

Abstract
Activated immune cells undergo a metabolic switch to aerobic glycolysis akin to the Warburg effect, presenting a potential therapeutic target in autoimmune disease. Dimethyl fumarate, a derivative of the Krebs cycle intermediate fumarate, is an immunomodulatory drug used to treat multiple sclerosis and psoriasis. Although its therapeutic mechanism remains uncertain, it covalently modifies cysteine residues in a process termed “succination.” Here, we show that dimethyl fumarate succinates and inactivates the catalytic cysteine of the glycolytic enzyme GAPDH both in vitro and in vivo. It thereby downregulates aerobic glycolysis in activated myeloid and lymphoid cells, which mediates its anti-inflammatory effects. Our findings provide mechanistic insight into immune modulation by dimethyl fumarate and represent a proof of concept that aerobic glycolysis is a therapeutic target in autoimmunity.
Pro-inflammatory stimuli induce a metabolic switch in both myeloid and lymphoid cells, leading to a Warburg-like up-regulation of aerobic glycolysis that regulates the balance between inflammatory and regulatory immune phenotypes (1, 2). Classically activated macrophages and effector lymphocytes such as T-helper (Th) 1 and Th17 cells require glycolysis for their survival, differentiation, and effector functions (3–9), whereas oxidative metabolism favors the differentiation of alternatively activated (M2) macrophages and regulatory T (Treg) cells (10, 11).
Dimethyl fumarate (DMF) is a serendipitously discovered immunomodulatory drug used to treat psoriasis and multiple sclerosis (MS) (12). Although its mechanisms of action remain incompletely understood, it is known to covalently modify cysteine residues in a process termed “succination” (not to be confused with lysine succinylation) (fig. S1) (13, 14). DMF succinates kelch-like ECH-associated protein 1 (KEAP1), which activates nuclear factor (erythroid-derived 2)-related factor 2 (Nrf2) to produce anti-oxidant and anti-inflammatory effects that nonetheless fail to fully account for the drug’s actions (15). Endogenous fumarate also succinates proteins, with a primary target being the active-site cysteine of the glycolytic enzyme glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (16). In this study, we show that DMF and its clinically relevant metabolite monomethyl fumarate (MMF) target GAPDH and inactivate its enzyme activity, both in vitro and after oral treatment in mice and humans. In turn, GAPDH inhibition downregulates aerobic glycolysis in myeloid and lymphoid cells, preventing immune activation and shifting the balance between inflammatory and regulatory cell types.
To determine whether GAPDH was succinated by DMF and MMF, we performed liquid chromatography–tandem mass spectrometry (LC–MS/MS). Treatment of recombinant human GAPDH with MMF led to monomethyl succination (2-monomethyl succinyl-cysteine) at its active-site cysteine (Cys-152 in human) and cysteines 156 and 247, whereas DMF produced a combination of dimethyl (2-dimethyl succinyl-cysteine) and monomethyl succination at the same cysteines (table S1 and fig. S2). Neither of these modifications was observed at any cysteine in vehicle-treated GAPDH. In mice, oral treatment with DMF led to both monomethyl and dimethyl succination exclusively of the active-site cysteine (Cys-150 in mouse) of GAPDH purified from the spleen and brain, with no such modifications observed on other cysteines or in vehicle-treated mice (Fig. 1A and table S1). Although only the monomethyl form of the drug is detected in serum (17), our finding that dimethyl succination occurred after oral administration was consistent with prior work (18, 19). Monomethyl succination of GAPDH Cys-152 was also identified in peripheral blood mononuclear cells (PBMCs) from MS patients treated with DMF (Fig. 1B and table S1) but not on other cysteines or in PBMCs from MS patients not treated with DMF. GAPDH succination by endogenous fumarate occurred physiologically, as we identified modification by fumarate (2-succinylcysteine) at the active-site cysteine in vehicle-treated mice and human healthy controls (fig. S3).
Fig. 1.
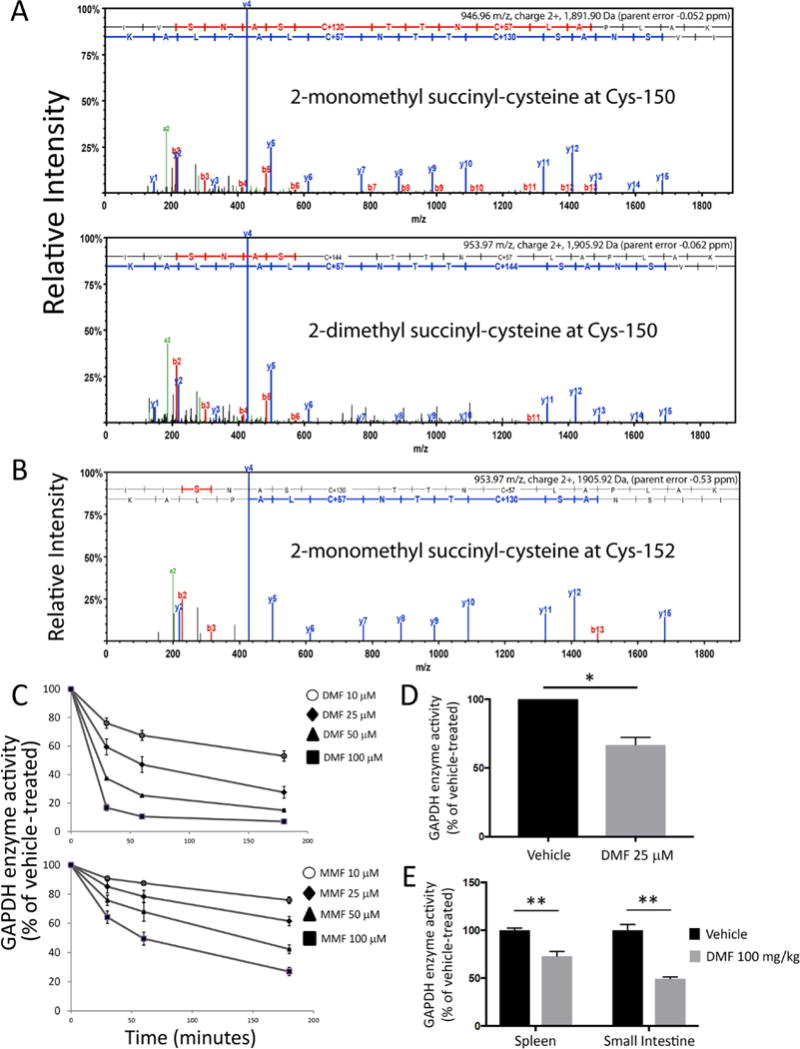
DMF and MMF succinate and inactivate GAPDH in vitro and after oral treatment. (A) Representative LC-MS/MS spectra demonstrating covalent modification of the catalytic cysteine (Cys-150 in mouse) by either monomethyl (2-monomethyl succinyl-cysteine, +130 Da) or dimethyl (2-dimethyl succinyl-cysteine, +144 Da) fumarate in GAPDH immunoprecipitated from splenic lysates of mice treated orally with 100 mg/kg DMF daily for five days. Because samples were reduced and treated with iodoacetate, non-succinated cysteines were modified with carbamidomethyl (Carb, +57 Da). Pooled samples were analyzed from two vehicle-treated and two DMF-treated animals. (B) Representative LC-MS/MS spectrum demonstrating monomethyl succination of the catalytic cysteine (Cys-152 in human) of GAPDH immunoprecipitated from PBMC lysates of MS patients treated with DMF for three months. Pooled samples were analyzed from three DMF-treated and two non-DMF-treated patients. (C) Dose- and time-dependent inactivation of GAPDH enzyme activity in vitro. Recombinant GAPDH was treated with the indicated drug concentrations or vehicle alone. Aliquots were removed at the specified time points, followed by enzyme activity assay. Data were pooled from four experiments performed in duplicate and represent mean ± SEM for each time point. Associated Kitz–Wilson plots and kinetic parameters are shown in fig. S4C. (D) Peritoneal macrophages were treated overnight with DMF. Cells lysates were used for GAPDH enzyme activity assay. Data represent mean ± SEM of three experiments performed in duplicate. (E) Mice were treated with DMF 100 mg/kg daily for five days by oral gavage. On Day 5, mice were sacrificed, and lysates from spleen and small intestine were used for GAPDH enzyme activity assay. Data represent mean ± SEM of five mice per group, with assays run in triplicate. Da = daltons. *P < 0.05 and **P < 0.01 by two-tailed Student’s t-test.
Covalent modification of its catalytic cysteine should irreversibly inactivate GAPDH. We found that both DMF and MMF decreased the catalytic activity of recombinant GAPDH in a dose- and time-dependent manner (Fig. 1C). This inhibition was irreversible, as desalting failed to restore activity (fig. S4A). Additionally, this inhibition was mediated by active-site binding of DMF/MMF, as the drug effect was blocked by pre-incubation with tenfold excess of GAPDH substrates (fig. S4B). GAPDH inhibition was biphasic, with an initial fast phase and a secondary slow phase. We calculated the kinetics of inhibition for both phases using the Kitz–Wilson method (20) (fig. S4C). GAPDH activity was similarly inhibited in cultured mouse peritoneal macrophages (mPMs) treated overnight with 25 μM DMF (Fig. 1D). In mice, oral treatment with DMF decreased GAPDH activity measured from both the spleen and small intestine (Fig. 1E). This effect was particularly profound in the small intestine, which may be relevant given the role of the gut immune system in autoimmune disorders such as MS (21).
We next asked whether GAPDH inhibition by DMF impacted aerobic glycolysis in activated immune cells. Using lactate production as a proxy measure, co-treatment with DMF significantly impaired glycolysis in mPMs stimulated in vitro for 24 hours with 1 μg/ml lipopolysaccharide (LPS) (Fig. 2A). MMF had a similar effect but with lower potency. The influence on glycolysis was not due to cytotoxicity (fig. S5). Measurements of the extracellular acidification rate (ECAR) revealed a similar inhibition of aerobic glycolysis by DMF and MMF in LPS-stimulated mPMs (Fig. 2B). In activated mouse and human CD4+ lymphocytes, DMF and MMF decreased basal glycolysis, with an even greater effect on maximal glycolytic capacity (Fig. 2C and fig. S6). In LPS-stimulated mPMs, treatment with DMF produced a blockade in glycolytic flux at GAPDH (Fig. 2D and fig. S7), providing evidence that GAPDH inactivation mediated the downregulation of glycolysis by DMF.
Fig. 2.
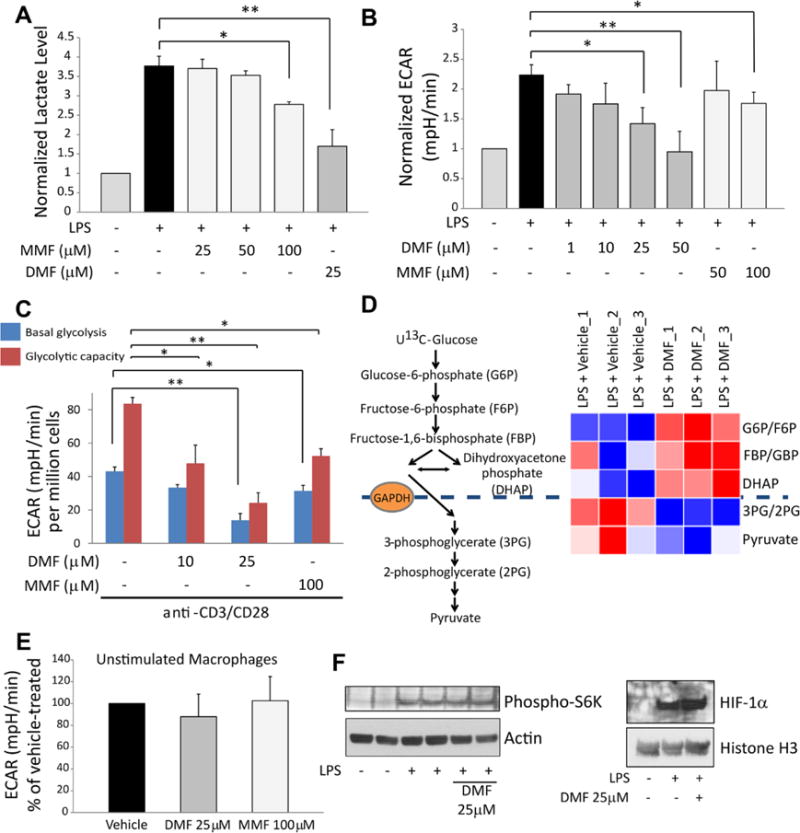
GAPDH inactivation by DMF and MMF inhibits glycolysis in activated, but not resting, macrophages and lymphocytes. (A) Mouse peritoneal macrophages (mPMs) were treated with LPS ± DMF or MMF for 24 hours followed by measurement of lactate (a proxy measure of glycolysis) in culture media by colorimetric assay. Data represent mean ± SEM of three experiments performed in duplicate. (B) mPMs were treated as in (A), and glycolysis was measured as extracellular acidification rate (ECAR) using a Seahorse extracellular flux analyzer. Data represent mean ± SEM of five experiments performed in quadruplicate. (C) Glycolysis was measured via Seahorse extracellular flux analyzer in mouse naive CD4+ lymphocytes activated overnight with anti-CD3/CD28 antibodies ± DMF/MMF. Data represent mean ± SEM of four experiments performed in triplicate. (D) mPMs were stimulated with LPS for 24 hours ± 25 μM DMF, in triplicate. Cells were then labeled with U13C-glucose, and 13C-labeling of glycolytic intermediates was measured from lysates via LC-MS. Heat map shows blockade of glycolytic flux at the level of GAPDH. (E) DMF/MMF had no effect on glycolysis in unstimulated mPMs, measured as ECAR. Data represent mean ± SEM of four experiments performed in quadruplicate. (F) Representative immunoblots showing no effect of DMF on phospho-S6K (a marker of mTOR activity) (N = two experiments performed in duplicate) or HIF-1α levels (N = three experiments) in LPS-stimulated mPMs. Data are quantified in fig. S8. *P < 0.05 and **P < 0.01 by one-way analysis of variance (ANOVA) with Dunnett’s multiple comparison.
DMF had no effect on glycolysis in unstimulated mPMs (Fig. 2E), raising the possibility that DMF acts not only on GAPDH but also on the signaling pathway required for glycolytic upregulation. However, DMF had no effect on the activity of mechanistic target of rapamycin (mTOR, as measured via p70-S6 kinase phosphorylation) or levels of hypoxia-inducible factor 1-α (HIF-1α) (Fig. 2F and fig. S8). This selective inhibition of glycolysis in LPS-stimulated mPMs is consistent with recent evidence that GAPDH only becomes a rate-limiting enzyme when glycolysis is upregu-lated in the setting of Warburg physiology (22–24), as it is in cancer and activated immune cells. This likely explains why DMF is not generally toxic.
We also examined whether DMF impacted oxidative phosphorylation (OXPHOS). DMF increased OXPHOS in mPMs under both resting and LPS-stimulated conditions (fig. S9A). The inhibition of glycolysis did not depend on the upregulation of OXPHOS (fig. S9, B and C).
We next asked whether inhibition of GAPDH and aerobic glycolysis mediated the immunologic actions of DMF. We first addressed this question in macrophages. We reproduced previous findings that both DMF and glycolytic blockade prevent classical macrophage activation (fig. S10, A to D) (3, 25) and then determined that the inhibition of cytokine production by DMF was unrelated to its effects on OXPHOS (fig. S10E). We next measured the production of IL-1β under low (0.5 mM) or high (10 mM) glucose concentrations and found that DMF was much less effective in the presence of high glucose (Fig. 3A), suggesting that its anti-inflammatory effect can be overcome by driving glycolysis higher with saturating concentrations of glucose. DMF augmented the IL-4-induced expression of arginase-1 (Arg-1), a marker of M2 alternative activation (fig. S10F), but the relative importance of DMF effects on aerobic glycolysis versus OXPHOS in promoting alternative activation was not examined.
Fig. 3.

Inhibition of GAPDH and aerobic glycolysis mediates anti-inflammatory effects of DMF in macrophages. (A) mPMs were treated with LPS ± DMF for 24 hours in either limiting (0.5 mM) or saturating (10 mM) concentrations of glucose, followed by measurement of IL-1β secretion by ELISA. Data represent mean ± SEM of three experiments performed in singlet or duplicate. (B) Treating LPS-stimulated mPMs with 30 μM heptelidic acid, a selective GAPDH inhibitor, replicated the effect of DMF on IL-1β secretion. Data represent mean ± SEM of three experiments performed in singlet or duplicate. (C and D) Representative immunoblots from three experiments showing that heptelidic acid replicated the effects of DMF on iNOS expression (C) and nuclear translocation of NF-κB (D) in LPS-stimulated mPMs. Data are quantified in fig. S11. (E) Overexpression of wild-type GAPDH (GAPDH-WT), but not Cys-150 mutant (GAPDH-C150S), mitigated the effect of DMF on IL-1β secretion in mPMs. Data represent the mean of two experiments performed in duplicate. ns = non-significant. *P < 0.05 and **P < 0.01 by two-tailed Student’s t-test (A) and one-way ANOVA with Dunnett’s multiple comparison (B and E).
Heptelidic acid (also known as koningic acid) is a GAPDH inhibitor, which binds the active site and covalently modifies the catalytic cysteine (23, 26). The treatment of mPMs with heptelidic acid replicated the effects of DMF on IL-1β secretion (Fig. 3B), inducible nitric oxide synthase (iNOS) expression (Fig. 3C and fig. S11A), and nuclear trans-location of nuclear factor-κB (NF- κB) (Fig. 3D and fig. S11B). Conversely, the overexpression of wild-type GAPDH, but not catalytically inactive GAPDH mutated at Cys-150, mitigated the effect of DMF on IL-1β production (Fig. 3E and fig. S12). Thus, the immunologic actions of DMF were replicated by GAPDH inhibition and reversed by increasing GAPDH expression.
We next examined the effects of DMF and MMF on lymphocyte differentiation and function. We activated mouse naïve CD4+ T cells under Th1-, Th17-, or Treg-cell-polarizing conditions for four days ± DMF or MMF, with treatment at the start of polarization. Consistent with known effects of glycolytic blockade (7, 8), DMF disproportionately impacted the survival of Th1 and Th17 versus Treg cells (Fig. 4A and fig. S13A). Similarly, DMF/MMF inhibited both differentiation and cytokine production under Th1- and Th17-polarizing conditions, an effect replicated by heptelidic acid (Fig. 4, B to D, and fig. S13B). As reported with HIF-1α deficiency and 2-deoxyglucose (11), DMF promoted Treg-cell differentiation under Treg-polarizing conditions (Fig. 4E and fig. S13B), and both DMF and MMF reciprocally inhibited Th17 and promoted Treg cell development under Th17-polarizing conditions (Fig. 4F). When added after three days of polarization, DMF, MMF, and low-dose heptelidic acid had no effect on viability (fig. S14A) or differentiation (fig. S14B) of Th1 or Th17 cells. However, all three drugs inhibited the expression of IFNγ and IL-17 (Fig. 4G), suggesting an effect on cytokine production independent of survival and differentiation.
Fig. 4.
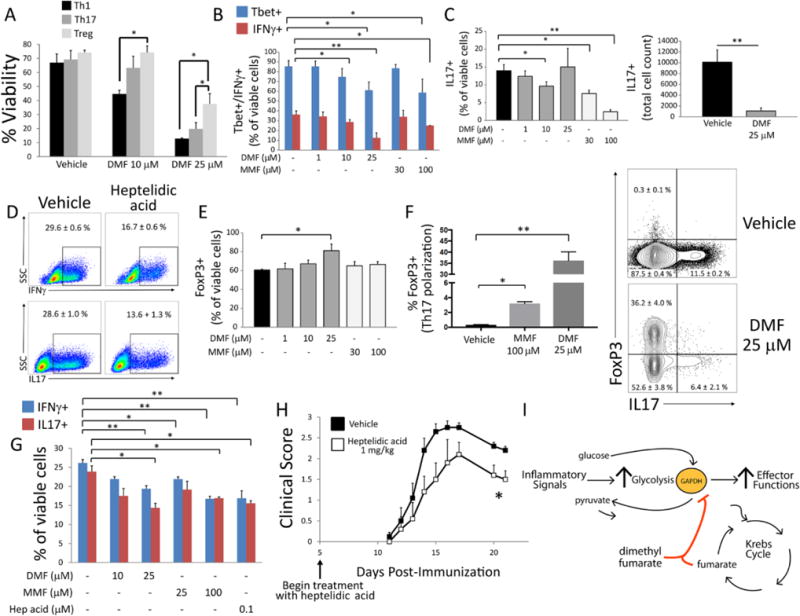
DMF and MMF differentially impact survival, differentiation, and effector function of metabolically distinct lymphocyte subsets. (A to F) Mouse naïve CD4+ lymphocytes were activated for four days with anti-CD3/CD28 antibodies under Th1-, Th17-, or Treg-cell-polarizing conditions. Cells were treated with indicated doses of DMF, MMF, or heptelidic acid on day 0 and assayed by flow cytometry on day 4. (A) DMF disproportionately decreased survival under Th1- and Th17-cell vs. Treg-cell polarizing conditions, as assessed by LIVE/DEAD aqua stain. Data represent mean ± SEM of three experiments performed in duplicate or triplicate. (B and C) DMF/MMF decreased the proportion of Tbet+ and IFNγ+ cells under Th1-cell-polarizing conditions (B) and of IL-17+ cells under Th17-cell-polarizing conditions (C, left). DMF (25 μM) produced variable results under Th17-cell-polarizing conditions, likely due to high toxicity at that dose, but nonetheless caused a significant decrease in total IL-17+ cell count (C, right). Data represent mean ± SEM of three experiments performed in duplicate or triplicate. (D) Representative flow cytometric plots demonstrating that low-dose (0.5 μM) heptelidic acid replicated the effect of DMF/MMF on IFNγ and IL-17 expression under Th1- and Th17-cell-polarizing conditions, respectively. Toxicity limited the testing of higher doses. Values represent mean ± SEM of a triplicate experiment. (E) In contrast to effects on Th1 and Th17 cells, DMF increased the proportion of FoxP3+ cells under Treg-cell-polarizing conditions. Data represent mean ± SEM of three experiments performed in duplicate or triplicate. (F) DMF/MMF produced a reciprocal increase in FoxP3+ cells under Th17-cell-polarizing conditions. Bar graph (left) and representative flow cytometric plot (right) from a triplicate experiment. Data represent mean ± SEM. (G) Mouse naïve CD4+ lymphocytes were activated under Th1- or Th17-cell-polarizing conditions for three days and then treated overnight with the indicated drug. Expression of IFNγ and IL-17 was then assessed by flow cytometry. Data represent mean ± SEM of a triplicate experiment. (H) Daily I.P. treatment with heptelidic acid attenuated the course of EAE. Data were pooled from five mice per group and represent mean ± SEM for each time point. (I) Proposed model of immune modulation by DMF, which may exploit a physiologic negative feedback function of endogenous fumarate. *P < 0.05 and **P < 0.01 by one-way ANOVA with Tukey’s multiple comparison (A); one-way ANOVA with Dunnett’s multiple comparison (B, C, E–G); and Mann–Whitney U test (H).
Because GAPDH binding to mRNA underlies post-transcriptional regulation of cytokine production (9), we tested the effect of GAPDH succination on RNA binding (fig. S15). Pre-treatment with DMF (or heptelidic acid) decreased GAPDH–RNA binding, as reported with other modifications of the active-site cysteine (27). This effect was small, however, as a similar decrease was produced by a tenfold lower concentration of NAD+. Thus, the alteration of such binding does not appear to underlie the immunologic actions of DMF.
Finally, to ascertain whether GAPDH inhibition produced anti-inflammatory actions in vivo, we examined the effect of heptelidic acid in experimental autoimmune encephalomyelitis (EAE), a mouse model of MS, and found that it attenuated the disease in these mice (Fig. 4H).
By demonstrating that a known immunomodulatory drug acts by inhibiting aerobic glycolysis, our findings provide a proof of concept that metabolism is a viable therapeutic target in autoimmunity. They may also explain important observations of DMF therapy in patients. DMF differentially impacts distinct lymphocyte subsets, producing lymphopenia that selectively depletes highly glycolytic effector T cells while sparing oxidative naïve T cells and Treg cells (28, 29). Our findings suggest that the inhibition of aerobic glycolysis underlies these selective effects. It is also notable that DMF is simply a derivative of fumarate, which is a metabolic intermediate of the Krebs cycle, lying downstream of glycolysis in cellular energy production. We hypothesize that fumarate-induced inactivation of GAPDH represents an endogenous negative feedback loop. DMF—a more cell-permeable and electrophilic derivative of fumarate—may simply exploit this physiologic pathway to produce its immunologic actions (Fig. 4I). It must be noted, however, that additional targets of succination (in addition to KEAP1) are likely relevant to both the therapeutic and toxic effects of the drug.
Supplementary Material
Acknowledgments
We thank J. Liu and J. Stivers for insights regarding enzyme kinetics experiments. We are grateful to Z. Zhou, W. Chen, and A. Hoke for providing access to the Seahorse extracellular flux analyzer and support with its use. We thank L. DeVine and R. Cole from the Johns Hopkins Mass Spectrometry Core Facility for their assistance and helpful discussion. We are grateful to B. Wipke and R. Scannevin for insightful discussion and for providing a protocol for preparation of DMF suspension. We thank B. Paul for help with mPM isolation and GAPDH–RNA binding, C. Darius for drawing blood samples, and J. Bo for preparation of PBMCs.
Funding:
Funding for M.D.K. was provided by the NINDS (R25 grant, RFA-NS-12-003), National Multiple Sclerosis Society (NMSS) – American Academy of Neurology (AAN) (Clinician Scientist Development Award, FAN 17107-A-1), and Conrad N. Hilton Foundation (Marilyn Hilton Bridging Award). Funding for P.B. was provided by the AAN (John F. Kurtzke Clinician Scientist Development Award), NMSS (Career Transition Award, TA-1503-03465), and Race to Erase MS (Young Investigator Award). V.P. and N.P. were supported by the CPRIT Core Facility Support Award (RP170005), NCI Cancer Center Support Grant (P30CA125123), and intramural funds from the Dan L. Duncan Cancer Center (DLDCC). N.P. was also supported by the American Cancer Society (127430-RSG-15-105-01-CNE) and NIH (R01CA220297 and R01CA216426). Funding for P.A.C. was provided by the NINDS (R37NS041435). Funding for S.H.S. was provided by USPHS grant MH18501.
Footnotes
Competing interests:
P.A.C. has received research funding in the past from Biogen, the company that sells DMF (trade name Tecfidera) as a therapy for MS. He received a consulting honorarium from Biogen in 2015 for work related to the compound opicinumab. The other authors declare no competing interests.
Author contributions:
M.D.K., P.B., P.A.C, and S.H.S. contributed to overall project design. M.D.K., P.B., P.M.K., V.P., N.P., and A.M.S. performed the research. M.D.K., P.B., P.M.K., V.P., and N.P., analyzed the data. M.D.K. and P.B. prepared the figures. M.D.K. and S.H.S. wrote the manuscript. P.B., P.A.C., and N.P. edited the manuscript.
Data and materials availability:
Data in this paper are presented and/or tabulated in the main text and supplementary materials.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38:633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LAJ. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cham CM, Gajewski TF. Glucose availability regulates IFN-γ production and p70S6 kinase activation in CD8+ effector T cells. J Immunol. 2005;174:4670–4677. doi: 10.4049/jimmunol.174.8.4670. [DOI] [PubMed] [Google Scholar]
- 5.Cham CM, Driessens G, O’Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38:2438–2450. doi: 10.1002/eji.200838289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, Green DR. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–882. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20:61–72. doi: 10.1016/j.cmet.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerriets VA, Kishton RJ, Nichols AG, Macintyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME, Haeberli L, Huck C, Turka LA, Wood KC, Hale LP, Smith PA, Schneider MA, MacIver NJ, Locasale JW, Newgard CB, Shinohara ML, Rathmell JC. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest. 2015;125:194–207. doi: 10.1172/JCI76012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang CH, Curtis JD, Maggi LB, Jr, Faubert B, Villarino AV, O’Sullivan D, Huang SC-C, van der Windt GJW, Blagih J, Qiu J, Weber JD, Pearce EJ, Jones RG, Pearce EL. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006;4:13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linker RA, Haghikia A. Dimethyl fumarate in multiple sclerosis: Latest developments, evidence and place in therapy. Ther Adv Chronic Dis. 2016;7:198–207. doi: 10.1177/2040622316653307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Linker RA, Lee DH, Ryan S, van Dam AM, Conrad R, Bista P, Zeng W, Hronowsky X, Buko A, Chollate S, Ellrichmann G, Brück W, Dawson K, Goelz S, Wiese S, Scannevin RH, Lukashev M, Gold R. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 14.Blewett MM, Xie J, Zaro BW, Backus KM, Altman A, Teijaro JR, Cravatt BF. Chemical proteomic map of dimethyl fumarate-sensitive cysteines in primary human T cells. Sci Signal. 2016;9:rs10. doi: 10.1126/scisignal.aaf7694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BAC, Sobel RA, Wipke BT, Steinman L, Scannevin RH, Zamvil SS. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc Natl Acad Sci USA. 2016;113:4777–4782. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blatnik M, Frizzell N, Thorpe SR, Baynes JW. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by fumarate in diabetes: Formation of S-(2-succinyl)cysteine, a novel chemical modification of protein and possible biomarker of mitochondrial stress. Diabetes. 2008;57:41–49. doi: 10.2337/db07-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Litjens NH, Burggraaf J, van Strijen E, van Gulpen C, Mattie H, Schoemaker RC, van Dissel JT, Thio HB, Nibbering PH. Pharmacokinetics of oral fumarates in healthy subjects. Br J Clin Pharmacol. 2004;58:429–432. doi: 10.1111/j.1365-2125.2004.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rostami-Yazdi M, Clement B, Schmidt TJ, Schinor D, Mrowietz U. Detection of metabolites of fumaric acid esters in human urine: Implications for their mode of action. J Invest Dermatol. 2009;129:231–234. doi: 10.1038/jid.2008.197. [DOI] [PubMed] [Google Scholar]
- 19.Peng H, Li H, Sheehy A, Cullen P, Allaire N, Scannevin RH. Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. J Neuroimmunol. 2016;299:35–44. doi: 10.1016/j.jneuroim.2016.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Parsons ZD, Gates KS. Redox regulation of protein tyrosine phosphatases: Methods for kinetic analysis of covalent enzyme inactivation. Methods Enzymol. 2013;528:129–154. doi: 10.1016/B978-0-12-405881-1.00008-2. [DOI] [PubMed] [Google Scholar]
- 21.Kamada N, Seo SU, Chen GY, Núñez G. Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol. 2013;13:321–335. doi: 10.1038/nri3430. [DOI] [PubMed] [Google Scholar]
- 22.Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, Locasale JW. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife. 2014;3:03342. doi: 10.7554/eLife.03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberti MV, Dai Z, Wardell SE, Baccile JA, Liu X, Gao X, Baldi R, Mehrmohamadi M, Johnson MO, Madhukar NS, Shestov AA, Chio IIC, Elemento O, Rathmell JC, Schroeder FC, McDonnell DP, Locasale JW. A predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017;26:648–659.e8. doi: 10.1016/j.cmet.2017.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio IIC, Giannopoulou EG, Rago C, Muley A, Asara JM, Paik J, Elemento O, Chen Z, Pappin DJ, Dow LE, Papadopoulos N, Gross SS, Cantley LC. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Michell-Robinson MA, Moore CS, Healy LM, Osso LA, Zorko N, Grouza V, Touil H, Poliquin-Lasnier L, Trudelle AM, Giacomini PS, Bar-Or A, Antel JP. Effects of fumarates on circulating and CNS myeloid cells in multiple sclerosis. Ann Clin Transl Neurol. 2015;3:27–41. doi: 10.1002/acn3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakai K, Hasumi K, Endo A. Identification of koningic acid (heptelidic acid)-modified site in rabbit muscle glyceraldehyde-3-phosphate dehydrogenase. Biochim Biophys Acta. 1991;1077:192–196. doi: 10.1016/0167-48389190058-8. [DOI] [PubMed] [Google Scholar]
- 27.Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD+-binding region (Rossmann fold) J Biol Chem. 1995;270:2755–2763. doi: 10.1074/jbc.270.6.2755. [DOI] [PubMed] [Google Scholar]
- 28.Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neuroinflamm. 2015;2:e76. doi: 10.1212/NXI.0000000000000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gross CC, Schulte-Mecklenbeck A, Klinsing S, Posevitz-Fejfár A, Wiendl H, Klotz L. Dimethyl fumarate treatment alters circulating T helper cell subsets in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2015;3:e183. doi: 10.1212/NXI.0000000000000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–674. doi: 10.1038/ncb1268. [DOI] [PubMed] [Google Scholar]
- 31.Calabresi PA, Allie R, Mullen KM, Yun SH, Georgantas RW, 3rd, Whartenby KA. Kinetics of CCR7 expression differ between primary activation and effector memory states of TH1 and TH2 cells. J Neuroimmunol. 2003;139:58–65. doi: 10.1016/S0165-57280300127-9. [DOI] [PubMed] [Google Scholar]
- 32.Shevchenko A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen P, Shevchenko A, Boucherie H, Mann M. Linking genome and proteome by mass spectrometry: Large-scale identification of yeast proteins from two dimensional gels. Proc Natl Acad Sci USA. 1996;93:14440–14445. doi: 10.1073/pnas.93.25.14440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 34.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 35.van der Windt GJ, Chang CH, Pearce EL. Measuring bioenergetics in T cells using a Seahorse extracellular flux analyzer. Curr Protoc Immunol. 2016;113:16B.1–16B.14. doi: 10.1002/0471142735.im0316bs113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vantaku V, Donepudi SR, Ambati CR, Jin F, Putluri V, Nguyen K, Rajapakshe K, Coarfa C, Battula VL, Lotan Y, Putluri N. Expression of ganglioside GD2, reprogram the lipid metabolism and EMT phenotype in bladder cancer. Oncotarget. 2017;8:95620–95631. doi: 10.18632/oncotarget.21038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin F, Thaiparambil J, Donepudi SR, Vantaku V, Piyarathna DWB, Maity S, Krishnapuram R, Putluri V, Gu F, Purwaha P, Bhowmik SK, Ambati CR, von Rundstedt FC, Roghmann F, Berg S, Noldus J, Rajapakshe K, Gödde D, Roth S, Störkel S, Degener S, Michailidis G, Kaipparettu BA, Karanam B, Terris MK, Kavuri SM, Lerner SP, Kheradmand F, Coarfa C, Sreekumar A, Lotan Y, El-Zein R, Putluri N. Tobacco-specific carcinogens induce hypermethylation, DNA adducts, and DNA damage in bladder cancer. Cancer Prev Res. 2017;10:588–597. doi: 10.1158/1940-6207.CAPR-17-0198. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Piyarathna DWB, Rajendiran TM, Putluri V, Vantaku V, Soni T, von Rundstedt FC, Donepudi SR, Jin F, Maity S, Ambati CR, Dong J, Gödde D, Roth S, Störkel S, Degener S, Michailidis G, Lerner SP, Pennathur S, Lotan Y, Coarfa C, Sreekumar A, Putluri N. Distinct lipidomic landscapes associated with clinical stages of urothelial cancer of the bladder. Eur Urol Focus. 2017;17:30107–30114. doi: 10.1016/j.euf.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.