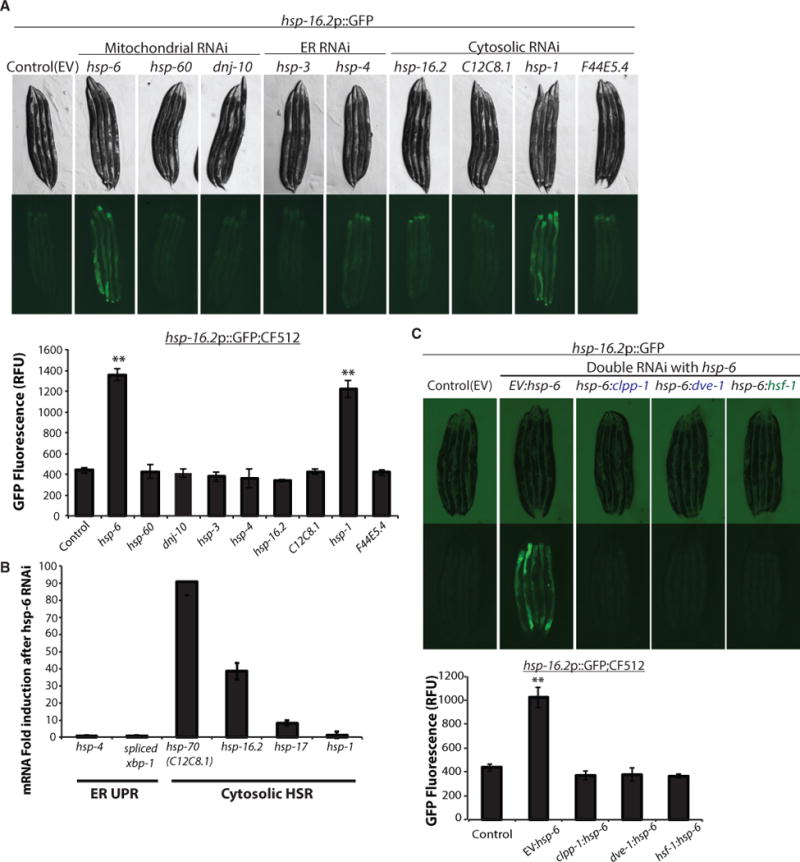
(A) hsp-6 and hsp-1 RNAi-induced cytosolic small heat shock protein expression. hsp-16.2p::GFP induction was measured by COPAS biosorter (bottom) (mean ± SD of three biological repeats; **p ≤ 0.01). EV, empty vector control RNAi.
(B) qPCR of UPRER and cytosolic HSR genes after hsp-6 RNAi (mean ± SD of three biological repeats).
(C) Cytosolic HSR induced by hsp-6 RNAi was dependent on both hsf-1 and UPRmt components. hsp-16.2p::GFP induction was measured by COPAS biosorter (bottom) (mean ± SD of three biological repeats; **p ≤ 0.001).
See also Figure S1 and Table S1.
Because the MCSR encompassed both the mitochondrial and cytosolic stress responses, we tested whether the canonical mediators of these pathways were required for the MCSR. While we found that the upregulation of the heat stress sentinel, hsp-16.2, during the MCSR required hsf-1 as expected, surprisingly, the transcription factor dve-1, a major regulator of the UPRmt, as well as the mitochondrial matrix protease clpp-1 were also essential for this response (Figure 1C). In addition, ubl-5, haf-1, and atfs-1 (Figure S1B), all well-known members of the canonical UPRmt pathway, were required for the MCSR. Collectively, these data indicate that disruption of mitochondrial proteostasis caused by mitochondrial chaperone hsp-6 knockdown plays a specific role in affecting the cytosolic protein-folding environment of the cell through the activation of both the UPRmt and the cytosolic heat shock response.
Orthologs of hsp-6, such as mortalin, have been found to have critical roles in the import and folding of nuclear-encoded mitochondrial matrix proteins (Becker et al., 2012; Horst et al., 1997). Therefore, induction of the cytosolic heat shock response caused by hsp-6 RNAi treatment could be due to reduced mitochondrial import, resulting in the accumulation of misfolded mitochondrial proteins in the cytosol, leading to the eventual induction of the HSR in addition to the UPRmt. To test this possibility, we examined the effect of reduction of mitochondrial protein import complex components on the cytosolic heat shock response. Proteins targeted for the mitochondria are unfolded upon translocation across the TOM (outer mitochondrial membrane) and TIM (inner mitochondrial membrane) channels (Wiedemann et al., 2004). We tested whether RNAi against any of the annotated components of the import machinery resulted in the MCSR. While knockdown of all the components of the import machinery we tested were able to induce UPRmt, only hsp-6 RNAi was capable of inducing the HSR (Figure S1C). Additionally, we found that steady-state cytosolic, pre-import levels of mitochondrial proteins, including Nuo-2 and Hsp60, remained unaltered by hsp-6 RNAi treatment (Figure S1D). Moreover, analysis of the insoluble cytosolic proteome of hsp-6 RNAi-treated animals suggested that the global level of insoluble proteins in the cytosol was decreased rather than increased, while proteasome activity was unaffected by hsp-6 RNAi (Figures S1E and S1F). Taken together, these data suggest that the reduction of mtHSP70 has a distinct effect on the HSR that is independent of mitochondrial import or proteasome activity.
DVE-1 and HSF-1 Co-regulate Fat Metabolism
Because the induction of the MCSR due to hsp-6 RNAi required both hsf-1 and dve-1, key transcription factors required for the HSR and UPRmt, respectively, we asked which gene sets are coordinately regulated by both factors. We performed microarray analyses and identified that the expression of 187 genes was altered by hsp-6 RNAi in comparison to control animals (Table S2A). Interestingly, 98 of these 187 genes were regulated by either hsf-1 or dve-1 (Figure 2A; Table S2A). More importantly, the vast majority (66/98) of hsp-6 RNAi-dependent changes in gene expression required both dve-1 and hsf-1, consistent with our earlier observations of the requirement of dve-1 and hsf-1 for MCSR induction (Figure 1C). To facilitate analysis of this data in more detail, we analyzed the gene expression data from the microarray analysis for enriched Gene Ontology biological process (GOBP) terms with LRPath (see STAR Methods) (Table S1B). Ten representative GOBP terms were enriched in both dve-1 and hsf-1 co-regulated genes that were altered by hsp-6 RNAi (Figure 2B; Table S1C). Interestingly, genes involved in lipid biosynthetic processes were enriched, in addition to genes involved in the responses to different stressors (immune response, inorganic substances, and endogenous stress) and genes affecting the function of the translation machinery as expected (Lindquist, 1980, 1981; Miller et al., 1979). qPCR analyses of individual genes involved in lipid synthesis confirmed their upregulation in hsp-6 RNAi-treated worms (Figure 2C). These genes were not upregulated by either cco-1 knockdown or heat stress, suggesting that the response is distinct from the canonical UPRmt and HSR (Figures S2A and S2B). Also, ectopic expression of dve-1 or hsf-1 failed to induce these lipogenic genes (data not shown). Because lipid species are intriguing candidates for signaling molecules between organelles, we hypothesized that specific pathways involved in the lipid biosynthesis process may be required for cross-compartmental communication between the mitochondria and cytosol to induce the MCSR.
Figure 2. Microarray Analysis Suggests the DVE-1- and HSF-1-Dependent Gene Regulation of Fat Metabolism.
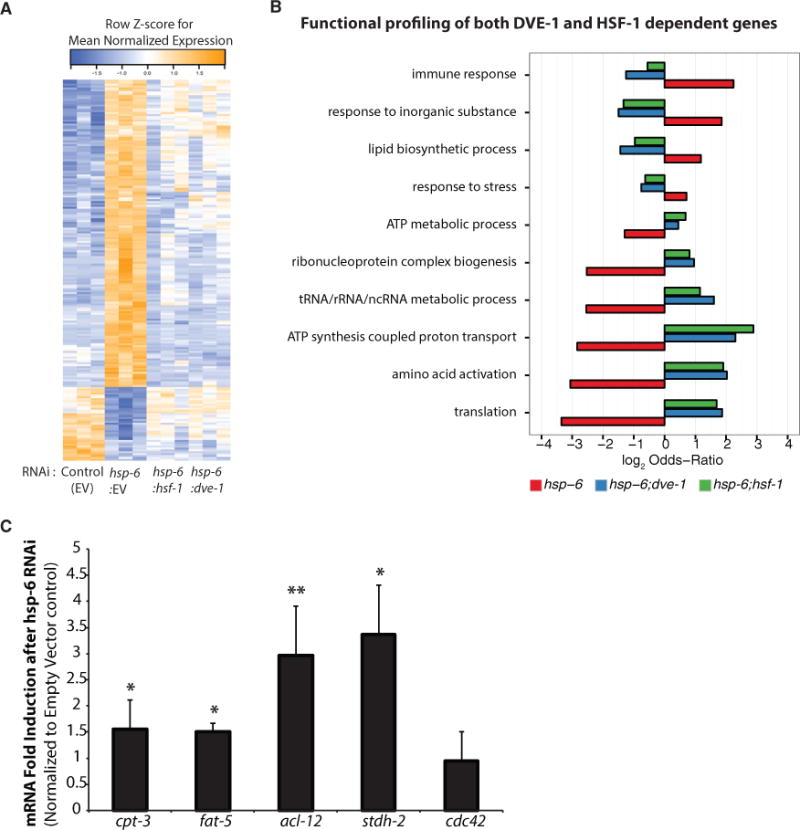
(A) Heatmap of normalized gene expression for 187 genes differentially expressed in hsp-6 RNAi relative to empty vector (EV) control.
(B) DVE-1- and HSF-1-dependent Gene Ontology (GO) biological process terms enriched in hsp-6 RNAi relative to EV. 30 GO terms were clustered to identify the 10 representative terms shown.
(C) Expression of the lipid synthesis genes from microarray experiments was verified by qPCR after hsp-6 RNAi (mean ± SD of three biological repeats; *p ≤ 0.05, **p ≤ 0.01).
See also Figure S2 and Table S2.
Consistent with the microarray and qPCR analysis, hsp-6 RNAi-treated animals also exhibited metabolic dysfunction, including the aberrant accumulation of lipid stores. hsp-6 RNAi-treated animals had higher incorporation of Nile red dye and displayed higher triglyceride levels (Figure 3A). Electron microscopy revealed that hsp-6 RNAi-treated animals contained more lipid droplets, the primary storage organelle for fats in the intestine, which were also larger in size (Figures 3B and S3A). This increase in lipid storage elicited by hsp-6 RNAi was also dependent upon dve-1 and hsf-1 activities and distinct compared to other mitochondrial stresses or heat stress (Figures 3C, S3B, and S3C). Together, these data indicate that dve-1 and hsf-1 not only were required in concert to induce the MCSR but also worked together to alter lipid metabolism of the animals.
Figure 3. Mitochondrial HSP70, hsp-6, Knockdown Leads to an Increase in Fat Storage.
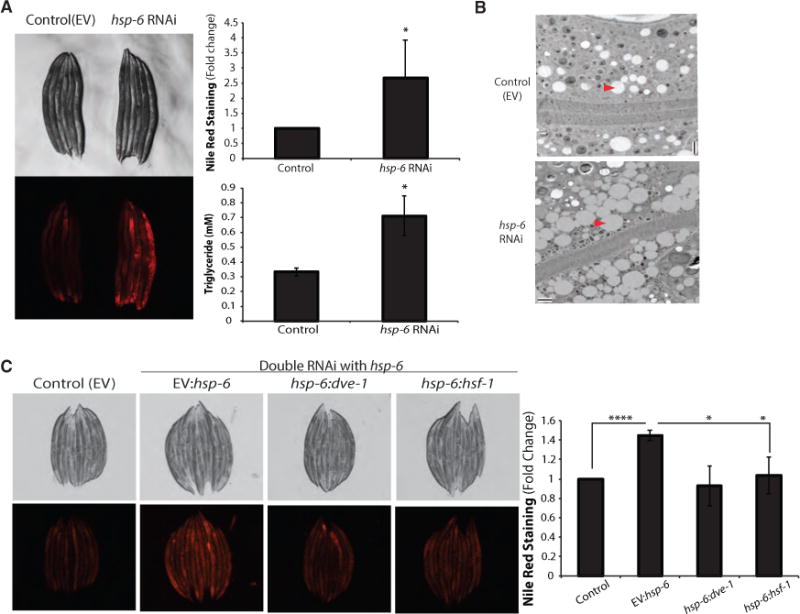
(A) hsp-6 RNAi-treated worms showed increase in fat content. Nile red staining was quantified by COPAS biosorter (mean ± SD of three biological repeats). Triglyceride content was measured after hsp-6 RNAi (mean ± SD of three biological repeats; *p ≤ 0.05).
(B) Electron microscopy showed increased number of lipid droplets in the intestine of hsp-6 RNAi-treated worms(scale bar represents 2 μm, longitudinal section). Arrowheads indicate the lipid droplets.
(C) Nile red staining on fixed worms after double RNAi. Nile red staining was quantified by COPAS biosorter (mean ± SD of four biological repeats; *p ≤ 0.05, ****p ≤ 0.0001).
Note that the Nile red staining intensities were RNAi-dose dependent (full hsp-6 RNAi in A versus half hsp-6 RNAi in C). See also Figure S3.
Fat Synthesis Is Required for Cytosolic Chaperone Induction
The dramatic change in lipid biosynthetic gene expression and lipid accumulation in hsp-6 RNAi-treated animals suggests a concerted change in both chaperone and metabolic gene expression that is dependent upon both hsf-1 and dve-1. We thus predicted that lipid disturbances might directly affect the MCSR, or that a lipid species might serve as a signal to communicate mitochondrial stress to induce the MCSR. To test whether induction of cytosolic chaperones may be mediated by fat accumulation, we reduced expression of each of the major enzymes that are essential for fatty acid biosynthesis and evaluated the induction of cytosolic small heat shock protein hsp-16.2 expression. pod-2 RNAi, which targets the homolog of mammalian acetyl-coenzyme A (acetyl-CoA) carboxylase (acc1), is predicted to decrease malonyl-CoA, a substrate for fatty acid synthesis and a regulator of fatty acid β-oxidation, triglycerides (Mao et al., 2006), and lipid accumulation in adipose tissues (Mao et al., 2009). Reduced expression of the fatty acid synthase (fasn) fasn-1 is similarly predicted to reduce fatty acid biosynthesis, resulting in the accumulation of malonyl-CoA and inhibition of fatty acid β-oxidation (Fritz et al., 2013) (Figure 4A).
Figure 4. Reducing Fat Synthesis Blocks Cytosolic Response, and Inhibiting CPT Activity Induces Cytosolic Stress Response.
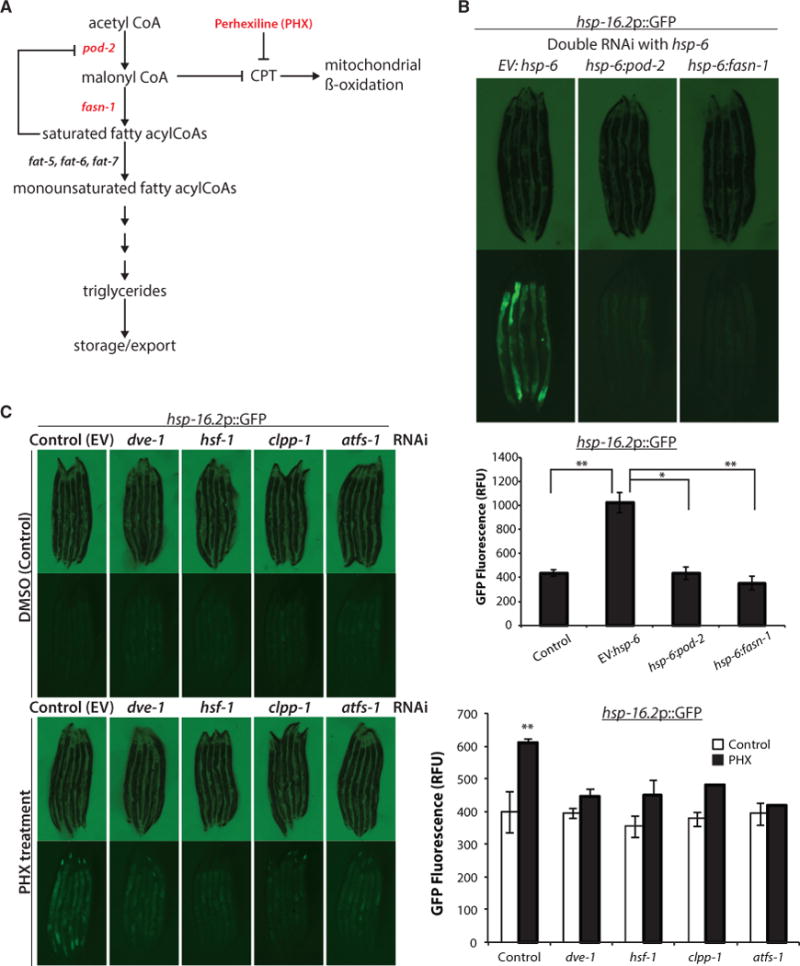
(A) Diagram showing C. elegans genes involved in the fat storage pathway. pod-2, acyl-CoA carboxylase; fasn-1, fatty acid synthase; fat-5, fat-6, and fat-7, delta-9 fatty acid desaturase; CPT, carnitine palmitoyltransferase.
(B) Knocking down two enzymes involved in fat synthesis inhibited cytosolic response. (Continued from Figure 1C; notethat the image of control worms are from Figure 1C.) hsp-16.2p::GFP reporter induction was measured by COPAS biosorter (bottom) (mean ± SD of three biological repeats; *p ≤ 0.05, **p ≤ 0.01).
(C) CPT inhibitor PHX-treated hsp-16.2p::GFP;CF512 reporter worms showed elevated levels of GFP that were inhibited by hsf-1 and UPRmt components. GFP induction was measured by COPAS biosorter (right) (mean ± SD of three biological repeats; **p ≤ 0.01).
See also Figure S4.
Remarkably, treatment of hsp-6 RNAi treated worms with secondary RNAi against either pod-2 or fasn-1 blocked induction of cytosolic hsp-16.2 expression (Figure 4B). Simultaneously, either pod-2 or fasn-1 RNAi blocked fat accumulation induced by hsp-6 RNAi as measured by triglyceride quantification (Figure S4A). In contrast, pod-2 and fasn-1 RNAi did not suppress hsp-16.2 expression upon heat shock treatment (Figure S4B), supporting the hypothesis that pod-2 and fasn-1 mediate a hsf-1 function in lipid biosynthesis for MCSR induction that is distinct from the canonical role of hsf-1 in the heat stress response.
Ectopic Fat Accumulation Induces the MCSR
Reduced fat accumulation blocks induction of the MCSR, indicating that fat accumulation is necessary for the MCSR. We asked whether fat accumulation was sufficient to induce the MCSR. Inhibition of fatty acid oxidation promotes fatty acid biosynthesis and the accumulation of lipids (Ashrafi, 2007). To block fatty acid oxidation, we treated animals with the carnitine palmitoyltransferase (CPT) inhibitor perhexiline (PHX), an inhibitor of fatty acid oxidation (Figure 4A) (Kennedy et al., 1996). As expected from our genetic results, PHX treatment increased fat accumulation and specifically induced cytosolic hsp-16.2 expression (Figure 4C). PHX-induced activation of the MCSR appears identical to hsp-6 RNAi treatment; RNAi of the UPRmt components (dve-1, clpp-1, and atfs-1), as well as hsf-1, reduced the induction of the MCSR upon PHX treatment (Figure 4C). Furthermore, PHX not only promoted fatty acid accumulation and induced the HSR (Figure S4C) but also moderately induced UPRmt (Figure S4D). While hsp-6 RNAi reduces oxygen consumption, PHX treatment did not have an added effect on hsp-6 RNAi-treated animals, suggesting that hsp-6 RNAi primarily affects mitochondrial respiration through its effects on fatty acid oxidation (Figure S4E).
We asked whether the changes in fatty acid oxidation were conserved in a mammalian system. To test this, we measured rates of respiration in populations of 293T cells in the presence or absence of mtHSP70. We observed that the resting respiration of mtHSP70 siRNA-treated 293T cells in the FAO media (see STAR Methods) was not significantly different from those treated with scrambled small interfering RNA (siRNA) (Figure S4F, BSA-added cells). Intriguingly, however, the respiratory capacity of cells following mtHSP70 siRNA treatment was greatly impaired when the fatty acid oxidation substrate was given, (Figure S4F, PALM-added cells). Taken together, we conclude that knockdown of mtHSP70 results in major defects in fatty acid oxidation.
Cardiolipin and Ceramide Mediate the MCSR
Our data suggest a requirement for lipid signaling in the induction of the MCSR. To identify the types of lipids that accumulate upon mtHSP70 knockdown, we carried out lipidomic analyses on hsp-6 RNAi-treated animals. In this experiment, we found hsp-6 RNAi caused widespread alterations in lipid content (Table S3). Among these changes, levels of ether lipids, phospholipids, and precursors of phosphatidylglycerol were significantly increased (Table S3). Intriguingly, and in stark contrast to the upregulation of these groups of lipids, ceramide levels were decreased (Table S3).
Previously, we had observed an upregulation of expression in acl-12, an ortholog of human lpgat1 (lysophosphatidylglycerol acyltransferase 1), upon hsp-6 RNAi treatment (Figure 2C). In humans, LPGAT1 functions to convert lysophosphatidic acid to phosphatidylglycerol, a precursor of cardiolipin (Yang et al., 2004). Cardiolipin is a mitochondrial phospholipid involved in mitochondrial dynamics, cristae organization, mitochondrial protein biogenesis, respiratory supercomplex assembly and function, apoptosis, and mitophagy (Lu and Claypool, 2015). Importantly, cardiolipins, whose lipid profiles would be grouped within many of the species seen upregulated in our lipidomic analysis, are also inhibitors of ceramide synthesis, which was downregulated in response to hsp-6 RNAi. We thus hypothesized that a modulation in cardiolipin levels might be critical for the induction of the MCSR. Using nonyl acridine orange staining, we found that hsp-6 RNAi indeed resulted in the accumulation of cardiolipin (Figure 5A). Importantly, this effect was blocked by the additional treatment of animals with cardiolipin synthase (crls-1) RNAi (Figure 5A). crls-1 RNAi was also sufficient to block induction of the MCSR in animals with decreased hsp-6 expression, suggesting that cardiolipin is necessary for MCSR induction (Figure 5B).
Figure 5. Cardiolipin Synthesis Is Required for MCSR Induction, and Inhibiting Ceramide Synthesis Resulted in MCSR Induction.
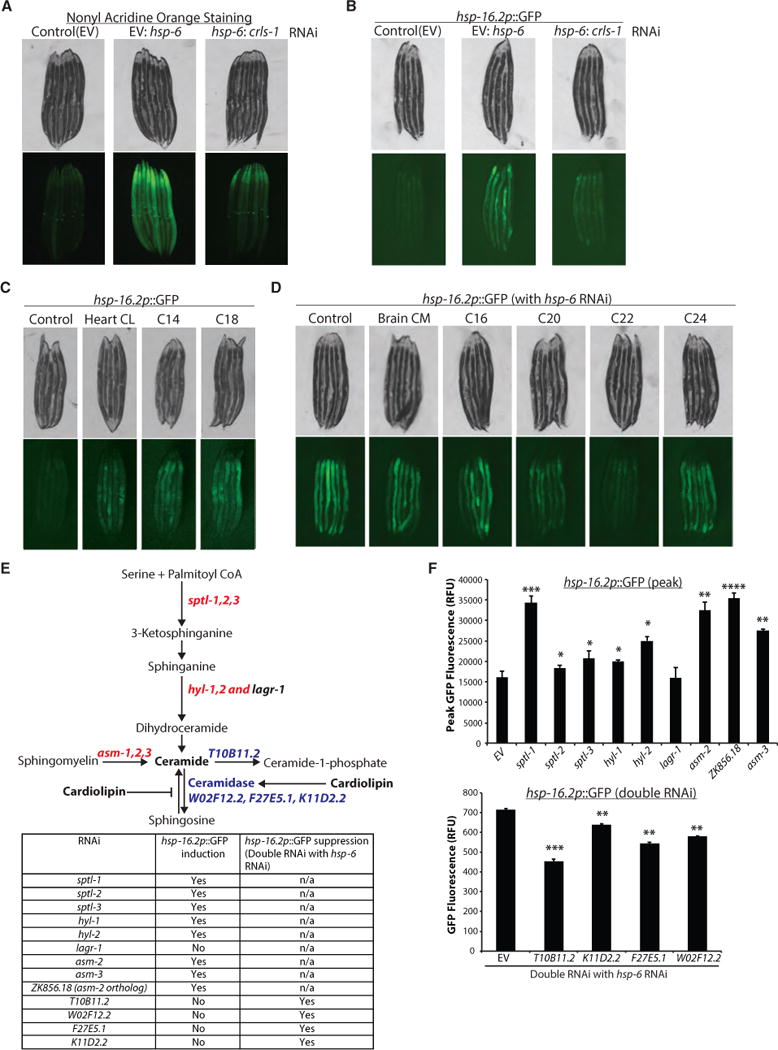
(A) Nonyl acridine orange staining showed that hsp-6 RNAi induced cardiolipin accumulation, while cardiolipin synthase (crls-1) RNAi in addition to hsp-6 RNAi blocked cardiolipin accumulation in wild-type worms.
(B) hsp-16.2p::GFP induction upon hsp-6 RNAi was inhibited by crls-1 RNAi.
(C) Cardiolipin-fed hsp-16.2p::GFP reporter worms showed increased GFP signal. Control, 0.5% methanol; Heart CL, cardiolipin purified from the bovine heart; C14, C14:0 cardiolipin; C18, C18:1 cardiolipin.
(D) Ceramide-fed hsp-16.2p::GFP reporter worms showed inhibition of MCSR upon hsp-6 RNAi. Control, 0.5% methanol; Brain CM, ceramide purified from the porcine brain; C16, C16 ceramide; C20, C20 ceramide; C22, C22 ceramide; C24, C24 ceramide.
(E) Diagram of the ceramide synthesis pathway. RNAi of enzymes written in red induced hsp-16.2 reporter, and RNAi of enzymes written in blue reduced MCSR induction. List of enzymes that were knocked down and the RNAi result are summarized in the table.
(F) Quantification of hsp-16.2p::GFP reporter induction and suppression (mean ± SD of three biological repeats; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001). hsp-16.2p::GFP reporter induction in the top panel shows the peak GFP signals from the individual worms, and the hsp-16.2p::GFP reporter suppression in the bottom panel shows the suppression of an hsp-6 RNAi-induced MCSR (double RNAi was applied at a one-to-one ratio).
See also Figure S5 and Table S3.
We tested whether exogenous cardiolipin was sufficient to drive expression of the MCSR. We found that ectopic feeding of cardiolipins to animals (Figure S5A) was sufficient to moderately induce hsp-16.2 expression (Figure 5C). In addition, cardiolipins were able to mildly turn on the hsp-6 reporter (Figure S5B). In contrast, Nile red staining remained unaffected by crls-1 RNAi (Figure S5C). These data suggested that although a wide range of lipids accumulates after mtHSP70 knockdown, cardiolipins play a critical role in the induction of the MCSR.
As mentioned above, cardiolipin is a potent inhibitor of the reverse activity of ceramidase (ceramide synthesis) (El Bawab et al., 2001; Okino et al., 2003), suggesting that increased levels of cardiolipin may result in reduced ceramide levels. Because of the link between cardiolipins and reduced ceramide levels, we hypothesized that ceramides could function to inhibit MCSR induction. Moreover, lipidomic analyses indicated reduced ceramide levels in hsp-6 knockdown worms (Table S3). We treated animals with ceramides across a range of carbon chain lengths and analyzed MCSR induction. We observed that specific ceramides were able to block the hsp-6 RNAi-induced MCSR (Figure 5D). C20 ceramide was able to partially block the MCSR, and C22 ceramide completely inhibited MCSR induction, suggesting that cardiolipin accumulation functions, in part, to affect the MCSR by inhibiting ceramide synthesis. The inhibition of the MCSR by C20 and C22 ceramide was specific, as C20 and C22 ceramide treatment did not affect UPRmt caused by cco-1 knockdown (Figure S5D). Ceramide did not induce the stress reporters tested in the absence of additional perturbations (Figure S5D). Ceramide treatment also did not affect the compartment-specific upregulation of the UPRER by tunicamycin treatment or of HSR by acute heat shock (Figure S5D).
Finally, we treated animals with RNAi against each of the enzymes in the ceramide synthesis pathway (Figure 5E). In these analyses, we found that RNAi against enzymes involved in synthesizing ceramide were sufficient to induce the MCSR even in the absence of additional genetic perturbations (Figures 5E, 5F, and S5E). In contrast, knockdown of enzymes involved in catabolizing ceramide compromised MCSR induction upon hsp-6 RNAi (Figures 5E, 5F, and S5E). Collectively, these data suggest that ceramides are necessary and sufficient to specifically inhibit the MCSR.
mtHSP70 Knockdown Improves Cytosolic Protein Homeostasis in C. elegans and Human Cells
Our data indicate that perturbing mitochondrial protein homeostasis by knocking down hsp-6 activates a cytosolic proteinfolding response and upregulates cytosolic chaperone genes through the activity of ceramide. We tested whether cytosolic protein homeostasis improves when MCSR is induced. To this end, we took advantage of a polyglutamine proteotoxicity model in C. elegans in which YFP is fused to 35 repeats of a polyQ expansion and targeted for the cytosol of body wall muscle cells (Morley et al., 2002). These animals exhibit an age-onset accumulation of polyQ aggregates in the muscle cells and subsequent motility defects.
We found that hsp-6 RNAi slowed the progression of motility defects in polyQ-expressing animals (Figure 6A). Consistently, these worms also accumulated fewer polyQ-YFP puncta (comparable to those of daf-2 RNAi-treated worms) than control animals (Figure 6A). Analysis of cytosolic protein aggregation, using filter trap methods revealed that hsp-6 RNAi and PHX treatments resulted in decreased levels of aggregated polyQ proteins (Figure 6B). This result demonstrates that the induction of the MCSR upon hsp-6 knockdown or pharmacological treatment plays a beneficial role in protein homeostasis of the cytosol and reduces proteotoxicity.
Figure 6. Cytosolic Stress Response after Mitochondrial HSP70 Knockdown Improved Cytosolic Protein Homeostasis in polyQ-Expressing C. elegans.
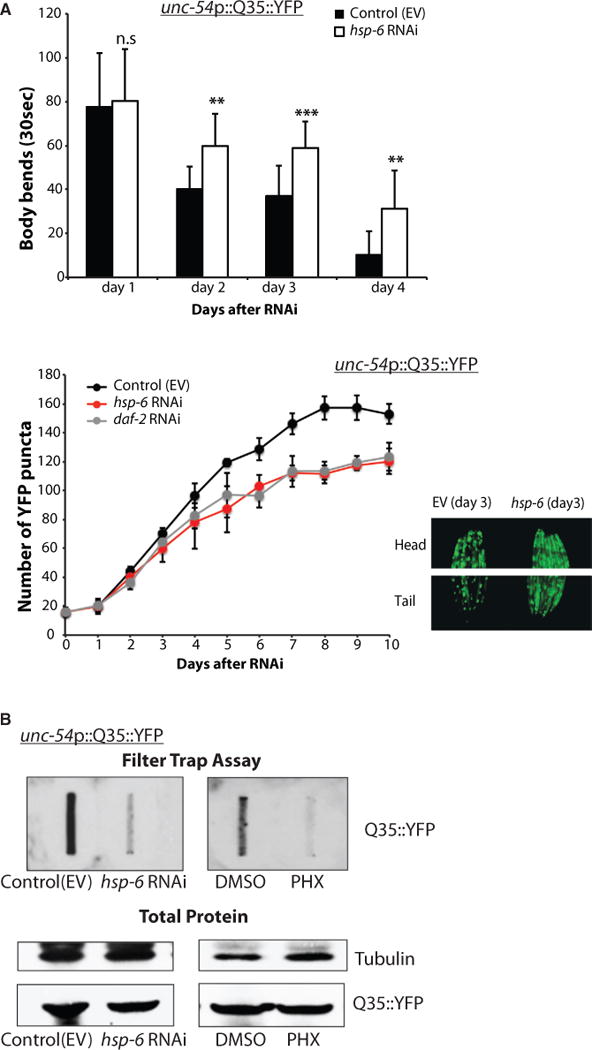
(A) (Top) AM140 worms expressing polyQ (Q35::YFP) in body wall muscle cells were tested for motility assay after RNAi treatment. Number of body bends was measured for 30 s in M9 buffer (mean ± SD of three biological repeats; **p ≤ 0.01, ****p ≤ 0.0001). (Bottom) The number of YFP puncta of AM140 worms is shown over time. The images of YFP puncta in the head and tail showed more aggregated YFP puncta in control worms and more soluble YFP signaling in hsp-6 RNAi- treated worms.
(B) hsp-6 RNAi- or PHX-treated AM140 worms showed fewer aggregates via filter trap assay.
To test whether our findings in C. elegans were conserved in a mammalian system, we created human primary fibroblast cell lines that express different lengths of polyglutamine repeats in Huntingtin exon 1 (Figure 7A). Cells with 78 polyglutamine repeats (Q78) accumulated aggregated huntingtin protein (Htt), while cells with 25 polyglutamine repeats (Q25) or GFP alone did not accumulate aggregates (Figure 7B). Interestingly, mtHSP70 protein levels increased with increasing polyglutamine length (Figure 7C) suggesting that polyQ aggregates induce the UPRmt. However, the UPRmt induced by polyQ aggregates in this cell system did not appear to alleviate cytosolic protein homeostasis, as Q78 cells still accumulated polyQ aggregates (Figure 7B). Consistent with our earlier results, we hypothesized that proteotoxic protection may require the MCSR. We found that mtHSP70 knockdown or PHX treatment of the human primary fibroblasts induced expression of cytosolic HSP70, similar to the results from C. elegans (Figure S6A). Furthermore, when we knocked down the mtHSP70 using siRNA, we observed a stark reduction in polyQ aggregates (Figure 7B). Similar to the results from C. elegans, PHX treatment also reduced polyglutamine protein aggregation in the primary human fibroblast cell lines (Figure 7C). In addition, triglyceride levels after PHX treatment were increased (Figure S6B). Similarly, the mRNA levels of many lipid synthesis genes were increased after mtHSP70 knockdown, consistent with our findings in C. elegans (Figure S6C). Moreover, double knockdown of mtHSP70 with lipid synthesis enzymes (acc1 or fas) compromised proteostasis and the cells were no longer able to reduce polyQ aggregates, consistent with the C. elegans findings (Figure S6D). These results indicate that the MCSR and its protective management of polyQ aggregates are conserved from C. elegans to human cells.
Figure 7. The MCSR Improved Cytosolic Protein Homeostasis in polyQ-Expressing Human Primary Fibroblasts.
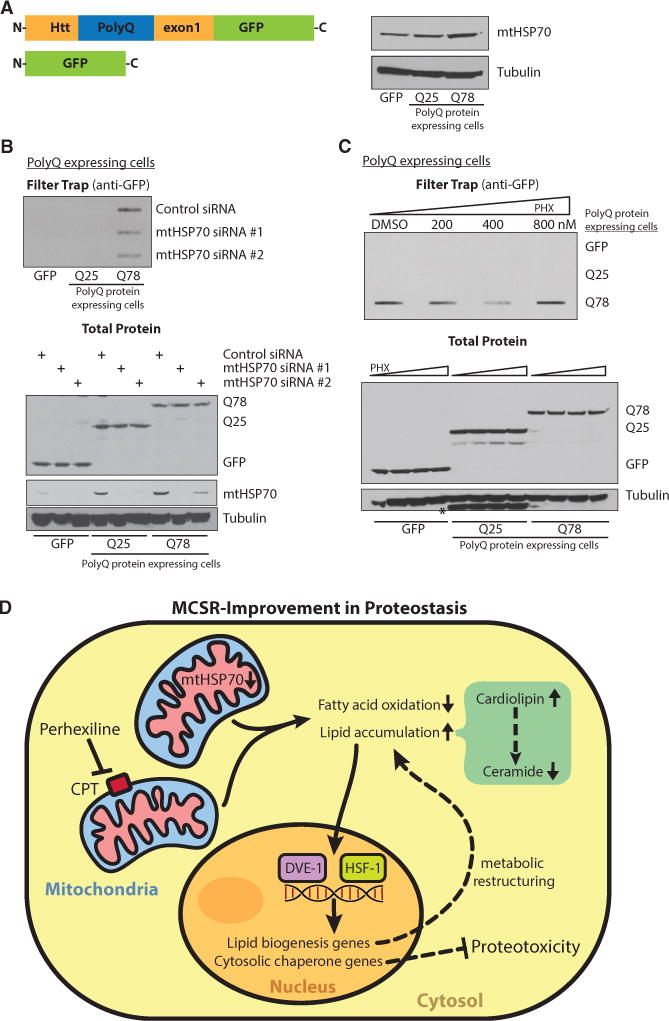
(A) PolyQ-expressing human primary fibroblasts were established and showed increased mtHSP70 expression with a longer polyQ tract by western blotting.
(B) mtHSP70 siRNA-treated cells showed fewer aggregates via filter trap assay (upper panel). Lower panel shows even expression of different-polyQ-length proteins, knockdown level of mtHSP70 after siRNA transfection, and loading control.
(C) PHX treated cells also showed less aggregates on filter trap at 400 nM (upper panel). Lower panel shows even expression of different-polyQ-length proteins and loading control. *Q25 bands from the previous probing.
(D) Proposed mechanism of MCSR regulation; mtHSP70 reduction or CPT inhibitor PHX can serve as UPRmt inducers and shift the fat metabolism pathway to the fat storage pathway. Stressed mitochondria (shown in pink) would alter fatty acid metabolism. Changes in lipid balance may serve as a cytosolic signal to turn on the cytosolic response to improve cytosolic protein homeostasis. In this, cardiolipin serves as an activator of the MCSR by reducing the level of an MCSR inhibitor, ceramide. DVE-1 and HSF-1 seem to cooperate to induce the cytosolic response upon mitochondrial perturbation.
See also Figure S6.
DISCUSSION
We find that mitochondrial perturbation caused by knocking down mtHSP70 induces a distinct stress response that not only turns on the UPRmt but also activates the HSR and is co-regulated by both dve-1 and hsf-1. We have termed this stress response the MCSR (Figure 7D). In summary, our results indicate that mtHSP70 knockdown or CPT inhibition can send a retrograde signal to the nucleus to turn on the MCSR to improve protein homeostasis in both the mitochondria and cytosol (Figure 7D). We thus propose a model in which ceramide serves as an inhibitor of MCSR under non-stressed conditions. In the presence of stress, such as the reduction of mtHSP70 expression or possibly other forms of severe mitochondrial dysfunction, cardiolipins and other lipids accumulate, acting as inhibitors of ceramide synthesis and shifting the metabolic state of the cell, thereby promoting the induction of the MCSR. Since hsp-16.2 reporter induction by cardiolipin feeding was dependent on both dve-1 and hsf-1 (Figure S5F), it is possible that cardiolipin accumulation serves as an initial signal to activate the MCSR and amplify the lipid biosynthesis signal to maintain the MCSR response (Figure 7D). Regardless, these results suggest a unique mechanism for facilitating crosstalk between mitochondrial and cytosolic stress responses via the re-structuring of fat metabolism. The MCSR is not merely a combination of the UPRmt and HSR but has distinct signaling inputs that require fat accumulation and a dedicated transcriptional circuit that overlaps with both UPRmt and HSR but also has distinct features.
Recent work found that C24 ceramide was required for mitochondrial immune surveillance (Liu et al., 2014). We hypothesize that different types of ceramide will have specific functions in mitochondrial stress signaling. In this work, key metabolic nodes, such as β-oxidation and lipid accumulation, are central for the coordination of the MCSR. We find that fat accumulation is both necessary and sufficient for induction of the MCSR in genetic and pharmacological models. Previously, losses in lipid membrane homeostasis caused by deficiencies in choline have been shown to trigger a unique stress response involving the upregulation of both cytoplasmic HSR and UPRER (Thibault et al., 2012). A loss of mtHSP70 in adult C. elegans may cause similar specific changes in membrane structure, triggering a stress response in the cytoplasm. In keeping with this hypothesis, the cytosolic chaperone HSP-16.2 can help to protect the mitochondrial membrane system during stress (Bellyei et al., 2007). Alternatively, however, the broad upregulation of cytosolic HSR components evident in our work, as well as the converse lack of upregulation of UPRER components, suggests that the MCSR is distinct from a general membrane stress response and might be triggered by causes such as a large change in the protein-folding landscape of the cytoplasm, gross changes in cellular lipid composition, or the presence of as little as a single lipid species or moiety.
Supporting the idea that the MCSR is generated by either a specific lipid signal or a change in metabolic state, we also observe that although mtHSP70 knockdown elicits the MCSR, general dysfunction in mitochondrial import and function cannot. RNAi against multiple mitochondrial ETC components and mitochondrial proteostasis genes turn on the UPRmt yet do not activate the MCSR. Many of these perturbations also disturb mitochondrial morphology and would be predicted to alter lipid synthesis pathways. These data instead suggest a specific role for mtHSP70 in coordinating cytosolic and mitochondrial homeostasis. However, we do not yet know why the MCSR is specific to the loss of mtHSP70. One important possibility we cannot exclude is that a quantitative titration of mitochondrial dysfunction (not readily discernible by the multiple RNAi analyses performed here), as opposed to a qualitative type of dysfunction, is capable of eliciting the MCSR. Alternatively, mtHSP70 may have an additional role in the cytosol, since mtHSP70 is detected in both cytosolic and mitochondrial fractions (albeit to a much less extent in the cytosol; Figure S1D). In the future, it will be critical to develop pharmacologic or genetic methods by which mtHSP70 can be specifically deleted from either the cytosol or mitochondria.
Previous work pharmacologically targeting mtHSP70 and HSP60 via tetrafluoroethylcystein also suggested that loss of function of these mitochondrial chaperones could trigger the upregulation of cytosolic HSR proteins (Ho et al., 2006). These chaperones are downstream of the heat shock transcription factor HSF-1, which is required for the MCSR. Multiple roles for HSF-1 in a response to metabolic stress have been previously reported. For example, reduced insulin/IGF-1 signaling (IIS) activates HSF-1, causing an upregulation of many of its target proteins in response to changes in nutrient availability (Hsu et al., 2003; Morley et al., 2002). Many of HSF-1’s downstream targets, heat shock proteins (HSPs), are found upregulated during caloric restriction (Steinkraus et al., 2008). Both of the metabolic sensors AMPK and Sirt1 have also been reported to directly activate HSF-1 through post-translational modifications (Dai et al., 2015; Westerheide et al., 2009). Finally, mitochondrial reactive oxygen species (ROS) have been suggested to affect HSF-1’s DNA binding and transcriptional activity (Nishizawa et al., 1999). These examples support a model in which an intricate relationship between master regulators of metabolism and the coordination of cytosolic stress response activation works in careful concert to ensure homeostasis within the cell.
Inhibition of mitochondrial enzymes such as CPT promotes fat accumulation by preventing fatty acids from entering mitochondria for β-oxidation. These inhibitors are used to treat patients with chronic heart failure, since CPT inhibitors switch the energy source from fatty acid to glucose by reducing fatty acid oxidation, improving the efficiency of energy metabolism in cardiac muscle cells. We find that mtHSP70 knockdown or CPT inhibition (via PHX treatment) reduced proteotoxicity by polyQ aggregates (Figures 6 and 7). Because cytosolic HSP70 reduces polyQ aggregation by promoting its degradation (Wang et al., 2013) and small HSPs stimulate disaggregation of amyloid aggregates (Duennwald et al., 2012), treatments capable of inducing HSP70 activity are attractive candidates for therapeutics against neurodegenerative diseases such as Huntington’s. Going forward, it will be critical to further study other CPT inhibitors in pathologic conditions involving cellular protein aggregation. In our mammalian cell culture experiments, increasing lengths of polyQ slightly induced mtHSP70, implying that the UPRmt is turned on (Figure 7). However, UPRmt induction under these conditions was insufficient to prevent accumulation of protein aggregates, indicating that these cells need extra help to turn on the proper cellular response to fight huntingtin aggregation, such as the MCSR.
If a lipid signal is emitted from the mitochondria to the cytoplasm, how might such a moiety have evolved? Mitochondria have bacterial ancestors, which contained their own HSP70 molecules. Perhaps loss of proteostasis within a bacterium, by reduction of HSP70 or overburdening of the bacterial HSP70 system, can generate a lipid molecule for communication of proteostasis imbalance to neighboring bacterium, thus inducing their own stress response for the betterment of the bacteria. We found that cardiolipin and ceramide are involved in modulating the MCSR. Further investigation on how fat accumulation turns on the cytosolic response will be the next important step to understand this signaling cascade. Perhaps these results provide a new therapeutic avenue to harness changes in fatty acid metabolism for therapeutic interventions of protein-misfolding diseases and revisit the ancestors of mitochondria.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| NDUFS3 | Abcam | Ab14711; RRID: AB_301429 |
| mtHSP70 | Abcam | Ab82591; RRID: AB_1860633 |
| Alpha-Tubulin | Sigma-Aldrich | T6074, RRID:AB_477582 |
| GFP | Roche | 11814460001; RRID: AB_390913 |
| Cytosolic HSP70/HSP72 | Enzo life | ADI-SPA-810-D; RRID: AB_2039260 |
| HSP60 | University of Iowa-Developmental Studies Hybridoma Bank | HSP60 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Perhexiline | Sigma-Aldrich | SML-0120 |
| Nile Red | Sigma-Aldrich | N3013 |
| Nonyl Acridine Orange | Invitrogen | A-1372 |
| Ceramides (C16) | Avanti Polar Lipids, Inc. | 860516P |
| Ceramide (C20) | Avanti Polar Lipids, Inc. | 860520P |
| Ceramide (C22) | Avanti Polar Lipids, Inc. | 860501P |
| Ceramide (C24) | Avanti Polar Lipids, Inc. | 860524P |
| Natural Ceramide | Avanti Polar Lipids, Inc. | 860052P |
| Cardiolipins (C14) | Avanti Polar Lipids, Inc. | 750332P |
| Cardiolipin (C18) | Avanti Polar Lipids, Inc. | 710335P |
| Natural Cardiolipin | Avanti Polar Lipids, Inc. | 840012P |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Triglyceride quantification Kit | Bio Vision | K622-100 |
|
| ||
| Deposited Data | ||
|
| ||
| Raw and analyzed microarray data | This paper | GEO: GSE83722 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human: HEK293 cells | Life Technologies | R70007 |
| Human: primary normal human dermal fibroblasts (NHDF) | Lonza | CC-2511 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| C. elegans: SJ4100 (zcIs13[hsp-6p::GFP]) | Caenorhabditis Genetics Center (CGC) | N/A |
| C. elegans: SJ4058 (zcIs9[hsp-60p::GFP]) | CGC | N/A |
| C. elegans: CL2070 (dvIs[hsp-16.2p::GFP]) | CGC | N/A |
| C. elegans: CF512 (fer-15(b26);fem-1(hc17)) | CGC | N/A |
| C. elegans: AM140 (rmls132[unc-54p::Q35::YFP]) | CGC | N/A |
| C. elegans: SJ4197 (zcIs39 [dve-1p::dve-1::GFP]) | CGC | N/A |
| C. elegans: AGD710 (N2, uthls235 [sur-5p::hsf-1, myo-2p::tomato] | (Baird et al., 2014) | N/A |
| C. elegans: N2 | CGC | N/A |
| C. elegans: AGD919 (dvIs[hsp-16.2p::GFP]; fer-15(b26);fem-1(hc17)) | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLenti6.3/V5-DEST | Proteostasis Therapeutics | N/A |
|
| ||
| Sequence-Based Reagents | ||
|
| ||
| On-TARGETplus siRNAs: hspa9 #1 | Ambion | J-057872-05 |
| On-TARGETplus siRNAs: hspa9 #2 | Ambion | J-057872-06 |
| On-TARGETplus siRNAs: acc1 #1 | Ambion | J-004551-06 |
| On-TARGETplus siRNAs: acc1 #2 | Ambion | J-004551-07 |
| On-TARGETplus siRNAs: fas #1 | Ambion | J-003954-11 |
| On-TARGETplus siRNAs: fas l#2 | Ambion | J-003954-12 |
| On-TARGETplus siRNAs: non-targeting siRNA #1 | Ambion | D-001810-01 |
| Primers for qPCR (human genes): listed in Table S4 | This paper | N/A |
| Primers for qPCR (C. elegans genes): listed in Table S4 | This paper | N/A |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author, Andrew Dillin (dillin@berkeley.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
SJ4100 (zcIs13[hsp-6p::GFP]), SJ4058 (zcIs9[hsp-60p::GFP]), CL2070 (dvIs[hsp-16.2p::GFP]), SJ4500 (zcIs4[hsp-4p::GFP]), (CF512 (fer-15(b26);fem-1(hc17)), AM140 (rmls132[unc-54p::Q35::YFP]), SJ4197 (zcIs39 [dve-1p::dve-1::GFP]), AGD710 (N2, uthls235 [sur-5p::hsf-1, myo-2p::tomato] and N2 wild-type were obtained from the Caenorhabditis Genetics Center. CL2070 strain was crossed with CF512 strain to generate a temperature-sensitive sterile reporter strain AGD919 (dvIs[hsp-16.2p::GFP]; fer-15(b26);fem-1(hc17)). RNAi screenings were done with AGD919.
Cell Culture and Maintenance
Adult human dermal fibroblasts (Lonza) were cultured in DMEM, 10% FBS and 1× GlutaMAX (GIBCO) supplemented with blasticidin (when appropriate, 2 ug/mL for selection and 1 ug/mL for maintenance of transduced cell lines). Viral particles carrying the different lengths of polyglutamine and eGFP control were made using the 3rd generation system in HEK293T cells. The viral transduction was done overnight in 8 ug/mL polybrene (Millipore). Selection was started 36 hr after the transduction for 7 days or until all the cells were eGFP positive.
Plasmids
Exon1 of the human huntingtin gene with different lengths of polyglutamine fused to eGFP in the C-terminal end or eGFP control were cloned into the pLenti6.3/V5-DEST vector (plasmids were a gift from Proteostasis Therapeutics).
siRNA Transfection
The following siRNAs were tested for depleting the indicated genes in the human primary fibroblasts or HEK cells: hspa9 siRNA (#1 and #2), acc1 siRNA (#1 and #2), fas siRNA (#1 and #2) (Ambion). Scrambled siRNA with no known mammalian homology (non-targeting siRNA #1 (Ambion) was used as negative control. Double siRNA treatment was performed by mixing two different siRNAs indicated at 1:1 ratio. Cells were transfected with the siRNAs using JetPrime according to the manufacturer’s manual and then harvested after 48 hr. Control vector-transfected cells were used as controls for all the experiments.
METHOD DETAILS
RNAi Treatment and Quantification of GFP Induction
Bacterial feeding of RNAi experiments were conducted during adulthood to exclude target genes’ function during developmental stage. Synchronized eggs were harvested by bleaching and nematodes were grown on plates with E. coli OP50 until they reach early adulthood before transferred to RNAi plates. Day 1 adult worms are then grown on the RNAi plates with E. coli HT115 that carried the RNAi construct for 3 days at 20°C unless indicated otherwise in the figure legends. In case of patchy induction of GFP expression, we used the ‘‘peak height’’ of GFP signal for quantification. Worms were imaged then using a fluorescent microscope for GFP induction or applied to COPAS Biosorter (Union Biometrica) to quantify the level of GFP induction. The temperature-sensitive sterile strains were grown at 15°C until they were gravid adults. Then, the cohorts were shifted to 25°C. Day 1 adult worms were transferred to RNAi plates for 2 days before taking images. 15-20 worms were used to take microscope images and at least 300 worms were used for COPAS biosorter analysis. Three independent biological repeats were performed.
For the double RNAi treatment, E. coli carrying the indicated RNAi constructs are mixed 1:1 ratio.
RNAi Screening: Mitochondrial Import Machinery Components
A list of genes for RNAi were obtained from a previous study (Ichishita et al., 2008). AGD919 (dvIs[hsp-16.2p::GFP]; fer-15(b26); fem-1(hc17)) eggs were synchronized by bleaching and grown on E. coli OP50 plates until they reach day 1 adult at 25°C. Day 1 adult worms were transferred to E. coli HT115 RNAi plates and grown for 2 more days before the analysis. Then, GFP induction was measured using ImageExpress (Molecular Devices) to look for the worms with more than a 2-fold increase in GFP expression compared to the control worms. All experiments were independently repeated three times using at lease 300 worms each time.
Microarray Analysis
At lease 10,000 synchronized N2 worms were grown on E. coli HT115 RNAi plates from day 1 adult for 3 days. Plates were washed-off with M9 every other day to get rid of eggs and larvae. Worms were harvested after 3 days on RNAi plates to isolate RNA for microarray. Raw expression data files were obtained for three replicates each of N2 worms treated with hsp-6 RNAi, hsp-6/hsf-1 RNAi, hsp-6/dve-1 RNAi and empty vector (EV) with the Affymetrix C. elegans Genome Array. All microarray analysis was performed with Bioconductor (Gentleman et al., 2004). Briefly, standard data quality validation as suggested by Affymetrix was carried out with the ‘simpleaffy’ package, followed by ‘affyPLM’, which identified no problematic chips. The raw data were preprocessed according to the GC-RMA method (implemented in ‘gcrma’), which performs probe-sequence- based background adjustment, quantile normalization, and utilizes a robust multi-chip average to summarize information into single expression measurements for each probeset (Irizarry et al., 2003). Before statistical testing, the data were submitted to a non-specific filter (via the package ‘genefilter’) that removed probesets with an expression interquartile range smaller than 0.5. To identify genes that were significantly differentially expressed between conditions, linear modeling and empirical Bayes analysis was performed using the ‘limma’ package (Ritchie et al., 2015). Limma computes an empirical Bayes adjustment for the t test (moderated t-statistic), which is more robust than the standard two-sample t test comparisons. To correct for multiple testing, Benjamin and Hochberg’s method to control for false discovery rate was used (Benjamini and Hochberg, 1995). Genes with an adjusted p-value of 0.05 or smaller and a fold-change in expression larger than two-fold were considered differentially expressed. Ward’s minimum variance method was used to cluster normalized expression values for genes differentially expressed in hsp-6 RNAi versus EV.
Functional Enrichment Testing
Microarray analysis expression data was used to test for enriched Gene Ontology Biological Process terms (Ashburner et al., 2000) with LRPath (Sartor et al., 2009), a logistic regression-based gene set enrichment method. LRpath related the odds of gene set membership with the significance of differential expression (p-values from limma). GO terms with an FDR of less than 1e-03 were deemed significant. Directional LRpath tests were used to distinguish between upregulated and downregulated terms. First, GO terms enriched in hsp-6 RNAi versus EV comparison were identified. Then, GO terms dependent on DVE-1 and HSF-1 were identified as those enriched in hsp-6; dve-1 RNAi versus EV; hsp-6 RNAi and hsp-6; hsf-1 RNAi versus EV; hsp-6 RNAi comparisons but with opposite regulation pattern. Representative GO terms were identified by clustering similar terms semantically with REVIGO (Supek et al., 2011), using a similarity cutoff (SimRel) of 0.5.
qPCR
Total RNA was harvested from at least 500 worms using Qiazol reagent (QIAGEN). RNA was purified using an RNeasy mini column (QIAGEN) and cDNA was synthesized using the QuantiTect reverse transcription kit (QIAGEN). According to the manufacturer’s manual, SybrGreen quantitative RT-PCR experiments were performed using an ABI Prism7900HT (Applied Biosystems), and data were analyzed using the comparative 2ΔΔCt method. pmp-3 and cdc-42 were used as housekeeping control genes for the analysis. Experiments were done with three biological repeats.
Nile Red Staining and Nonyl Acridine Orange Staining
200-300 worms were washed off from plates with M9 for fixing. Briefly, worms were fixed with freshly made 0.5% paraformaldehyde and frozen in liquid nitrogen immediately. Worms underwent two freeze thaw cycles prior to complete thawing on ice and removal of the fixation solution. M9 with Nile Red was added (1 μg/ml in final concentration) to the worms prior to staining for 15-30 min. Worms were washed once with M9 before images were taken immediately. For the quantification of staining, we used COPAS biosorter using an RFP filter. For staining the cardiolipin contents, we used Nonyl Acridine Orange (NAO, Invitrogen). After fixation and washing, 10 μM of NAO solution is added for 15-30 min. Prior to taking pictures (GFP filter), worms were washed with M9 once to get rid of extra NAO in the solution. Broken worms after fixation were excluded when taking microscope images. For the COPAS biosorter analysis, broken worms were excluded by filtering the extinction and TOF.
Electron Microscopy
300-500 worms grown on E. coli HT115 carrying RNAi, were loaded into specimen carriers and fixed using high pressure freezing (Balzers HPM 010 High Pressure Freezer), freeze substituted in 1.0% osmium tetroxide, and 0.1% uranyl acetate in acetone at −90°C, and then slowly warmed to −10°C and washed with pure acetone. Worms were embedded in increasing concentrations of Epon resin at room temperature, transferred to flat bottom embedding capsules in pure resin, and cured at 65°C for 48h. Serial sections were cut at 70cnm, and placed onto formvar coated mesh copper grids. These were imaged using the FEI Tecnai 12 Transmission electron microscope.
Mitochondrial Fractionation
8,000-10,000 animals were treated with indicated RNAi and washed off from plates. Worms were homogenized in mitochondrial isolation buffer (210 mM Mannitol, 70 mM Sucrose, 0.1 mM EDTA, 5 mM Tris-HCl, pH 7.4, and 1× protease inhibitor cocktail). Worm debris and nuclei were eliminated by centrifuging lysates for 15 min at 800 g. Supernatants containing mitochondria were then pelleted for 15 min at 12,000 g (saving the supernatants for cytosolic fractions). The mitochondrial pellet was washed with mitochondrial isolation buffer three times. 40 μg of proteins from each fraction (mitochondria and cytosol) was loaded onto the SDS-PAGE gel for Western blotting analysis.
Filter Trap Assay and Western Blotting Experiments
100 μg of protein samples were applied on to cellulose acetate membrane with 0.22 μm pore size (Schlechtes + Schule), assembled in vacuum slot blotter (Bio-Dot, Bio-Rad). Membrane was washed with 0.2% SDS five times on the blotter and subjected to antibody incubation for detecting aggregated protein retained on the membrane. Briefly, membrane was incubated with anti-GFP antibody (1:3000 dilution in 5% milk in PBS, Roche) overnight in a cold room. Membrane was washed with PBST (PBST with 0.05% Tween-20) for three times, then incubated with secondary antibody (donkey anti-mouse antibody conjugated with HRP, 1:5000 dilution in 5% milk in PBS, Jackson Immuno Research). Membranes were washed with PBST three times and exposed to film after applying ECL solutions (Pierce) to visualize the protein bands.
For SDS-PAGE, 20-40 μg of protein samples were loaded on 4%-12% bis-tris SDS gel (Invitrogen). The gel was transferred to nitrocellulose membrane (GE) using XCell II blot module (Invitrogen). Then, membranes were incubated with antibodies and exposed to film as described above. NDUFS3 (Abcam), mtHSP70 (Abcam), α-Tubulin (Sigma), GFP (Roche), and cytosolic HSP70 (HSP70/HSP72, Enzo life), HSP60 antibody supernatant (University of Iowa) were used to probe the membrane.
The Western blot bands intensity was measured using ImageJ software.
SDS-Insoluble Protein Isolation
Isolation of SDS-insoluble protein from RNAi treated worms were performed as previously described with modifications (Reis-Rodrigues et al., 2012). Briefly, up to 5000 animals that were treated with RNAi were collected and washed with M9 media. Worm pellet was resuspended in PBS containing protease inhibitor cocktail (Roche) and sonicated on ice. Then the lysates were centrifuged for 10 min at 3000 g to remove cell debris. The same amount of proteins from each sample was then centrifuged for 15 min at 16,000 g and washed three times (saving the supernatants as PBS buffer-soluble proteins). The pellet was resuspended in PBS containing 1% SDS to extract SDS-soluble proteins and was centrifuged for 15 min at 16,000 g (saving the supernatants as SDS-soluble proteins). The pellets were then resuspended in 6M GnHCl (60 min at 30°C) to extract detergent-insoluble proteins. Samples were diluted and loaded onto the SDS-PAGE gel for silver staining.
Proteasome Activity Assay
The in vitro assay of 26S proteasome activities was performed using a fluorogenic peptide substrate. Lysates were centrifuged at 10,000g for 10cmin at 4c°C. Approximately 15-25cμg of total protein of worm lysates were transferred to a 96-well microtiter plate (BD Falcon), and the fluorogenic substrate was then added to lysates. To measure the chymotrypsin-like activity of the proteasome we used Suc-Leu-Leu-Val-Tyr-AMC (Enzo). Fluorescence (380cnm excitation, 460cnm emission) was monitored on a microplate fluorometer (Infinite M1000, Tecan) every 1cmin for 2chours at 20c°C.
Oxygen Consumption Rate Measurement
Oxygen consumption of whole worms was measured using the Seahorse XF96 (Agilent Technologies). Worms were washed-off from plates with M9 to remove residual bacteria. Then, 50 worms (10 worms/well/100 μl) were transferred to the microplate. 50 μl of M9 was added to the wells and the oxygen consumption rate was measured. All experiments were repeated at least three times.
Lipidomics Sample Preparation
50,000 eggs were bleached onto the NGM plates before transferred to the RNAi plates at day1 adults. Worms were then collected 48 hr after RNAi treatments and washed with M9 for three times. Worm pellets were snap frozen with Liquid N2 for further processing. Total five biological repeats were collected for lipid extraction and followed previous protocol described (Benjamin et al., 2013).
Preparation of Palmitate-BSA Conjugate
Palmitate was conjugated to BSA as described (Seahorse protocols). Briefly, sodium palmitate was solubilized in 150 mM sodium chloride by heating up to 70°C in a water bath. Fat-free bovine serum albumin (FA-BSA) that was obtained from Sigma-Aldrich was dissolved in phosphate buffered saline (PBS) and warmed up to 37°C. Solubilized palmitate was added to BSA at 37°C with continuous stirring. The conjugated palmitate-BSA was aliquoted and stored at − 20°C. Palmitate-BSA conjugate was used to assess oxidation of exogenous fatty acid.
Fatty Acid Oxidation Measurement of Cellular Respiration
OCR measurement was performed using the Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies). HEK293 cells were plated in XF96 cell culture plates coated with poly-d-lysine (Sigma Aldrich) at 2 × 104 cells/well the day prior to the experiment. Cells were equilibrated with the substrate-limited medium for 1 hr (DMEM, 0.5 mM Glucose, 1 Mm Glutamax, 0.5 mM carnitine, 1% FBS, pH 7.4). Cells were then washed with the FAO medium (111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.2 mM Na2HPO4, 2 mM MgCl2, 5 mM HEPES, 0.5 mM carnitine, 0.72 M glucose, pH 7.2) and incubated in the FAO medium in a 37°C non-CO2 incubator for 45 min immediately before XF assay. Wells were assessed one of each treatment: BSA (5 nM), BSA (5 nM) and etomoxir (40 μM BSA-Palmitate (20 nM), BSA-Palmitate (5 nM) and etomoxir (40 μM), all mixed with the FAO medium. All cells were probed with the XF Cell Mito Stress test (Agilent Technologies), which consists of serial treatments with oligomycin (1 μM), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) (0.2 μM) and rotenone/antimycin-A (0.5 μM). These compounds were prepared in the FAO medium and were injected from the reagent ports automatically to the wells at the time indicated.
Perhexiline Treatment
PHX (Sigma) powder was dissolved in DMSO (100 mM). The stock was diluted to 1 mM and 100 μl was spotted on the E. coli OP50 plates or HT115 RNAi plates and dried before transferring the worms. Worms were transferred to new PHX plates every other day until harvested for following experiment. For the PHX treatment with multiple numbers of RNAi (Figure 4C), worms were transferred to the S-media containing HT115 RNAi bacteria, 100 μM of PHX and 1 mM of IPTG in a 96-well plate for 48 hr.
For the cell culture experiments, cells were grown to 90% confluent and washed with PBS. Indicated concentrations of PHX were diluted in the cell growth media with serum and replaced in cell culture dishes. After 48 hr, cells were washed with PBS and harvested for following experiments.
Motility Assay
Synchronized AM140 worms were transferred to the RNAi plates at day 1 of adulthood. Worms were washed-off with M9 to get rid of the eggs and larvae and transferred to new plates every other day. 15-20 worms were transferred to the M9 solution on an empty plate to take video for 30 s. Body bends were counted for 30 s for each worm; a total 12-15 worms were counted for body bends. All experiments were done in three biological repeats.
Feeding Lipids to Worms
Indicated cardiolipins and ceramides were purchased from the Avanti Polar Lipids, Inc. Lyophilized lipids were dissolved in Methanol with sonication. Ceramides were diluted to 100 mg/ml and spotted on top of the OP50 plates or RNAi plates. Worms were transferred to Ceramide-spotted plates at L4 stage and the images were taken after 48 hr. Cardiolipin was fed to bacteria for 2-4 hr (OP50 or RNAi construct-carrying HT115) with final concentration of 100 μg/ml. Then the bacteria were spotted on plates before transferring the worms. L4 worms were then transferred to the cardiolipin spotted plates and images were taken after 48 hr.
Heat Shock Treatment
100 worms were grown up to day 1 adults and underwent heat shock at 34°C for 2 hr, then recovered at 20°C for overnight before taking images. For the Nile Red Staining experiment, worms were recovered for 48 hr.
QUANTIFICATION AND STATISTICAL ANALYSIS
Triglyceride Quantification
To quantify triglyceride content, we used a Triglyceride quantification kit (Bio Vision) and followed the manufacturer’s protocol. Briefly, worms were harvested from plates and washed with M9 three times. Worms were homogenized with 5% NP-40 in water using a glass Dounce homogenizer to extract mostly the intestinal tissues by checking the lysates under a dissecting microscope. Worms looked like empty shells once the intestinal tissues were extracted out. The samples were heated slowly at 90°C for 5 min and cooled down to room temperature. Heating was repeated once more prior to centrifugation of the samples for 2 min to bring down debris. Supernatant was removed and mixed with enzymes provided in the kit according to the manufacturer’s manual. Measurement was done using microplate reader M1000 (TECAN) at Ex/Em = 535/590 nm. Calculations were done after subtracting 0 standard readings as a background.
Sample Triglyceride control (C) = B/V × D nmol/μl or mM
Where: B is the amount of triglyceride from standard curve (nmol)
V is the sample volume added into the reaction well (μl)
D is the sample dilution factor
For the mammalian cell culture samples, cells were washed with cold PBS and homogenized with 5% NP-40 in water using a syringe with 22G needle, followed by the procedure shown above. All experiments were done with three biological repeats.
Quantification
Western blots were quantified using the ImageJ software.
GFP reporter expression was quantified using the COPAS Biosorter. At least 300 adult worms were used for each biological sample. Larvae or broken worms were excluded by filtering the extinction and TOF from raw data.
Statistics
Statistical parameters, including the exact value of n and descriptive statistics (mean ± SD) and statistical significance are reported in the method details, figures and the figure legends: Number of worms used for analyses are reported in the method details. Mean of three independent biological replicates were shown with standard deviation (SD) and P values were calculated using a standard Student’s t test. Statistical significance is indicated (*: p < 0.05, **: p < 0.01, ***: p < 0.0001) in the figures and figure legends.
Lipidomic analysis was performed with five independent biological replicates and mean values were shown with standard mean of the error (mean ± SEM).
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is NCBI GEO: GSE83722.
Supplementary Material
Highlights.
Distinct disruption of mitochondrial proteostasis activates the MCSR
Fatty acid biosynthesis is required for the MCSR
MCSR requires the stress-responsive transcription factors dve-1 and hsf-1
MCSR ameliorates polyQ-mediated toxicity and can be targeted with small molecules
Acknowledgments
We thank Dr. Vidhya Ramachandran for COPAS biosort analysis. We thank Drs. Vidhya Ramachandran and Kristan Steffan for critical comments. We thank Dr. Derek Joyce for proteasome activity analysis. This work was supported by funds from the Howard Hughes Medical Institute, the NIH (grants R37AG024365 and R01ES021667), and the Glenn Foundation for Medical Research (to A.D. and S.W.) and Jane Coffin Childs Memorial Funds for medical research (H.-E.K.). A.D. is a cofounder of Proteostasis Therapeutics, Inc. and Mitobridge, Inc. and declares no financial interest related to this work.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.08.027.
AUTHOR CONTRIBUTIONS
H.-E.K. and A.D. designed research; H.-E.K. performed research and analyzed data; A.R.G. contributed to microarray data analysis; M.S.S. constructed mammalian cell lines; R.A.K. and D.K.N. contributed to lipidomic analysis; J.D. contributed to qPCR of mammalian cell culture experiments; C.E.R. contributed to fatty acid oxidation measurement of mammalian cells; M.S. contributed to the processing of electron microscope samples; E.K. contributed to RNAi screening experiments; and H.-E.K., S.W., and A.D. wrote the paper.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. The Gene Ontology Consortium Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K. Obesity and the regulation of fat metabolism. WormBook. 2007:1–20. doi: 10.1895/wormbook.1.130.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird NA, Douglas PM, Simic MS, Grant AR, Moresco JJ, Wolff SC, Yates JR, Manning G, Dillin A. HSF-1-mediated cytoskelet al integrity determines thermotolerance and life span. Science. 2014;346:360–363. doi: 10.1126/science.1253168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker T, Böttinger L, Pfanner N. Mitochondrial protein import: from transport pathways to an integrated network. Trends Biochem Sci. 2012;37:85–91. doi: 10.1016/j.tibs.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Bellyei S, Szigeti A, Boronkai A, Pozsgai E, Gomori E, Melegh B, Janaky T, Bognar Z, Hocsak E, Sumegi B, Gallyas F., Jr Inhibition of cell death by a novel 16.2 kD heat shock protein predominantly via Hsp90 mediated lipid rafts stabilization and Akt activation pathway. Apoptosis. 2007;12:97–112. doi: 10.1007/s10495-006-0486-x. [DOI] [PubMed] [Google Scholar]
- Benjamin DI, Cozzo A, Ji X, Roberts LS, Louie SM, Mulvihill MM, Luo K, Nomura DK. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc Natl Acad Sci USA. 2013;110:14912–14917. doi: 10.1073/pnas.1310894110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57:289–300. [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–366. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Dai S, Tang Z, Cao J, Zhou W, Li H, Sampson S, Dai C. Suppression of the HSF1-mediated proteotoxic stress response by the metabolic stress sensor AMPK. EMBO J. 2015;34:275–293. doi: 10.15252/embj.201489062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- Duennwald ML, Echeverria A, Shorter J. Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol. 2012;10:e1001346. doi: 10.1371/journal.pbio.1001346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bawab S, Birbes H, Roddy P, Szulc ZM, Bielawska A, Hannun YA. Biochemical characterization of the reverse activity of rat brain ceramidase. A CoA-independent and fumonisin B1-insensitive ceramide synthase. J Biol Chem. 2001;276:16758–16766. doi: 10.1074/jbc.M009331200. [DOI] [PubMed] [Google Scholar]
- Fritz V, Benfodda Z, Henriquet C, Hure S, Cristol JP, Michel F, Carbonneau MA, Casas F, Fajas L. Metabolic intervention on lipid synthesis converging pathways abrogates prostate cancer growth. Oncogene. 2013;32:5101–5110. doi: 10.1038/onc.2012.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes CM, Ron D. The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci. 2010;123:3849–3855. doi: 10.1242/jcs.075119. [DOI] [PubMed] [Google Scholar]
- Ho HK, Jia Y, Coe KJ, Gao Q, Doneanu CE, Hu Z, Bammler TK, Beyer RP, Fausto N, Bruschi SA, Nelson SD. Cytosolic heat shock proteins and heme oxygenase-1 are preferentially induced in responseto specificand localized intramitochondrial damage bytetrafluoroe-thylcysteine. Biochem Pharmacol. 2006;72:80–90. doi: 10.1016/j.bcp.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Horst M, Oppliger W, Rospert S, Schönfeld HJ, Schatz G, Azem A. Sequential action of two hsp70 complexes during protein import into mitochondria. EMBO J. 1997;16:1842–1849. doi: 10.1093/emboj/16.8.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Hu F, Liu F. Mitochondrial stress: a bridge between mitochondrial dysfunction and metabolic diseases? Cell Signal. 2011;23:1528–1533. doi: 10.1016/j.cellsig.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Gottschling DE. An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast. Nature. 2012;492:261–265. doi: 10.1038/nature11654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem. 2008;143:449–454. doi: 10.1093/jb/mvm245. [DOI] [PubMed] [Google Scholar]
- Indiveri C, Iacobazzi V, Tonazzi A, Giangregorio N, Infantino V, Convertini P, Console L, Palmieri F. The mitochondrial carnitine/acylcarnitine carrier: function, structure and physiopathology. Mol Aspects Med. 2011;32:223–233. doi: 10.1016/j.mam.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovaisaite V, Mouchiroud L, Auwerx J. The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol. 2014;217:137–143. doi: 10.1242/jeb.090738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- Kim D, Ouyang H, Li GC. Heat shock protein hsp70 accelerates the recovery of heat-shocked mammalian cells through its modulation of heat shock transcription factor HSF1. Proc Natl Acad Sci USA. 1995;92:2126–2130. doi: 10.1073/pnas.92.6.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirstein-Miles J, Morimoto RI. Caenorhabditis elegans as a model system to study intercompartmental proteostasis: Interrelation of mitochondrial function, longevity, and neurodegenerative diseases. Dev Dyn. 2010;239:1529–1538. doi: 10.1002/dvdy.22292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist S. Varying patterns of protein synthesis in Drosophila during heat shock: implications for regulation. Dev Biol. 1980;77:463–479. doi: 10.1016/0012-1606(80)90488-1. [DOI] [PubMed] [Google Scholar]
- Lindquist S. Regulation of protein synthesis during heat shock. Nature. 1981;293:311–314. doi: 10.1038/293311a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chang A. Heat shock response relieves ER stress. EMBO J. 2008;27:1049–1059. doi: 10.1038/emboj.2008.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature. 2014;508:406–410. doi: 10.1038/nature13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YW, Claypool SM. Disorders of phospholipid metabolism: an emerging class of mitochondrial disease due to defects in nuclear genes. Front Genet. 2015;6:3. doi: 10.3389/fgene.2015.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, DeMayo FJ, Li H, Abu-Elheiga L, Gu Z, Shaikenov TE, Kordari P, Chirala SS, Heird WC, Wakil SJ. Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc Natl Acad Sci USA. 2006;103:8552–8557. doi: 10.1073/pnas.0603115103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Yang T, Gu Z, Heird WC, Finegold MJ, Lee B, Wakil SJ. aP2-Cre-mediated inactivation of acetyl-CoA carboxylase 1 causes growth retardation and reduced lipid accumulation in adipose tissues. Proc Natl Acad Sci USA. 2009;106:17576–17581. doi: 10.1073/pnas.0909055106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Xuong NH, Geiduschek EP. A response of protein synthesis to temperature shift in the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1979;76:5222–5225. doi: 10.1073/pnas.76.10.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa J, Nakai A, Matsuda K, Komeda M, Ban T, Nagata K. Reactive oxygen species play an important role in the activation of heat shock factor 1 in ischemic-reperfused heart. Circulation. 1999;99:934–941. doi: 10.1161/01.cir.99.7.934. [DOI] [PubMed] [Google Scholar]
- Okino N, He X, Gatt S, Sandhoff K, Ito M, Schuchman EH. The reverse activity of human acid ceramidase. J Biol Chem. 2003;278:29948–29953. doi: 10.1074/jbc.M303310200. [DOI] [PubMed] [Google Scholar]
- Reis-Rodrigues P, Czerwieniec G, Peters TW, Evani US, Alavez S, Gaman EA, Vantipalli M, Mooney SD, Gibson BW, Lithgow GJ, Hughes RE. Proteomic analysis of age-dependent changes in protein solubility identifies genes that modulate lifespan. Aging Cell. 2012;11:120–127. doi: 10.1111/j.1474-9726.2011.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Sartor MA, Leikauf GD, Medvedovic M. LRpath: a logistic regression approach for identifying enriched biological groups in gene expression data. Bioinformatics. 2009;25:211–217. doi: 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40:141–148. doi: 10.1016/j.tibs.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinkraus KA, Smith ED, Davis C, Carr D, Pendergrass WR, Sutphin GL, Kennedy BK, Kaeberlein M. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7:394–404. doi: 10.1111/j.1474-9726.2008.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavaria M, Gabriele T, Kola I, Anderson RL. A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones. 1996;1:23–28. doi: 10.1379/1466-1268(1996)001<0023:ahsgtt>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault G, Shui G, Kim W, McAlister GC, Ismail N, Gygi SP, Wenk MR, Ng DTW. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol Cell. 2012;48:16–27. doi: 10.1016/j.molcel.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannuvel K, Renard P, Raes M, Arnould T. Functional and morphological impact of ER stress on mitochondria. J Cell Physiol. 2013;228:1802–1818. doi: 10.1002/jcp.24360. [DOI] [PubMed] [Google Scholar]
- Veatch JR, McMurray MA, Nelson ZW, Gottschling DE. Mitochondrial dysfunction leads to nuclear genome instability via an iron-sulfur cluster defect. Cell. 2009;137:1247–1258. doi: 10.1016/j.cell.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wey S, Zhang Y, Ye R, Lee AS. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid Redox Signal. 2009;11:2307–2316. doi: 10.1089/ars.2009.2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. J Biol Chem. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cao J, Shi Y. Identification and characterization of a gene encoding human LPGAT1, an endoplasmic reticulum-associated lysophosphatidylglycerol acyltransferase. J Biol Chem. 2004;279:55866–55874. doi: 10.1074/jbc.M406710200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.