

Abstract
N-methyl-D-aspartate receptors (NMDARs) are ionotropic glutamate receptors that mediate excitatory synaptic transmission and have been implicated in numerous neurological disorders. NMDARs typically comprise two GluN1 and two GluN2 subunits. The four GluN2 subtypes (GluN2A-GluN2D) have distinct functional properties and gene expression patterns, which contribute to diverse functional roles for NMDARs in the brain. Here, we present a series of GluN2C/2D-selective negative allosteric modulators built around a N-aryl benzamide (NAB) core. The prototypical compound, NAB-14, is >800-fold selective for recombinant GluN2C/GluN2D over GluN2A/GluN2B in Xenopus oocytes and has an IC50 value of 580 nM at recombinant GluN2D-containing receptors expressed in mammalian cells. NAB-14 inhibits triheteromeric (GluN1/GluN2A/GluN2C) NMDARs with modestly reduced potency and efficacy compared to diheteromeric (GluN1/GluN2C/GluN2C) receptors. Site-directed mutagenesis suggests that structural determinants for NAB-14 inhibition reside in the GluN2D M1 transmembrane helix. NAB-14 inhibits GluN2D-mediated synaptic currents in rat subthalamic neurons and mouse hippocampal interneurons, but has no effect on synaptic transmission in hippocampal pyramidal neurons, which do not express GluN2C or GluN2D. This series possesses some drug-like physical properties and modest brain permeability in rat and mouse. Altogether, this work identifies a new series of negative allosteric modulators that are valuable tools for studying GluN2C- and GluN2D-containing NMDAR function in brain circuits, and suggests that the series has the potential to be developed into therapies for selectively modulating brain circuits involving the GluN2C and GluN2D subunits.
Keywords: NMDA receptor, NMDAR, glutamate receptor, GluN2C, GluN2D, negative allosteric modulator, NAB-14
INTRODUCTION
N-methyl-D-aspartate (NMDA) receptors are essential for normal brain function and have been associated with numerous neurological disorders, including intellectual disabilities, epilepsy, ischemia, psychiatric illnesses, and neurodegenerative disorders.1–4 NMDARs are ionotropic glutamate receptors that mediate a slow, calcium-permeable current that contributes to excitatory synaptic transmission in the brain. NMDAR activation requires binding of the co-agonists glutamate and glycine as well as depolarization to remove extracellular Mg+2 from the channel pore. These tetrameric receptors typically contain two glycine-binding GluN1 subunits and two glutamate-binding GluN2 subunits. The four GluN2 subunits (GluN2A-GluN2D) have markedly different functional properties and distinct expression patterns in the central nervous system.5 For example, GluN2C and GluN2D have enhanced glutamate potency and reduced sensitivity to voltage-dependent Mg+2 block compared to GluN2A and GluN2B, which may allow GluN2C- and GluN2D-containing NMDARs to be activated by ambient glutamate at resting membrane potential. In addition, the unique and restricted expression patterns of GluN2C and GluN2D make them intriguing targets for modulating particular neuron types or brain circuits. In the adult hippocampus and cerebral cortex, GluN2A and GluN2B are expressed in principal neurons and interneurons, whereas GluN2C and GluN2D are expressed only in interneurons.6–8 GluN2C is also expressed in cerebellum, thalamus, amygdala, and olfactory bulb,9–11 and GluN2D is expressed in the basal ganglia, neuromodulatory brainstem nuclei, thalamus, and the deep cerebellar nuclei.6, 12–14 In addition, GluN2C and GluN2D mRNA are expressed in oligodendrocytes in the optic nerve, cerebellum, and corpus collosum.15–18 GluN2 subunit diversity provides an opportunity for therapeutically targeting specific NMDAR populations expressed in the brain circuits involved in neurological diseases.19, 20
Although the GluN2C and GluN2D subunits are expressed in vital brain circuits, the functional roles for GluN2C and GluN2D in the brain remain poorly understood compared to GluN2A and GluN2B. The recent development of GluN2C/2D-selective modulators has enabled the first pharmacological studies of GluN2C and GluN2D function in synaptic transmission, brain circuit function, and behavior. The GluN2C/2D-selective negative allosteric modulators DQP-1105 and QNZ46 have up to 50-fold selectivity for GluN2C/2D over GluN2A/2B.21–24 DQP-1105 and QNZ46 have been used to demonstrate that GluN2D regulates synaptic transmission in the subthalamic nucleus (STN), substantia nigra, striatum, and spinal cord,14, 25–29 and presynaptic GluN2C-containing NMDARs modulate gabaergic synaptic transmission in the suprachiasmatic nucleus.30 Studies with the GluN2C/2D-selective positive allosteric modulator CIQ, or the active enantiomer (+)-CIQ, have demonstrated a role for GluN2D in synaptic transmission or neuronal excitability in the striatum, STN, nucleus of the solitary tract, and hippocampal interneurons.8, 14, 31–34 In vivo studies with GluN2C/2D modulators suggest that GluN2C- or GluN2D-containing NMDARs are involved in fear conditioning, working memory, motor control, circadian timekeeping, and epilepsy.30, 35–38 These studies indicate that GluN2C/2D-containing receptors have essential roles in specific brain circuits and behaviors, and provide impetus for the continued development of more selective and drug-like GluN2C/2D-selective NMDAR modulators.
Here, we report the mechanism of action and structural determinants of a N-aryl benzamide (NAB) class of non-competitive NMDAR inhibitors selective for the GluN2C and GluN2D subunits. The prototypical compound, NAB-14, is >800-fold selective for GluN2C and GluN2D over GluN2A and GluN2B and has an IC50 value of 580 nM at recombinant GluN1/GluN2D receptors expressed in mammalian cells. NAB-14 inhibits both diheteromeric (GluN1/GluN2C/GluN2C) and triheteromeric (GluN1/GluN2A/GluN2C) NMDARs. Furthermore, we demonstrated that NAB-14 selectively inhibits recombinant GluN2C- and GluN2D-containing receptors in cultured neurons as well as synaptic currents mediated by GluN2D-containing receptors in the subthalamic nucleus and hippocampal interneurons. This study identifies a highly selective and brain-penetrant class of GluN2C/2D antagonists with utility as tools to study NMDAR function in native tissues and potential for further development toward therapeutics.
RESULTS AND DISCUSSION
Structure-activity relationship of the NAB series of NMDAR inhibitors
We have previously described a fluorescence-based assay that we used to screen approximately 100,000 compounds against NMDARs containing GluN1/GluN2C and GluN1/GluN2D, which sought to discover subunit-selective allosteric modulators for NMDARs.39, 40 These efforts identified compound 1, a negative allosteric modulator with IC50 values of 2.6 μM at GluN1/GluN2C and 1.4 μM at GluN1/GluN2D when tested on recombinant NMDARs in Xenopus oocytes (Figure 1a and Table 1). Compound 1 was >400-fold selective for GluN2C- and GluN2D-containing receptors over GluN2A- and GluN2B-containing receptors, but had low solubility in aqueous solution (20 μM) as determined by nephelometry.
Figure 1.
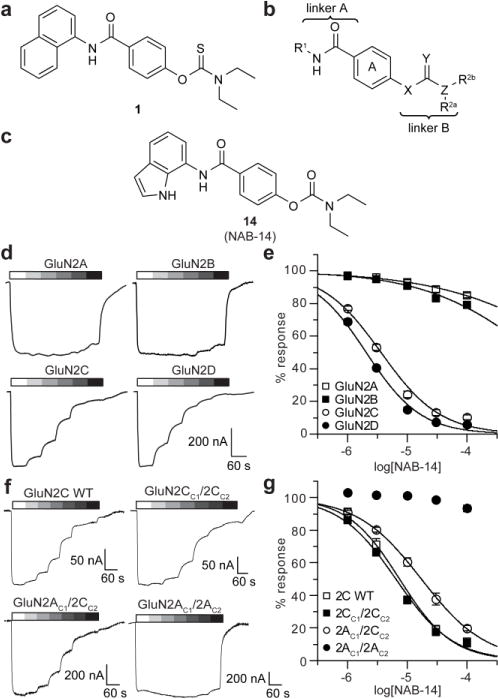
NAB-14 is a GluN2C/2D-selective NMDAR antagonist. The chemical structures are shown for (a) compound 1, (b) the regions evaluated in the NAB series structure-activity relationship, and (c) NAB-14. (d) Current responses to maximal concentrations of glutamate (100 μM) and glycine (30 μM) co-applied with increasing concentrations of NAB-14 were recorded by two-electrode voltage-clamp (TEVC) in Xenopus oocytes expressing GluN1 and GluN2A, GluN2B, GluN2C, or GluN2D. (e) Concentration-response data for NAB-14 were plotted as the percent of the maximal response to glutamate and glycine (mean ± s.e.m.) and fit by the Hill equation. (f) Representative current responses to 100 μM glutamate and 30 μM glycine co-applied with increasing concentrations of NAB-14 are shown for Xenopus oocytes expressing GluN1 and wild type (WT) GluN2C, GluN2CC1/2CC2, GluN2AC1/2CC2, or GluN2AC1/2AC2. (g) Concentration-response data for NAB-14 were plotted as the percent of the maximal glutamate and glycine response (mean ± s.e.m.) and fit by the Hill equation. The pIC50 values for 2AC1/2CC2 and 2CC1/2CC2 groups were compared by an F test [F (1,118) = 28.65, p < 0.001].
Table 1.
Optimization of N-aryl group (R1)
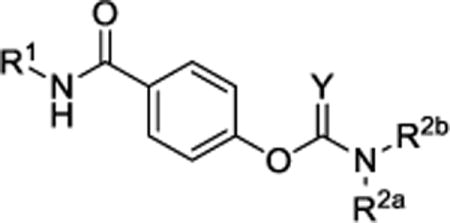
| ||||||||
|---|---|---|---|---|---|---|---|---|
| # | R1 | R2a & R2b | Y | IC50, mean (μM) pIC50, mean (95% CI) % control at 10 μM, mean (99% CI) |
N | |||
| GluN2A | GluN2B | GluN2C | GluN2D | |||||
| 1 |
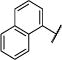
|
Et | S | 1070 −3.60(−4.98,−2.22) 82 (69,96) |
2320 −2.98 (−3.71,−2.25) 85 (76,93) |
2.6 −5.65 (−5.80,−5.50) 27 (17,36) |
1.4 −5.91 (−6.10,−5.72) 16 (5,28) |
7–10 |
| 2 |
|
Et | O | 83 (61,106) | 172 −3.82 (−4.00,−3.64) 82 (77,86) |
6.7 −5.21 (−5.33,−5.10) 35 (27,42) |
5.0 −5.38 (−5.49,−5.27) 27 (21,33) |
7–21 |
| 3 |
|
Et | S | 95 (25,164) | 100 (85,114) | 54 −4.27 (−4.42,−4.11) 87 (78,97) |
68 −4.23 (−4.23,−3.94) 75 (71,80) |
3–5 |
| 4 |
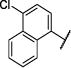
|
Et | S | 98 (81,116) | 89 (76,100) | 556 −3.51(−4.21,−2.82) 87 (76,97) |
148 −3.94 (−4.29,−3.59) 93 (89,97) |
4–6 |
| 5 |
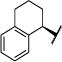
|
Et | O | 99 (57,119) | 93 (86,100) | 96 (87,104) | 96 (75,117) | 4–7 |
| 6 |
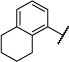
|
Et | O | 104 (102,106) | 272 −3.60 (−3.82,−3.39) 89 (85,93) |
32 −4.51 (−4.57,−4.45) 74 (70,79) |
19 −4.75 (−4.89,−4.62) 66 (55,76) |
4–8 |
| 7 |
|
Et | O | 95 (90,100) | 1030 −2.99 (−3.07,−2.90) 87 (85,88) |
62 −4.22 (−4.37,−4.07) 69 (61,76) |
48 −4.37 (−4.57,−4.17) 76 (56,96) |
4-8 |
| 8 |
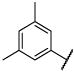
|
Et | O | 100 (98,102) | 98 −4.02 (−4.22,−3.82) 86 (80,91) |
18 −4.95 (−5.26,−4.64) 47 (27,67) |
8.2 −5.11 (−5.24,−4.98) 43 (31,54) |
4–9 |
| 9 |
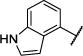
|
Et | O | 98 (79,117) | 91 (79,103) | 8.4 −5.09 (−5.26,−4.91) 46 (29,62) |
11 −5.02 (−5.23,−4.81) 50 (33,67) |
3–8 |
| 10 |
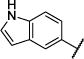
|
iPr | O | 100 (77,124) | 92 (80,105) | 134 −3.91 (−4.25,−3.57) 73 (68,79) |
65 −4.25 (−4.39,−4.11) 78 (68,88) |
4–14 |
| 11 |
|
iPr | O | 139 −3.87 (−4.10,−3.65) 78 (66,89) |
181 −3.76 (−3.98,−3.54) 72 (63,80) |
28 −4.60 (−4.77,−4.44) 66 (48,85) |
30 −4.55 (−4.68,−4.41) 69 (49,89) |
4-10 |
| 12 |
|
Et | O | 88 (57,119) | 100 (75,125) | 93 (85,101) | 80 (18,141) | 4 |
| 13 |
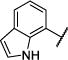
|
Et | S | 94 (60,127) | 559 −3.26 (−3.32,−3.19) 94 (89,98) |
17 −4.84 (−5.06,−4.61) 54 (36,72) |
6.1 −5.26 (−5.41,−5.10) 36 (28,45) |
4–10 |
| 14 |
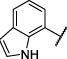
|
Et | O | 5170 −2.36 (−2.58,−2.13) 93 (88,98) |
3010 −2.69 (−2.97,2.41) 91 (87,95) |
3.7 −5.45 (−5.54,5.37) 24 (18,31) |
2.2 −5.68 (−5.74,−5.61) 15 (10,20) |
12 |
Current responses to 100 μM glutamate and 30 μM glycine co-applied with increasing concentrations of compound were recorded in Xenopus oocytes expressing GluN1 with GluN2A, GluN2B, GluN2C, or GluN2D. The data were fit by the Hill equation, unless the 99% CI of the mean percent maximal response at 10 μM included 100%.
The structure-activity relationship for the NAB series of NMDAR inhibitors was probed via the synthesis of 48 novel compounds with modifications to 5 regions: the left aromatic region (R1), the carbamothioate or carbamate substituents (R2a and R2b), the center phenyl ring (A), and the two linkers (Figure 1b, Tables 1 and 2, Supplementary Tables 1–5). Replacement of the sulfur in the carbamothioate of compound 1 with an oxygen, as in carbamate 2, improved the solubility of the compound in aqueous solution to 60 μM, but decreased both potency and selectivity for GluN2C- and GluN2D-containing receptors (Table 1). A series of analogs with modifications at R1 were evaluated for their ability to selectively inhibit GluN2C- and GluN2D-containing NMDARs (Table 1). Substituted naphthyl derivative compound 4 had minimal activity, and replacement with a phenyl ring in compound 8 decreased potency compared to compound 2. A series of analogs containing indole rings at R1 were synthesized to evaluate the effect of replacing the naphthyl moiety with a heterocycle. Linking an indole to the C-4 (9), C-5 (10) or C-6 (11 and 12) position resulted in decreased potency compared to compound 2. In contrast, linking an indole to the C-7 position of the carbamate, as in compound 14, maintained low micromolar potency at GluN2C- and GluN2D-containing receptors. Carbamate 14 had IC50 values of 3.7 μM at GluN2C and 2.2 μM at GluN2D and was >800-fold more potent at GluN2C and GluN2D compared to GluN2A and GluN2B (Figure 1c–e). Furthermore, the solubility of compound 14 was improved to 100 μM when initially dissolved in aqueous solution and 70 μM after 3 hr in aqueous solution at room temperature. A variety of substituents on the 7-amino linked indole were systematically tested while holding the carbamate constant, but these compounds resulted in loss of potency or inactivity (Supplementary Table 1).
Table 2.
Optimization of carbamate alkyl substituents.
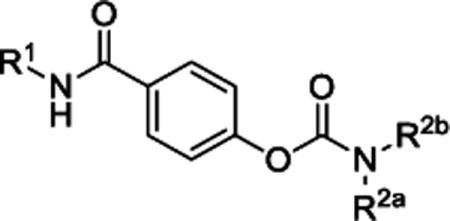
| ||||||||
|---|---|---|---|---|---|---|---|---|
| # | R1 | R2a | R2b | IC50, mean (μM) pIC50, mean (95% CI) % control at 10 μM, mean (99% CI) |
N | |||
| GluN2A | GluN2B | GluN2C | GluN2D | |||||
| 2 |
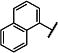
|
Et | Et | 83 (61,106) | 172 −3.82 (−4.00,−3.64) 82 (77,86) |
6.7 −5.21 (−5.33,−5.10) 35 (27,42) |
5.0 −5.38 (−5.49, −5.27) 27 (21,33) |
7-21 |
| 14 |
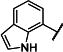
|
Et | Et | 5170 −2.36 (−2.58,−2.13) 93 (88,98) |
3010 −2.69 (−2.97,2.41) 91 (87,95) |
3.7 −5.45 (−5.54,5.37) 24 (18,31) |
2.2 −5.68 (−5.74, −5.61) 15 (10,20) |
12 |
| 15 |
|
Me | Me | 99 (65,132) | 900 −3.07 (−3.21,−2.93) 95 (91,99) |
95 (89,100) | 197 −3.73 (−3.86, −3.60) 86 (80,92) |
4-8 |
| 16 |
|
Me | Et | 98 (72,124) | 86 (68,103) | 71 −4.17 (−4.28,−4.05) 85 (82,89) |
52 −4.31 (−4.44, −4.18) 81 (78,84) |
4-8 |
| 17 |
|
iPr | iPr | 95 (89,102) | 2610 −3.75 (−3.98,−3.53) 85 (79,92) |
18 −4.99 (−5.25,−4.72) 41 (27,55) |
5.6 −5.30 (−5.44, −5.16) 31 (21,40) |
12-14 |
| 18 |
|
CH2CH=CH2 | 99 (91,106) | 893 −3.13 (−3.65,−2.80) 90 (85,96) |
68 −4.39 (−4.70,−4.09) 69 (59,80) |
49 −4.35 (−4.53,-4.18) 67 (61,73) |
4-8 | |
| 19 |
|
-CH2CH2CH2CH2- | 93 (56,131) | 90 (68,113) | 170 −3.82 (−4.24,−3.41) 88 (86,91) |
267 −3.59 (−3.79,−3.39) 88 (80,95) |
4 | |
| 20 |
|
-CH2CH2CH2CH2CH2- | 94 (80,109) | 98 (86,110) | 92 (70,115) | NF 86 (80,91) | 4-8 | |
| 21 |
|
(CH2)3CH3 | 100 (94,106) | 98 (96,105) | 95 (94,100) | NF 93 (88,99) | 4 | |
Current responses to 100 μM glutamate and 30 μM glycine co-applied with increasing concentrations of compound were recorded in Xenopus oocytes expressing GluN1 with GluN2A, GluN2B, GluN2C, or GluN2D. The data were fit by the Hill equation, unless the 99% CI of the mean percent maximal response at 10 μM included 100%. NF: the 99% CI did not include 100%, but the data could not be fit by the Hill equation. Data for compounds 2 and 14, shown in Table 1, are included here for comparison.
We subsequently evaluated the effect of modifying the alkyl substituents on the carbamate (R2a and R2b; Table 2). Shortening the carbamate of compound 2 to a N,N-dimethyl group (15) greatly reduced activity, and replacing one N-ethyl on the 7-amino linked indole (14) with N-methyl as in compound 16 reduced potency at GluN2C- and GluN2D-containing NMDARs. Extension of the carbamate to N,N-diisopropyl as in compound 17 caused a modest decrease in potency and selectivity compared to compound 2 as well as a decrease in solubility to 20 μM in aqueous solution. Analogs containing lengthier linear substituents (18–21) such as N,N-diallyl (18) and N,N-dibutyl (21) groups resulted in only minimal activity. These data suggest that the binding pocket may prefer bulky and branched hydrophobic carbamates. Modifications to the center phenyl ring and the two linkers produced no discernible improvement in efficacy or potency (Supplementary Tables 2–5). We concluded that carbamate 14, hereafter referred to as NAB-14, had the best combination of efficacy, potency, selectivity, and solubility. NAB-14 was used to evaluate the structural determinants, mechanism of action, and utility of this class of tool compounds.
Selectivity of NAB-14 for NMDAR subtypes
We first evaluated the specificity of NAB-14 for NMDARs over several other classes of ion channels that are expressed in the nervous system. The ion channels were expressed in Xenopus oocytes and current responses to maximal agonist concentrations were recorded in the absence and presence of 20 μM NAB-14. The current responses of other ionotropic glutamate receptors including α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and kainate receptors were not affected by NAB-14 (Table 3). Further, there were no significant effects of 20 μM NAB-14 on responses of glycine, GABAA, serotonin, nicotinic, and purinergic receptors expressed in Xenopus oocytes (Table 3).
Table 3.
NAB-14 has minimal off-target effects at ligand-gated ion channels.
| receptor | % control response (20 μM NAB-14) | N | p-value |
|---|---|---|---|
| GluA1 | 101 ± 2.8 | 9 | 0.997 |
| GluA2 | 101 ± 1.9 | 5 | 0.999 |
| GluK2 | 101 ± 3.2 | 10 | 0.954 |
| GluK2/GluK4 | 101 ± 1.9 | 5 | 0.999 |
| GluK2/GluK5 | 96.5 ± 4.6 | 4 | 0.999 |
| α4β2-nACh | 97.3 ± 3.9 | 3 | 0.999 |
| α7-nACh | 89.6 ± 4.2 | 6 | 0.584 |
| α1β2γδ-nACh | 100 ± 3.3 | 4 | 0.999 |
| 5-HT3A | 94.5 ± 4.5 | 7 | 0.999 |
| α1β2γ2-GABAA | 107 ± 2.8 | 6 | 0.998 |
| ρ-GABAA | 96.6 ± 0.8 | 5 | 0.999 |
| α1-Glycine | 103 ± 2.2 | 6 | 0.999 |
| P2X2 | 93.1 ± 1.6 | 5 | 0.662 |
The control current responses to agonist and the responses to agonist with 20 μM NAB-14 [% control response (mean ± s.e.m.)] for all groups were compared by one-way ANOVA [F(13,84) = 2.054, p = 0.028]. Post hoc Bonferroni tests compared the control and 20 μM NAB-14 groups for each receptor type.
Native NMDAR tetramers can contain two different GluN2 subunits (e.g. GluN2A and GluN2C), and it has been suggested that these triheteromeric receptors are prevalent in the brain.41–45 Therefore, we evaluated the activity of NAB-14 at triheteromeric receptors using a previously described recombinant system that allows for selective surface expression of NMDARs containing GluN1/GluN2A/GluN2C.46, 47 The GluN2 subunits were tagged with C-terminal peptides containing complimentary coiled-coil domains and an endoplasmic reticulum (ER) retention signal (hereafter referred to as C1 and C2). Triheteromeric NMDARs containing GluN2 subunits with complementary C1- and C2-tagged subunits traffic to the surface, whereas diheteromers containing two C1-tagged subunits or two C2-tagged subunits are retained in the ER. Concentration-response relationships for NAB-14 were acquired in Xenopus oocytes expressing GluN1 with C1- and C2-tagged GluN2 subunits. The IC50 value for NAB-14 at triheteromeric GluN1/2AC1/2CC2 receptors was 15 μM [pIC50 = −4.82 (−5.07, −4.57), N = 8–13], which was significantly greater than the IC50 value at diheteromeric GluN1/2CC1/2CC2 receptors [5.1 μM, pIC50 = −5.29 (−5.35, −5.22), N = 8–14, Figure 1f,g]. NAB-14 (100 μM) inhibited GluN1/2AC1/2CC2 to 19.9 ± 1.6% of the maximal response to glutamate and glycine; whereas, GluN1/2CC1/2CC2 receptors were inhibited to 11.7 ± 1.4% of the maximal response (N = 8, t-test, p = 0.002). To ensure that the system promoted selective trafficking of GluN1/2AC1/2CC2 triheteromeric receptors, we had two additional control groups expressing mutant GluN2 subunits that cannot bind glutamate (2AC1/2C-RKTIC2 or 2A-RKTIC1/2CC2).46 These control experiments demonstrated that 95.8% (mean of all experimental days) of the total current response was produced by triheteromeric GluN1/2AC1/2CC2 receptors (Supplementary Figure 3). While the potency of NAB-14 was significantly reduced at triheteromeric NMDARs compared to diheteromers, our data indicate that NAB-14 does effectively inhibit NMDARs containing only one GluN2C subunit. The ability to inhibit triheteromeric receptors is an important characteristic of this class of GluN2C/2D-selective NMDAR modulators because native GluN2C and GluN2D subunits are often expressed with GluN2A and GluN2B subunits, respectively.25, 48–51
In addition to GluN2 subunit diversity, there are eight splice variants of the GluN1 subunit (GluN1-1a/b, 2a/b, 3a/b, 4a/b) that can be included in NMDAR tetramers.5 These variants exhibit different expression profiles and confer distinct biophysical and pharmacological properties on the NMDAR.52–55 Neither the efficacy nor potency of NAB-14 differed significantly between the eight splice variants when co-expressed with GluN2D (Supplementary Figure 4 and Supplementary Table 6), suggesting that the diverse GluN1 splice variant expression across brain regions will not affect NAB-14 inhibition.
NAB-14 is a non-competitive antagonist
To evaluate the mechanism of action for NAB-14, we first determined whether NAB-14 acted as a competitive antagonist using recombinant NMDARs expressed in Xenopus oocytes. Concentration- response curves for glutamate and glycine were obtained at GluN1/GluN2D receptors in the absence and presence of 3 μM NAB-14. NAB-14 did not significantly affect glutamate potency [EC50: 0.61 μM, pEC50 = −6.22 (−6.29, −6.14) vs. 0.71 μM, pEC50 = −6.15 (−6.36, −5.93), N = 7-8] or glycine potency [EC50: 0.16 μM, pEC50 = −6.81 (−6.91, −6.70) vs. 0.15 μM, pEC50 = −6.82 (−6.92, −6.72), N = 7-10; Supplementary Figure 5]. Furthermore, concentration-response curves for NAB-14 were not significantly altered when the glutamate concentration was increased from 100 μM to 500 μM or the glycine concentration was increased from 30 μM to 150 μM (Supplementary Figure 5 and Supplementary Table 7). Together, these data suggest that NAB-14 does not compete with glutamate or glycine for binding, nor does the binding of NAB-14 produce an allosteric change that detectably alters glutamate or glycine potency. In addition, NAB-14 activity at GluN2D-containing receptors was unaffected by holding potential or magnesium concentration (Supplementary Figure 6), suggesting that NAB-14 does not bind in the channel pore. Finally, the potency of NAB-14 was not altered by changes in pH, suggesting that it is unaffected by proton inhibition (Supplementary Figure 6). Altogether, these data suggest that NAB-14 is a negative allosteric modulator of NMDARs that acts in a purely noncompetitive fashion.
Structural determinants of NAB-14 activity reside in the GluN2D M1 transmembrane helix
Given the finding that NAB-14 is an allosteric modulator, a series of GluN2A-GluN2D chimeras were used to identify the structural determinants of NAB-14 action. The amino-terminal domain as well as portions of the agonist binding domain and transmembrane domain of GluN2D were inserted into GluN2A, and NAB-14 inhibition was tested in oocytes expressing the chimeric receptors. Replacement of the M1 transmembrane helix of GluN2A with that of GluN2D resulted in significant inhibition by NAB-14, which was comparable to inhibition of wild type GluN2D-containing receptors (Supplementary Figure 7). To determine if specific residues in this region mediated GluN2 subunit selectivity, we identified the residues that differed between GluN2A/2B and GluN2C/2D, and then used site-directed mutagenesis to exchange the GluN2A and GluN2D residues (Figure 2a). Mutating Cys590 in GluN2D to the analogous GluN2A amino acid (Leu565) led to a significant reduction in NAB-14 activity (52.6 ± 5.0% of control compared to 14.1 ± 3.0% of control for wild type GluN2D, Figure 2b,c). Mutating Leu565 in GluN2A to cysteine was sufficient to transfer NAB-14 activity to the GluN2A subunit (34.7 ± 3.4% of control compared to 94.3 ± 4.4% of control for wild type GluN2A). These data suggest that Cys590, which is located in the middle of the transmembrane M1 helix, is a critical residue for the GluN2 subunit selectivity of NAB-14.
Figure 2.
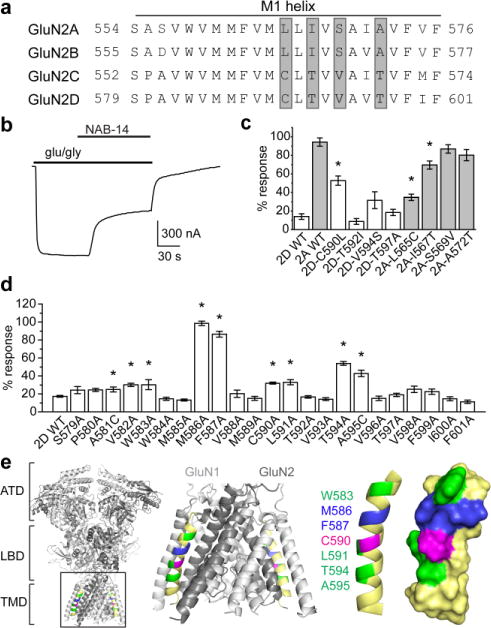
Structural determinants of NAB-14 reside in the M1 transmembrane helix. (a) A sequence alignment of the M1 transmembrane helix across rat GluN2 subunits shows four residues that differ between GluN2A/2B and GluN2C/2D (gray shading). Site-directed mutagenesis was used to switch these GluN2A and GluN2D residues and to mutate all M1 residues to alanine or cysteine. (b) A representative trace depicts the current response to 100 μM glutamate and 30 μM glycine recorded by TEVC in Xenopus oocytes in the absence and presence of 10 μM NAB-14. (c,d) The peak amplitudes of current responses were measured and expressed as the percent of the maximal response to glutamate and glycine. The data were analyzed by one-way ANOVA and post hoc (c) Bonferroni tests or (d) Dunnett’s tests. GluN2A mutants were compared to 2A WT, and GluN2D mutants were compared to 2D WT [c: F(9,42) = 38.155, p < 0.001; d: F(23,243) = 95.155, p < 0.001; *p < 0.05; see Supplementary Table 12 for mean comparison p-values]. (e) A homology model of the GluN1/GluN2D63 receptor based on crystal structures of the GluN1/GluN2B receptor64, 65 shows the GluN2 M1 helix in yellow and illustrates the residues that affect NAB-14 activity when mutated; blue residues resulted in a nearly complete loss of inhibition and green residues modestly affected inhibition at 10 μM NAB-14, and Cys590 in magenta was involved in GluN2 selectivity as shown by the effects of switching the analogous GluN2A and GluN2D residues at this position in panel c.
To further analyze the structural determinants for NAB-14 activity in the M1 region, we tested the efficacy of 10 μM NAB-14 on GluN2D mutant subunits wherein each residue of the pre-M1 region and M1 helix was mutated to alanine, or to cysteine if the endogenous GluN2D residue was alanine.56 Several mutations in the M1 region affected NAB-14 activity, with the Met586Ala mutation producing the most robust effect as it abolished activity of 10 μM NAB-14 (98.4 ± 2.5% of control; Figure 2d). The mutated residues that affected NAB-14 activity largely aligned on one side of the M1 helix (Figure 2e), and we also detected a modest reduction in NAB-14 activity by one pre-M1 mutation (Phe574Ala, Supplementary Figure 8). Interestingly, the structural determinants of NAB-14 activity in the M1 helix are shared with the GluN2C/2D-selective positive allosteric modulator CIQ;56, 57 this raises the possibility that the M1 transmembrane helix is a site for bidirectional subunit-selective modulation of the NMDAR. While these data identify structural determinants of NAB-14 activity, we cannot determine if NAB-14 binds to this region. It is also possible that the M1 helix is responsible for downstream actions during channel gating that are disrupted by NAB-14 acting elsewhere on the receptor, such as the agonist binding domain or linkers connecting the agonist binding domain to the transmembrane domain.
Kinetic mode of action of NAB-14 at GluN1/GluN2D receptors in mammalian cells
To better understand the mechanism of action for NAB-14, the association and dissociation time courses were evaluated in mammalian cells. A rapid solution exchange system was used to apply agonists and NAB-14 to HEK cells transiently expressing GluN1/GluN2D, and current responses were recorded by whole-cell voltage-clamp. The time course for onset of NAB-14 inhibition could be fit by a single exponential function and was dependent on the inhibitor concentration (Figure 3a). The association rate, kon, was determined to be (1.6 ± 0.1) × 105 M−1s−1 (Figure 3b). Recovery from NAB-14 inhibition was fit by a single exponential function and was independent of inhibitor concentration; the dissociation rate, koff, was determined to be 0.20 ± 0.01 s−1. Using the association and dissociation rates, the apparent affinity (KD) of NAB-14 for GluN1/GluN2D receptors was determined to be 1.2 μM.
Figure 3.

Association and dissociation kinetics of NAB-14. (a) Glutamate (100 μM) and glycine (30 μM) plus increasing concentrations of NAB-14 were co-applied to HEK cells transiently expressing GluN1/GluN2D using a rapid solution exchange system, as shown in a representative current response recorded by whole-cell voltage-clamp. (b) The current responses during NAB-14 association and dissociation were fit with single exponentials. The inverse of these time constants were plotted vs. NAB-14 concentration and fit by linear regression to determine the rate constants KON, the slope of the 1/τON line, and KOFF, the y-intercept of the 1/τOFF line. (c) Glycine plus increasing concentrations of NAB-14 were applied to HEK cells, then glutamate was applied for either 15 s or 5 ms in the continued presence of glycine and NAB-14. (d) Concentration-response data from 15 s (peak and steady-state) and 5 ms (peak) applications were plotted. pIC50 values from the fitted curves were compared by an F-test [F(2,72) = 31.50, p <0.001]. (e) NAB-14 concentration-response data were acquired for responses to five NMDA/glycine pressure pulses applied to HEK cells at 1 Hz. The respresentative trace shows responses for control and 3 μM NAB-14. (f) The peak amplitude of the 1st and 5th responses were measured, and pIC50 values from the fitted curves were compared by an F-test [F(1,20) = 8.86, p = 0.008].
The IC50 value for NAB-14 at GluN1/GluN2D receptors in HEK cells was 580 nM [pIC50 = −6.23 (−6.29, −6.18); Supplementary Figure 9], which was decreased (more potent) compared to the IC50 value observed in oocytes (2.2 μM; Table 1). To determine if this potency increase in mammalian cells was an attribute of this series or specific to NAB-14, we also acquired concentration-response data for compounds 22 and 34, which had IC50 values of 6.4 μM and 5.7 μM at GluN1/GluN2D receptors in oocytes, respectively (Supplementary Tables 1 and 2). Indeed, these analogs also showed increased potency in HEK cells; the IC50 values of compounds 22 and 34 were 1.7 μM [pIC50 = −5.78 (−5.87, −5.70)] and 1.2 μM [pIC50 = −5.92 (−6.01, −5.84)], respectively (Supplementary Figure 9).
At synapses, NMDARs are activated by vesicular release of glutamate that is rapidly cleared from the synaptic cleft, a process that takes only a few milliseconds. Some antagonists have shown agonist-dependent binding to NMDARs. That is, agonist must bind to the receptor before the antagonist can bind, suggesting that agonist-dependent antagonists may not effectively bind and inhibit activated NMDARs during a single synaptic event.22, 23 To test the agonist-dependence of NAB-14 binding, we used the rapid solution exchange system to first apply glycine and NAB-14 to HEK cells expressing GluN1/GluN2D, then glutamate was applied in the continuous presence of glycine and NAB-14 (Figure 3c). The application of glutamate resulted in an immediate peak response that relaxed to a steady-state level of inhibition. The IC50 of NAB-14 at the peak current response was 1.9 μM [pIC50 = −5.72 (−5.84, −5.61)], which was slightly higher than the IC50 at the steady-state current response [580 nM, pIC50 = −6.24 (−6.30, −6.17); Figure 3d). The IC50 value for NAB-14 inhibition of the response to a 5 ms glutamate pulse was 1.7 μM [pIC50 = −5.78 (−5.87, −5.69)]. Overall, the fitted concentration-response curves were significantly different [F(2,72) = 31.50, p < 0.001], suggesting that NAB-14 inhibits the steady-state response with enhanced potency compared to the peak response. The maximal levels of NAB-14 inhibition at 30 μM were significantly different between the steady-state and peak responses (2.0 ± 0.7% and 12.7 ± 2.0% of control, respectively; p = 0.005, paired t-test). A similar experiment was performed to test whether NAB-14 activity was dependent on glycine binding, wherein glycine was rapidly applied in the continuous presence of glutamate and NAB-14. Glycine binding did not significantly affect the potency or efficacy of NAB-14 at GluN1/GluN2D receptors (Supplementary Figure 10). These data suggest that NAB-14 can bind GluN1/GluN2D receptors without glycine or glutamate present, but that glutamate binding may increase the affinity of NAB-14 for the receptor.
Given these results, we next tested how NAB-14 would affect NMDARs activated by a train of brief stimuli, as may occur during a series of synaptic events. We applied a train of 5 brief (3–20 ms) pressure pulses of NMDA and glycine to HEK cells transiently expressing GluN1/GluN2D. Increasing concentrations of NAB-14 were bath-applied, and the 5 pulses were delivered at 1 Hz every 30 s (Figure 3e). The IC50 for NAB-14 inhibition was 3.5 μM [pIC50 = −5.45 (−5.59, −5.32)] for the first pulse and 2.2 μM [pIC50 = −5.66 (−5.76, −5.57)] for the fifth pulse (Figure 3f). The inhibition at 30 μM NAB-14 was 18.9 ± 3.2% of control for the first pulse and 7.2 ± 3.2% of control for the fifth pulse (p < 0.001, paired t-test). These data suggest that NAB-14 inhibits GluN2D-containing NMDARs activated by brief agonist pulses, albeit with marginally reduced potency and efficacy compared to prolonged agonist applications. Additionally, NAB-14 had no activity at GluN1/GluN2A receptors expressed in HEK cells (Supplementary Figure 11). Altogether, we interpret these findings as evidence that glutamate binding to the agonist binding domain induces a conformational change that modestly enhances NAB-14 activity, perhaps by increasing its affinity for the putative binding site in the GluN2D M1 helix. Therefore, this series of antagonists will likely inhibit single synaptic events, and may be more efficacious during high frequency synaptic activity.
In vivo exposure studies for the NAB series of GluN2C/2D-selective modulators
Plasma and brain concentrations of NAB-14 as well as compounds 1, 2, and 17 were measured following an oral dose of 20 mg/kg in mice and rats (Table 4). NAB-14 and compound 2 showed higher systemic exposure in both mice and rats compared to compounds 1 and 17. Where systemic exposure could be obtained, all four compounds were detected in the brain suggesting brain penetrance in general for this class of the compounds. Consistent with the high non-specific binding in brain tissue, the estimated free (unbound) concentrations in the brain were all below 5 nM at the tested dose except for compound 2 in rats, which had a free concentration of 76 nM. All compounds were found to be rapidly metabolized in vivo with an elimination half-life less than 1 h. This instability was also found in vitro in mice and rat plasma, where values for free fractions could not be obtained due to instability. Although the brain penetration of the most selective compound NAB-14 was limited, it is important that the main scaffold of this series of compounds allows for brain penetration, which is promising for future development of this series as a tool for in vivo experiments or drug therapies.
Table 4.
In vivo exposure of NAB compounds in mice and rats.
| # | Species | Peak plasma | t1/2 | Brain:plasma | fu, brain (%) | Peak brain free (nM) | |
|---|---|---|---|---|---|---|---|
| (ng/mL) | (nM) | ||||||
| 1 | Mouse Rat |
<LOQ 28 |
<LOQ 74 |
NA < 30 min |
NA 3.0 |
0.6 0.6 |
<LOQ 1.3 |
|
| |||||||
| 2 | Mouse Rat |
146 909 |
403 2500 |
< 30 min 1 hr |
0.58 1.7 |
1.8 1.8 |
4.2 76 |
|
| |||||||
| 14 | Mouse Rat |
470 692 |
1338 1969 |
< 30 min < 1 hr |
0.16 0.13 |
1.5 1.5 |
3.2 3.8 |
|
| |||||||
| 17 | Mouse Rat |
39 <LOQ |
100 <LOQ |
< 30 min NA |
2.1 NA |
0.5 0.5 |
1.0 <LOQ |
LOQ: limit of quantification, fu: fraction unbound.
NAB-14 inhibits GluN2C/2D-containing receptors in cultured neurons and brain slices
To test whether NAB-14 was selective for GluN2C/2D-containing NMDARs in neurons, cultured rat cortical neurons were transiently transfected with recombinant GluN2C or GluN2D, and NMDARs were activated by pressure-applied pulses of NMDA and glycine. First, we demonstrated that GluN2C and GluN2D were not endogenously expressed in these cells by testing the effect of CIQ (20 μM), a GluN2C/2D-selective positive allosteric modulator.57 CIQ increased agonist-evoked responses in GluN2C-expressing neurons to 145 ± 5.9% of control (N = 5), but had no significant effect on responses in neurons transfected with GFP alone (98 ± 1.8% of control, N = 5). NAB-14 (20 μM) inhibited agonist-evoked responses in GluN2C- and GluN2D-expressing neurons to 54 ± 4.8% and 69 ± 6.3% of control responses, respectively (Figure 4a,b). NAB-14 did not affect agonist-evoked current responses in neurons transfected with GFP alone (101 ± 0.7% of control). To test the efficacy of NAB-14 on native GluN2D-containing NMDARs, we activated NMDARs on subthalamic neurons in acute slices of the STN with pressure-applied NMDA and glycine. NAB-14 significantly inhibited agonist-evoked currents in the STN to 59 ± 2.8% of control (Figure 4c–e). Together, these results indicate that NAB-14 inhibits native GluN2D-containing receptors in brain slices with no effect on native GluN2A- or GluN2B-containing NMDARs in cultured cortical neurons.
Figure 4.
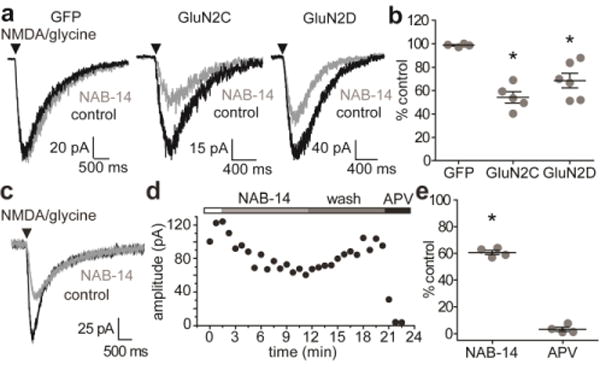
NAB-14 inhibits GluN2C/2D-containing NMDARs in cultured neurons and brain slices. (a) Cultured rat cortical neurons were dissociated and transfected with GFP, GFP and GluN2C, or GFP and GluN2D, and then cultured for 2-5 days. NMDAR current responses were evoked by pressure pulses of NMDA and glycine and recorded by whole-cell voltage-clamp in the absence and presence of NAB-14 (20 μM). (b) The peak amplitude of the current responses were measured and compared between the three transfection groups by one-way ANOVA and post hoc Dunnett’s test [F(2,14) = 16.257, p < 0.001, *GluN2C: p < 0.001, *GluN2D: p = 0.005]. (c,d) NMDAR responses were evoked with pressure pulses of NMDA and glycine at 30 s intervals in acute slices of the rat STN. 20 μM NAB-14 was bath-applied after stable control responses were obtained, then washed out, and 400 μM D,L-APV was applied to ensure the response was NMDAR-mediated. (e) The peak amplitudes of responses in NAB-14 and D,L-APV were plotted as the percent of the control response, and the amplitudes in control and NAB-14-treated conditions were compared by paired t-test (*p < 0.001).
NAB-14 modulates NMDAR EPSCs in subthalamic neurons and hippocampal interneurons
GluN2D-containing receptors contribute to excitatory postsynaptic currents (EPSCs) in the STN, and the slow deactivation rate of GluN2D-containing NMDARs controls the time course of excitatory synaptic transmission in the STN.14 Therefore, we hypothesized that NAB-14 inhibition would decrease the amplitude and accelerate the deactivation time course of STN EPSCs. NMDAR-mediated EPSCs were evoked by electrical stimulation in rat brain slices and pharmacologically isolated by application of CNQX (10 μM), an AMPA and kainate receptor antagonist (Figure 5a). NAB-14 (10 μM) reduced the peak amplitude of evoked EPSCs to 55 ± 3.0% of control, which was significantly different from the vehicle-treated slices (100 ± 2.6%, Figure 5b,c). The total charge transfer of EPSCs, which is measured by taking the integral of the evoked current response, was reduced to 45 ± 2.8% of control, whereas the charge transfer in the vehicle group was 95 ± 2.8% of the control responses (Figure 5b,c). To determine whether inhibition by NAB-14 affected EPSC deactivation time course, the decay of evoked EPSCs were fit with a two-component exponential function, and the fold-change in weighted time constants (τW) relative to control were compared between vehicle and NAB-14 groups. NAB-14 significantly reduced the τW of EPSC deactivation, suggesting that inhibition of GluN2D-containing NMDARs by NAB-14 accelerates the EPSC deactivation time course in the STN (Figure 5b,c). The individual time constants for the two exponential components used to calculate τW are reported in Supplementary Table 8. These data are consistent with our previous results14 demonstrating that DQP-1105, a GluN2C/D-selective negative allosteric modulator from another class of molecules, affects the amplitude and deactivation of STN EPSCs, and thus, strengthen the conclusion that GluN2D-containing receptors control the time course of excitatory synaptic transmission in the STN.
Figure 5.
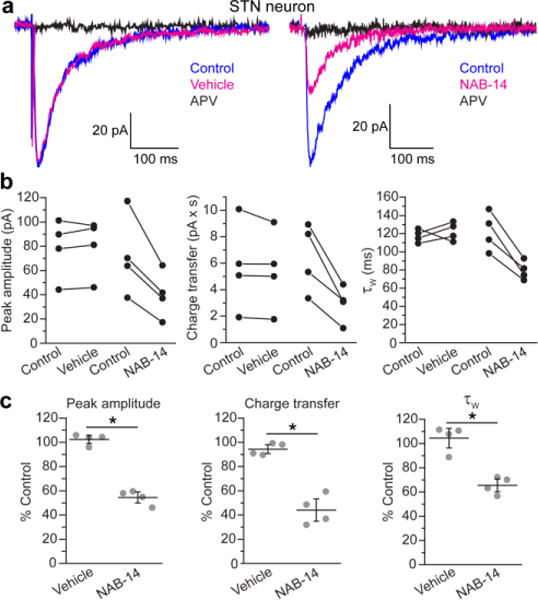
NAB-14 inhibits EPSCs in subthalamic neurons. (a) EPSCs were evoked in rat brain slices of the STN, and the NMDAR component was isolated by application of CNQX (10 μM) and bicuculline (10 μM). EPSCs recorded by whole-cell voltage-clamp (hold = −40 mV) are shown for representative neurons in aCSF (control), vehicle (0.1% DMSO), and D,L-APV (100 μM) conditions as well as control, NAB-14 (10 μM), and D,L-APV (100 μM) conditions. (b) Paired values for EPSC peak amplitude, charge transfer, and τW were plotted for each neuron under control and treated (vehicle or NAB-14) conditions. (c) Values were plotted as the percent of the control response for each cell (gray circles) with the group mean ± s.e.m. Data were compared between vehicle and NAB-14 groups by unpaired t-tests. The significance level was corrected for family-wise error: α = 0.05/3 = 0.016 (*p < 0.001).
Recently, we showed that the GluN2C/2D-selective positive allosteric modulator CIQ potentiated NMDAR-mediated ESPCs in wild-type hippocampal interneurons, but not GluN2D-deficient interneurons, suggesting that hippocampal interneurons express functional GluN2D-containing synaptic receptors and not GluN2C7, 8 Here, we tested whether negative modulation of GluN2D-containing NMDARs affected excitatory synaptic transmission at the CA3 Schaffer collateral inputs to CA1 hippocampal interneurons. We also examined CA3-CA1 EPSCs in pyramidal neurons, which do not express GluN2C or GluN2D. The Schaffer collaterals were electrically stimulated, and we measured the peak amplitude, charge transfer, and deactivation time constants of pharmacologically isolated NMDAR-EPSCs (Figure 6a). The membrane properties of interneurons are provided in Supplementary Table 9. NAB-14 decreased the peak amplitude and charge transfer of interneuron EPSCs to 59 ± 9.9% and 63 ± 9.7% of control, respectively (Figure 6b). Pyramidal neuron EPSC amplitude and charge transfer in the presence of NAB-14 were 100 ± 3.5% and 105 ± 6.2% of control EPSCs, respectively (Figure 6c). In addition, NAB-14 decreased τW for interneuron EPSCs from 150 ± 12 ms to 101 ± 14 ms, whereas τW for pyramidal neuron EPSCs was 111 ± 12 ms for control EPSCs and 118 ± 9.8 ms in the presence of NAB-14 (Figure 6b,c). The effects of NAB-14 on EPSC amplitude, charge transfer, and τW were significantly different between interneurons and pyramidal neurons (Figure 6d). The time constants for the slow and fast components of the EPSC deactivation time course are reported in Supplementary Table 10. These data advance our understanding of the role for GluN2D in the hippocampus by demonstrating that GluN2D-containing NMDARs control the time course of excitatory synaptic transmission in hippocampal interneurons. NAB-14 (10 μM) may have fully inhibited diheteromeric GluN1/2D receptors based on the potency established in our concentration-response curves in HEK cells, but NAB-14 activity at triheteromeric GluN1/2A/2D and GluN1/2B/2D has not yet been tested. NAB-14 effectively inhibited GluN1/2A/2C triheteromeric receptors, but with reduced potency, and it is possible that NAB-14 potency is similarly reduced at GluN2D-containing triheteromeric receptors. Therefore, the NAB-14-insensitive current in subthalamic neurons and hippocampal interneurons may be mediated primarily by receptors containing only GluN1, GluN2A, and GluN2B, or a substantial portion of the residual current may be mediated by triheteromeric GluN2D-containing receptors.
Figure 6.
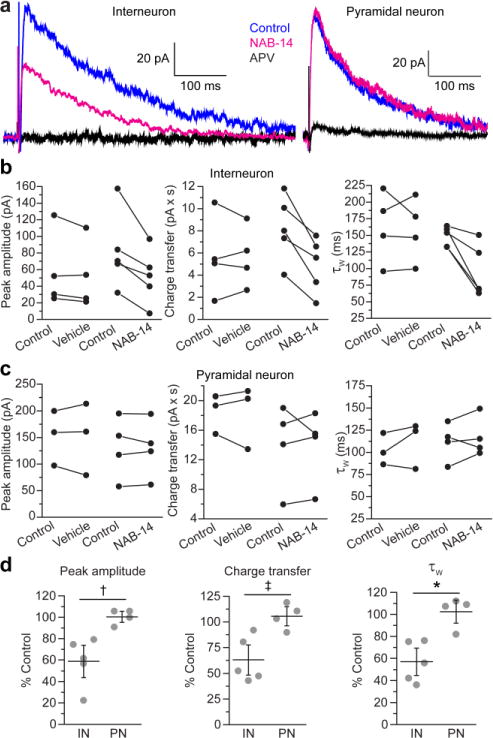
GluN2D-containing NMDARs mediate synaptic transmission at CA3-CA1 synapses in hippocampal interneurons. (a) Electrically evoked NMDAR EPSCs were recorded from CA1 interneurons or pyramidal neurons by whole-cell voltage-clamp (hold = +40 mV) in aCSF with NBQX (10 μM) and bicuculline (10 μM). EPSCs were recorded in aCSF (control), aCSF containing 0.1% DMSO (vehicle) or 10 μM NAB-14, and 100 μM D,L-APV. The peak amplitude, charge transfer, and τW were measured for (b) interneuron and (c) pyramidal neuron EPSCs, and the paired values for control and vehicle or NAB-14 conditions were plotted for each neuron. (d) The data were normalized to baseline and plotted as the percent of the control responses for each cell (gray circles) with the mean ± s.e.m. The effect of NAB-14 on each parameter was compared between interneuron (IN) and pyramidal neuron (PN) groups by unpaired t-tests. The significance level was corrected for family-wise error: α = 0.05/3 = 0.016 (†p = 0.009, ‡p = 0.011, and *p = 0.005).
Our findings identify NAB-14 as a tool for selectively modulating excitation of gabaergic hippocampal interneurons, while leaving excitatory input to glutamatergic pyramidal neurons unaffected, which could prove useful in studies aimed at understanding and modulating the balance of excitation and inhibition in the hippocampus. An interesting result from a qualitative assessment of our results with NAB-14 in the STN versus hippocampal interneurons is that NAB-14 affected STN EPSC parameters with little variation, whereas interneuron EPSCs were affected to varying degrees. Our STN findings are consistent with previous work using the GluN2C/2D-selective antagonist DQP-1105, which reliably inhibited all STN EPSCs by similar magnitudes.14 In addition, the effects of CIQ, a GluN2C/2D-selective modulator on EPSCs in hippocampal interneurons were variable in that only a subpopulation of interneuron EPSCs were potentiated by CIQ.8 These findings are consistent with the hypothesis that a majority, or perhaps all, of the STN neurons highly express GluN2D, whereas GluN2D expression levels vary across interneurons in CA1 hippocampus. In situ hybridization studies support this hypothesis as GluN2D mRNA was detected in a subpopulation of interneurons, but it remains unclear whether GluN2D expression is specific to particular subclasses of interneurons.7, 8 Why GluN2D is beneficial, or perhaps even necessary, for the function of some interneurons, but not others, remains a fundamental unanswered question. Elucidating how GluN2D modulation affects hippocampal circuit function could shed light on the role for GluN2 subunit diversity in the brain and advance strategies for modulating hippocampal function in animal models of disease.
In summary, we have developed a series of highly selective GluN2C/2D negative allosteric modulators with sub-micromolar potency in mammalian cells. Compound NAB-14 inhibits GluN2C- and GluN2D-containing NMDARs in neurons, with no activity at native GluN2A- and GluN2B-containing receptors. Importantly, NAB-14 inhibits triheteromeric GluN1/GluN2A/GluN2C receptors, which suggests that NAB-14 may inhibit all receptors containing one or two GluN2C or GluN2D subunits in the brain. Structural determinants for NAB-14 selectivity reside in the M1 transmembrane helix, which suggests that the compound either binds near to this region or that this region is necessary for the downstream actions of the compound during gating. Further work will be necessary to precisely define the binding site for these compounds. NAB-14 inhibits GluN2D-mediated synaptic transmission in the STN and CA1 interneurons, and this work strengthens the assertion that GluN2D has important roles in controlling the magnitude and time course of excitatory synaptic transmission in the brain. In addition, this series has some promising drug-like properties including absorption into plasma after oral dosing and penetration of the blood-brain-barrier, indicating that these compounds have the potential to be further developed towards therapies for neurological diseases. This novel class of compounds provides advances in both potency and selectivity over available GluN2C/2D-selective antagonists, making these modulators excellent tools for studying the function of GluN2C and GluN2D in the brain and examining how NMDAR modulation in a subset of neurons affects neural circuit function in the healthy brain as well as disease models.
METHODS
Synthetic chemistry
The detailed synthetic schemes are described in the Supporting Information. Compounds for which a synthetic route is not described were purchased from commercial vendors. The purity of purchased compounds was determined by the suppliers, via HPLC or NMR, to be greater than 90%. All reagents were purchased from commercial suppliers and used without additional purification. All dry organic solvents were obtained from a Glass Contour System. Reaction progress was monitored via thin layer chromatography (TLC) using pre-coated glass plates (silica gel 60 F254, 0.25 mm). Purification via flash column chromatography was performed using a Teledyne ISCO Combiflash Companion with pre-packaged Teledyne RediSep disposable normal phase silica columns. Proton and carbon NMR spectra were recorded on an INOVA-400 (400 MHz), VNMRS 400 (400 MHz), INOVA-600 (600 MHz), VNMRS 400 (400 MHz), Unity-600 (600 MHz), or Mercury 300 Vx (300 MHz). All spectra were referenced to the residual solvent peak, with chemical shifts reported in parts per million. The Emory University Mass Spectrometry Center obtained mass spectra on a VG 70-S Nier Johnson or JEOL instrument. The purity of all final compounds was found to be ≥ 95% unless otherwise noted.
cDNA constructs
The following rat NMDAR cDNAs were used: GluN2A (D13211), GluN2B (U11419), GluN2C (M91563), GluN2D (L31611), GluN1-1a, -1b, -2a, -2b, -3a, -3b, -4a, -4b (U08261, U08263, U08262, U08264, U08265, U08266, U08267, U08268).53 The triheteromeric C1- and C2-tagged GluN2A and GluN2C cDNAs were described in Khatri et al., 2014. The GluN2A-D chimeras and point mutations for the structural determinant studies were previously described.56, 57 All NMDAR cDNAs were in the pCI-Neo vector except GluN2C and GluN2D, which were in an unpublished vector provided by Peter Seeburg.58 For the off-target experiments, plasmids containing cDNA for GABAA (α1β2γ2 and ρ1), glycine (α1), serotonin (5-HT3A), nicotinic acetylcholine receptor (α1β1δγ, α2β4, α4β3), and purinergic (P2X2) receptors were provided by Drs. Heinemann (Salk), Weiss (Univ. of Texas, San Antonio), Papke (Univ. of Florida), and Hume (Univ. of Michigan).
Two-electrode voltage-clamp recordings in Xenopus oocytes
cRNA was synthesized by in vitro transcription according to the manufacturer’s specifications (Ambion). Xenopus laevis oocytes were injected with 10 ng of cRNA and incubated at 15°C in Barth’s solution consisting of (in mM) 88 NaCl, 1 KCl, 24 NaHCO3, 10 HEPES, 0.82 MgSO4, 0.33 Ca(NO3)2, and 0.91 CaCl2 and supplemented with 100 μg/mL gentamycin, 40 μg/mL streptomycin, and 50 μg/mL penicillin. The oocytes for triheteromeric experiments were injected with 2 ng of cRNA and incubated at 19°C. TEVC recordings were made from Xenopus oocytes 2-7 days post-injection of cRNA with recording conditions as described previously.54 Triheteromeric receptor recordings were always performed 2 days post-injection of cRNA. To prevent a gradual increase in current response over the course of the experiment, some oocytes expressing GluN1/GluN2A were injected with 50 nl of 2 mM K-BAPTA. Concentration-response curves for test compounds were generated by applying a maximally effective concentration of glutamate (100 μM) and glycine (30 μM), followed by variable concentrations of test compound up to 100 μM. Test compounds were prepared as 20 mM stock solutions in DMSO, and diluted to the final concentration in recording solution. DMSO content was 0.05-0.5% (vol/vol). 1-10 mM 2-hydroxypropyl-β-cyclodextrin was added to the recording solution for Xenopus oocyte recordings to ensure that the compounds remained in solution. No detectable effect on NMDAR response was observed for 2-hydroxypropyl-β-cyclodextrin alone.
For testing off-target effects on non-NMDARs, currents were evoked with respective agonists in the absence and presence of 20 μM NAB-14. Currents from GluA1-2 and GluK1-2 receptors were evoked with 100 μM glutamate; oocytes expressing GluK2 were incubated in concanavalin A (1 mg/ml) for 10 min prior to recording. GluK2/GluK4 and GluK2/GluK5 currents were evoked with 100 μM AMPA. The following receptors were tested at the agonist concentration listed: GABAA α1β2γ2 (20 μM GABA), GABAA ρ1 (1 μM GABA), glycine α1 (50 μM glycine), 5-HT3A (1 μM serotonin), nicotinic acetylcholine α1β1δγ (1 μ M acetylcholine), α4β2 (10 μM acetylcholine), α7 (300 μM acetylcholine), and P2X2 (9 μM ATP).
Nephelometry
Compound solubility in aqueous solution was determined using nephelometry. Briefly, the compounds were dissolved in DMSO (20 mM) and diluted to eight different concentrations between 1.75–200 μM in buffer containing (in mM) 150 NaCl, 10 HEPES, 3 KCl, and 0.5 CaCl2 and incubated for 0 min, 5 min, 30 min, 1 hr, or 3 hr at room temperature. Forward light scattering of the solutions was measured using a microplate nephelometer (NEPHELOstar, BMG Labtech). The maximum solubility was determined as the intersection point of a segmental regression fit of the data. Solubility of each compound was measured over two independent experiments with three replicates in each experiment, and the estimated maximal solubility was reported as the average of the two experiments.
Cell culture and transfection
HEK-293 cells (CRL 1573, ATCC) were cultured in DMEM with GlutaMax-I supplemented with 10% fetal bovine serum (FBS), 10 U/ml penicillin, and 10 μg/ml streptomycin. The cells were plated on coverslips coated with 0.1 mg/ml poly-D-lysine approximately 48 hr before the experiment, and transfected with rat GluN1-1a and GluN2D cDNA plasmids using the calcium phosphate method 24 hr prior to the experiment. After transfection, the cells were cultured with 200 μM D,L-amino-5-phosphonovaleric acid (D,L-APV) and 200 μM 7-chlorokyneurenic acid in the culture medium.
Primary cortical neurons were isolated from E18 Sprague Dawley rat embryos of both sexes (Charles River Labs) as described previously.59 These procedures were approved by the Emory University Institutional Animal Care and Use Committee, and were performed in accordance with state and federal Animal Welfare Acts and the policies of the Public Health Service. The dissociated neurons were transfected using the Amaxa Nucleofector with preset program O-3 and the Rat Neuron Nucleofection reagent (Lonza) as per manufacturer instructions. Briefly, cortical neurons were pelleted by centrifugation, and then resuspended in nucleofection reagent (3 × 106 cells/100 μl reagent) plus 2 μg of cDNA [0.5 μg pMax-GFP cDNA (Lonza) and 1.5 μg rat GluN2C or GluN2D cDNA]. Neurons were recovered in Roswell Park Memorial Institute (RPMI) media containing 10% horse serum for 15 min at 37°C, and then plated on acid-treated glass coverslips coated with 0.5 mg/ml poly-D-lysine in DMEM supplemented with 10% FBS. After 1 hr, the media was changed to RPMI media supplemented with GlutaMax-I and B-27 supplement (Invitrogen). Twenty-four hours later, the media was changed to normal neuronal culture media: NeuroBasal media supplemented with GlutaMax-I and B-27 supplement. Neurons were cultured for 2-5 days after transfection.
Whole-cell voltage-clamp recordings from cultured cells
HEK-293 cells and cultured neurons were recorded at a holding potential of −60 mV using an Axopatch 200B amplifier (Molecular Devices, Union City, CA) at room temperature. Recording electrodes (3–4 MΩ) were made from thin wall glass micropipettes pulled using a vertical puller (Narishige) and filled with internal solution that contained (in mM) 110 D-gluconate, 110 CsOH, 30 CsCl, 5 HEPES, 4 NaCl, 0.5 CaCl2, 2 MgCl2, 5 BAPTA, 2 Na-ATP, and 0.3 Na-GTP (pH 7.35, adjusted with CsOH). For HEK cells, the extracellular recording solution that contained (in mM) 150 NaCl, 10 HEPES, 3 KCl, 0.5 CaCl2, 0.01 EDTA, and 30 mM D-mannitol (pH 7.4 with NaOH). For cultured neurons, the extracellular recording solution contained (in mM) 150 NaCl, 10 HEPES, 3 KCl, 1 CaCl2, 20 mM D-mannitol, and 10 mM glucose (pH 7.4 with NaOH). Rapid solution exchange was achieved with a two-barrel theta-glass pipette controlled by a piezoelectric translator (Burleigh Instruments) on HEK cells lifted from the coverslip. The open tip solution exchange had a 10–90% rise times of < 1 ms. A Picospritzer II (Parker Hannifin) was used to evoke NMDAR currents by pressure applying brief pulses (4–12 psi; 3–50 ms) of NMDA (1 mM) and glycine (0.5 mM) through a borosilicate glass tube (3.5 MΩ). Three to ten stable control measurements were obtained at 30 s intervals for either single pulses or five pulses delivered at 1 Hz. NMDAR modulators were bath-applied in external solution while currents were continuously evoked by pressure application of agonists. The NMDAR competitive antagonist D,L-APV (400 μM) was bath-applied to determine if the current responses were entirely NMDAR-mediated. Series resistance was monitored during recordings, and was typically 10-20 MΩ; if the series resistance changed more than 20% during the recording, then the cell was excluded.
Brain slice preparation and whole-cell voltage-clamp recordings
Sprague Dawley rats (aged P15–P20) of both sexes were used to study acute STN slices, and C57Bl/6J mice (aged P14–P21) of both sexes were used to study acute hippocampal slices. These procedures were approved by the Emory University Institutional Animal Care and Use Committee, and were performed in accordance with state and federal Animal Welfare Acts and the policies of the Public Health Service. For STN slices, rats were deeply anesthetized with an overdose of inhaled isoflurane and transcardially perfused with ice-cold sucrose-based aCSF solution that contained (in mM) 230 sucrose, 24 NaHCO3, 10 glucose, 3 KCl, 10 MgSO4, 1.25 NaH2PO4, and 0.5 CaCl2 saturated with 95% O2/5% CO2. The brain was rapidly removed, hemisected, and glued to the stage of a vibratome (Leica VT1200S). Sagittal brain slices (250 μm) were cut in ice-cold sucrose-based aCSF solution. Slices were incubated at room temperature in NaCl-based aCSF solution that contained (in mM) 130 NaCl, 24 NaHCO3, 10 glucose, 3 KCl, 3 MgSO4, 1.25 NaH2PO4, and 1 CaCl2 that was saturated with 95% O2/5% CO2 for at least 1 h before use. STN recordings were performed at 32°C in extracellular aCSF that contained (in mM) 130 NaCl, 24 NaHCO3, 10 glucose, 3 KCl, 0.2 MgSO4, 2.5 CaCl2, and 1.25 NaH2PO4 saturated with 95% O2/5% CO2. Recording electrodes were filled with (in mM) 120 Cs-methanesulfonate, 15 CsCl, 10 tetraethylammonium chloride, 10 HEPES, 8 NaCl, 3 Mg-ATP, 1.5 MgCl2, 1 QX-314, 0.3 Na-GTP, and 0.2 EGTA, pH 7.3. For hippocampal slices, mice were deeply anesthetized with an overdose of inhaled isoflurane and decaptitated, and then the brain was removed, hemisected, and glued to the vibratome stage (Leica VT1200S). Horizontal brain slices (300 μm) were cut in ice-cold sucrose-based aCSF as above, and incubated in NaCl-based aCSF as above at 34°C for 30 min, and then room temperature for at least 30 min. Hippocampal slice recording conditions were the same as the STN recordings above, but with 1.5 mM MgSO4. All recordings were made using an Axopatch 200B amplifier (Molecular Devices), digitized at 20 kHz using Axon pClamp10 software, and filtered at 2 kHz using an eight-pole Bessel filter (−3 dB; Frequency Devices).
A Picospritzer II was used to evoke NMDAR current responses with pressure pulses (4–12 psi; 3–20 ms) of NMDA (1 mM) and glycine (0.5 mM) through a borosilicate glass tube (3.5 MΩ). During the picospritzer recordings, the external solution was supplemented with 0.5 μM tetrodotoxin, 10 μM bicuculline, and 5 μM nimodipine. NMDAR modulators or vehicle were bath-applied in aCSF for 5–10 min while currents were continuously evoked by pressure application of NMDA and glycine. Then, the slice was washed with the control aCSF solution, and the NMDAR competitive antagonist D,L-APV (400 μM) was bath-applied. EPSCs were evoked by injecting 50–500 μA of current for 0.1 ms using a bipolar tungsten-stimulating electrode (FHC) placed near the internal capsule fibers rostral to the STN or near the CA3 fibers in the CA1 stratum radiatum. Voltage-clamp recordings were performed at −40 mV for the STN and +40 mV for the hippocampus. EPSCs were evoked every 30 s, and the external aCSF was supplemented with 10 μM bicuculline methiodide and 10 μM NBQX or 10 μM CNQX. NMDAR modulators were applied as described for NMDA/glycine pressure pulses and D,L-APV (100 μM) was applied at the end of each experiment to determine if the current response was NMDAR-mediated. Series resistance was monitored throughout the experiment, and was typically 8-20 MΩ. Current responses were corrected for series resistance offline using ChanneLab (Synaptosoft).60 If the series resistance changed >20% during the experiment, then the cell was excluded.
Compound NAB-14 does not adhere to polyethylene tubing when moving at a high rate (1-5 ml/min) through large diameter tubing as is the case for all of our electrophysiological recordings. However, when NAB-14 (20 μM) was dissolved in PBS with 0.1% DMSO and run through small diameter polyethylene tubing at a very slow rate (0.1 μl/min), as would be used to administer drug into the brain parenchyma for in vivo studies, no NAB-14 was present in the solution expelled from the tubing as analyzed by HPLC. We concluded that NAB-14 can adhere to the polyethylene tubing given sufficient time, and thus, we washed all tubing and chambers between experiments with ethanol.
In vivo exposure studies
Measurements of test compound concentrations in plasma and brain following administration of compounds 1, 2, 17, and NAB-14 were performed in mice and rats. Groups of mice (male C57BL/6, 18-22 g, Charles River Lab., UK) and rats (male Wistar, 225-250 g, Charles River Lab., UK) received an oral dose of 20 mg/kg. All test compounds were solubilized in PEG400 dosed in a volume of 10 ml/kg and 5 ml/kg for mice and rats, respectively. Samples were collected at 0.5, 1, and 3 hr after administration (2-3 animals per time point) from plasma and brain after being subject to isoflurane anesthesia. Blood samples were taken in EDTA-coated glass tubes by cardiac puncture followed by decapitation and removal of the brains. Isolation of the plasma from whole blood was achieved by centrifugation (10 min at 4 °C). Brain homogenate was prepared by homogenizing the whole brain with 70% acetonitrile (1:4 v/v). Quantitative bioanalysis was performed using ultra-performance liquid chromatography (UPLC) (Waters, Milford, MA) coupled to tandem mass spectrometry (Sciex 4000; AB Sciex, Foster City, CA). The lower limit of quantification (LOQ) was 1 ng/ml in plasma and 5 ng/g in brain for all test compounds. Ethical permission for the in vivo procedures was granted by the Danish Animal Experiments Inspectorate, and all animal procedures were performed in compliance with Directive 2010/63/EU of the European Parliament and of the Council, and with Danish Law and Order regulating animal experiments.
The free fraction of the test compounds in mouse and rat plasma and brain tissue were determined in vitro at 37°C using equilibrium dialysis as described.61 The assay was performed using a test compound concentration of 1 μM incubated for 5 h. Free (unbound) brain concentrations in vivo were derived by multiplying the measured total brain concentration with the free fraction.
Data analysis
Concentration-response data was analyzed using OriginPro 9.0 or GraphPad 5.0. For inhibition concentration-response curves, the inhibitory response evoked by test compounds was given as a percentage of the initial response to glutamate and glycine alone. Data for individual cells were fit with the Hill equation:
| (1) |
where N is the Hill slope, [I] is the inhibitor concentration, and minimum is the minimum response predicted for saturating concentrations of inhibitor. Minimum was fixed to 0 unless stated otherwise. For agonist concentration-response curves, the data for individual cells were fit with:
| (2) |
where EC50 is the concentration of agonist that produces half of the Maximal Response and N is the Hill slope. Fitted EC50 and IC50 values as well as pEC50 and pIC50 from individual cells were used to calculate the mean EC50/IC50 values and mean pEC50/pIC50 values with the 95% CI, which is reported as the lower and upper confidence levels (LCL,UCL). For the graphical representation, the data were normalized to the maximum response, averaged across all cells, and fit with the Hill equation.
Current responses from whole-cell voltage-clamp recordings were analyzed using ChanneLab (Synaptosoft). The time courses for onset and recovery of inhibition with test compounds were best measured by a single exponential fit. EPSC deactivation time courses were measured with a two-component exponential fit of the current response decay from the peak and were reported as time constants for the fast and slow components, the percent amplitude of the fast component, and a weighted time constant (τW).
The deactivation time constants of EPSCs were calculated by fitting the following equation to the data:
Where τFAST is the fast deactivation time constant, τSLOW is the slow deactivation time constant, AmpFAST is the current amplitude of the fast deactivation component, and AmpSLOW is the current amplitude of the slow deactivation component. Weighted deactivation time constants (τW) were calculated using the following equation:
Statistical Analysis
Prospective power analyses were performed using GPower 3.162 to determine sample sizes necessary for detecting large effects (0.8–1.0) at a power of 0.80 for all cell-based experiments. For experiments using acute brain slices, pilot data showed a very large effect size for NAB-14 in subthalamic neurons; therefore, sample size was determined for an effect size of 2.0. The data were analyzed and graphs were prepared using OriginPro 9.0 or GraphPad 5.0. Data were tested for normality with the Kolmogorov-Smirnov test, and for homogeneity of variances with Levene’s test. Group comparisons were made using a t-test (paired or unpaired, as necessary), or one-way ANOVA and post hoc tests as indicated in the figure legends. The alpha level was set at 0.05 for all experiments and corrected for multiple pairwise comparisons and familywise error rate as stated in the figure or table legends. Data were presented as the mean ± s.e.m. EC50 and IC50 values were reported as the mean EC50/IC50 and mean pEC50/pIC50 (LCL,UCL of the 95% CI) and statistical comparisons were computed using F tests comparing the pEC50 or pIC50 values in GraphPad. For the SAR, the 99% CI of the percent maximal response at 10 μM for each compound was determined, and, if the 99% CI included 100%, then the data were not fit with the Hill equation.
Supplementary Material
Acknowledgments
The authors thank the Emory Chemical Biology Discovery Center as well as Phuong Le and Jing Zhang for excellent technical assistance.
FUNDING SOURCES
NIH: NS036654 and NS065371 (SFT), F32-NS086368 and T32-DA015040 (SAS), F31-NS071802 (TMA), The Michael J. Fox Foundation (SFT).
Footnotes
SUPPORTING INFORMATION
Supplemental Chemistry Methods: 12 Schemes, General Chemistry Procedures, and Chemical Synthesis
AUTHOR CONTRIBUTIONS
SAS, KMV, TMA, SSZ, CH, SLS, DSM, HSJ, DCL, and SFT designed the research. SAS, KMV, TMA, SSZ, JDO, CH, SJM, SLS, and CAM performed experiments and analyzed data. SAS, KMV, TMA, SSZ, SLS, CH, DSM, HSJ, and SFT wrote the paper.
CONFLICT OF INTEREST
SFT is a consultant for Boehringer-Ingleheim Pharma GmbH and Janssen Pharmaceuticals Inc., is the principle investigator on a research grant from Janssen Pharmaceuticals Inc. to Emory University School of Medicine, is a member of the SAB for Sage Therapeuetics, and is co-founder of NeurOp Inc. DCL is a member of the Board of Directors for NeurOp Inc. DCL, SFT, SLS, DSM, TMA, SSZ, and CAM are co-inventors on Emory-owned intellectual property associated with allosteric modulators of NMDAR function.
References
- 1.Parsons MP, Raymond LA. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron. 2014;82:279–293. doi: 10.1016/j.neuron.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 2.Gonzalez J, Jurado-Coronel JC, Avila MF, Sabogal A, Capani F, Barreto GE. NMDARs in neurological diseases: a potential therapeutic target. Int J Neurosci. 2014 doi: 10.3109/00207454.2014.940941. [DOI] [PubMed] [Google Scholar]
- 3.Mellone M, Gardoni F. Modulation of NMDA receptor at the synapse: promising therapeutic interventions in disorders of the nervous system. Eur J Pharmacol. 2013;719:75–83. doi: 10.1016/j.ejphar.2013.04.054. [DOI] [PubMed] [Google Scholar]
- 4.Yuan H, Low CM, Moody OA, Jenkins A, Traynelis SF. Ionotropic GABA and Glutamate Receptor Mutations and Human Neurologic Diseases. Molecular pharmacology. 2015;88:203–217. doi: 10.1124/mol.115.097998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, Hansen KB, Yuan H, Myers SJ, Dingledine R. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 7.von Engelhardt J, Bocklisch C, Tonges L, Herb A, Mishina M, Monyer H. GluN2D-containing NMDA receptors-mediate synaptic currents in hippocampal interneurons and pyramidal cells in juvenile mice. Frontiers in cellular neuroscience. 2015;9:95. doi: 10.3389/fncel.2015.00095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perszyk RE, DiRaddo JO, Strong KL, Low CM, Ogden KK, Khatri A, Vargish GA, Pelkey KA, Tricoire L, Liotta DC, Smith Y, McBain CJ, Traynelis SF. GluN2D-Containing N-methyl-d-Aspartate Receptors Mediate Synaptic Transmission in Hippocampal Interneurons and Regulate Interneuron Activity. Molecular pharmacology. 2016;90:689–702. doi: 10.1124/mol.116.105130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goebel DJ, Poosch MS. NMDA receptor subunit gene expression in the rat brain: a quantitative analysis of endogenous mRNA levels of NR1Com, NR2A, NR2B, NR2C, NR2D and NR3A. Brain Res Mol Brain Res. 1999;69:164–170. doi: 10.1016/s0169-328x(99)00100-x. [DOI] [PubMed] [Google Scholar]
- 10.Karavanova I, Vasudevan K, Cheng J, Buonanno A. Novel regional and developmental NMDA receptor expression patterns uncovered in NR2C subunit-beta-galactosidase knock-in mice. Mol Cell Neurosci. 2007;34:468–480. doi: 10.1016/j.mcn.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- 12.Standaert DG, Testa CM, Young AB, Penney JB., Jr Organization of N-methyl-D-aspartate glutamate receptor gene expression in the basal ganglia of the rat. J Comp Neurol. 1994;343:1–16. doi: 10.1002/cne.903430102. [DOI] [PubMed] [Google Scholar]
- 13.Wenzel A, Villa M, Mohler H, Benke D. Developmental and regional expression of NMDA receptor subtypes containing the NR2D subunit in rat brain. J Neurochem. 1996;66:1240–1248. doi: 10.1046/j.1471-4159.1996.66031240.x. [DOI] [PubMed] [Google Scholar]
- 14.Swanger SA, Vance KM, Pare JF, Sotty F, Fog K, Smith Y, Traynelis SF. NMDA Receptors Containing the GluN2D Subunit Control Neuronal Function in the Subthalamic Nucleus. J Neurosci. 2015;35:15971–15983. doi: 10.1523/JNEUROSCI.1702-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burzomato V, Frugier G, Perez-Otano I, Kittler JT, Attwell D. The receptor subunits generating NMDA receptor mediated currents in oligodendrocytes. J Physiol. 2010;588:3403–3414. doi: 10.1113/jphysiol.2010.195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X, Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK. NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature. 2006;439:988–992. doi: 10.1038/nature04474. [DOI] [PubMed] [Google Scholar]
- 17.Salter MG, Fern R. NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature. 2005;438:1167–1171. doi: 10.1038/nature04301. [DOI] [PubMed] [Google Scholar]
- 18.Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santangelo RM, Acker TM, Zimmerman SS, Katzman BM, Strong KL, Traynelis SF, Liotta DC. Novel NMDA receptor modulators: an update. Expert Opin Ther Pat. 2012;22:1337–1352. doi: 10.1517/13543776.2012.728587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monaghan DT, Irvine MW, Costa BM, Fang G, Jane DE. Pharmacological modulation of NMDA receptor activity and the advent of negative and positive allosteric modulators. Neurochem Int. 2012;61:581–592. doi: 10.1016/j.neuint.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosley CA, Acker TM, Hansen KB, Mullasseril P, Andersen KT, Le P, Vellano KM, Brauner-Osborne H, Liotta DC, Traynelis SF. Quinazolin-4-one derivatives: A novel class of noncompetitive NR2C/D subunit-selective N-methyl-D-aspartate receptor antagonists. J Med Chem. 2010;53:5476–5490. doi: 10.1021/jm100027p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hansen KB, Traynelis SF. Structural and mechanistic determinants of a novel site for noncompetitive inhibition of GluN2D-containing NMDA receptors. J Neurosci. 2011;31:3650–3661. doi: 10.1523/JNEUROSCI.5565-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Acker TM, Yuan H, Hansen KB, Vance KM, Ogden KK, Jensen HS, Burger PB, Mullasseril P, Snyder JP, Liotta DC, Traynelis SF. Mechanism for noncompetitive inhibition by novel GluN2C/D N-methyl-D-aspartate receptor subunit-selective modulators. Molecular pharmacology. 2011;80:782–795. doi: 10.1124/mol.111.073239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Acker TM, Khatri A, Vance KM, Slabber C, Bacsa J, Snyder JP, Traynelis SF, Liotta DC. Structure-activity relationships and pharmacophore model of a noncompetitive pyrazoline containing class of GluN2C/GluN2D selective antagonists. J Med Chem. 2013;56:6434–6456. doi: 10.1021/jm400652r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang Z, Gibb AJ. Mg2+ block properties of triheteromeric GluN1-GluN2B-GluN2D NMDA receptors on neonatal rat substantia nigra pars compacta dopaminergic neurones. J Physiol. 2014;592:2059–2078. doi: 10.1113/jphysiol.2013.267864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hildebrand ME, Pitcher GM, Harding EK, Li H, Beggs S, Salter MW. GluN2B and GluN2D NMDARs dominate synaptic responses in the adult spinal cord. Sci Rep. 2014;4:4094. doi: 10.1038/srep04094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pearlstein E, Gouty-Colomer LA, Michel FJ, Cloarec R, Hammond C. Glutamatergic synaptic currents of nigral dopaminergic neurons follow a postnatal developmental sequence. Front Cell Neurosci. 2015;9:210. doi: 10.3389/fncel.2015.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tozzi A, de Iure A, Bagetta V, Tantucci M, Durante V, Quiroga-Varela A, Costa C, Di Filippo M, Ghiglieri V, Latagliata EC, Wegrzynowicz M, Decressac M, Giampa C, Dalley JW, Xia J, Gardoni F, Mellone M, El-Agnaf OM, Ardah MT, Puglisi-Allegra S, Bjorklund A, Spillantini MG, Picconi B, Calabresi P. Alpha-Synuclein Produces Early Behavioral Alterations via Striatal Cholinergic Synaptic Dysfunction by Interacting With GluN2D N-Methyl-D-Aspartate Receptor Subunit. Biol Psychiatry. 2016;79:402–414. doi: 10.1016/j.biopsych.2015.08.013. [DOI] [PubMed] [Google Scholar]
- 29.Wu YN, Johnson SW. Memantine selectively blocks extrasynaptic NMDA receptors in rat substantia nigra dopamine neurons. Brain Res. 2015;1603:1–7. doi: 10.1016/j.brainres.2015.01.041. [DOI] [PubMed] [Google Scholar]
- 30.Brancaccio M, Patton AP, Chesham JE, Maywood ES, Hastings MH. Astrocytes Control Circadian Timekeeping in the Suprachiasmatic Nucleus via Glutamatergic Signaling. Neuron. 2017;93:1420–1435 e1425. doi: 10.1016/j.neuron.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Feng ZJ, Chergui K. Allosteric modulation of GluN2C/GluN2D-containing NMDA receptors bidirectionally modulates dopamine release: implication for Parkinson’s disease. Br J Pharmacol. 2014;171:3938–3945. doi: 10.1111/bph.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vance KM, Rogers RC, Hermann GE. NMDA receptors control vagal afferent excitability in the nucleus of the solitary tract. Brain Res. 2015;1595:84–91. doi: 10.1016/j.brainres.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, Feng ZJ, Chergui K. GluN2D-containing NMDA receptors inhibit neurotransmission in the mouse striatum through a cholinergic mechanism: implication for Parkinson’s disease. J Neurochem. 2014;129:581–590. doi: 10.1111/jnc.12658. [DOI] [PubMed] [Google Scholar]
- 34.Feng ZJ, Zhang X, Chergui K. Allosteric modulation of NMDA receptors alters neurotransmission in the striatum of a mouse model of Parkinson’s disease. Exp Neurol. 2014;255:154–160. doi: 10.1016/j.expneurol.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Suryavanshi PS, Ugale RR, Yilmazer-Hanke D, Stairs DJ, Dravid SM. GluN2C/GluN2D subunit-selective NMDA receptor potentiator CIQ reverses MK-801-induced impairment in prepulse inhibition and working memory in Y-maze test in mice. Br J Pharmacol. 2014;171:799–809. doi: 10.1111/bph.12518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogden KK, Khatri A, Traynelis SF, Heldt SA. Potentiation of GluN2C/D NMDA receptor subtypes in the amygdala facilitates the retention of fear and extinction learning in mice. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2014;39:625–637. doi: 10.1038/npp.2013.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamoto H, Kamegaya E, Sawada W, Hasegawa R, Yamamoto T, Hagino Y, Takamatsu Y, Imai K, Koga H, Mishina M, Ikeda K. Involvement of the N-methyl-D-aspartate receptor GluN2D subunit in phencyclidine-induced motor impairment, gene expression, and increased Fos immunoreactivity. Mol Brain. 2013;6:56. doi: 10.1186/1756-6606-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lozovaya N, Gataullina S, Tsintsadze T, Tsintsadze V, Pallesi-Pocachard E, Minlebaev M, Goriounova NA, Buhler E, Watrin F, Shityakov S, Becker AJ, Bordey A, Milh M, Scavarda D, Bulteau C, Dorfmuller G, Delalande O, Represa A, Cardoso C, Dulac O, Ben-Ari Y, Burnashev N. Selective suppression of excessive GluN2C expression rescues early epilepsy in a tuberous sclerosis murine model. Nat Commun. 2014;5:4563. doi: 10.1038/ncomms5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hansen KB, Mullasseril P, Dawit S, Kurtkaya NL, Yuan H, Vance KM, Orr AG, Kvist T, Ogden KK, Le P, Vellano KM, Lewis I, Kurtkaya S, Du Y, Qui M, Murphy TJ, Snyder JP, Brauner-Osborne H, Traynelis SF. Implementation of a fluorescence-based screening assay identifies histamine H3 receptor antagonists clobenpropit and iodophenpropit as subunit-selective N-methyl-D-aspartate receptor antagonists. J Pharmacol Exp Ther. 2010;333:650–662. doi: 10.1124/jpet.110.166256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman SS, Khatri A, Garnier-Amblard EC, Mullasseril P, Kurtkaya NL, Gyoneva S, Hansen KB, Traynelis SF, Liotta DC. Design, synthesis, and structure-activity relationship of a novel series of GluN2C-selective potentiators. J Med Chem. 2014;57:2334–2356. doi: 10.1021/jm401695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Al-Hallaq RA, Conrads TP, Veenstra TD, Wenthold RJ. NMDA di-heteromeric receptor populations and associated proteins in rat hippocampus. J Neurosci. 2007;27:8334–8343. doi: 10.1523/JNEUROSCI.2155-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luo J, Wang Y, Yasuda RP, Dunah AW, Wolfe BB. The majority of N-methyl-D-aspartate receptor complexes in adult rat cerebral cortex contain at least three different subunits (NR1/NR2A/NR2B) Molecular pharmacology. 1997;51:79–86. doi: 10.1124/mol.51.1.79. [DOI] [PubMed] [Google Scholar]
- 43.Rauner C, Kohr G. Triheteromeric NR1/NR2A/NR2B receptors constitute the major N-methyl-D-aspartate receptor population in adult hippocampal synapses. J Biol Chem. 2011;286:7558–7566. doi: 10.1074/jbc.M110.182600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- 45.Tovar KR, McGinley MJ, Westbrook GL. Triheteromeric NMDA receptors at hippocampal synapses. J Neurosci. 2013;33:9150–9160. doi: 10.1523/JNEUROSCI.0829-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen KB, Ogden KK, Yuan H, Traynelis SF. Distinct functional and pharmacological properties of Triheteromeric GluN1/GluN2A/GluN2B NMDA receptors. Neuron. 2014;81:1084–1096. doi: 10.1016/j.neuron.2014.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khatri A, Burger PB, Swanger SA, Hansen KB, Zimmerman S, Karakas E, Liotta DC, Furukawa H, Snyder JP, Traynelis SF. Structural determinants and mechanism of action of a GluN2C-selective NMDA receptor positive allosteric modulator. Molecular pharmacology. 2014;86:548–560. doi: 10.1124/mol.114.094516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brickley SG, Misra C, Mok MH, Mishina M, Cull-Candy SG. NR2B and NR2D subunits coassemble in cerebellar Golgi cells to form a distinct NMDA receptor subtype restricted to extrasynaptic sites. J Neurosci. 2003;23:4958–4966. doi: 10.1523/JNEUROSCI.23-12-04958.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones S, Gibb AJ. Functional NR2B- and NR2D-containing NMDA receptor channels in rat substantia nigra dopaminergic neurones. J Physiol. 2005;569:209–221. doi: 10.1113/jphysiol.2005.095554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pina-Crespo JC, Gibb AJ. Subtypes of NMDA receptors in new-born rat hippocampal granule cells. J Physiol. 2002;541:41–64. doi: 10.1113/jphysiol.2001.014001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dubois CJ, Lachamp PM, Sun L, Mishina M, Liu SJ. Presynaptic GluN2D receptors detect glutamate spillover and regulate cerebellar GABA release. J Neurophysiol. 2016;115:271–285. doi: 10.1152/jn.00687.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Traynelis SF, Hartley M, Heinemann SF. Control of proton sensitivity of the NMDA receptor by RNA splicing and polyamines. Science. 1995;268:873–876. doi: 10.1126/science.7754371. [DOI] [PubMed] [Google Scholar]
- 53.Hollmann M, Boulter J, Maron C, Beasley L, Sullivan J, Pecht G, Heinemann S. Zinc potentiates agonist-induced currents at certain splice variants of the NMDA receptor. Neuron. 1993;10:943–954. doi: 10.1016/0896-6273(93)90209-a. [DOI] [PubMed] [Google Scholar]
- 54.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rumbaugh G, Prybylowski K, Wang JF, Vicini S. Exon 5 and spermine regulate deactivation of NMDA receptor subtypes. J Neurophysiol. 2000;83:1300–1306. doi: 10.1152/jn.2000.83.3.1300. [DOI] [PubMed] [Google Scholar]
- 56.Ogden KK, Traynelis SF. Contribution of the M1 transmembrane helix and pre-M1 region to positive allosteric modulation and gating of N-methyl-D-aspartate receptors. Molecular pharmacology. 2013;83:1045–1056. doi: 10.1124/mol.113.085209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mullasseril P, Hansen KB, Vance KM, Ogden KK, Yuan H, Kurtkaya NL, Santangelo R, Orr AG, Le P, Vellano KM, Liotta DC, Traynelis SF. A subunit-selective potentiator of NR2C- and NR2D-containing NMDA receptors. Nat Commun. 2010;1:90. doi: 10.1038/ncomms1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuner T, Schoepfer R. Multiple structural elements determine subunit specificity of Mg2+ block in NMDA receptor channels. J Neurosci. 1996;16:3549–3558. doi: 10.1523/JNEUROSCI.16-11-03549.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Swanger SA, He YA, Richter JD, Bassell GJ. Dendritic GluN2A synthesis mediates activity-induced NMDA receptor insertion. J Neurosci. 2013;33:8898–8908. doi: 10.1523/JNEUROSCI.0289-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Traynelis SF. Software-based correction of single compartment series resistance errors. J Neurosci Methods. 1998;86:25–34. doi: 10.1016/s0165-0270(98)00140-x. [DOI] [PubMed] [Google Scholar]
- 61.Redrobe JP, Jorgensen M, Christoffersen CT, Montezinho LP, Bastlund JF, Carnerup M, Bundgaard C, Lerdrup L, Plath N. In vitro and in vivo characterisation of Lu AF64280, a novel, brain penetrant phosphodiesterase (PDE) 2A inhibitor: potential relevance to cognitive deficits in schizophrenia. Psychopharmacology (Berl) 2014;231:3151–3167. doi: 10.1007/s00213-014-3492-7. [DOI] [PubMed] [Google Scholar]
- 62.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–191. doi: 10.3758/bf03193146. [DOI] [PubMed] [Google Scholar]
- 63.Li D, Yuan H, Ortiz-Gonzalez XR, Marsh ED, Tian L, McCormick EM, Kosobucki GJ, Chen W, Schulien AJ, Chiavacci R, Tankovic A, Naase C, Brueckner F, von Stulpnagel-Steinbeis C, Hu C, Kusumoto H, Hedrich UB, Elsen G, Hortnagel K, Aizenman E, Lemke JR, Hakonarson H, Traynelis SF, Falk MJ. GRIN2D Recurrent De Novo Dominant Mutation Causes a Severe Epileptic Encephalopathy Treatable with NMDA Receptor Channel Blockers. Am J Hum Genet. 2016;99:802–816. doi: 10.1016/j.ajhg.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karakas E, Furukawa H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science. 2014;344:992–997. doi: 10.1126/science.1251915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee CH, Lu W, Michel JC, Goehring A, Du J, Song X, Gouaux E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014;511:191–197. doi: 10.1038/nature13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.