

Abstract
A series of diverse small molecules have been designed and synthesized through structure-based drug design by taking advantage of fragment merging and elaboration approaches. Compounds ZL0420 (28) and ZL0454 (35) were identified as potent and selective BRD4 inhibitors with nanomolar binding affinities to bromodomains (BDs) of BRD4. Both of them can be well docked into the acetyl-lysine (KAc) binding pocket of BRD4, forming key interactions including the critical hydrogen bonds with Asn140 directly and Tyr97 indirectly via a H2O molecule. Both compounds 28 and 35 exhibited submicromolar potency of inhibiting the TLR3-dependent innate immune gene program, including ISG54, ISG56, IL-8, and Groβ genes in cultured human small airway epithelial cells (hSAECs). More importantly, they also demonstrated potent efficacy reducing airway inflammation in a mouse model with low toxicity, indicating a proof of concept that BRD4 inhibitors may offer the therapeutic potential to block the viral-induced airway inflammation.
Keywords: bromodomains, bromodomain-containing protein 4 (BRD4), structure-based drug design, immune response genes, airway inflammation
Graphical abstract
1. Introduction
Bromodomain-containing protein 4 (BRD4) belongs to the family of bromodomain and extra-terminal domain (BET) proteins which also includes BRD2, BRD3, and BRDT. Similar to other BET protein members, BRD4 contains two bromodomains at its N-terminal that can recognize acetylated lysine (KAc) residues [1]. Each bromodomain of BRD4 comprises a left-handed bundle of four helices (αZ, αA, αB and αC) linked by the inter-helical ZA loop and BC loop which constitute the active KAc-binding pocket [2]. Bromodomain-containing proteins (BCPs) act as KAc readers of modified histones mediating signaling pathways of gene regulatory networks [3]. Disturbing the interaction between BET protein bromodomains and acetylated lysine represents a very promising therapeutic target for human diseases including cancer and inflammations [4–6]. Discovery and development of BET inhibitors have attracted increasing attention since the BET family is considered as the most druggable target proteins among BCPs for regulating cellular epigenetics [7]. Based on the diverse structures, currently available BET inhibitors under the investigation can be classified as azepines (e.g. (+)-JQ1, 1, Fig. 1) [8], quinazolin-4(3H)-one (e.g. RVX-208, 2) [9], pyridones (e.g. ABBV-075, 3) [10], 3,5-dimethylisoxazoles (e.g. I-BET151, 4) [11], tetrahydroquinolines (e.g. I-BET726, 5) [12], 4-acylpyrroles (e.g. XD14, 6) [13], and others [3]. Among these compounds, compound 1 is one of the first reported BET inhibitors that was most extensively used as a research tool in the field. Compound 2 is a well-developed BET inhibitor that has been advanced into Phase III human clinical trials for cardiovascular diseases [14]. In addition, more than a dozen of other BET inhibitors including compound 3 are at the different phases of clinical trials [15,16]. Nevertheless, none of these compounds have yet been approved by FDA. Meanwhile, most of them are pan-BET inhibitors lacking selectivity for individual BET family members [17,18]. As the most heavily investigated member of BET family, BRD4 represents a very promising therapeutic target. Developing potent and highly selective BRD4 inhibitors for more applications is in an urgent need.
Fig. 1.
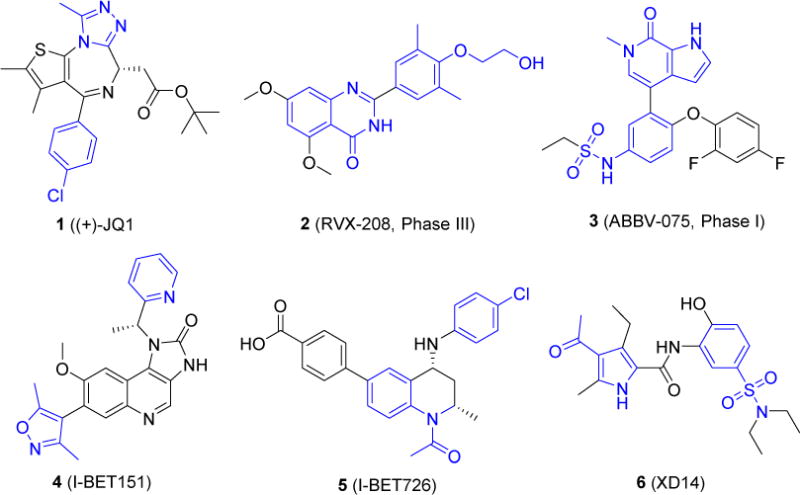
Structures of representative BET inhibitors. Some privileged fragments that are important for binding with BRD4 are highlighted in blue.
As part of our ongoing drug discovery efforts, we are interested in developing novel potent and selective BRD4 inhibitors as potential therapeutics for acute airway inflammation and chronic inflammation-associated airway remodeling as well as other inflammatory diseases. Our team has observed the essential role of BRD4 in NF-κB mediated epithelial-mesenchymal transition (EMT) in an in vitro model of airway epithelial cells and in a murine pulmonary fibrosis model in vivo [19]. To identify new and more selective BRD4 inhibitors, we initiated our structure-based drug design by analyzing the crystal structures of available BRD4 inhibitors with BRD4 BD1 domain exemplified with co-complex of 1 as depicted in Fig. 2a. Most inhibitors mimic acetyl-lysine, and occupy the central hydrophobic cavity and anchored by hydrogen bonds with Asn140 directly and Tyr97 indirectly via a water molecule [20]. BRD4 inhibitors can often extend to the WPF shelf, a hydrophobic region of ZA loop that includes Trp/Pro/Phe motif, and ZA channel (notably Pro82 to Leu91 in BRD4 BD1) via a substituted phenyl ring [21]. These hydrophobic interactions are also crucial for BRD4 binding affinities. Thus, we have summarized a pharmacophore model as two key aromatic rings attached with a proper linker. The substituents or hetero atoms of the head aromatic ring form the critical hydrogen bonds with Asn140 as well as Tyr97, and the tail aromatic ring with substituents interacts with the hydrophobic WPF shelf and ZA channel (Fig. 2b). By utilizing a deconstruction and reconstruction approach [22,23], we took advantage of druglike fragments from available inhibitors and drug libraries as the critical head and tail moieties as well as the linker scaffold for the structure and fragment-based drug design through fragment merging and elaboration [24,25]. For example, some privileged fragments of inhibitors 1, 4, 5 and 6 highlighted in Fig. 1 have been considered in the design of the head and tail binding partners.
Fig. 2.
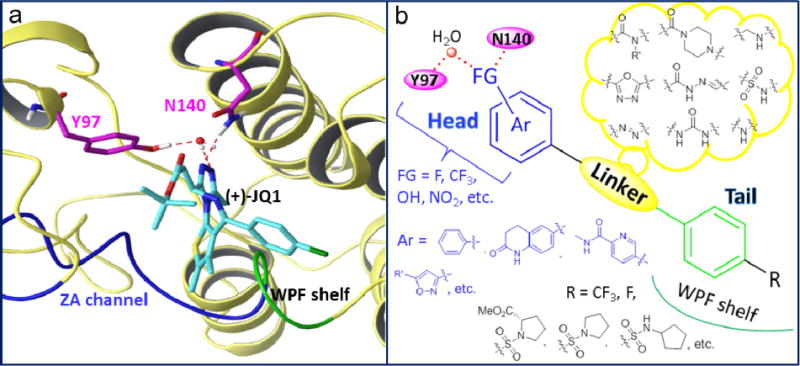
a) Co-complex crystal structure of 1 with BRD4 BD1 (PDB code: 3MXF) as an example. b) Proposed pharmacophore model and design of new BRD4 inhibitors in this effort.
2. Results and discussion
2.1. Chemistry
New compounds 11~16 have been initially designed with benzenesulfonamide as the tail pharmacophore inspired by a privileged scaffold of compound 6, and their synthetic route was outlined in Scheme 1. With starting material 4-nitrobenzenesulfonyl chloride (7) and methyl L-prolinate (8), key intermediate 9 was obtained in the presence of Hünig’s base DIPEA. Reduction of 9 with Zn dust and NH4Cl gave compound 10 in a quantitative yield. Intermediate 10 was coupled with various acids in the presence of HBTU and DIPEA to generate compounds 11 and 12. Through acylation reaction of 10 with benzoyl chlorides, compounds 13-14 were readily achieved. New compound 15 was produced via reductive amination reaction of 10 with the treatment of 4-hydrobenzaldehyde in the presence of NaBH3CN. Intermediate 10 reacted with 5-amino-2-methylphenol assisted by tert-butyl nitrite leading to compound 16 in a yield of 78% [26].
Scheme 1.
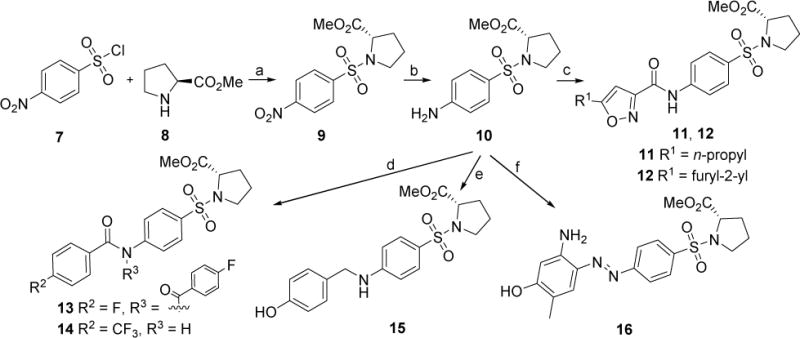
Synthetic route of newly designed compounds 11~16. Reagents and conditions: (a) DIPEA, DCM, 0~5 °C, 89%; (b) Zn, NH4Cl, EtOH/H2O, 80 °C, quant.; (c) 5-propylisoxazole-3-carboxylic acid (for 11) or 5-(furan-2-yl)isoxazole-3-carboxylic acid (for 12), HBTU, DIPEA, DCM, 0~5 °C. 68% for 11; 86% for 12; (d) 4-fluorobenzoyl chloride, pyridine (for 13) or 4-trifluorobenzoyl chloride (for 14), DCM, 0~5 °C, 67% for 13, quant. for 14; (e) 4-hydroxybenzaldehyde, NaBH3CN, MeOH, 80 °C, 85%; (f) 5-amino-2-methylphenol, tert-butyl nitrite, 38% HCl (aq.), K2CO3, MeOH/CH3CN/H2O, 0 °C, 78%.
New compounds 19, 22 and 23 were designed by taking isoxazole ring as the head moiety and its hetero atoms were expected to form the critical hydrogen bonds with Asn140 and Tyr97. The synthetic route to access these compounds was out lined in Scheme 2. Starting material ethyl 5-(tert-butyl)isoxazole-3-carboxylate (17) was hydrolyzed to the acid 18, followed by the coupling with 1-(4-fluorophenyl)piperazine in the presence of HBTU and DIPEA to produce compound 19 in a yield of 81%. 17 was also hydrazinolyzed with NH2NH2 leading to intermediate 20 with subsequent coupling with various benzaldehydes to yield new compounds 21 and 22. Cyclization of compound 21 in the presence of I2 and K2CO3 in DMSO resulted in compound 23 [27].
Scheme 2.
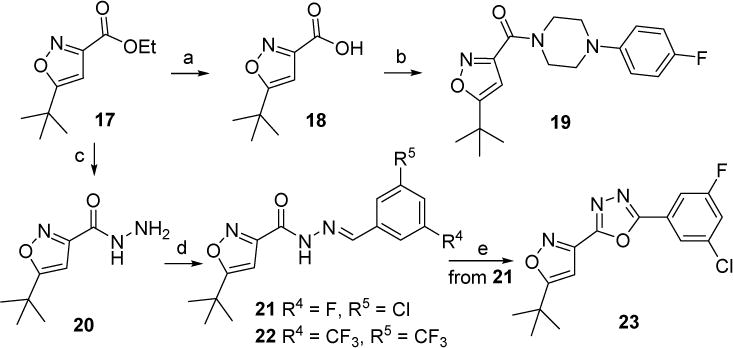
Synthetic route of new compounds 19, 22 and 23. Reagents and conditions: (a) LiOH·H2O, CH3OH/H2O, rt., 95%; (b) 1-(4-fluorophenyl) piperazine, HBTU, DIPEA, DCM, 81%; (c) NH2NH2, EtOH, reflux, used directly for the next step; (d) 3-chloro-5-fluorobenzaldehyde (for 21) or 3,5-bis(trifluoromethyl)benzaldehyde (for 22), EtOH, rt. 44% for 21 (two steps). 59% for 22 (two steps); (e) I2, K2CO3, DMSO, 110 °C, 13%.
3,4-Dihydroquinolin-2(1H)-one was also utilized to design new molecules 26~30 and 33~35 given that this moiety may play both roles of head and tail pharmacophores. The according synthesis was outlined in Scheme 3. 6-Nitro-3,4-dihydroquinolin-2(1H)-one 24 was reduced by Zn dust with NH4Cl to give the critical intermediate 6-amino-3,4-dihydroquinolin-2(1H)-one 25. Building block 25 was sulfonized in the presence of the base of Et3N to give compounds 26 and 27. Amine 25 was also treated with 5-amino-2-methylphenol in the presence of tert-butyl nitrite to produce compound 28. C-N coupling of 25 with 5-bromo-N-methylpicolinamide in the presence of the catalyst of Pd(OAc)2 led to compound 29. Compound 30 with a urea linker was obtained through the addition of amine 25 to 1-isocyanato-4-(trifluoromethyl)benzene. 5-Amino-2-methylphenol 31 was then taken as the head moiety, and reacted with several sulfonamide substituted aniline 32 in the presence of tert-butyl nitrite to afford new compounds 33-35 in a yield of 67%-92%.
Scheme 3.
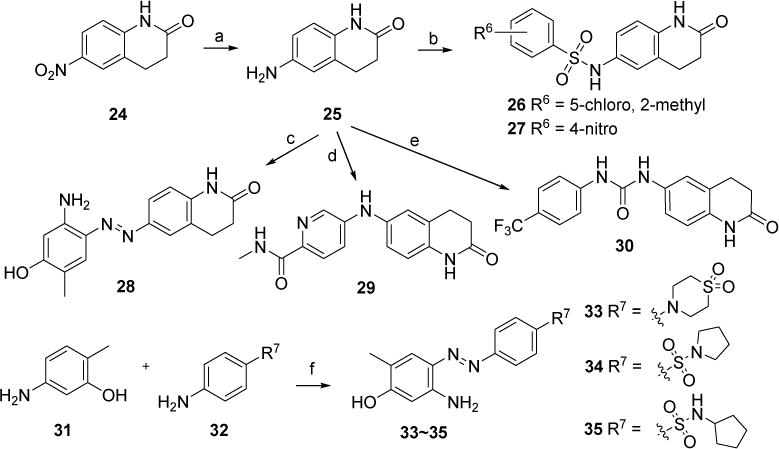
Synthetic route of new compounds 26~30 and 33~35. Reagents and conditions: (a) Zn, NH4Cl, MeOH/H2O, 80 °C, quant. (b) 5-chloro-2-methoxybenzenesulfonyl chloride (for 26) or 4-nitrobenzenesulfonyl chloride (for 27), Et3N, DMF, rt., 29% for 26, 13% for 27; (c) 5-amino-2-methylphenol, tert-butyl nitrite, 38% HCl (aq.), K2CO3, MeOH/CH3CN/H2O, 0°C, 81%. (d) 5-bromo-N-methylpicolinamide, Pd(OAc)2, xantphos, Cs2CO3, 1,4-dioxane, 110 °C, 63%. (e) 1-isocyanato-4-(trifluoromethyl)benzene, DCM, rt., 41%; (f) tert-butyl nitrite, 38% HCl (aq.), K2CO3, MeOH/CH3CN/H2O, 0 °C, 67% for 33, 92% for 34, 81% for 35.
2.2. Biology
2.2.1. Evaluation of innate immune response genes in cellular assays
All the newly designed and synthesized compounds were evaluated in a cellular assay system first (Table 1) given that a number of reported BRD4 inhibitors are lack of satisfactory cellular activities due to limited drug properties, thereby hindering their further preclinical development. This strategy allows us to prioritize our inhibitors and exclude those compounds that are unable to penetrate cell membrane at the early evaluation stage. The in vitro efficacies of all new molecules were first determined by polyinosinic:polycytidylic acid (poly(I:C))-induced expression of innate immune response genes in human small airway epithelial cells (hSAECs) which have many characteristics of representative primary lower-airway epithelial cells [28]. Administered in the extracellular medium, poly(I:C) is a potent and selective agonist of the Toll-Like Receptor 3 (TLR3) pattern recognition receptor that mediates the innate response program to RNA virus infection. The use of this synthetic viral pattern enables a robust and reproducible assay to screen BRD4 inhibitors, and quantify their IC50 values. hSAECs were first preincubated with 10 μM of test compounds overnight, followed by poly(I:C) addition into culture medium at 10 g/mL for another 4 h (the time when maximal innate immune gene expression is reached). The total RNA was extracted and quantitative real-time polymerase chain reaction (Q-RT-PCR) was performed to determine their inhibitory effect on poly(I:C) induced innate immune genes including ISG54, ISG56, IL-8 and Groβ expression in hSAECs. Percentages of inhibition (%) were calculated based on the control (poly(I:C) induced and without administration of compounds). Afterwards, IC50 values of most promising compounds were determined from their inhibitory curves at 8 concentrations of each inhibitor using curve-fitting algorithms. Compound (+)-JQ1 was also included as the positive control for comparison. It was found that among compounds 11~16 with sulfonyl-L-prolinate attached to the tail moiety, compound 13 taking 4-flurophenyl as the head moiety and substituted amide as the linker, showed the most promising inhibitory activity. All the inhibitory rates of compound 13 regulating immune genes’ expression are around 90%. With an unsubstituted amide as a linker, compound 14 completely lost its activity. Compound 16 with 2-amino-4-hydroxy-5-methylphenyl as the head and diazo as a linker has a moderate inhibitory effect (60% to 64%). For compounds 19, 22 and 23 taking tert-butyl isoxazole as the head, 19 and 23 with constrained linkers displayed better inhibitory activities. The inhibitory effect on ISG54 and ISG56 expression of compound 23 reached to 99%. Compounds 26~30 employed 3,4-dihydroquinolin-2(1H)-one as their tail pharmacophore, and diverse heads including substituted phenyl rings and pyridines and different linkers (e.g. sulfonamide, diazo, NH and urea) all showed acceptable inhibitory activities. Interestingly, compound 28, with 2-amino-4-hydroxy-5-methylphenyl as the head and diazo as a linker again, exhibited very impressive inhibitory activity (94% to 95%). We kept the head and linker intact, and modified several different tail pharmacophores (e.g. 33~35). Both 34 and 35, with a substituent of sulfonamide attached to the tail phenyl ring, displayed promising inhibitory activity, while 33 with a sulfone group in the ring was found inactive.
Table 1.
Effects of newly designed and synthesized molecules on TLR3 agonist-induced expression of innate immune genes in vitro.
| Compds | %a
|
|||
|---|---|---|---|---|
| ISG54 | ISG56 | IL-8 | Groβ | |
| (+)-JQ1 | 91 | 90 | 90 | 91 |
| 11 | −62 | −73 | −61 | −57 |
| 12 | −114 | −93 | −86 | −98 |
| 13 | 92 | 92 | 88 | 88 |
| 14 | 3.6 | 2.7 | −1.0 | 5.7 |
| 15 | −9.0 | −17 | −5.0 | −12 |
| 16 | 61 | 64 | 60 | 61 |
| 19 | 94 | 94 | 88 | 82 |
| 22 | 22 | 21 | 48 | 48 |
| 23 | 99 | 99 | 98 | 98 |
| 26 | 57 | 56 | 53 | 56 |
| 27 | −21 | −29 | −32 | −37 |
| 28 | 95 | 95 | 94 | 94 |
| 29 | 29 | 32 | 56 | 52 |
| 30 | 92 | 92 | 48 | 48 |
| 33 | 20 | 20 | –b | – |
| 34 | 85 | 85 | 81 | 81 |
| 35 | 93 | 93 | 99 | 99 |
Concentration of compounds: 10 μM. Inhibitory rates (%) are reported as the geometric mean derived from three independent measurements except where indicated.
Not tested.
Based on the preliminary data, compounds 23, 28 and 35 were selected to calculate their IC50 values (Table 2). Compounds 28 and 35 showed submicromolar IC50 values of 0.49~0.86 μM against all these innate immune genes, which are 15~20-fold more potent than 23. Also, both of them are more potent than positive control compound (+)-JQ1, a widely used tool compound, and compound RVX-208, the most advanced Phase III clinical candidate, which inhibited the production of immune genes with IC50 values of 1.38 to 3.85 μM.
Table 2.
IC50 values of selected compounds of inhibiting TLR3 agonist-induced expression of innate immune genes in hSAECs.
| Compds | IC50, μMa
|
|||
|---|---|---|---|---|
| ISG54 | ISG56 | IL-8 | Groβ | |
| (+)-JQ1 | 1.38 | 1.63 | 1.51 | 1.49 |
| RVX-208 | 2.63 | 2.74 | 3.85 | 3.73 |
| 23 | 7.9 | 7.7 | 4.3 | 3.9 |
| 28 | 0.49 | 0.51 | 0.53 | 0.58 |
| 35 | 0.81 | 0.86 | 0.73 | 0.79 |
IC50 values are reported as the mean derived from three independent measurements. Each one is generated from at least 8 different concentrations.
2.2.2. Binding affinities and selectivity
Binding affinities of compounds 28 and 35 for BRD4 were evaluated using a commercially available time resolved (TR)-fluorescence resonance energy transfer (FRET) assay (Table 3) [8,29,30]. BRD4 specificity of both compounds was also confirmed through comparing with close BET family members BRD2, BRD3 and BRDT as well as non-BET CREB binding protein (CBP). Compound 28 exhibited potent BRD4 inhibitory activities, with IC50 values of 27 nM against BRD4 BD1 and 32 nM against BRD4 BD2, respectively. Compound 35 displayed similar activity and did not show preference between BD1 and BD2 of BRD4 either. However, both of them have 30~60-fold BRD4 selectivity over its close family member BRD2 (IC50 values of 0.77~1.8 μM), 50~90-fold selectivity over BRD3 (IC50 values of 2.2 ~2.5 μM), and 70~120-fold over BRDT (IC50 values of 2.8 ~3.3 μM) as well as more than 200-fold over non-BET protein CBP (IC50 values of > 10 μM), while the positive control (+)-JQ1 is non-specific among BET members and RVX-208 is BD2 selective. Off-target effects of compound 35 were also evaluated through the Eurofins Cerep panel assays of various receptors and enzymes including kinases such as IKK (Supporting Information, Table S1). No significant off-target effects were observed at the tested concentration of 10 μM.
Table 3.
Binding affinities of compounds 28 and 35 with BRD4 and the selectivity over its BET family members BRD2, BRD3, BRDT as well as non-BET protein CBP.a
| BDs\Compds | (+)-JQ1 | RVX-208 | 28 | 35 |
|---|---|---|---|---|
| BRD4 BD1 (IC50, nM) | 92 | 1,142 | 27 | 49 |
| BRD4 BD2 (IC50, nM) | 62 | 135 | 32 | 32 |
| BRD2 BD1 (IC50, nM) | 78 | 5,780 | 803 | 772 |
| BRD2 BD2 (IC50, nM) | 52 | 251 | 1,736 | 1,836 |
| BRD3 BD1 (IC50, nM) | 81 | 3,962 | 2,275 | 2,493 |
| BRD3 BD2 (IC50, nM) | 69 | 203 | 2,193 | 2,241 |
| BRDT BD1 (IC50, nM) | 183 | 4,836 | 3,183 | 3,292 |
| BRDT BD2 (IC50, nM) | 217 | 708 | 2,781 | 3,082 |
| CBP (IC50, nM) | 9,600 | > 10,000 | > 10,000 | > 10,000 |
Binding affinity was measured using an TR-FRET assay with the isolated bromodomain. Reported as mean of at least two separate assay runs.
2.3. Predicted binding modes of compounds 28 and 35 with BRD4 BD1
Computational docking studies of compounds 28 and 35 with the first bromodomain of human BRD4 protein complex (PDB code: 4NUD) as well as BRD2 BD1 (4A9M) using Schrödinger Small-Molecule Drug Discovery Suite were conducted (Fig. 3 and Figure S1). Both compounds 28 and 35 can well occupy the KAc binding pocket. The OH group of compound 35 forms critical hydrogen bonds with Asn140 directly and Tyr97 indirectly via a H2O molecule. NH2 group of the head also interacts with Asn140 via a water molecule. Besides, the N atom of diazo linker interacts with Pro82 through another water molecule. The fragment cyclopentylbenzenesulfonamide extends to the hydrophobic WPF shelf and ZA channel, and importantly, it forms a T shape л-л interaction with Trp81. For the docking result of 35 with BRD2 BD1, there are not that many critical interactions observed (Figure S1), and this may explain their difference of binding affinities and the selectivity. The overlay analysis of 28 and 35 demonstrated that their binding poses are strikingly similar (Fig. 3b).
Fig. 3.
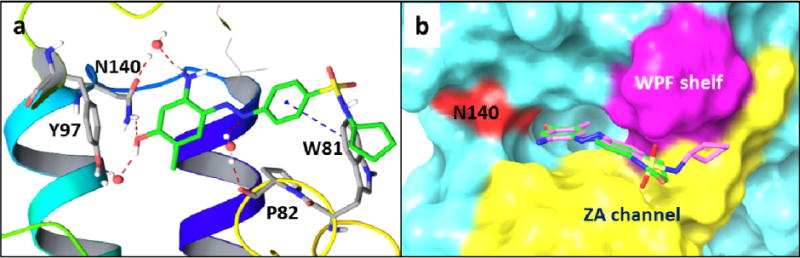
a) Docking result of 35 with BRD4 BD1 (PDB code: 4NUD); b) Overlay analysis of 35 (pink) and 28 (green) docked into BRD4 BD1.
2.4. In vivo pharmacokinetic profiles of 28 and 35
On the basis of their combination of cellular potency and binding affinities, compounds 28 and 35 were further evaluated for their in vivo metabolic profile in rats. As shown in Table 4, a relatively high clearance and low exposures were observed with oral bioavailability not favorable for both compounds. In contrast, intravenous administration gave an excellent drug exposure (high AUC value) and moderate half-life. Thus, in the subsequent in vivo efficacy study, intraperitoneal (i.p.) injection was administered for mice instead of oral gavage. An alternative drug delivery method through nanoparticles is currently under investigation to improve their metabolic stability, and the findings will be reported in due course.
Table 4.
Pharmacokinetic parameters of compounds 28 and 35.a
| Compounds | 28 | 35 | |
|---|---|---|---|
| IV (10 mg/kg) |
t1/2b (h) | 1.2 | 0.83 |
| AUC0-tc (ng·h/mL) | 14,700 | 11,400 | |
| VSSd (L/kg) | 0.864 | 1.125 | |
| CLe (mL/min/kg) | 11.5 | 14.7 | |
|
| |||
| PO (20 mg/kg) |
Cmaxf (ng/mL) | 80 | 36 |
| AUC0-t (ng·h/mL) | 450 | 104 | |
Compounds were formulated in 10% DMSO/60% PEG-400/30% Saline.
half-life.
Exposure over test time.
Volume of distribution at steady state.
Total clearance.
Maximum plasma concentration.
2.5. In vivo evaluations of compounds 28 and 35 in TLR3 mediated acute airway inflammatory murine model
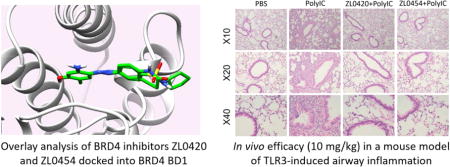
With two promising BRD4 inhibitors compound 28 and 35 available in hand, we then performed in vivo efficacy evaluations in our established murine model of TLR3 mediated acute airway inflammation [28]. Intranasal administration of poly(I:C)-induced a substantial increase of total cells and neutrophils into the airway fluids, and cytokine expression in the lung tissue. These changes were more effectively blocked by BRD4 inhibitors 28 and 35 than positive control (+)-JQ1 or RVX-208 (Fig. 4a), at the dose of 10 mg/kg (via i.p.). Histology analyses of lung sections in the poly(I:C)-treated mice with/without BRD4 inhibitors are also demonstrated in Fig. 4b. Compounds 28 and 35 displayed higher efficacy and almost completely blocked the profound accumulation of neutrophils around the small and medium sized airways induced by poly(I:C) administration. In addition, daily administration of 35 from 1-50 mg/kg in groups of n = 5 mice over one month each had no apparent toxic effects observed on body weight, hematological measures (WBC, RBC, platelets), liver function (albumin, globulin, ALK, ALT), and renal function (CRE, BUN) (Figure S2), indicating that they are much safer and better tolerated than pan-BET inhibitors (e.g. JQ1). No discernable effects on hepatic, renal or pulmonary tissues derived from compound 35-treated mice were observed during histological examination.
Fig. 4.
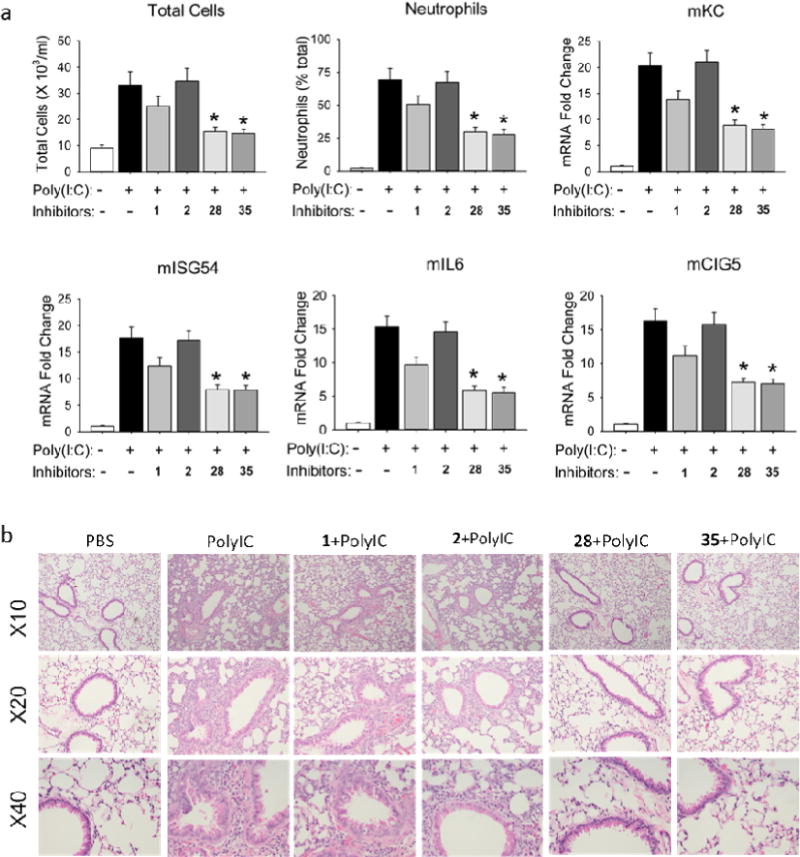
BRD4 inhibitors 28 and 35 effectively blocked TLR3 agonist-induced acute airway inflammation in vivo. Positive controls (+)-JQ1 and RVX-208 were also tested for comparison.
3. Conclusions
In summary, we have identified two novel BRD4 inhibitors ZL0420 (28) and ZL0454 (35) via a structure-based drug design approach, which are more potent and selective than known inhibitors (+)-JQ1 and RVX-208. These compounds are capable of significantly blocking the TLR3-dependent expression of innate immune genes ISG54, ISG56, IL-8, and Groβ in hSAECs and display nanomolar binding affinities for BDs of BRD4 protein with good selectivity over that of the related BRD2 homolog. Molecular docking revealed their classical binding modes with the critical interactions identified between the ligand and the target protein. Significant in vivo efficacy in standardized murine model of TLR3 agonist-induced airway inflammation with low toxicity further confirmed their therapeutic potential as a proof of concept for the treatment of the viral-induced airway inflammation.
4. Experimental section
4.1. Chemistry
All commercially available starting materials and solvents were reagent grade, and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063-0.200 mm (70-230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 (1H, 600 MHz; 13C, 150 MHz) spectrometer or Bruker-300 (1H, 300 MHz; 13C, 75 MHz). 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: Nano ESI spray voltage was 1.8 kV; Capillary temperature was 275 °C and the resolution was 60,000; Ionization was achieved by positive mode. Melting points were determined on a hot stage apparatus and are uncorrected. Purity of final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/VIS). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm); flow rate 0.5 mL/min; UV detection at 270 and 254 nm; linear gradient from 30% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min followed by 30 min of the last-named solvent. All the new compounds have a purity of >95% prior to the submission for the biological studies.
4.1.1. Methyl ((4-(5-propylisoxazole-3-carboxamido)phenyl)sulfonyl)-L-prolinate (11)
To a solution of methyl L-prolinate (1.66 g, 10 mmol) and DIPEA (2.58 g, 20 mmol) in 40 mL of DCM, 4-nitrobenzenesulfonyl chloride (2.22 g, 10 mmol) was added at 0 °C. After stirring at rt. for 2 h, the solution was extracted with DCM (50 mL×2). The organic layer was washed with 1 N NaHSO4 (aq.), saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (hexane/EtOAc = 10/1 to 5/1) to give the intermediate 9 (2.8 g, 89%) as a white solid. 1H NMR (300 MHz, CDCl3) δ 8.39 (d, J = 8.9 Hz, 2H), 8.10 (d, J = 8.9 Hz, 2H), 4.48 (dd, J = 8.5 Hz, 3.7 Hz, 1H), 3.73 (s, 3H), 3.47 (dd, J = 7.0 Hz, 6.2 Hz, 2H), 2.26 – 1.87 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 172.14, 144.68, 128.67, 124.20, 60.57, 52.54, 48.32, 30.96, 24.74.
To a solution of 9 (2.68 g, 8.54 mmol) in EtOH (40 mL), NH4Cl (4.50 g, 85.4 mmol) in H2O (20 mL) and Zn dust (3.33 g, 51.2 mmol) were added. After refluxing at 80 °C for 1 h, the solution was filtered to remove Zn dust. After cooled to rt., the solution was extracted with EtOAc (50 mL×2). The organic layer was washed with saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (hexane/EtOAc = 3/1) to give 10 (2.2 g, quant.) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 8.7 Hz, 2H), 6.68 (d, J = 8.7 Hz, 2H), 4.34 (s, 2H), 4.26 – 4.17 (m, 1H), 3.71 (s, 3H), 3.52 – 3.40 (m, 1H), 3.31 – 3.19 (m, 1H), 1.97 (dt, J = 9.2 Hz, 6.5 Hz, 3H), 1.72 (d, J = 4.9 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.93, 151.20, 129.57, 125.39, 113.98, 60.37, 52.41, 48.55, 30.90, 24.64.
To a solution of 5-propylisoxazole-3-carboxylic acid (58 mg, 0.375 mmol) and methyl ((4-aminophenyl)sulfonyl)-L-prolinate 10 (70 mg, 0.25 mmol) in 5 mL of DCM, HBTU (284 mg, 0.75 mmol) and DIPEA (220 μL, 1.25 mmol) were added. After stirring at rt. overnight, the mixture was extracted with DCM (20 mL×3). The organic layer was washed with 1 N NaHSO4 (aq.), saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by PTLC (DCM/MeOH = 40/1) to give the desired product 11 (40 mg, 38%) as a pale yellow solid. m.p.: 142-143 °C. 1H NMR (300 MHz, CDCl3) δ 8.83 (s, 1H), 7.87(dd, J = 8.9 Hz, 4H), 6.55 (s, 1H), 4.33 (dd, J = 7.6 Hz, 4.2 Hz, 1H), 3.73 (s, 3H), 3.51 (dd, J = 9.5 Hz, 6.9 Hz, 1H), 3.41 – 3.28 (m, 1H), 2.82 (t, J = 7.5 Hz, 2H), 2.10 – 1.95 (m, 3H), 1.79 (dd, J = 14.7 Hz, 7.3 Hz, 3H), 1.03 (t, J = 7.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 176.36, 172.54, 158.35, 157.28, 141.12, 133.84, 128.89, 119.70, 100.75, 60.40, 52.45, 48.42, 30.91, 28.67, 24.66, 20.84, 13.56. HRMS (ESI) m/z calcd for C19H24N3O6S [M + H]+, 422.1386; found, 422.1398.
4.1.2. Methyl ((4-(5-(furan-3-yl)isoxazole-3-carboxamido)phenyl)sulfonyl)-L-prolinate (12)
Compound 12 was prepared in 84% yield taking 2-(5-(furan-2-yl)isoxazol-3-yl)-2-oxoacetic acid as a key intermediate by a procedure similar to that used to synthesize compound 11. The title compound was obtained as a pale yellow solid. m.p.: 186-188 °C. 1H NMR (300 MHz, CDCl3) δ 8.83 (s, 1H), 7.98 – 7.83 (m, 4H), 7.62 (s, 1H), 7.02 (d, J = 3.4 Hz, 1H), 6.96 (s, 1H), 6.65 – 6.52 (m, 1H), 4.35 (dd, J = 7.7 Hz, 4.2 Hz, 1H), 3.74 (s, 3H), 3.52 (dd, J = 9.4 Hz, 7.0 Hz, 1H), 3.42 – 3.30 (m, 1H), 2.13 – 1.93 (m, 3H), 1.81 (dd, J = 6.8 Hz, 4.8 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 172.54, 163.83, 158.62, 156.65, 145.05, 142.16, 140.96, 134.06, 128.93, 119.79, 112.20, 111.79, 98.64, 60.41, 52.47, 48.43, 30.93, 24.67. HRMS (ESI) m/z calcd for C20H20N3O7S [M + H]+, 446.1022; found, 446.1032.
4.1.3. Methyl ((4-(4-fluoro-N-(4-fluorobenzoyl)benzamido)phenyl)sulfonyl)-L-prolinate (13)
To a solution of 10 (50 mg, 0.176 mmol) and pyridine (97 mg, 1.23 mmol) in 5 mL of DCM, 4-fluorobenzoyl chloride (139 mg, 0.88 mmol) was added. After stirring at rt. overnight, the mixture was extracted with DCM (30 mL×2). The organic layer was washed with 1 N NaHSO4 (aq.), saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (DCM/MeOH = 100/1 to 50/1) to give the desired product (60 mg, 67%) as a pale yellow solid. m.p.: 78-80 °C. 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 8.7 Hz, 2H), 7.75 (dd, J = 8.9 Hz, 5.2 Hz, 4H), 7.30 (d, J = 8.7 Hz, 2H), 7.06 (t, J = 8.6 Hz, 4H), 4.35 (dd, J = 8.1 Hz, 3.9 Hz, 1H), 3.68 (s, 3H), 3.54 – 3.31 (m, 2H), 2.08 – 1.80 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 172.31, 171.86, 167.05, 163.65, 143.97, 137.59, 132.03, 131.91, 130.12, 128.88, 127.96, 116.32, 116.02, 60.40, 52.42, 48.40, 30.91, 24.62. HRMS (ESI) m/z calcd for C26H23F2N2O6S [M + H]+, 529.1245; found, 529.1260.
4.1.4. Methyl ((4-(4-(trifluoromethyl)benzamido)phenyl)sulfonyl)-L-prolinate (14)
To a solution of 10 (50 mg, 0.176 mmol) and Et3N (53 mg, 0.528 mmol) in 5 mL of DCM, 4-(trifluoromethyl)benzoyl chloride (73 mg, 0.352 mmol) was added. After stirring at rt. overnight, the mixture was extracted with DCM (30 mL×2). The organic layer was washed with 1 N NaHSO4 (aq.), saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column chromatography (DCM/MeOH = 100/1 to 50/1) to provide the desired product (80 mg, quant.) as a pale yellow solid. m.p.: 182-184 °C. HPLC purity 99.4% (tR = 19.27 min). 1H NMR (300 MHz, CDCl3) δ 8.03 (d, J = 7.9 Hz, 2H), 7.85 (dd, J = 23.2 Hz, 8.5 Hz, 4H), 7.73 (d, J = 8.0 Hz, 2H), 4.25 (d, J = 7.3 Hz, 1H), 3.70 (s, 3H), 3.47 (d, J = 6.0 Hz, 1H), 3.29 (t, J = 7.1 Hz, 1H), 1.88 (m, J = 68.6, 4.7 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 172.75, 165.37, 142.43, 137.69, 133.41, 132.94, 128.57, 127.99, 125.62, 120.24, 60.44, 52.46, 48.48, 30.88, 24.60. HRMS (ESI) m/z calcd for C20H20F3N2O5S [M + H]+, 457.1045; found, 457.1053.
4.1.5. Methyl ((4-((4-hydroxybenzyl)amino)phenyl)sulfonyl)-L-prolinate (15)
To a solution of methyl ((4-aminophenyl)sulfonyl)-L-prolinate (10) (70 mg, 0.25 mmol) and 4-hydroxybenzaldehyde (38 mg, 0.25 mmol) MeOH (5 mL), NaBH3CN (32 mg, 0.5 mmol) was added. After refluxing for 30 min, the mixture was extracted with DCM (20 mL×3). The organic layer was washed with saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated to give a crude solid, which was then purified with PTLC to give the desired product (35 mg, 85% after recovery of starting material) as a pale yellow solid with 40 mg of the starting material 10 recovered. m.p.: 151-153 °C. 1H NMR (300 MHz, CDCl3) δ 7.60 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.3 Hz, 2H), 6.81 (d, J = 8.3 Hz, 2H), 6.61 (d, J = 8.7 Hz, 2H), 4.28 – 4.17 (m, 3H), 3.70 (s, 3H), 3.45 (dd, J = 9.5, 6.8 Hz, 1H), 3.24 (dd, J = 11.6 Hz, 4.7 Hz, 1H), 2.07 – 1.86 (m, 3H), 1.78 – 1.65 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 173.15, 156.15, 151.75, 129.54, 129.05, 128.85, 124.10, 115.59, 111.81, 60.34, 52.43, 48.50, 47.00, 30.88, 24.63. HRMS (ESI) m/z calcd for C19H23N2O5S [M + H]+, 391.1328; found, 391.1335.
4.1.6. Methyl (E)-((4-((2-amino-4-hydroxy-5-methylphenyl)diazenyl)phenyl)sulfonyl)-L-prolinate (16)
To a solution of 10 (114 mg, 0.4 mmol) and HCl (concentrated aq, 160 μL, 2.4 mmol) in MeOH (3 mL) and CH3CN (3 mL) at 0 °C (pre-cooled for 15 min) under N2, isopentyl nitrite (48 μL, 0.4 mmol) was added over 15 min and then the mixture was stirred at 0 °C for 45 min to get a solution. At the same time, 5-amino-2-methylphenol (50 mg, 0.4 mmol) and K2CO3 (276 mg, 2.0 mmol) were dissolved in MeOH (1 mL) and H2O (8 mL) and degassed for 15 min. To the reaction mixture, the previous solution was added over 15 min and stirred at 0 °C for 1 h. After the completion of the reaction, 10% HCl was added to adjust pH = 1 and then NaHCO3 (saturated aq.) was used to adjust pH = 8. The solution was extracted with DCM (30 mL×2). The organic layer was washed with brine and dried over anhydrous Na2SO4. The resulting solution was filtered and concentrated to give a crude solid, which was further purified by silica gel column chromatography (DCM/CH3OH = 50/1 to 30/1) to give the desired product (130 mg, 78%) as a red solid. m.p.: 98-100 °C (decomposition). 1H NMR (300 MHz, CDCl3) δ 7.87 (s, 3H), 7.53 (s, 1H), 6.48 – 6.12 (m, 2H), 4.31 (t, J = 5.9 Hz, 1H), 3.74 (s, 3H), 3.52 (s, 1H), 3.38 – 3.22 (m, 1H), 2.14 (d, J = 7.6 Hz, 2H), 2.07 – 1.87 (m, 4H), 1.80 – 1.67 (m, 1H). 13C NMR (75 MHz, CDCl3) δ 173.01, 160.53, 156.10, 144.10, 136.04, 132.18, 131.67, 128.47, 122.06, 116.08, 101.13, 60.58, 52.68, 48.68, 30.96, 24.68, 15.05. HRMS (ESI) m/z calcd for C19H23N4O5S [M + H]+, 419.1389; found, 419.1397.
4.1.7. (5-(tert-Butyl)isoxazol-3-yl)(4-(4-fluorophenyl)piperazin-1-yl)methanone (19)
Ethyl 5-(tert-butyl)isoxazole-3-carboxylate 17 (1,000 mg, 5.1 mmol) and LiOH (638 mg, 15.3 mmol) were dissolved in 30 mL MeOH and 10 mL H2O, respectively. Then the solution was stirred at rt. for 1 hr. In an ice-cooled bath, 1 N Na2SO4 was added, and the mixture was extracted by EtOAc. The organic layer was separated and washed with brine. After drying with anhydrous Na2SO4, the solution was concentrated to give the intermediate 18 (818 mg, 95%) as a light yellow oil. 1H NMR(300 MHz, CDCl3) δ 10.64 (s, 1H), 6.42 (s, 1H), 1.37 (s, 9H).
To a solution of 18 (70 mg, 0.41 mmol) and 1-(4-fluorophenyl)piperazine (74 mg, 0.41 mmol) in 5 mL DCM, HBTU (395 mg, 1.23 mmol) and DIEA (271 mg, 2.1 mmol) were added. The mixture was stirred at rt. for 18 h. The mixture was washed with 1 N Na2SO4, saturated NaHCO3 and brine. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (hexane/EtOAc=10/1 to 5/1) to obtain the desired product (107 mg, 81%) as a white solid. m.p.: 93-95 °C. 1H NMR(300 MHz, CDCl3) δ 7.06 – 6.97 (m, 2H), 6.97 – 6.88 (m, 2H), 6.31 (s, 1H), 4.12 – 4.03 (m, 2H), 4.01 – 3.93 (m, 2H), 3.24 – 3.13 (m, 4H), 1.40 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 181.83, 159.82, 159.25, 158.18, 118.72, 118.62, 115.87, 115.57, 99.92, 51.06, 50.52, 46.91, 42.56, 32.88, 28.80.
4.1.8. (E)-N′-(3,5-Bis(trifluoromethyl)benzylidene)-5-(tert-butyl)isoxazole-3-carbohydrazide (22)
Methyl 5-(tert-butyl)isoxazole-3-carboxylate (100 mg, 0.5 mmol) and NH2NH2 (76 mg, 1.5 mmol) were dissolved in 5 mL EtOH and refluxed for 2 h. 3-Chloro-5-fluorobenzaldehyde (402 mg, 2.5 mmol) was then added and the solution was allowed to stir at rt. for 2 h. Then the solution was concentrated and extracted with DCM. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (hexane/EtOAc=20/1 to 10/1) to obtain 21 (72 mg, 44% for two steps) as a white solid. 1H NMR (300 MHz, CDCl3) δ 9.98 (s, 1H), 8.26 (s, 1H), 7.57 (s, 1H), 7.43 (d, J = 7.9 Hz, 1H), 7.15 (d, J = 6.5 Hz, 1H), 6.57 (s, 1H), 1.40 (s, 9H). 13C NMR (75 MHz, CDCl3) δ 183.65, 164.37, 157.27, 155.56, 146.48, 136.54, 135.47, 123.77, 118.41, 118.08, 112.89, 112.58, 98.80, 33.06, 28.75.
Methyl 5-(tert-butyl)isoxazole-3-carboxylate (100 mg, 0.5 mmol) and NH2NH2 (76 mg, 1.5 mmol) were dissolved in 5 mL EtOH and refluxed for 2 h. 3,5-Bis(trifluoromethyl)benzaldehydebenzaldehyde (605 mg, 2.5 mmol) was then added and the solution was allowed to stir at rt. for 2 h. Then the solution was concentrated and extracted with DCM. After drying over anhydrous Na2SO4, the solution was concentrated and purified with silica gel column (Hexane/EtOAc=20/1 to 10/1) to obtain 22 (120 mg, 59% for two steps) as a white solid. m.p.: 169-171 °C. 1H NMR (300 MHz, CDCl3) δ 10.13 (s, 1H), 8.50 (s, 1H), 8.23 (s, 2H), 7.92 (s, 1H), 6.59 (s, 1H), 1.41 (s, 9H). 13C NMR(75 MHz, CDCl3) δ 183.79, 157.19, 155.72, 145.91, 135.69, 133.04, 132.59, 132.14, 131.63, 127.46, 124.73, 123.84, 121.14, 98.82, 33.08, 28.73. HRMS (ESI) m/z calcd for C17H16F6N3O2 [M + H]+, 408.1147; found, 408.1142.
4.1.9. 2-(5-(tert-Butyl)isoxazol-3-yl)-5-(3-chloro-5-fluorophenyl)-1,3,4-oxadiazole (23)
Compound 21 (56 mg, 0.173 mmol), I2 (53 mg, 0.21 mmol) and K2CO3 (72 mg, 0.52 mmol) were dissolved in DMSO and stirred at 100 °C for 1 h. Then the solution was poured into ice water and saturated Na2S2O3 was added. The mixture was extracted with DCM, and the organic layer was dried over Na2SO4 and purified by silica gel column to obtain 23 (7 mg, 13%) as a white solid. m.p.: 190-193 °C (decomposition). 1H NMR (300 MHz, CDCl3) δ 8.02 (s, 1H), 7.83 (ddd, J = 8.5, 2.3, 1.4 Hz, 1H), 7.38 – 7.31 (m, 1H), 6.70 (s, 1H), 1.46 (s, 9H). HRMS (ESI) m/z calcd for C15H13N3FClNaO2 [M + Na]+, 344.0578; found, 344.0568.
4.1.10. 5-Chloro-2-methoxy-N-(2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)benzenesulfonamide (26)
6-Nitro-3,4-dihydroquinolin-2(1H)-one (1.0 g, 5.2 mmol) was dissolved in 40 mL of EtOH, and then NH4Cl (2.76 g, 52 mmol) in 20 mL of H2O and Zn dust (2.37 g, 36.4 mmol) were added. After refluxing at 80 °C for 1 h, the mixture was filtered to remove Zn dust. The filtration was concentrated to give 25 (1.8 g, including partial NH4Cl) as a gray solid. 1H NMR (300 MHz, DMSO-d6) δ 9.67 (s, 1H), 6.55 (d, J = 8.2 Hz, 1H), 6.42 – 6.31 (m, 2H), 2.70 (t, J = 7.5 Hz, 2H), 2.33 (dd, J = 8.5 Hz, 6.5 Hz, 2H).
To a solution of 25 (70 mg, 0.43 mmol) and Et3N (87 mg, 0.86 mmol) in 5 mL DMF, 5-chloro-2-methoxybenzenesulfonyl chloride (156 mg, 0.65 mmol) was added. After stirring at rt. for 1 h, the mixture was poured into 20 mL of ice-water. The precipitate was filtered to get the desired product (45 mg, 29%) as a white solid. m.p.: 253-255 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 9.97 (s, 1H), 9.85 (s, 1H), 7.61 (s, 1H), 7.24 (dd, J = 10.2, 6.8 Hz, 2H), 6.94 – 6.79 (m, 2H), 6.67 (d, J = 8.4 Hz, 1H), 3.91 (s, 3H), 2.81 – 2.71 (m, 2H), 2.36 (t, J = 7.5 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.38, 155.70, 135.72, 134.90, 133.70, 131.74, 129.59, 128.53, 127.45, 124.68, 124.05, 121.63, 120.67, 115.80, 115.36, 57.00, 30.58, 25.25. HRMS (ESI) m/z calcd for C16H16N2O4SCl [M + H]+, 367.0519; found, 367.0506.
4.1.11. 4-Nitro-N-(2-oxo-1,2,3,4-tetrahydroquinolin-6-yl)benzenesulfonamide (27)
Compound 27 was prepared from 4-nitrobenzenesulfonyl chloride in 13% yield by a procedure similar to that used to prepare compound 26. The title compound was obtained as a yellow solid. m.p.: 242-246 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.36 (d, J = 8.7 Hz, 2H), 7.94 (d, J = 8.8 Hz, 2H), 6.89 (s, 1H), 6.79 (s, 1H), 6.69 (d, J = 8.4 Hz, 1H), 2.78 (d, J = 7.1 Hz, 2H), 2.37 (dd, J = 8.5, 6.6 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.34, 149.99, 136.06, 128.73, 124.86, 115.84, 30.60, 25.23. HRMS (ESI) m/z calcd for C15H14N3O5S [M + H]+, 348.0654; found, 348.0646.
4.1.12. (E)-6-((2-Amino-4-hydroxy-5-methylphenyl)diazenyl)-3,4-dihydroquinolin-2(1H)-one (28)
Compound 28 was prepared from 5-amino-2-methylphenol in 81% yield by a procedure similar to that used to prepare compound 16. The title compound was obtained as a red solid. m.p.: 208-210 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 10.25 (s, 1H), 7.69 – 7.56 (m, 2H), 7.36 (s, 1H), 6.93 (d, J = 8.3 Hz, 1H), 6.25 (s, 1H), 2.95 (t, J = 7.4 Hz, 2H), 2.47 (s, 2H), 2.03 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 170.64, 160.39, 148.35, 145.73, 139.21, 130.98, 126.96, 124.67, 121.85, 120.73, 115.83, 114.51, 101.07, 30.75, 25.31, 15.66. HRMS (ESI) m/z calcd for C16H17N4O2 [M + H]+, 297.1352; found, 297.1342.
4.1.13. 5-((4-(N-Cyclopentylsulfamoyl)phenyl)amino)-N-methylpicolinamide (29)
To a solution of 5-bromo-N-methylpicolinamide (80 mg, 0.37 mmol) and 6-amino-3,4-dihydroquinolin-2(1H)-one (50 mg, 0.31 mmol) in 5 mL 1,4-dioxane, Pd(OAc)2 (18 g, 0.08 mmol), Cs2CO3 (202 mg, 0.62 mmol) and xantphos (90 mg, 0.16 mmol) were added. The mixture was allowed to reflux at 110 °C overnight. Then the mixture was filtered, poured into H2O and extracted by DCM. The organic layer was washed with saturated NaHCO3 (aq.), brine and dried over anhydrous Na2SO4. The resulting solution was evaporated, and the residue was purified by silica gel column (DCM/MeOH = 50:1) to give the desired product (58 mg, 63%) as a white solid. m.p.: 276-278 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.58 (d, J = 2.5 Hz, 1H), 8.37 (s, 1H), 8.19 (s, 1H), 7.86 – 7.74 (m, 1H), 7.44 – 7.32 (m, 1H), 6.99 (d, J = 9.0 Hz, 2H), 6.84 (d, J = 3.6 Hz, 1H), 2.85 (s, 2H), 2.78 (s, 3H), 2.44 (s, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.31, 165.02, 144.05, 140.36, 135.99, 135.56, 134.07, 125.25, 123.17, 120.24, 119.83, 119.50, 116.31, 30.81, 26.25, 25.43. HRMS (ESI) m/z calcd for C16H17N4O2 [M + H]+, 297.1352; found, 297.1345.
4.1.14. 1-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)-3-(4-(trifluoromethyl)phenyl)urea (30)
1-Isocyanato-4-(trifluoromethyl)benzene (131 mg, 0.7 mmol) and 6-amino-3,4-dihydroquinolin-2(1H)-one (95 mg, 0.6 mmol) were mixed together and stirred at 50°C overnight. Then the solution was concentrated and purified by silica gel column (DCM/CH3OH = 100/1 to 50/1) to give 30 (85 mg, 41%) as a white solid. m.p.: 258-260 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 9.98 (s, 1H), 9.01 (s, 1H), 8.61 (s, 1H), 7.63 (q, J = 9.0 Hz, 4H), 7.31 (s, 1H), 7.18 (d, J = 7.2 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 2.85 (t, J = 7.4 Hz, 2H), 2.42 (t, J = 7.4 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 170.36, 152.83, 144.04, 134.03, 133.79, 126.83, 126.51, 126.47, 126.42, 124.47, 123.24, 122.71, 122.28, 121.86, 121.44, 119.06, 118.21, 115.68, 30.86, 25.54. HRMS (ESI) m/z calcd for C17H15N3O2F3 [M + H]+, 350.1116; found, 350.1111.
4.1.15. (E)-4-(4-((2-Amino-4-hydroxy-5-methylphenyl)diazenyl)phenyl)thiomorpholine 1,1-dioxide (33)
Compound 33 was prepared in 87% yield taking 4-(4-aminophenyl)thiomorpholine 1,1-dioxide as the key intermediate by a procedure similar to that used to prepare compound 16. The title compound was obtained as a yellow solid. m.p.: 214-216 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 7.70 (d, J = 8.9 Hz, 2H), 7.10 (d, J = 9.0 Hz, 2H), 6.63 (s, 2H), 6.24 (s, 1H), 3.88 (s, 4H), 3.15 (s, 4H), 2.03 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 159.79, 148.30, 146.13, 145.41, 130.95, 126.92, 123.36, 115.83, 113.92, 101.02, 50.46, 46.90, 15.66. HRMS (ESI) m/z calcd for C17H21N4O3S [M + H]+, 361.1334; found, 361.1328.
4.1.16. (E)-5-Amino-2-methyl-4-((4-(pyrrolidin-1-ylsulfonyl)phenyl)diazenyl)phenol (34)
Compound 34 was prepared in 92% yield taking 4-(pyrrolidin-1-ylsulfonyl)aniline as the key intermediate by a procedure similar to that used to prepare compound 16. The title compound was obtained as a dark solid. m.p.: 177-179 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 8.05 (d, J = 8.5 Hz, 2H), 7.84 (d, J = 8.7 Hz, 2H), 7.75 (t, J = 12.8 Hz, 1H), 6.38 (s, 1H), 3.16 (t, J = 6.6 Hz, 4H), 2.04 (s, 3H), 1.65 (dd, J = 8.1, 5.1 Hz, 4H). 13C NMR (75 MHz, DMSO-d6) δ 166.60, 134.11, 131.06, 129.04, 120.43, 100.11, 48.31, 25.18, 16.02. HRMS (ESI) m/z calcd for C17H21N4O3S [M + H]+, 361.1334; found, 361.1330.
4.1.17. (E)-4-((2-Amino-4-hydroxy-5-methylphenyl)diazenyl)-N-cyclopentylbenzenesulfonamide (35)
Compound 35 was prepared in 81% yield taking 4-amino-N-cyclopentylbenzenesulfonamide as the key intermediate by a procedure similar to that used to prepare compound 16. The title compound was obtained as a red solid. m.p.: 160-162 °C (decomposition). 1H NMR (300 MHz, DMSO-d6) δ 8.03 (d, J = 7.2 Hz, 2H), 7.85 (d, J = 8.7 Hz, 2H), 7.68 (d, J = 6.6 Hz, 1H), 6.42 (d, J = 6.3 Hz, 1H), 3.42 (d, J = 6.1 Hz, 1H), 2.05 (s, 3H), 1.65 – 1.48 (m, 4H), 1.44 – 1.24 (m, 4H). 13C NMR (75 MHz, DMSO-d6) δ 166.55, 139.65, 130.75, 128.27, 119.99, 100.15, 54.95, 32.90, 23.27, 16.10. HRMS (ESI) m/z calcd for C18H23N4O3S [M + H]+, 375.1538; found, 375.1530.
4.2. Cell culture
Immortalized human small airway epithelial cells (hSAECs) were previously described [31,32]. hSAECs were grown in SAGM small airway epithelial cell growth medium (Lonza, Walkersville, MD) in a humidified atmosphere of 5% CO2. Poly(I:C) was obtained from Sigma (St. Louis, MO) and used at 10 μg/mL in cell culture. JQ1 was purchased from Cayman Chemical (Ann Arbor, Michigan) and RVX208 was purchased from Tocris. Compounds were solubilized in DMSO and added at the indicated concentrations.
4.3. Quantitative Real-Time PCR (Q-RT-PCR)
For gene expression analyses, 1 μg of RNA was reverse transcribed using Super Script III as previously described. One μL of cDNA product was amplified using SYBR Green Supermix (Bio-Rad) and indicated gene-specific primers. The reaction mixtures were subjected to 40 cycles of 15 s at 94 °C, 60 s at 60 °C, and 1 min at 72 °C in an iCycler (BioRad). Quantification of relative changes in gene expression was calculated using the ΔΔCt method and expression as the fold change between experimental and control samples was normalized to internal control cyclophilin (PPIA).
4.4. In vitro efficacy of BRD4 inhibitors on poly(I:C) induced innate immune response
hSAECs were first pretreated with a series final concentrations of BRD4 inhibitors from 0.01 nM to 100 μM for 24 hours and were then added poly(I:C) at 10 μg/mL for another 4 hours prior to harvesting the cells. The harvested cells were first washed with PBS twice and then the total RNA was extracted using acid guanidinium phenol extraction (Tri Reagent; Sigma). The total RNA was further reverse-transcribed for gene expression analysis by Q-RT-PCR. The inhibitory effect of BRD4 inhibitors on poly(I:C)-induced innate immune gene expression was compared with that of poly(I:C) alone and inhibitory percentage of each treatment was obtained. For compounds 23, 28 and 35, in vitro efficacy of these BRD4 inhibitors on poly(I:C) induced innate immune response were presented as the IC50 values of these compounds. Compounds were dissolved in DMSO and further diluted at cell culture medium to appropriate concentrations.
4.5. Time-resolved fluorescence energy transfer (TR-FRET) assays
384 well plate-based commercial TR-FRET Assay kits (Cayman Chemical, Ann Arbor, Michigan) were used to determine the binding ability of tested BRD4 inhibitors to the BRD4 and BRD2 bromodomains (BD) using the two recombinant BRD4 BDs or BRD2 BDs by time-resolved fluorescence energy transfer (TR-FRET) assays. A series of concentrations of BRD4 inhibitors from 0.01 nM to 100 μM were added into a 384 well test plate and mixed with other reaction components based on the instructions from vendor followed by incubation 1h at room temperature. The commercially available BRD inhibitors JQ1 and RVX208 were used as the controls. The plates were read in time-resolved format by exciting the sample at 340 nm and reading emissions at 620 and 670 nm, using a 100 μs delay and a 500 μs window at a Tecan M1000 pro reader. A plot of the TR-FRET ratio (670 nm emission/620 nm emission versus inhibitor concentration on semi-log axes results in a sigmoidal dose-response curve typical of competitive assays. These data were further calculated out with the IC50 values of tested BRD4 inhibitors to the bromodomains of BRD2 and BRD4 as well as other relevant target proteins, respectively.
4.6. In vivo efficacy of BRD4 inhibitors on poly(I:C)-induced acute airway inflammation
Animal experiments were performed according to the NIH Guide for Care and Use of Experimental Animals and approved by the University of Texas Medical Branch (UTMB) Animal Care and Use Committee (approval no. 1312058A). Male C57BL6/J mice (12 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed under pathogen-free conditions with food and water ad libitum. C57BL/6 mice were pre-treated in the absence or presence of the indicated BRD4 inhibitors [10 mg/kg body weight, via the intraperitoneal route] one day prior to poly(I:C) stimulation. The next day, animals were given another dose of BRD4 inhibitor immediately followed by intranasal (i.n.) administration of phosphate-buffered saline (PBS, 50 μL) or poly(I:C) (300 μg dissolved in 50 μL PBS). One day later, the mice were euthanized. The bronchoalveolar lavage fluid (BALF) and lung tissues of treated mice were collected for further analysis. Compounds were first dissolved in DMSO and further diluted in 10% hydroxypropyl β-cyclodextrin in PBS to appropriate concentration prior to intraperitoneal administration.
4.7. Evaluation of airway inflammation
Cellular recruitment into the airway lumen was assessed in the bronchoalveolar lavage fluid (BALF). Lungs were perfused twice with 1 mL of sterile PBS (pH 7.4) to obtain the BALF. Total cell counts were determined by trypan blue staining 50 μL of BALF and counting viable cells using a hemocytometer. Differential cell counts were performed on cytocentrifuge preparations (Cytospin 3; Thermo Shandon, Pittsburgh, Pa) stained with Wright-Giemsa. A total of 300 cells were counted per sample using light microscopy. Formalin-fixed lungs were embedded in paraffin, sectioned at a 4 μm thickness, and stained with hematoxylin and eosin or Masson’s trichrome. Microscopy was performed on a NIKON Eclipse Ti System.
4.8. Molecular docking studies
The docking study was performed with Schrödinger Small-Molecule Drug Discovery Suite. The crystal structure of BRD4 BD1 (PDB code: 4NUD) was downloaded from RCSB PDB Bank and prepared with Protein Prepared Wizard. During this step, hydrogens were added, crystal waters were removed while water molecules around the KAc pocket were kept, and partial charges were assigned using the OPLS-2005 force field. The 3D structures of ZL0420 and ZL0454 were created with Schrödinger Maestro, and the initial lowest energy conformations were calculated with LigPrep. For all dockings, the grid center was chosen on the centroid of included ligand of PDB structure KAc site and a 24 × 24 × 24 Å grid box size was used. All dockings were employed with Glide using the XP protocol. Docking poses were incorporated into Schrödinger Maestro for a visualization of ligand-receptor interactions and overlay analysis.
Supplementary Material
Highlights.
Structure-based drug design led to discovery of potent BRD4 inhibitors.
28 and 35 show nanomolar BRD4 binding affinities.
28 and 35 exhibit good BRD4 selectivity over other BET family members.
28 and 35 display submicromolar inhibition against immune gene expression.
In vivo efficacy of 28 and 35 blocking airway inflammation has been confirmed.
Acknowledgments
This work was supported, in part, by NIH grants (NIAID AI062885, UL1TR001439, P30 DA028821), UTMB Technology Commercialization Program, and Sanofi Innovation Awards (iAwards) as well as Fellowship award entitled “Drug Discovery of BRD4 Inhibitors for the Treatment of Inflammatory Bowel Disease” (Identifier: 548813) from the Crohn’s & Colitis Foundation of America (ZL). Core laboratory support was provided by the UTMB Histopathology Core. We want to thank Drs. Lawrence C. Sowers at the Department of Pharmacology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance, and Dr. Xuemei Luo at UTMB mass spectrometry core with funding support from UT system proteomics network for the HRMS analysis.
Abbreviations
- BRD4
bromodomain-containing protein 4
- BET
bromodomain and extra-terminal domain
- KAc
acetylated lysine
- BCP
bromodomain-containing proteins
- EMT
epithelial-mesenchymal transition
- DIPEA
N,N-diisopropylethylamine
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- Pd(OAc)2
palladium(II) acetate
- hSAECs
human small airway epithelial cells
- poly(I:C)
polyinosinic:polycytidylic acid
- TR-FRET
time resolved-fluorescence resonance energy transfer. ISG, interferon stimulated gene
- IL-8
interleukin-8
- Groβ
growth-regulated protein β
- qRT-PCR
quantitative real-time polymerase chain reaction
- TLR3
toll-like receptor 3
- CREB
cAMP responsive element binding protein
- CBP
CREB binding protein
- CL
clearance
- IV
intravenous
- PO
per os
- Vss
volume of distribution at steady state
- AUC
area under the curve
- PEG
polyethylene glycol
- i.p.
intraperitoneal
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://XXXX.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Notes
The authors declare no competing financial interest.
References
- 1.Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature. 1999;399:491–496. doi: 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- 2.Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, Gingras AC, Arrowsmith CH, Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Z, Wang P, Chen H, Wold EA, Tian B, Brasier AR, Zhou J. Drug Discovery Targeting Bromodomain-Containing Protein 4. J Med Chem. 2017;60:4533–4558. doi: 10.1021/acs.jmedchem.6b01761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 6.Romero FA, Taylor AM, Crawford TD, Tsui V, Cote A, Magnuson S. Disrupting Acetyl-Lysine Recognition: Progress in the Development of Bromodomain Inhibitors. J Med Chem. 2016;59:1271–1298. doi: 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- 7.Vidler LR, Brown N, Knapp S, Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J Med Chem. 2012;55:7346–7359. doi: 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Picaud S, Wells C, Felletar I, Brotherton D, Martin S, Savitsky P, Diez-Dacal B, Philpott M, Bountra C, Lingard H, Fedorov O, Muller S, Brennan PE, Knapp S, Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci U S A. 2013;110:19754–19759. doi: 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bui MH, Lin X, Albert DH, Li L, Lam LT, Faivre EJ, Warder SE, Huang X, Wilcox D, Donawho CK, Sheppard GS, Wang L, Fidanze S, Pratt JK, Liu D, Hasvold L, Uziel T, Lu X, Kohlhapp F, Fang G, Elmore SW, Rosenberg SH, McDaniel KF, Kati WM, Shen Y. Preclinical Characterization of BET Family Bromodomain Inhibitor ABBV-075 Suggests Combination Therapeutic Strategies. Cancer Res. 2017;77:2976–2989. doi: 10.1158/0008-5472.CAN-16-1793. [DOI] [PubMed] [Google Scholar]
- 11.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478:529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gosmini R, Nguyen VL, Toum J, Simon C, Brusq JM, Krysa G, Mirguet O, Riou-Eymard AM, Boursier EV, Trottet L, Bamborough P, Clark H, Chung CW, Cutler L, Demont EH, Kaur R, Lewis AJ, Schilling MB, Soden PE, Taylor S, Walker AL, Walker MD, Prinjha RK, Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J Med Chem. 2014;57:8111–8131. doi: 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- 13.Lucas X, Wohlwend D, Hugle M, Schmidtkunz K, Gerhardt S, Schule R, Jung M, Einsle O, Gunther S. 4-Acyl pyrroles: mimicking acetylated lysines in histone code reading. Angew Chem Int Ed Engl. 2013;52:14055–14059. doi: 10.1002/anie.201307652. [DOI] [PubMed] [Google Scholar]
- 14.Nicholls SJ, Puri R, Wolski K, Ballantyne CM, Barter PJ, Brewer HB, Kastelein JJ, Hu B, Uno K, Kataoka Y, Herrman JP, Merkely B, Borgman M, Nissen SE. Effect of the BET Protein Inhibitor, RVX-208, on Progression of Coronary Atherosclerosis: Results of the Phase 2b, Randomized, Double-Blind, Multicenter, ASSURE Trial. Am J Cardiovasc Drugs. 2016;16:55–65. doi: 10.1007/s40256-015-0146-z. [DOI] [PubMed] [Google Scholar]
- 15.Sarthy A, Li L, Albert DH, Lin X, Scott W, Faivre E, Bui MH, Huang X, Wilcox DM, Magoc T, Buchanan FG, Tapang P, Sheppard GS, Wang L, Fidanze SD, Pratt J, Liu D, Hasvold L, Hessler P, Uziel T, Lam L, Rajaraman G, Fang G, Elmore SW, Rosenberg SH, McDaniel K, Kati W, Shen Y. Abstract 4718: ABBV-075, a novel BET family bromodomain inhibitor, represents a promising therapeutic agent for a broad spectrum of cancer indications. Cancer Res. 2016;76:4718–4718. [Google Scholar]
- 16.Shapiro GI, Dowlati A, LoRusso PM, Eder JP, Anderson A, Do KT, Kagey MH, Sirard C, Bradner JE, Landau SB. Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC) Mol Cancer Ther. 2016;14:A49. [Google Scholar]
- 17.Raux B, Voitovich Y, Derviaux C, Lugari A, Rebuffet E, Milhas S, Priet S, Roux T, Trinquet E, Guillemot JC, Knapp S, Brunel JM, Fedorov AY, Collette Y, Roche P, Betzi S, Combes S, Morelli X. Exploring Selective Inhibition of the First Bromodomain of the Human Bromodomain and Extra-terminal Domain (BET) Proteins. J Med Chem. 2016;59:1634–1641. doi: 10.1021/acs.jmedchem.5b01708. [DOI] [PubMed] [Google Scholar]
- 18.Andrieu G, Belkina AC, Denis GV. Clinical trials for BET inhibitors run ahead of the science. Drug Discov Today Technol. 2016;19:45–50. doi: 10.1016/j.ddtec.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian B, Zhao Y, Sun H, Zhang Y, Yang J, Brasier AR. BRD4 mediates NF-kappaB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am J Physiol Lung Cell Mol Physiol. 2016;311:L1183–L1201. doi: 10.1152/ajplung.00224.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung CW, Coste H, White JH, Mirguet O, Wilde J, Gosmini RL, Delves C, Magny SM, Woodward R, Hughes SA, Boursier EV, Flynn H, Bouillot AM, Bamborough P, Brusq JM, Gellibert FJ, Jones EJ, Riou AM, Homes P, Martin SL, Uings IJ, Toum J, Clement CA, Boullay AB, Grimley RL, Blandel FM, Prinjha RK, Lee K, Kirilovsky J, Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54:3827–3838. doi: 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- 22.Chen H, Zhou X, Wang A, Zheng Y, Gao Y, Zhou J. Evolutions in fragment-based drug design: the deconstruction-reconstruction approach. Drug Discov Today. 2015;20:105–113. doi: 10.1016/j.drudis.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Zhou X, Gao Y, Chen H, Zhou J. Chapter 8: Fragment-based drug design: strategic advances and lessons learned. In: Chackalamannil S, Rotella D, Ward SE, editors. Comprehensive Medicinal Chemistry III. Elsevier; Oxford: 2017. pp. 212–232. [Google Scholar]
- 24.Erlanson DA, Fesik SW, Hubbard RE, Jahnke W, Jhoti H. Twenty years on: the impact of fragments on drug discovery. Nat Rev Drug Discov. 2016;15:605–619. doi: 10.1038/nrd.2016.109. [DOI] [PubMed] [Google Scholar]
- 25.Simmons KJ, Chopra I, Fishwick CW. Structure-based discovery of antibacterial drugs. Nat Rev Microbiol. 2010;8:501–510. doi: 10.1038/nrmicro2349. [DOI] [PubMed] [Google Scholar]
- 26.Zhang G, Plotnikov AN, Rusinova E, Shen T, Morohashi K, Joshua J, Zeng L, Mujtaba S, Ohlmeyer M, Zhou MM. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J Med Chem. 2013;56:9251–9264. doi: 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu W, Huang G, Zhang Y, Liu H, Dong L, Yu X, Li Y, Chang J. I2-mediated oxidative C-O bond formation for the synthesis of 1,3,4-oxadiazoles from aldehydes and hydrazides. J Org Chem. 2013;78:10337–10343. doi: 10.1021/jo401751h. [DOI] [PubMed] [Google Scholar]
- 28.Tian B, Patrikeev I, Ochoa L, Vargas G, Belanger KK, Litvinov J, Boldogh I, Ameredes BT, Motamedi M, Brasier AR. NF-kappaB Mediates Mesenchymal Transition, Remodeling, and Pulmonary Fibrosis in Response to Chronic Inflammation by Viral RNA Patterns. Am J Respir Cell Mol Biol. 2017;56:506–520. doi: 10.1165/rcmb.2016-0259OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung M, Philpott M, Muller S, Schulze J, Badock V, Eberspacher U, Moosmayer D, Bader B, Schmees N, Fernandez-Montalvan A, Haendler B. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem. 2014;289:9304–9319. doi: 10.1074/jbc.M113.523019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian B, Liu Z, Yang J, Sun H, Zhao Y, Wakamiya M, Chen H, Rytting E, Zhou J, Brasier AR. Novel Molecular Antagonists of the Bronchiolar Epithelial NFkB-Bromodomain-Containing Protein 4 (BRD4) Pathway in Viral-induced Airway Inflammation. Cell Rep. 2017 doi: 10.1016/j.celrep.2018.03.106. accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, Peyton M, Zou Y, Kurie JM, DiMaio JM, Milchgrub S, Smith AL, Souza RF, Gilbey L, Zhang X, Gandia K, Vaughan MB, Wright WE, Gazdar AF, Shay JW, Minna JD. Immortalization of Human Bronchial Epithelial Cells in the Absence of Viral Oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 32.Tian B, Li X, Kalita M, Widen SG, Yang J, Bhavnani SK, Dang B, Kudlicki A, Sinha M, Kong F, Wood TG, Luxon BA, Brasier AR. Analysis of the TGFbeta-induced program in primary airway epithelial cells shows essential role of NF-kappaB/RelA signaling network in type II epithelial mesenchymal transition. BMC Genomics. 2015;16:529. doi: 10.1186/s12864-015-1707-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.