

ABSTRACT
Norovirus infections are a significant health and economic burden globally, accounting for hundreds of millions of cases of acute gastroenteritis every year. In the absence of an approved norovirus vaccine, there is an urgent need to develop antivirals to treat chronic infections and provide prophylactic therapy to limit viral spread during epidemics and pandemics. Toll-like receptor (TLR) agonists have been explored widely for their antiviral potential, and several are progressing through clinical trials for the treatment of human immunodeficiency virus (HIV) and hepatitis B virus (HBV) and as adjuvants for norovirus viruslike particle (VLP) vaccines. However, norovirus therapies in development are largely direct-acting antivirals (DAAs) with fewer compounds that target the host. Our aim was to assess the antiviral potential of TLR7 agonist immunomodulators on norovirus infection using the murine norovirus (MNV) and human Norwalk replicon models. TLR7 agonists R-848, Gardiquimod, GS-9620, R-837, and loxoribine were screened using a plaque reduction assay, and each displayed inhibition of MNV replication (50% effective concentrations [EC50s], 23.5 nM, 134.4 nM, 0.59 μM, 1.5 μM, and 79.4 μM, respectively). RNA sequencing of TLR7-stimulated cells revealed a predominant upregulation of innate immune response genes and interferon (IFN)-stimulated genes (ISGs) that are known to drive an antiviral state. Furthermore, the combination of R-848 and the nucleoside analogue (NA) 2′C-methylcytidine elicited a synergistic antiviral effect against MNV, demonstrating that combinational therapy of host modulators and DAAs might be used to reduce drug cytotoxicity. In summary, we have identified that TLR7 agonists display potent inhibition of norovirus replication and are a therapeutic option to combat norovirus infections.
KEYWORDS: Toll-like receptors, antiviral agents, immunomodulation, innate immunity, norovirus
INTRODUCTION
Human norovirus is recognized as a leading cause of viral gastroenteritis globally, with an estimated 699 million norovirus infections and 212,000 attributed deaths annually (1), mostly young children in developing countries (2). Norovirus infections cause diarrhea, vomiting, abdominal cramps, fever, and nausea, with symptoms typically lasting 3 days (3). The combination of prolonged viral shedding, a low infectious dose (4), and genetic diversity (5) makes norovirus a highly transmissible pathogen that commonly causes epidemics of gastroenteritis and less frequently pandemics, costing the world economy $60 billion annually (6). Without an approved vaccine, there is a clear unmet need for the development of norovirus antivirals, particularly as prophylactic agents for use during outbreaks and for the treatment of chronic norovirus infections.
Norovirus is a member of the Caliciviridae family of nonenveloped, positive-sense, single-stranded RNA viruses and belong to the Norovirus genus, which is further divided into seven genogroups (GI to GVII) (7, 8). GI, GII, and GIV infect humans, whereas the closely related murine norovirus (MNV) belongs to GV (9). MNV, like human noroviruses, has an open reading frame 1 (ORF1), encoding nonstructural proteins (NS1 to NS7), and ORF2 and ORF3, encoding the structural proteins VP1 and VP2, respectively (10). In addition, MNV contains a fourth ORF that reportedly encodes a putative virulence factor 1 (11).
Historically, it has been difficult to propagate human norovirus in cell culture (12), and thus, most of our understanding of norovirus replication and biology has been through MNV and feline calicivirus (FCV) studies (13). In recent years, some success in propagating human norovirus has been achieved using either the B-cell (14, 15) or intestinal enteroid (15) systems. However, these systems generate modest increases in viral replication, in contrast to the MNV model, which can generate >4-log-fold increases in viral titer (PFU/milliliter) (16). Since high levels of viral replication are best suited for antiviral studies, MNV remains the primary cell culture-based tool for norovirus antiviral discovery.
The innate immune response is an important antiviral system that plays an integral role in the recognition and elimination of intracellular pathogens. One key aspect of mammalian innate recognition involves the Toll-like receptors (TLRs), which lie within plasma, endosomal, and lysosomal membranes and initiate intracellular signaling pathways that culminate in the activation of transcription factors (17). Several important examples of these factors include nuclear factor-kappa B (NF-κB), activator protein-1 (AP-1) and interferon (IFN)-regulatory factors (IRFs) IRF3 and IRF7, which can alter the expression of genes involved in antiviral defense (18–20).
The use of TLR agonists as therapeutics is a rapidly growing area of research, with several compounds in the clinical and preclinical phases of development. For example, the TLR7 agonist imiquimod (R-837) is approved as a topical treatment for genital and perianal warts associated with human papillomavirus (HPV) infection (21, 22) and has been used to treat drug-resistant genital herpes simplex virus (HSV) infections (23). Vesatolimod (GS-9620) is another TLR7 agonist that has shown promise as a potent inhibitor of HIV in vitro (24) and hepatitis B virus (HBV) in chimpanzees (25). GS-9620 is currently in phase I and II clinical trials for treatment of HIV and HBV infections, respectively (26, 27). A benefit of using TLR7 agonists is that they pose a low risk for the development of antiviral resistance, which is commonly reported with direct-acting antivirals (DAAs) (28). These agonists bind to TLR7, causing homo- or heterodimerization and subsequent recruitment of the adaptor protein myeloid differentiation primary response protein 88 (MyD88) to Toll and interleukin-1 receptor (TIR) domains of TIR domain-containing proteins (20, 29). This results in the formation of a protein complex containing tumor necrosis factor (TNF) receptor-associated factor (TRAF) proteins 3 and 6, IRAK (interleukin-1 receptor-associated kinase) proteins 1 and 4, IκB kinase alpha (IKKα), and IRF7, among others, which ultimately results in the phosphorylation of IRF7 (20, 29). Translocation of p-IRF7 into the nucleus induces robust expression of IFN and inflammatory cytokine genes (20, 29).
The development of antivirals for the treatment of norovirus infections consists predominantly of DAAs that target the viral polymerase and the protease (30). In addition, there is an increase in research toward targeting features of the host to limit norovirus replication (31–34); however, the antiviral effects of TLR7 agonists against norovirus have not been reported. We hypothesized that TLR7 agonists would be capable of inhibiting norovirus replication since the innate immune response plays an essential role in the elimination of norovirus infections (35–37). We explored the antiviral effects of the TLR7 agonists loxoribine, R-837, Gardiquimod, GS-9620, and R-848 (Table 1), using both MNV infectious cell culture and the Norwalk virus replicon systems. We show that several of these agonists potently inhibit MNV infection and can be used to generate conditioned media (CM) capable of inhibiting the human Norwalk virus replicon. Moreover, we have measured the host transcriptional changes induced by loxoribine to show that TLR7 agonists induce strong expression of genes involved in the innate immune response that contribute to the antiviral effects observed. Overall, we show that TLR7 agonists may represent a new class of antivirals that might be used to limit the burden of norovirus-associated disease.
TABLE 1.
Chemical structures and classification of compounds used in this study

RESULTS
TLR7 agonists inhibit MNV infectivity.
The antiviral effects of TLR7 agonists against HPV, HSV, HIV, and HBV have recently been described; however, the antiviral potentials of these compounds against calicivirus infections have yet to be explored. We therefore investigated whether a range of these molecules inhibited norovirus replication using the infectious MNV cell culture and human norovirus replicon models. Five TLR7 agonists were chosen and initially tested for their antiviral effects, including R-848, Gardiquimod, GS-9620, R-837, and the prototype agonist, loxoribine (Table 1). The dose-responses of all agonists were examined using a MNV plaque reduction assay, and the inhibitory activity of each compound was calculated after 48 h relative to a mock control (dimethyl sulfoxide [DMSO] treatment) (Fig. 1A; see also Fig. S1 in the supplemental material). We observed that all agonists displayed dose-response inhibition of MNV infection with the following 50% effective concentrations (EC50s): 23.5 nM (R-848), 134.4 nM (Gardiquimod), 590.0 nM (GS-9620), 1.5 μM (R-837) (Fig. 1A), and 79.4 μM (loxoribine) (Fig. S1). R-848 was the most potent TLR7 agonist, while a 50-fold increase in concentration was required for effective inhibition of MNV infection by loxoribine (Fig. 1A).
FIG 1.

TLR7 agonists exhibit antiviral activity against MNV with minimal cytotoxic effects. (A) The antiviral effects of each TLR7 agonist against MNV were assessed in cell culture using a plaque reduction assay in RAW264.7 cells. Each compound was tested over eight concentrations (0.1 nM to 10 μM for R-848, 50 nM to 20 μM for imiquimod, and 10 nM to 20 μM for Gardiquimod and GS-9620) after incubation for 48 h. Plaques were enumerated and compared to those obtained in uninhibited DMSO controls to calculate the half-maximal effective concentration (EC50) of each compound. (B) The cytotoxic effects of each TLR7 agonist against RAW264.7 cells were assessed at concentrations that ranged between 0.05 and 50 μM using the CellTitre-Blue assay. All compounds retained >50% viability at 25 μM. Plotted data represent the means ± SEM for two (panel B) or three (panel A) independent experiments performed in triplicate.
Each TLR7 agonist (except loxoribine, due to poor potency) was then tested for its cytotoxic effects on RAW264.7 cells at a concentration range of 0.05 to 50 μM (Fig. 1B). At 25 μM, ≥60% of cells remained viable for all compounds tested; however, Gardiquimod and GS-9620 treatment at 50 μM resulted in significant cell death (>50%) (Fig. 1B). There was a >2-log-fold difference between EC50 and 50% cytotoxic concentration (CC50) values for R-848 and Gardiquimod, suggesting that these molecules have good therapeutic potential (Fig. 1). Therefore, we calculated the therapeutic index of each TLR7 agonist against MNV in RAW264.7 cells using the calculation CC50/EC50. Therapeutic indices were approximately 2,127:1, 134:1, 33:1, and 41:1 for R-848, Gardiquimod, R-837, and GS-9620, respectively. Overall, we found that TLR7 agonists inhibit MNV infection in a dose-dependent manner at low concentrations with minimal effects on cellular cytotoxicity.
R-848 is a potent inhibitor of MNV replication and viral protein synthesis.
Since TLR7 agonists can stimulate a broad host response, it is likely that more than one mechanism of action is involved in the antiviral activity against the MNV. To delineate the antiviral mechanism of TLR7 agonists against MNV in more detail, we examined the effect of the most potent TLR7 agonist tested, R-848, on viral genome replication and viral protein synthesis (Fig. 2). First, we measured inhibition of viral replication by R-848 (0.5 μM), using quantitative real-time PCR (RT-qPCR) to quantify MNV genomes at 12-h intervals over 48 h. Compared to untreated controls, R-848 inhibited MNV replication by 58.2% (standard error of the mean [SEM], ±6.1%), 67.2% (±2.5%), 74.5% (±0.7%), and 67.6% (±1.8%) following 12, 24, 36, and 48 h of incubation, respectively (Fig. 2A). MNV-infected cells were also treated with R-848 at concentrations ranging from 5 nM to 10 μM and incubated for 48 h to gain a second EC50 by quantifying RNA levels. This revealed an EC50 of 42.5 nM (Fig. 2B) compared to 23.5 nM obtained using the plaque reduction assay (Fig. 1A).
FIG 2.

R-848 displays potent inhibition of MNV genome replication and protein synthesis. For all panels, RAW264.7 infections were performed with an MOI of 0.05, and cells were incubated for 48 h. R-848 was tested at 0.5 μM unless otherwise stated. All calculations were performed relative to mock control, i.e., 0.05% (vol/vol) DMSO. (A) The inhibitory effect of R-848 on MNV replication was determined by quantification of viral genomes at 12-h intervals by RT-qPCR. Inhibition increased over time with a maximum level of inhibition (75.7%) recorded at 36 hpi. (B) To determine the EC50 of R-848 against MNV at the replication level, viral genomes were again quantified by RT-qPCR following incubation with eight different concentrations of R-848 and results were compared to those obtained in an uninhibited DMSO control (5 nM to 10 μM). R-848 treatment resulted in an EC50 of 42.5 nM. (C) To illustrate the inhibition of viral protein synthesis by R-848, the levels of MNV polymerase (NS7) were detected by Western blotting of lysates from infected cells. The nucleoside inhibitor 2′CM (10 μM) was used as a positive control, and the level of b-actin was also probed in samples for normalization. Inhibition of MNV protein synthesis is evident from the decreased band intensity observed with R-848 treatment. Graphed data in panels A and B show the means ± SEM for two independent experiments performed in either triplicate (panel A and C) or quadruplicate (panel B) reactions.
In addition to viral replication and infectivity, we analyzed the cytoplasmic levels of the nonstructural 7 (NS7) viral protein (the viral polymerase) to confirm whether its abundance was reduced by TLR7 agonist treatment. Densitometry analysis of NS7 levels detected by Western blotting showed that viral protein synthesis was attenuated 100.0% with nucleoside inhibitor 2′-C methylcytidine (2′CM) treatment (10 μM) and 84.0% with R-848 treatment (0.5 μM) for 48 h (Fig. 2C). Together, these data show that R-848 treatment potently decreased MNV replication and protein production, two features essential to the norovirus replication cycle.
R-848 does not inhibit MNV RdRp activity.
Previous work has illustrated that the TLR7 agonist Gardiquimod inhibits the HIV reverse transcriptase in vitro at low micromolar concentrations (38). Since we observed a decrease in MNV genome replication following R-848 treatment (Fig. 2A and B), we aimed to determine if it was attributable to inhibition of the MNV RNA-dependent RNA polymerase (RdRp) using a previously described RdRp transcription assay with poly(C) RNA as the template (39). When tested in vitro, the MNV RdRp was not inhibited by R-848 and retained 91.2% (SEM, ±4.61%), 95.9% (±6.75%), and 101.7% (±3.99%) activity at R-848 concentrations of 10, 50, and 100 μM, respectively (Fig. 3A).
FIG 3.

R-848 is synergistic with the nucleoside analogue 2′CM and induces cellular secretion of antiviral molecules. Several assays were performed to further probe the antiviral nature of TLR7 agonists. (A) First, the effect of R-848 on the in vitro transcriptional activity of the MNV RdRp was measured using a quantitative fluorescent assay. R-848 was tested at 10, 50, and 100 μM, and transcriptional activity was compared to that in mock-treated samples (DMSO). No effect on RdRp activity was observed with the addition of R-848. (B) The combined inhibitory effects of the TLR7 agonist R-848 (0 to 40 nM) and the nucleotide inhibitor 2′CM (0 to 3 μM) were tested over a range of combinations against MNV in cell culture using the plaque reduction assay. A dose-response matrix was generated and analyzed for synergism using SynergyFinder. The ZIP mode synergy score is presented as the average of all δ-scores across the dose-response landscape, and a δ-score of >0 indicates synergism. 2′CM and R-848 display a synergistic antiviral effect against MNV. (C) To determine if R-848-treated cells produce soluble antiviral molecules, CM from R-848-treated cells was screened for anti-MNV activity using the plaque reduction assay. CM used at a final concentration of 50% (vol/vol) displayed >50% inhibition of MNV with an absence of cytotoxic effects. Data were analyzed using an unpaired t test. n.s (not significant), P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. All data shown are the means ± SEM from three independent experiments with either duplicate (panel B) or triplicate (panels A and C) reactions.
The combination of R-848 and 2′CM displays a synergistic antiviral effect against MNV.
To explore whether R-848 and the nucleoside inhibitor 2′CM exhibit a synergistic antiviral effect against MNV, we measured plaque reduction with a range of drug combinations over several concentrations (Fig. 3B). Inhibition at each combination dose was analyzed with SynergyFinder to obtain a zero-interaction potency (ZIP) synergy score of 9.673 (Fig. 3B). This value indicates a moderately synergistic interaction between R-848 and 2′CM.
R-848-stimulated RAW264.7 CM has antiviral properties against MNV.
To demonstrate whether TLR7 agonists induce the production of antiviral molecules that inhibit norovirus, we tested CM (50%, vol/vol) from R-848-stimulated RAW264.7 cells for inhibition of MNV infection using a plaque assay (Fig. 3C). Cytotoxicity of the RAW264.7 CM was also measured at 50% (vol/vol). At this concentration, we observed that the CM displayed >50% inhibition of MNV plaque formation without cytotoxicity (Fig. 3C). These results suggested that R-848 stimulation of the host cell involves the generation of antiviral molecules that are responsible for inhibition of MNV infection and replication.
TLR7 agonists upregulate the expression of IFN-stimulated genes (ISGs) and innate genes important for the control of viral infection.
We next examined the transcriptome of TLR7-stimulated RAW264.7 cells to underpin the gene expression changes that induce these antiviral secretions. Sequencing data from our previous study of loxoribine-treated RAW264.7 cells (40) were analyzed to characterize the ontology of differentially expressed genes (DEGs) (n = 1,891/12,113) compared to untreated cells (see Table S1 in the supplemental material). The five most significant ontological clusters overrepresented by DEGs included those involved in immune system process (n = 85/1,891), innate immune response (n = 70/1,891), cellular response to lipopolysaccharide (n = 40/1,891), positive regulation of IKK/NF-κB signaling (n = 33/1,891), and negative regulation of viral genome replication (n = 16/1,891) (Fig. 4). To validate our RNA-sequencing analysis, we compared the expression changes of 12 genes involved in the innate immune response by RT-qPCR (see Fig. S2 in the supplemental material). Linear regression analysis confirmed that a good correlation existed between the values obtained with the two techniques (Pearson's r = 0.94; P value < 0.001) and thus validated our analysis.
FIG 4.

Gene ontology of DEGs from TLR7 agonist-stimulated RAW264.7 cells. Gene ontology analysis was performed on genes found to be differentially expressed in the loxoribine transcriptome data set (n = 1,891). Up- and downregulated genes were analyzed separately using DAVID (gray and black bars, respectively), and the ontological terms that relate to either gene set were further refined by the removal of redundant terms using REVIGO. Ontological terms that remained are plotted based on their level of statistical significance.
In addition, we also explored the expression changes of a subset of 76 ISGs known to have a pronounced role in the control of viral infection (41, 42). Of the 76 ISGs queried, most were significantly upregulated (n = 74/76) (false-discovery rate [FDR]-adjusted P value, <0.01), and a select group also had >10-fold increase in expression (n = 30/74) following loxoribine treatment (Fig. 5A). To determine whether RAW264.7 cell treatment (48 h) with loxoribine (1 mM) or R-848 (0.5 μM) demonstrated consistent patterns of gene expression, we performed RT-qPCR assays for seven innate genes. We show that all seven genes displayed upregulation in gene expression with both agonists (Fig. 5B). Together these findings demonstrated that TLR7 agonist stimulation of RAW264.7 macrophages induced powerful expression of innate genes, a key feature of the antiviral environment that limits MNV replication.
FIG 5.

TLR7 agonists upregulate the expression of ISGs and innate genes important to viral defense. (A) Transcriptomic analyses were performed on an RNA-sequencing data set generated from RAW264.7 cells treated with the TLR7 agonist loxoribine (1 mM) for 12 h. ISGs associated with viral infection were queried for expression changes (n = 76), and a large proportion of these genes were significantly upregulated (n = 74/76). ISGs in black have a higher level of statistical significance than do ISGs in gray. (B) Transcriptional profiling of RAW264.7 cells treated with R-848 (0.5 μM) or loxoribine (1 mM) for 48 h was performed by quantifying expression changes in genes that encode transcription factors (Irf7 and Stat1), chemokines (Il1b, Il6, and Il12a), and two prominent IFN-mediated antiviral molecules (Oas2 and Mx1). Expression levels for each gene were calculated relative to mock treatment using the ΔΔCT method and compared between experimental conditions. To determine whether the gene expression changes following loxoribine and R-848 treatment were significantly different from one another, ΔCT values of both agonist treatments were analyzed using an unpaired t test with Welch's correction. Results represent the means ± SEM from two independent experiments, performed in triplicate. n.s (not significant), P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
R-848 treatment does not inhibit the Norwalk virus replicon or induce innate gene expression.
To determine whether TLR7 agonists also inhibit human norovirus replication, we measured the effect of R-848 against the human Norwalk virus strain GI.1 replicon. HG23s were treated with increasing concentrations of R-848 (ranging from 1 to 100 μM) for 72 h. Following treatment, RNA was extracted, and RT-qPCR was used to quantify norovirus replicon RNA levels. R-848 exhibited inhibition of the Norwalk virus replicon in a dose-dependent manner (Fig. 6A), albeit with limited potency compared to that seen with MNV (Fig. 2A and B). We observed a 54% (SEM, ±4.1%) reduction in replicon RNA with R-848 treatment at 100 μM (Fig. 6A); however, increased cytotoxic effects were also observed at this concentration, with 72.2% (SEM, ±2.9%) of cells remaining viable (Fig. 6A). To address the limited potency of R-848 against the Norwalk virus replicon, we determined whether R-848 stimulated innate immunity in HG23s and the parental Huh7 cell line, as seen with RAW264.7 macrophages. HG23s and Huh7 cells were treated with 1 and 10 μM R-848 for 48 h, and changes in expression of three critical innate response genes (Il6, Isg15, and Ifnb) were quantified using RT-qPCR (Fig. 6B) and compared to mock treatment. Following treatment with R-848, none of the genes displayed >3-fold increase in transcript abundance in Huh7 or HG23 cells (Fig. 6B).
FIG 6.

Direct R-848 treatment is ineffective against the Norwalk replicon and fails to stimulate innate gene expression. (A) HG23s were treated with eight concentrations of R-848 over a period of 72 h followed by RNA extraction and quantification of replicon RNA by RT-qPCR. The number of Norwalk genomes detected under each condition was normalized based on the abundance of the b-actin gene transcript. In addition, R-848-treated HG23 cells were also assessed for cytotoxic effects. One-way analysis of variance (ANOVA) was used to measure the significance of differences between viral genomes measured in the DMSO control relative to each of the R-848 doses. (B) To quantify the level of innate gene expression following TLR7 stimulation, both HG23 and the parental Huh7 cell lines were treated with R-848 at 1 μM and 10 μM. Fold changes for Il6, Isg15, and Ifnb were measured by RT-qPCR (ΔΔCT method) and calculated relative to mock control (DMSO). ΔCT values were analyzed for significant differences between the various treatment concentrations for each cell type by using two-way ANOVA. All plotted data represent the means ± SEM from two independent experiments performed in triplicate. n.s (not significant), P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
R-848-stimulated THP-1 CM displays antiviral activity against the Norwalk replicon.
To explore whether CM generated from R-848-stimulated THP-1 cells resulted in inhibition of the Norwalk replicon, we tested its antiviral effects at a concentration of 50% (vol/vol) using RT-qPCR. Following 48 h of incubation, THP-1 CM generated from R-848 stimulation at 1 μM and 10 μM resulted in 17.1% (SEM, ±6.4%) and 36.3% (SEM, ±2.6%) inhibition of the replicon, respectively (Fig. 7). A similar dose-dependent trend was observed at 72 h of CM incubation, with 45.7% (SEM, ±1.1%) (1 μM R-848) and 59.9% (SEM, ±2.2%) (10 μM R-848) inhibition of replicon replication (Fig. 7). In addition, the level of cytotoxicity was minimal and >80% viability was observed under each treatment condition (Fig. 7).
FIG 7.
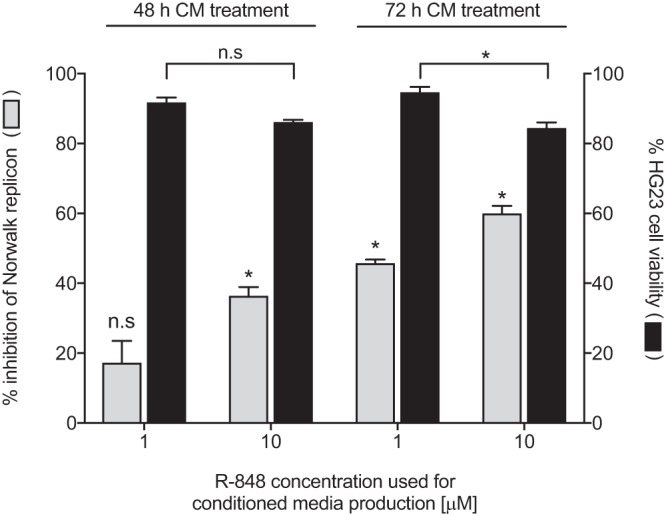
CM generated from R-848-stimulated THP-1 cells inhibits the Norwalk replicon. THP-1 cells were treated for 12 h with R-848 at 1 or 10 μM to generate CM, which was then tested at a concentration of 50% (vol/vol) against the Norwalk replicon for either 48 or 72 h. Norwalk replicon RNA was quantified by RT-qPCR and normalized to the b-actin gene transcript using the ΔΔCT method. The percent inhibition of replication was calculated relative to mock control (DMSO). ΔCT values for each CM treatment were compared to the values for the mock treatment, and differences were analyzed for significance using an unpaired t test with Welch's correction. The cytotoxic effects of R-848-generated CM were also assessed for each treatment condition. One-way ANOVA statistical analysis was used to measure any significant difference in cytotoxicity between the R-848 CM dosages. All plotted data represent the means ± SEM from two independent experiments performed in triplicate. n.s. (not significant), P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
DISCUSSION
Small-molecule agonists of TLRs are currently used in cancer immunotherapy and as adjuvants for vaccine delivery (43, 44). In recent years, however, there has been increased focus on using TLR agonists as a therapeutic option to treat viral infections (45). One important example has been the use of TLR7 and TLR9 agonists in combination with antiretroviral therapy (ART) as a part of the “kick-and-kill” eradication strategy for HIV-infected patients (27, 46, 47). This therapy is designed to increase the replication of latent HIV reservoirs and promote greater immune recognition of replicating HIV to facilitate viral elimination by a combination of the host response and the antiretroviral drug component (48, 49). Given the success with HIV treatment and given the lack of antiviral agents for norovirus infections, we investigated the effects of five TLR7 agonists (Table 1) against the MNV in vitro cell culture model and the effects of the most-potent agonist, R-848, against the Norwalk virus replicon. Our rationale was that TLR7 agonists might activate the innate immune response, resulting in inhibition of MNV and human Norwalk virus replication. Our data show that TLR7 agonists have the potential to be a therapeutic option to reduce the global burden of norovirus infections.
Agonists R-837, R-848, Gardiquimod, and GS-9620 inhibited MNV replication with EC50s in the range of 23.5 nM to 1.5 μM (Fig. 1A). The potent inhibition displayed by these compounds is akin to the nanomolar and low micromolar inhibition observed against HIV and hepatitis C virus (HCV) infections treated with TLR7 agonists in vitro (24, 50). We also performed cytotoxicity analyses, since the pharmacokinetic profile of any antiviral compound is an important factor in clinical trial progression. Significant cellular toxicity (>50%) was observed only at concentrations severalfold higher than the respective EC50 of each TLR7 agonist (Fig. 1B). However, cytotoxicity is only one measure of compound safety and adverse events of these compounds should be explored further, for example, in the MNV mouse model. Despite this, the therapeutic indices of the TLR7 agonists against MNV in the RAW264.7 cell line were 2,127:1, 134:1, 33:1, and 41:1 for R-848, Gardiquimod, R-837, and GS-9620, respectively. This trend fits well with the current published safety profile of GS-9620 (26, 51) and thus demonstrates that these compounds have the features desirable for a norovirus antiviral.
Previous studies on the effects of TLR agonists against RNA virus infections have demonstrated that treatment results in the production of proinflammatory cytokines that drive the inhibition of viral replication (24, 25, 50, 52). Given this knowledge, we aimed to ascertain the stages of the MNV replication cycle that were inhibited by TLR7 agonist treatment. When MNV-infected cells were treated with R-848 (0.5 μM), we observed greater than 50% inhibition of the expanding viral population as early as 12 hours postinfection (hpi) (Fig. 2A). The EC50 of R-848 was 42.5 nM when calculated using RNA genome levels (Fig. 2B), a concentration close to the value obtained using plaque reduction assays (23.5 nM) (Fig. 1A and 2B), which demonstrates consistency between assays. Moreover, we quantified the level of MNV RdRp (NS7) using Western blotting and showed that viral protein synthesis is attenuated following R-848 (0.5 μM) treatment after 48 h (Fig. 2C). Although we cannot conclusively infer that TLR7 agonists directly block MNV protein production, reductions in both RNA and protein levels are complementary and confirm that TLR7 stimulation inhibits norovirus replication.
Clinical approval of any drug therapy can be hampered by adverse reactions within patients, including toxicity. Immunomodulators, for example, can induce systemic inflammation and side effects that may prevent clinical trial progression (53). However, one way to reduce the toxicity of antivirals and to reduce the development of resistance is to combine drugs that target multiple facets of the host or viral replication that result in a combined or synergistic effect (54). The dual action of a TLR7 agonist and a nucleoside analogue (NA) may be an effective way to overcome off-target or toxic effects. First, we concluded that inhibition of MNV replication is not a result of direct RdRp inhibition (Fig. 3A) and thus any combination with an RdRp inhibitor would not result in antagonism. Thereafter, we showed that the combination of 2′CM (a nucleoside analogue) and R-848 had a synergistic inhibitory effect against MNV, with an average ZIP score of 9.672 over the dose-response matrix of varied combinations of these two drugs (Fig. 3B). These findings suggest that TLR7 agonists could be used in combination with novel norovirus NAs (39, 55–57) to reduce the dosage of each constituent compound and thereby limit cytotoxic effects.
We next sought to focus on the host changes that drive the antiviral effects observed. Transcriptomic analysis was performed to characterize the gene expression changes significantly altered by TLR7 agonist treatment in murine macrophages (Fig. 4). Overall, we show that upregulated genes are predominantly involved in the host innate immune response and viral defense (Fig. 4). Further to this, a small subset of key innate genes (n = 12) were also queried by RT-qPCR, and when these results were compared to our RNA-sequencing counts, a high level of correlation existed, providing support for these conclusions (Fig. S2). We then quantified the expression changes of a select group of 76 ISGs, including but not limited to Isg15, Rsad2, Oas2, Mb21d1, Irf1, Irf7, Dhx58, and Ifih1, all which have a role in viral interference and clearance (41, 42, 58). Of the ISGs examined, 74 were significantly upregulated and 14 of these had a >100-fold increase in transcript abundance (Fig. 5A). Many of these highly expressed genes are involved in the type I interferon response and encode chemokines (Ccl5), transcription factors (Irf7), receptor and signaling molecules (Rtp4), members of the Ifit (interferon-induced protein with tetratricopeptide) family (Ifit1, Ifit3), exonucleases (Isg20), nucleic acid sensors (Zbp1, Ddx60), GTPases (Mx1, Mx2), proteins involved in ISGylation (Isg15), RNase L activation (Oasl2), those with a multitude of described roles in the innate immune response (viperin [virus inhibitory protein, endoplasmic reticulum-associated, interferon-inducible]), and those with no described mechanism (Ifi44l). Given the extensive antiviral role that ISGs have been shown to exhibit, it is likely that these gene expression changes are a major contributing factor to the inhibition of norovirus displayed by TLR7 agonists.
Since our transcriptomic analysis was based on loxoribine, but the majority of the cell-based experiments were performed with the more-potent R-848, we compared expression changes of key innate genes (n = 7) by RT-qPCR. We illustrate that R-848 and loxoribine display a consistent trend in immune upregulation (Fig. 5B), which shows that TLR7 stimulation with different agonists results in the same antiviral effect.
Previous work has shown that CM from TLR7 agonist-stimulated cells has a potent antiviral activity against HBV (59), HCV (50), and HIV (24). These studies allude to the production of soluble molecules following TLR7 stimulation that display more-potent inhibition of viral replication than is seen with direct TLR7 agonist treatment. We studied the effects of CM generated by R-848 stimulation of RAW264.7 cells and measured a significant level of inhibition (>50%) of MNV plaque formation when tested at a concentration of 50% (vol/vol) (Fig. 3C). In line with previous studies of TLR7 stimulation of RAW264.7 cells (60), these data illustrate that the marked increase in gene expression induced with TLR7 agonist treatment results in the production of soluble antiviral molecules that generate an environment capable of norovirus inhibition. Furthermore, we show that TLR7 agonists can be used not only directly but also in an indirect manner (via CM) to limit norovirus replication.
Host-modulating compounds have previously been shown to inhibit the human Norwalk virus replicon and include deubiquitinase inhibitors, vitamin A, and recombinant interferon (31, 34, 36, 61). We show that direct treatment of Norwalk virus replicon-bearing cells with R-848 resulted in partial inhibition of replication, with ∼50% inhibition at 100 μM (Fig. 6A). However, this is >1,000-fold higher than the EC50 exerted against MNV infection (Fig. 1A and 2B). To explore the lack of potency of TLR agonists in the replicon system, mRNA abundance of three key innate genes was quantified following 48 h of treatment with R-848 (1 and 10 μM) in both HG23s and the parental cell line, Huh7 (Fig. 6B). When the expression changes induced by R-848 in RAW264.7 cells (Fig. 5B) are compared to those in the Huh7 and HG23 cell lines (Fig. 6B), there is a clear difference in the intensity of cellular innate stimulation. Genes Il6, Isg15, and Ifnb were upregulated in the range of 78.2- to 705.9-fold in RAW264.7 cells (Fig. 5B), whereas no greater than a 3-fold increase was measured in Huh7 and HG23 cells (Fig. 6B). These findings may reflect the low-level expression of TLRs in hepatic tissue (62) and/or the lack of cytokine production in response to TLR agonist stimulation observed in some cell lines of hepatocyte origin (63, 64).
However, the lack of potency displayed by R-848 against the human Norwalk replicon is perhaps not a true representation of the antiviral effects that occur within a host. Norovirus is thought to interact with many different intestinal cell types during infection, which have modest to high levels of TLR7 expression (B cells, macrophages, epithelial cells) and thus have the potential to be stimulated to induce an antiviral state capable of inhibiting norovirus replication. To test this hypothesis, THP-1 CM was tested at a 50% (vol/vol) concentration against the human Norwalk replicon for either 48 or 72 h (Fig. 7). We observed significant dose-dependent inhibition of replication at both time points. These data show that the lack of an antiviral effect with R-848 treatment of HG23 cells is likely a result of minimal TLR7 expression. We show that R-848-generated CM from other immune cells can stimulate the innate response of HG23s and inhibit the replicon by bypassing TLR7. Thus, an environment capable of preventing norovirus replication can be induced by R-848, when the agonist cannot work directly.
In conclusion, our results indicate that immunomodulatory compounds such as TLR7 agonists have the potential to be a future therapeutic option for the treatment of norovirus infections. The most potent agonists displayed effective viral inhibition in the low nanomolar range with minimal cytotoxicity and thus represent good candidates for antiviral therapy. We also show that TLR7 agonists might be used in combination with DAAs in a multitargeted approach, a treatment strategy widely successful at curing HCV infections. We postulate that TLR7 agonists may enhance the immune response to minimize the effects of acute viral infections but may also be useful for viral clearance of chronic norovirus sufferers and therefore could significantly reduce the burden of this disease.
MATERIALS AND METHODS
Cells, viruses, and compounds.
HG23 cells (Huh-7 origin) bearing the Norwalk virus replicon (36) (a gift from Kim Y. Green, NIAID, Bethesda, MD, USA) and murine macrophage RAW264.7 cells (kindly provided by Herbert W. Virgin, Washington University, St. Louis, MO, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO, USA), 100 U/ml penicillin-streptomycin (Life Technologies), and 2 mM Glutamax (Life Technologies). The THP-1 human monocyte cell line (a gift from Sheila Donnelly, University of Technology, Sydney, NSW, Australia) was maintained in RPMI 1640 (Sigma-Aldrich) with identical supplements. HG23 cells were also supplemented with 10 mM HEPES (Life Technologies) and 1 mg/ml Geneticin (Life Technologies). MNV strain CW1 was kindly provided by Herbert Virgin, and stocks were prepared as previously described (65). All experimental incubations were performed at 37°C with 5% CO2. The small-molecule TLR7 agonists resiquimod (R-848), vesatolimod (GS-9620), Gardiquimod, imiquimod (R-837), and loxoribine were purchased from Adipogen Life Sciences (San Diego, CA, USA). 2′-C methylcytidine (2′CM) and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich. Compounds were reconstituted in 100% dimethyl sulfoxide (DMSO) and stored at −20°C in single-use aliquots.
Generation of TLR7 agonist-induced CM.
To prepare CM of murine origin, 2.5 × 106 RAW264.7 cells were seeded into 10-cm culture dishes and incubated overnight. Cells were then treated with 5 μM R-848 and incubated for 12 h. Thereafter, medium was removed and cell monolayers were washed with phosphate-buffered saline (PBS) to remove residual compound. Fresh serum-free medium was then added, and cells were incubated for an additional 24 h. To prepare CM of human origin, 8 × 106 THP-1 cells were treated with PMA at a concentration of 100 nM in 10-cm culture dishes and incubated for 3 days to differentiate into macrophage-like cells. Medium was removed, and monolayers were washed with complete RPMI 1640 medium to remove excess PMA. Following a rest period of 3 days, the cells were treated with R-848 at 1 or 10 μM for 24 h in serum-free medium. All CM were filtered before storage at −80°C.
Cytotoxicity assays.
To assess cytotoxic effects, 20,000 RAW264.7 cells or 5,000 HG23 cells were seeded per well into 96-well plates and treated with each compound (0.05 to 50 μM) or 50% (vol/vol) CM for 48 or 72 h. Following incubation, cytotoxicity was determined using the CellTitre-Blue viability assay (Promega, Madison, WI, USA) as per the manufacturer's instructions, and fluorescence was measured on a FluoStar Optima microplate reader (BMG Labtech, Ortenberg, Germany).
Antiviral assays.
For all experiments, DMSO (vehicle only, 0.5% [vol/vol]) was used as a negative control and 2′CM (10 μM) as a positive control. All antiviral activity attributed to TLR7 agonists or CM was calculated relative to the negative control. To generate 50% effective concentration (EC50) values, each TLR7 agonist was tested over a range of eight concentrations (R-848, 0.1 nM to 10 μM; Gardiquimod and GS-9620, 10 nM to 20 μM; imiquimod, 50 nM to 20 μM; loxoribine, 1 μM to 1 mM) in antiviral assays.
Inhibition of MNV infectivity.
To test the antiviral effects of TLR7 agonists or CM against MNV infection, plaque reduction assays were performed as previously described (16, 66). Semisolid overlays contained TLR7 agonist, CM (50%, vol/vol), or combinations of R-848 (0 to 40 nM) and the nucleoside analogue 2′CM (0 to 3 μM). Plaques were enumerated to determine the EC50s or the percent inhibition of MNV infection. The percent inhibitions of MNV infection resulting from the combination dosing of R-848 and 2′CM were assessed for synergistic action using SynergyFinder (67) with the zero-interaction potency (ZIP) model (68), which generates synergy scores from a dose-response matrix.
R-848 inhibition of MNV replication.
RAW264.7 cells (20,000) were seeded into the wells of a 96-well plate and incubated overnight. The next day, monolayers were infected with MNV at a multiplicity of infection (MOI) of 0.05 for 1 h, followed by the addition of R-848 (0.5 μM) in a final volume of 150 μl, and incubated from 0 to 48 h (12-h intervals). In addition, R-848 was tested at a range of concentrations (1 nM to 10 μM) and incubated for 48 h. For both experiments, total RNA was extracted using the RNeasy minikit (Qiagen, Hilden, Germany) and MNV genomes were quantified using quantitative real-time PCR (RT-qPCR), as previously described (39).
Inhibition of MNV protein synthesis.
The effect of R-848 on viral protein production was examined by Western blot detection of the MNV NS7 polymerase. Briefly, 1 × 106 cells were seeded into the wells of a 12-well plate and incubated overnight. Medium was removed, and monolayers were infected with MNV (MOI, 0.05) for 1 h, followed by the addition of R-848 (0.5 μM) in a final volume of 1 ml, and cells were incubated for 48 h. Infected cells were lysed with radioimmunoprecipitation assay (RIPA) buffer containing protease/phosphatase inhibitor (Cell Signaling Technology, Danvers, MA, USA). Extracted protein (50 μg) was separated by SDS-PAGE (10%, wt/vol) and transferred to nitrocellulose membranes. Membranes were blocked with 5% (wt/vol) skim milk before incubation with rabbit polyclonal serum raised against MNV NS7 (1:8,000) (Life Technologies) in 5% bovine serum albumin (BSA) overnight at 4°C. Membranes were washed and probed with goat anti-rabbit horseradish peroxidase (HRP) in blocking solution (Santa Cruz, Dallas, TX, USA) at room temperature for 2 h. Protein detection was performed using Immobilon chemiluminescent HRP substrate (Millipore, Billerica, MA, USA).
Effect of R-848 on polymerase activity.
Recombinant MNV RNA-dependent RNA polymerase (RdRp) containing a C-terminal hexahistidine tag was expressed in Escherichia coli and purified by nickel affinity chromatography, as previously described (69). A fluorescent polymerase assay was performed using a homopolymeric poly(C) template (70) to measure R-848 inhibition of polymerase activity at 10, 50, and 100 μM.
Antiviral effects of R-848 and R-848-induced CM on the Norwalk virus replicon.
The antiviral effects of R-848 were tested against the Norwalk virus strain GI.1 replicon at eight concentrations between 1 and 100 μM or alternatively with a 50% (vol/vol) solution of CM generated by R-848 stimulation of THP-1 cells at 1 or 10 μM. HG23 cells were seeded (5,000 cells/well) in 96-well plates and incubated for 24 h, followed by the addition of R-848 or CM at the appropriate concentration. After 48 and/or 72 h, total RNA was isolated using TRIzol and the RNeasy kit (Qiagen), and norovirus genomes were quantified using methods described elsewhere (39, 55). The percent inhibition of Norwalk replication was calculated relative to the mock DMSO control using the ΔΔCT method (where CT is threshold cycle) (71) with b-actin as the reference gene.
RT-qPCR gene expression profiling of cells following TLR7 stimulation.
RAW264.7 cells (2 × 106) were treated with loxoribine (1 mM) or R-848 (0.5 μM) for 48 h. After incubation, RNA was extracted using TRIzol LS (Life Technologies), treated with RNase-free DNase (Qiagen), and then reverse transcribed using Superscript VILO (Life Technologies). To quantitate the relative expression changes of innate genes Irf7, Stat1, Il1b, Il6, IL12a, Oas2, and Mx1, RT-qPCR was performed using iTaq Universal SYBR green Supermix (Bio-Rad) with 2 μl of cDNA (diluted 10-fold) and 0.5 μM each primer (see Table S2 in the supplemental material). The following cycling conditions were used: 95°C denaturation for 5 min followed by 40 cycles of 95°C for 5 s, 55°C for 20 s, and 72°C for 20 s. Gene expression changes in TLR7 agonist-treated cells were compared to changes in mock treatment and normalized to Gapdh (encoding glyceraldehyde-3-phosphate dehydrogenase) using the ΔΔCT method (71). In a similar analysis, 5 × 104 Huh7 and HG23 cells were treated with 1 or 10 μM R-848 for 48 h. The method described above was used to measure the fold changes of innate genes Ifnb, Isg15, and Il6 with normalization to b-actin.
Transcriptomic profiling of loxoribine-treated cells.
RNA-sequencing reads obtained from RAW264.7 cells treated for 12 h with loxoribine (1 mM) or mock treated were analyzed to probe the cellular pathways altered by TLR7 agonists. Sequencing reads were trimmed with Trimmomatic (v0.32) (72) and mapped to the mm10 (UCSC) reference genome using TopHat (v2.0.14) (73), and transcript counts were performed with HTSeq (v0.5) (74). Expression analysis was performed using EdgeR (v3.18.1) (75), and genes were considered differentially expressed based on the following parameters: log CPM greater than or equal to 1, log FC greater than or equal to 1 or less than or equal to 1, and the FDR-adjusted P value of <0.01. Differentially expressed genes were submitted to DAVID for enrichment analysis (76, 77), and redundant ontology terms were removed using REVIGO (78). In addition, ISGs (n = 76) known to be involved in viral infection (41, 79) were queried for expression changes with TLR7 agonist treatment. To confirm the accuracy of our RNA-sequencing analysis, the transcript abundances of 12 genes (Il1b, Il6, Oas2, Ddx58, Cd40, Ifih1, Stat1, Dhx58, Tnfa, Ccl4, Cxcl11, Cxcl10) were also queried using RT-qPCR, and a correlation analysis was performed against RNA-sequencing data as previously described (40).
Statistical analysis.
All statistical calculations were performed using Graphpad Prism software (v7.0b). Data were analyzed either using one- or two-way ANOVA or using an unpaired t test with Welch's correction. All error bars depict standard errors of the means (SEM), and the following shorthand was used to indicate the level of significance: n.s., not significant (P > 0.05); *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Availability of data.
Expression data for RNA-sequencing reads obtained from RAW264.7 cells treated for 12 h with loxoribine (1 mM) or mock treated are available on the NCBI Short Reads Archive under accession numbers SRX2556753, SRX2556754, SRX2556755, SRX2556756, SRX2556761, SRX2556762, SRX2556763, and SRX2556764.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially funded by a National Health and Medical Research Council project grants (APP1083139 and 1123135). D.E.T., N.E.N., and J.H.L. acknowledge support through Australian Postgraduate Awards. J.H.L. acknowledges support from a Water Research Australia Postgraduate Scholarship.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02417-17.
REFERENCES
- 1.Pires SM, Fischer-Walker CL, Lanata CF, Devleesschauwer B, Hall AJ, Kirk MD, Duarte AS, Black RE, Angulo FJ. 2015. Aetiology-specific estimates of the global and regional incidence and mortality of diarrhoeal diseases commonly transmitted through food. PLoS One 10:e0142927. doi: 10.1371/journal.pone.0142927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 3.Graham DY, Jiang X, Tanaka T, Opekun AR, Madore HP, Estes MK. 1994. Norwalk virus infection of volunteers: new insights based on improved assays. J Infect Dis 170:34–43. doi: 10.1093/infdis/170.1.34. [DOI] [PubMed] [Google Scholar]
- 4.Teunis PF, Moe CL, Liu P, Miller ES, Lindesmith L, Baric RS, Le Pendu J, Calderon RL. 2008. Norwalk virus: how infectious is it? J Med Virol 80:1468–1476. doi: 10.1002/jmv.21237. [DOI] [PubMed] [Google Scholar]
- 5.Hansman GS, Natori K, Shirato-Horikoshi H, Ogawa S, Oka T, Katayama K, Tanaka T, Miyoshi T, Sakae K, Kobayashi S. 2006. Genetic and antigenic diversity among noroviruses. J Gen Virol 87:909–919. doi: 10.1099/vir.0.81532-0. [DOI] [PubMed] [Google Scholar]
- 6.Bartsch SM, Lopman BA, Ozawa S, Hall AJ, Lee BY. 2016. Global economic burden of norovirus gastroenteritis. PLoS One 11:e0151219. doi: 10.1371/journal.pone.0151219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroneman A, Vega E, Vennema H, Vinjé J, White PA, Hansman G, Green K, Martella V, Katayama K, Koopmans M. 2013. Proposal for a unified norovirus nomenclature and genotyping. Arch Virol 158:2059–2068. doi: 10.1007/s00705-013-1708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng D-P, Ando T, Fankhauser RL, Beard RS, Glass RI, Monroe SS. 2006. Norovirus classification and proposed strain nomenclature. Virology 346:312–323. doi: 10.1016/j.virol.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW. 2003. STAT1-dependent innate immunity to a Norwalk-like virus. Science 299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, Wang M, Wang K, Estes MK. 1993. Sequence and genomic organization of Norwalk virus. Virology 195:51–61. doi: 10.1006/viro.1993.1345. [DOI] [PubMed] [Google Scholar]
- 11.McFadden N, Bailey D, Carrara G, Benson A, Chaudhry Y, Shortland A, Heeney J, Yarovinsky F, Simmonds P, Macdonald A. 2011. Norovirus regulation of the innate immune response and apoptosis occurs via the product of the alternative open reading frame 4. PLoS Pathog 7:e1002413. doi: 10.1371/journal.ppat.1002413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karst SM, Zhu S, Goodfellow IG. 2015. The molecular pathology of noroviruses. J Pathol 235:206–216. doi: 10.1002/path.4463. [DOI] [PubMed] [Google Scholar]
- 13.Vashist S, Bailey D, Putics A, Goodfellow I. 2009. Model systems for the study of human norovirus biology. Future Virol 4:353–367. doi: 10.2217/fvl.09.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones MK, Grau KR, Costantini V, Kolawole AO, De Graaf M, Freiden P, Graves CL, Koopmans M, Wallet SM, Tibbetts SA. 2015. Human norovirus culture in B cells. Nat Protoc 10:1939–1947. doi: 10.1038/nprot.2015.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng X-L, Qu L. 2016. Replication of human noroviruses in stem cell–derived human enteroids. Science 353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wobus CE, Karst SM, Thackray LB, Chang K-O, Sosnovtsev SV, Belliot G, Krug A, Mackenzie JM, Green KY, Virgin HW IV. 2004. Replication of Norovirus in cell culture reveals a tropism for dendritic cells and macrophages. PLoS Biol 2:e432. doi: 10.1371/journal.pbio.0020432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blasius AL, Beutler B. 2010. Intracellular toll-like receptors. Immunity 32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 18.Honda K, Taniguchi T. 2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6:644–658. doi: 10.1038/nri1900. [DOI] [PubMed] [Google Scholar]
- 19.Kawai T, Akira S. 2007. Signaling to NF-κB by Toll-like receptors. Trends Mol Med 13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 20.Kawasaki T, Kawai T. 2014. Toll-like receptor signaling pathways. Front Immunol 5:461. doi: 10.3389/fimmu.2014.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hengge U, Esser S, Schultewolter T, Behrendt C, Meyer T, Stockfleth E, Goos M. 2000. Self-administered topical 5% imiquimod for the treatment of common warts and molluscum contagiosum. Br J Dermatol 143:1026–1031. doi: 10.1046/j.1365-2133.2000.03777.x. [DOI] [PubMed] [Google Scholar]
- 22.Moore RA, Edwards JE, Hopwood J, Hicks D. 2001. Imiquimod for the treatment of genital warts: a quantitative systematic review. BMC Infect Dis 1:3. doi: 10.1186/1471-2334-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilbert J, Drehs MM, Weinberg JM. 2001. Topical imiquimod for acyclovir-unresponsive herpes simplex virus 2 infection. Arch Dermatol 137:1015–1017. [PubMed] [Google Scholar]
- 24.Bam RA, Hansen D, Irrinki A, Mulato A, Jones GS, Hesselgesser J, Frey CR, Cihlar T, Yant SR. 2017. TLR7 agonist GS-9620 is a potent inhibitor of acute HIV-1 infection in human peripheral blood mononuclear cells. Antimicrob Agents Chemother 61:e01369-. doi: 10.1128/AAC.01369-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanford RE, Guerra B, Chavez D, Giavedoni L, Hodara VL, Brasky KM, Fosdick A, Frey CR, Zheng J, Wolfgang G. 2013. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 144:1508–1517. e10. doi: 10.1053/j.gastro.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gane EJ, Lim Y-S, Gordon SC, Visvanathan K, Sicard E, Fedorak RN, Roberts S, Massetto B, Ye Z, Pflanz S. 2015. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J Hepatol 63:320–328. doi: 10.1016/j.jhep.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 27.US National Institutes of Health, US National Library of Medicine. 2016. ClinicalTrials.gov. https://clinicaltrials.gov/ Accessed 30 October 2017.
- 28.Pawlotsky JM. 2011. Treatment failure and resistance with direct-acting antiviral drugs against hepatitis C virus. Hepatology 53:1742–1751. doi: 10.1002/hep.24262. [DOI] [PubMed] [Google Scholar]
- 29.Lester SN, Li K. 2014. Toll-like receptors in antiviral innate immunity. J Mol Biol 426:1246–1264. doi: 10.1016/j.jmb.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thorne L, Arias A, Goodfellow I. 2016. Advances toward a norovirus antiviral: from classical inhibitors to lethal mutagenesis. J Infect Dis 213:S27–S31. doi: 10.1093/infdis/jiv280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Hernandez MJ, Pal A, Gyan KE, Charbonneau M-E, Showalter HD, Donato NJ, O'Riordan M, Wobus CE. 2014. Chemical derivatives of a small molecule deubiquitinase inhibitor have antiviral activity against several RNA viruses. PLoS One 9:e94491. doi: 10.1371/journal.pone.0094491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vashist S, Urena L, Gonzalez-Hernandez MB, Choi J, de Rougemont A, Rocha-Pereira J, Neyts J, Hwang S, Wobus CE, Goodfellow I. 2015. Molecular chaperone Hsp90 is a therapeutic target for noroviruses. J Virol 89:6352–6363. doi: 10.1128/JVI.00315-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charbonneau M-E, Gonzalez-Hernandez MJ, Showalter HD, Donato NJ, Wobus CE, O'Riordan MX. 2014. Small molecule deubiquitinase inhibitors promote macrophage anti-infective capacity. PLoS One 9:e104096. doi: 10.1371/journal.pone.0104096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perry JW, Ahmed M, Chang K-O, Donato NJ, Showalter HD, Wobus CE. 2012. Antiviral activity of a small molecule deubiquitinase inhibitor occurs via induction of the unfolded protein response. PLoS Pathog 8:e1002783. doi: 10.1371/journal.ppat.1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang K-O, George DW. 2007. Interferons and ribavirin effectively inhibit Norwalk virus replication in replicon-bearing cells. J Virol 81:12111–12118. doi: 10.1128/JVI.00560-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang K-O, Sosnovtsev SV, Belliot G, King AD, Green KY. 2006. Stable expression of a Norwalk virus RNA replicon in a human hepatoma cell line. Virology 353:463–473. doi: 10.1016/j.virol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Changotra H, Jia Y, Moore TN, Liu G, Kahan SM, Sosnovtsev SV, Karst SM. 2009. Type I and type II interferons inhibit the translation of murine norovirus proteins. J Virol 83:5683–5692. doi: 10.1128/JVI.00231-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buitendijk M, Eszterhas SK, Howell AL. 2013. Gardiquimod: a Toll-like receptor-7 agonist that inhibits HIV type 1 infection of human macrophages and activated T cells. AIDS Res Hum Retroviruses 29:907–918. doi: 10.1089/aid.2012.0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eltahla AA, Lim KL, Eden J-S, Kelly AG, Mackenzie JM, White PA. 2014. Nonnucleoside inhibitors of norovirus RNA polymerase: scaffolds for rational drug design. Antimicrob Agents Chemother 58:3115–3123. doi: 10.1128/AAC.02799-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Enosi Tuipulotu D, Netzler NE, Lun JH, Mackenzie JM, White PA. 2017. RNA sequencing of murine norovirus-infected cells reveals transcriptional alteration of genes important to viral recognition and antigen presentation. Front Immunol 8:959. doi: 10.3389/fimmu.2017.00959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V. 2014. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505:691. doi: 10.1038/nature12862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse array of gene products are effectors of the type I interferon antiviral response. Nature 472:481. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dowling JK, Mansell A. 2016. Toll-like receptors: the swiss army knife of immunity and vaccine development. Clin Transl Immunol 5:e85. doi: 10.1038/cti.2016.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. 2007. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med 13:552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 45.Mifsud EJ, Tan AC, Jackson DC. 2014. TLR agonists as modulators of the innate immune response and their potential as agents against infectious disease. Front Immunol 5:79. doi: 10.3389/fimmu.2014.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Offersen R, Nissen SK, Rasmussen TA, Østergaard L, Denton PW, Søgaard OS, Tolstrup M. 2016. A novel Toll-like receptor 9 agonist, MGN1703, enhances HIV-1 transcription and NK cell-mediated inhibition of HIV-1-infected autologous CD4+ T cells. J Virol 90:4441–4453. doi: 10.1128/JVI.00222-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsai A, Irrinki A, Kaur J, Cihlar T, Kukolj G, Sloan DD, Murry JP. 2017. Toll-like receptor 7 agonist GS-9620 induces HIV expression and HIV-specific immunity in cells from HIV-infected individuals on suppressive antiretroviral therapy. J Virol 91:e02166-. doi: 10.1128/JVI.02166-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Halper-Stromberg A, Lu C-L, Klein F, Horwitz JA, Bournazos S, Nogueira L, Eisenreich TR, Liu C, Gazumyan A, Schaefer U. 2014. Broadly neutralizing antibodies and viral inducers decrease rebound from HIV-1 latent reservoirs in humanized mice. Cell 158:989–999. doi: 10.1016/j.cell.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Churchill MJ, Deeks SG, Margolis DM, Siliciano RF, Swanstrom R. 2016. HIV reservoirs: what, where and how to target them. Nat Rev Microbiol 14:55–60. doi: 10.1038/nrmicro.2015.5. [DOI] [PubMed] [Google Scholar]
- 50.Thomas A, Laxton C, Rodman J, Myangar N, Horscroft N, Parkinson T. 2007. Investigating Toll-like receptor agonists for potential to treat hepatitis C virus infection. Antimicrob Agents Chemother 51:2969–2978. doi: 10.1128/AAC.00268-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lopatin U, Wolfgang G, Tumas D, Frey CR, Ohmstede C, Hesselgesser J, Kearney B, Moorehead L, Subramanian GM, McHutchison JG. 2013. Safety, pharmacokinetics and pharmacodynamics of GS-9620, an oral Toll-like receptor 7 agonist. Antivir Ther 18:409–418. doi: 10.3851/IMP2548. [DOI] [PubMed] [Google Scholar]
- 52.Horsmans Y, Berg T, Desager JP, Mueller T, Schott E, Fletcher SP, Steffy KR, Bauman LA, Kerr BM, Averett DR. 2005. Isatoribine, an agonist of TLR7, reduces plasma virus concentration in chronic hepatitis C infection. Hepatology 42:724–731. doi: 10.1002/hep.20839. [DOI] [PubMed] [Google Scholar]
- 53.Engel AL, Holt GE, Lu H. 2011. The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev Clin Pharmacol 4:275–289. doi: 10.1586/ecp.11.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Foucquier J, Guedj M. 2015. Analysis of drug combinations: current methodological landscape. Pharmacol Res Perspect 3(3):e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rocha-Pereira J, Jochmans D, Debing Y, Verbeken E, Nascimento MS, Neyts J. 2013. The viral polymerase inhibitor 2′-C-methylcytidine inhibits Norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J Virol 87:11798–11805. doi: 10.1128/JVI.02064-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kolawole AO, Rocha-Pereira J, Elftman MD, Neyts J, Wobus CE. 2016. Inhibition of human norovirus by a viral polymerase inhibitor in the B cell culture system and in the mouse model. Antiviral Res 132:46–49. doi: 10.1016/j.antiviral.2016.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Netzler NE, Tuipulotu DE, Eltahla AA, Lun JH, Ferla S, Brancale A, Urakova N, Frese M, Strive T, Mackenzie JM. 2017. Broad-spectrum non-nucleoside inhibitors for caliciviruses. Antiviral Res 146:65–75. doi: 10.1016/j.antiviral.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 58.Schneider WM, Chevillotte MD, Rice CM. 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guo F, Han Y, Zhao X, Wang J, Liu F, Xu C, Wei L, Jiang J-D, Block TM, Guo J-T. 2015. STING agonists induce an innate antiviral immune response against hepatitis B virus. Antimicrob Agents Chemother 59:1273–1281. doi: 10.1128/AAC.04321-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmitz F, Mages J, Heit A, Lang R, Wagner H. 2004. Transcriptional activation induced in macrophages by Toll-like receptor (TLR) ligands: from expression profiling to a model of TLR signaling. Eur J Immunol 34:2863–2873. doi: 10.1002/eji.200425228. [DOI] [PubMed] [Google Scholar]
- 61.Lee H, Ko G. 2016. Antiviral effect of vitamin A on norovirus infection via modulation of the gut microbiome. Sci Rep 6:25835. doi: 10.1038/srep25835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zarember KA, Godowski PJ. 2002. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 63.De Creus A, Abe M, Lau AH, Hackstein H, Raimondi G, Thomson AW. 2005. Low TLR4 expression by liver dendritic cells correlates with reduced capacity to activate allogeneic T cells in response to endotoxin. J Immunol 174:2037–2045. doi: 10.4049/jimmunol.174.4.2037. [DOI] [PubMed] [Google Scholar]
- 64.Lichtman SN, Wang J, Lemasters JJ. 1998. LPS receptor CD14 participates in release of TNF-α in RAW 264.7 and peritoneal cells but not in Kupffer cells. Am J Physiol 275:G39–G46. [DOI] [PubMed] [Google Scholar]
- 65.McCartney SA, Thackray LB, Gitlin L, Gilfillan S, Virgin HW IV, Colonna M. 2008. MDA-5 recognition of a murine norovirus. PLoS Pathog 4:e1000108. doi: 10.1371/journal.ppat.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hyde JL, Gillespie LK, Mackenzie JM. 2012. Mouse norovirus 1 utilizes the cytoskeleton network to establish localization of the replication complex proximal to the microtubule organizing center. J Virol 86:4110–4122. doi: 10.1128/JVI.05784-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ianevski A, He L, Aittokallio T, Tang J. 2017. SynergyFinder: a web application for analyzing drug combination dose–response matrix data. Bioinformatics 33:2413–2415. doi: 10.1093/bioinformatics/btx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yadav B, Wennerberg K, Aittokallio T, Tang J. 2015. Searching for drug synergy in complex dose–response landscapes using an interaction potency model. Comput Struct Biotechnol J 13:504–513. doi: 10.1016/j.csbj.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bull RA, Hyde J, Mackenzie JM, Hansman GS, Oka T, Takeda N, White PA. 2011. Comparison of the replication properties of murine and human calicivirus RNA-dependent RNA polymerases. Virus Genes 42:16–27. doi: 10.1007/s11262-010-0535-y. [DOI] [PubMed] [Google Scholar]
- 70.Eltahla AA, Lackovic K, Marquis C, Eden J-S, White PA. 2013. A fluorescence-based high-throughput screen to identify small compound inhibitors of the genotype 3a hepatitis C virus RNA polymerase. J Biomol Screen 18:1027–1034. doi: 10.1177/1087057113489883. [DOI] [PubMed] [Google Scholar]
- 71.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 72.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Anders S, Pyl PT, Huber W. 2015. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang DW, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 78.Supek F, Bošnjak M, Škunca N, Šmuc T. 2011. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.