

Abstract
Trypanosoma brucei Tim17 (TbTim17), the single member of the Tim17/23/22 protein family, is an essential component of the translocase of the mitochondrial inner membrane (TIM). In spite of the conserved secondary structure, the primary sequence of TbTim17, particularly the N-terminal hydrophilic region, is significantly divergent. In order to understand the function of this region we expressed two N-terminal deletion mutants (Δ20 and Δ30) of TbTim17 in T. brucei. Both of these mutants of TbTim17 were targeted to mitochondria, however, they failed to complement the growth defect of TbTim17 RNAi cells. In addition, the import defect of other nuclear encoded proteins into TbTim17 knockdown mitochondria were not restored by expression of the N-terminal deletion mutants but complemented by knock-in of the full-length protein. Further analysis revealed that Δ20-TbTim17 and Δ30-TbTim17 mutants were not localized in the mitochondrial inner membrane. Analysis of the protein complexes in the wild type and mutant mitochondria by two-dimensional Blue-native/SDS-PAGE revealed that none of these mutants are assembled into the TbTim17 protein complex. However, FL-TbTim17 was integrated into mitochondrial inner membrane and assembled into TbTim17 complex. Co-immunoprecipitation analysis showed that unlike the FL-TbTim17, mutant proteins are not associated with the endogenous TbTim17 as well as its interacting partner TbTim62, a novel trypanosome specific Tim. Together, these results show that the N-terminal domain of TbTim17 plays unique and essential roles for its sorting and assembly into the TbTim17 protein complex.
Keywords: Mitochondria, Mitochondrial inner membrane, N-terminal mutants, protein import, protein assembly, Tim17, Trypanosoma brucei
1. Introduction
Trypanosoma brucei is a group of parasitic protozoa, which belong to the eukaryotic supergroup excavatae [1]. T. brucei causes a fatal disease, African trypanosomiasis, in human and livestock [1–3]. The disease is prevalent in sub-Saharan Africa as it is the habitat of the tsetse fly, the insect vector of this parasite. Trypanosomatids possess a single mitochondrion per cell with many unique characteristics [4,5]. Similar to other eukaryotes, the majority of mitochondrial proteins is encoded in the nuclear DNA and is required to be translocated after synthesis on the cytosolic ribosomes [6,7]. Elaborate machinery consisting of 4–5 multi-protein complexes, which are mostly studied in fungi, and later in mammals and plants, is responsible for import and sorting of proteins into different sub-mitochondrial locations. These complexes include the translocases of the mitochondrial outer (TOM) and inner (TIM) membranes [8,9], pre-sequence activated motor complex (PAM) [8–10], sorting and assembly complex of the mitochondrial β-barrel proteins (SAM) [11,12], and the mitochondrial inter-membrane space (IMS) assembly complex (MIA) [13,14]. In contrast, very little is known about the mitochondrial protein import apparatus in trypanosomatids. Recent studies revealed that these parasites harbor divergent translocases for mitochondrial proteins [15–17]. It has a non-canonical TOM complex (ATOM), consisting of trypanosome-specific components [18]. Instead of two TIM complexes, TIM23-17 and TIM22-54, with distinct substrate specificities in higher eukaryotes, trypanosomatids most likely have a single TIM capable to import a wide variety of proteins [19].
It has been shown in fungi to human that proteins destined to mitochondrial matrix generally possess an N-terminal targeting signal and are transported via the TIM23-17 complex. Some inner membrane (IM) proteins with additional sorting signal also take this route and are then laterally sorted into the lipid bilayer [8,9]. However, a large group of multi-spanning IM proteins, which does not have the N-terminal MTS, instead possess internal targeting signals, are translocated via the TIM22-54 complex [20,21]. The core components of the TIM23-17 complex are Tim23, Tim17, and Tim50 [22,23]. Tim23 dimer associates with Tim17 to form the twin-pore channel in the IM [24]. Tim50 acts as the receptor for the presequence-containing proteins and facilitates their translocation from the TOM to the TIM23-17 complex [25,26]. Tim23, Tim17, and the pore-forming unit of the TIM22-54 complex, Tim22, belong to the presequence and amino acid transport (PRAT) protein family, which are conserved from fungi to human [27,28]. In contrast, trypanosomatids possess a single member of this family, Tim17 [15,29]. We have shown previously that T. brucei Tim17 (TbTim17) is functionally closer to the fungal Tim17 than Tim23 [29]. TbTim17 is essential for the bloodstream and procyclic forms, the two major developmental forms of T. brucei [15,30]. TbTim17 is present in large molecular mass protein complexes and associates with some conserved proteins, such as Hsp70, and several novel proteins like TbTim62 and TbTim54 [15,16]. We have also showed that TbTim17 is directly involved in the import of the presequence-containing proteins, like the cytochrome oxidase subunit IV (COIV) [15]. TbTim17 knockdown significantly reduced the levels of the mitochondrial ADP/ATP carrier (AAC/MCP5), a highly abundant member of the mitochondrial carrier family [19,30]. Further evidence indicates that TbTim17 is involved in the import of MCPs [19]. TbTim17 is also required for the import of tRNAs into mitochondria [31]. However, it remains elusive how this single protein performs all these tasks.
In spite of significant divergence in its primary sequence, the predicted secondary structure of TbTim17 is overall conserved with other proteins of the Tim17/23/22 family. TbTim17 has 4 transmembrane (TM) domains in the center of the protein with a PRAT signature motif in the second to third TM domains [29,30]. Both the N- and C-terminal regions of TbTim17 are hydrophilic and are presumably exposed in the intermembrane space of the mitochondria. Similar to the Tim17, Tim23, and Tim22 in other systems, TbTim17 does not possess a cleavable N-terminal targeting signal and therefore, most likely depends on the internal targeting signals for its import into mitochondria. In comparison to Tim17 in fungi and mammals, the N-terminal hydrophilic region of TbTim17 is relatively long and the amino acid sequence of this region is mostly divergent. However, the function of this region of TbTim17 has not yet been tested. Here, we show that the N-terminal domain of TbTim17 is essential for proper sorting of this protein into the IM and is critical for its assembly into the TbTIM complex.
2. Materials and Methods
2.1. Strains and media
The procyclic form of Trypanosoma brucei 427 double resistant cell line (29–13) expressing the tetracycline repressor gene and T7RNA polymerase were grown in SDM-79 medium supplemented with 10% fetal bovine serum and the antibiotics (50 μg/ml hygromycin and 15μg/ml G418) [32,33]. Cell growth was assessed by inoculating the procyclic form at a cell density of 2 × 106/ml in fresh medium containing antibiotics in the presence or absence of doxycycline. Cells were counted at different growth time points with a Neubauer hemocytometer. The log of the cumulative cell number was plotted against time of incubation in culture.
2.2. Generation of the N-terminal deletion mutants of TbTim17 and other cell lines
For generation of the FL-, Δ20-, and Δ30-TbTim17 expression constructs the coding regions 1–476, 21–476, and 31–476 amino acid residues, respectively, were PCR amplified using T. brucei genomic DNA. The forward and reverse primers were designed to add restriction sites HindIII and XbaI at the 5′ends, respectively. The PCR products were cloned into the inducible expression vector pLEW 100-2X-myc [34]. All constructs were verified by sequencing. Plasmid DNA was linearized by Not I digestion and transfected into the T. brucei 29-13 parental cell line. Transfected cells, FL-TbTim17 overexpressed (OE), Δ20-Tim17OE, and Δ30-Tim17OE were selected by phleomycin (2.5 μg/ml). For generation of the constitutive expression (CE) constructs, the inserts from the pLEW 100-2X-myc constructs were excised by restriction digestion with HindIII and BamH1 and sub-cloned into the pHD1344 vector [29]. Constructs were verified by sequencing, linearized by NotI, and transfected to TbTim17 3′UTR RNAi cells. Transfected cells were selected by puromycin (1 μg/ml). TbTim17 3′UTR RNAi cells were developed previously, as described [29]. The ORF of the mitochondrial RNA binding protein2 (MRP2) was cloned into pLEW 100-2X-myc inducible expression vector and transfected to T. brucei to generate TbMRP2-2X-myc stable cell line resistant to blasticidin, as described [15]. TbTim17 KD/MRP2 cells were generated after transfection of the TbMRP2-2X-myc T. brucei with TbTim17 3′UTR RNAi construct. Transfected cells (TbTim17KD/MRP2) were selected by phleomycin and maintained in the presence of 4 antibiotics; hygromycin, G418, phleomycin, and blasticidin. These cells were further transfected with either FL-, Δ20-, or Δ30-TbTim17CE constructs and selected by puromycin to develop TbTim17KD/FL-Tim17CE/MRP2, TbTim17KD/Δ20-Tim17CE/MRP2, and TbTim17KD/Δ30-Tim17CE/MRP2 cell lines.
2.3. Subcellular fractionation
Fractionation of T. brucei procyclic cells was performed as described [15,16]. Briefly, 2 × 108 cells were re-suspended in 500 μl of SMEP buffer (250 mM sucrose, 20 mM MOPS/KOH, pH 7.4, 2 mM EDTA, 1 mM PMSF) containing 0.03% digitonin and incubated on ice for 5 min. The cell suspension was then centrifuged for 5 min at 6,800 × g at 4 °C. The resultant pellet was considered a crude mitochondrial fraction and the supernatant contained soluble cytosolic proteins.
2.4. Isolation and post-isolation treatments of mitochondria
Mitochondria were also isolated from the parasite after lysis via nitrogen cavitation in isotonic buffer as described [15,16]. The isolated mitochondria were stored at a protein concentration of ~10 mg/ml in SME buffer (250 mM sucrose, 20 mM MOPS/KOH, pH 7.4, 2 mM EDTA) containing 50% glycerol at −70°C. For limited proteinase K (PK) digestion, mitochondria in SME buffer (1 mg/ml) were treated with various concentration of PK (0–200 μg/ml) for 30 min on ice. After incubation, PK was inhibited by phenyl-methyl-sulphonyl-fluoride (PMSF, 2mM) and mitochondria were re-isolated by centrifugation at 10,000 × g at 4 °C for 10 min. For alkali extraction, isolated mitochondria (100 μg) from T. brucei were treated with 100 mM Na2CO3 (100 μl) at pH 11.5 for 30 min on ice [15]. The supernatant and pellet fractions were collected after centrifugation and analyzed by SDS-PAGE and immunoblotting.
2.5. In vivo mitochondrial protein import assay
TbTim17KD/MRP2, TbTim17KD/FL-Tim17CE/MRP2, TbTim17KD/Δ20-Tim17CE/MRP2, and TbTim17KD/Δ30-Tim17CE/MRP2 cells were induced with doxycycline for expression of MRP2-2X-myc as well as the double stranded RNAs for Tim17 3′UTR. At different time points after induction with doxycycline, cells were lysed and mitochondrial fractions were isolated. Equal amount of proteins from the mitochondrial fractions were analyzed by SDS-PAGE and immunobloting to assess the level of MRP2-2X-myc localized in the mitochondria.
2.6. SDS-PAGE and western bot analysis
Proteins from whole cells or isolated mitochondria were separated on a SDS-PAGE and immunoblotted with polyclonal antibodies for TbTim17 [30], T. brucei voltage-dependent anion channel protein (VDAC) [35], T. brucei serine/threonine protein phosphatase 5 (TbPP5) (36), Carnitine paimitoyltransferase (CPT) (30), cytochrome c1 (37), translocase of the inner mitochondrial membrane 62 (Tim62) (16), and heat shock protein 70 (mHsp70) [38] or monoclonal antibodies for trypanosome alternative oxidase (TAO) [39], and myc-epitope (abcam). Blots were developed with appropriate secondary antibodies and an enhanced chemiluminescence (ECL) kit (Pierce).
2.7. Blue-Native PAGE analysis
Mitochondrial proteins (100 μg) were solubilized in 50 μl of the ice-cold native buffer (50 mM BisTris, pH 7.2, 50 mM NaCl, 10% w/v glycerol, 1 mM PMSF, 1 μg/ml leupeptin and 1% digitonin) [15,16]. The solubilized mitochondrial proteins were clarified by centrifugation at 100,000 × g for 30 min at 4 °C. The supernatants were mixed with 2.5 μl of native PAGE G250 sample additive (Invitrogen) and were electrophoresed on a precast (4–16%) Bis-Tris polyacrylamide gel (Invitrogen), according to the manufacturer’s protocol. Protein complexes were detected by immunoblot analysis. Molecular size marker proteins apoferritin dimer (886 kDa) and apoferritin monomer (443 kDa), β-amylase (200 kDa), alcohol dehydrogenase (150 kDa), and bovine serum albumin (66 kDa) were electrophoresed on the same gel and visualized by Coomassie staining.
2.8. Co-immunoprecipitation (Co-IP)
Mitochondrial proteins (200 μg) isolated from T. brucei wild type (Tb 29-13), FL-TbTim17OE, Δ20-TbTim17OE, and Δ30-TbTim17OE were solubilized with native buffer (200 μl). Solubilized proteins were then subjected to immunoprecipitation with anti-myc conjugated Sepharose beads at 4 °C overnight. The beads were washed sequentially with the same buffer, bound proteins were eluted with 2X Laemmlie buffer and analyzed by SDS-PAGE and immunobloting.
3. Results
3.1. The N-terminal deletion mutants of TbTim17 are targeted to mitochondria
Bioinformatics analysis reveals that the primary sequence of the N-terminal domain of Tim17 in kinetoplastid parasites is more divergent than its counterparts in yeast and human (Fig. 1A). To investigate the role of this domain of TbTim17, we generated TbTim17 mutants by deleting the first 20 and 30 amino acid residues of this protein, Δ20- and Δ30-TbTim17, respectively. The mutant proteins were fused to the 2X-myc epitope tag at the C-terminus (Fig. 1B) in order to differentiate these from the endogenous TbTim17. A full-length (FL) construct of TbTim17 with a 2X-myc epitope at the C-terminus was generated similarly (Fig. 1B). We expressed these proteins in T. brucei from an inducible expression vector. Immunoblot analysis of proteins isolated from cells grown for 48 h in the presence and absence of doxycycline using the anti-myc antibody demonstrated that FL-TbTim17-2X-myc, Δ20-TbTim17-2X-myc and Δ30-TbTim17-2X-myc were expressed only after induction with doxycycline (Fig. 1C). The sizes of the FL- and Δ30-Tim17-2X-myc proteins were as expected, however, Δ20-TbTim17-2X-myc was slightly higher than the predicted size. This could be due to anomalous migration of the truncated hydrophobic proteins on SDS-PAGE. Similar banding pattern was observed when we expressed these proteins in vitro on reticulocyte extract (not shown) or expressed constitutively in T. brucei (Fig. 2). We did not observe any significant differences in cell growth due to forced expression of the FL-, Δ20-, and Δ30-TbTim17-2X-myc (Fig. 1D).
Fig. 1.
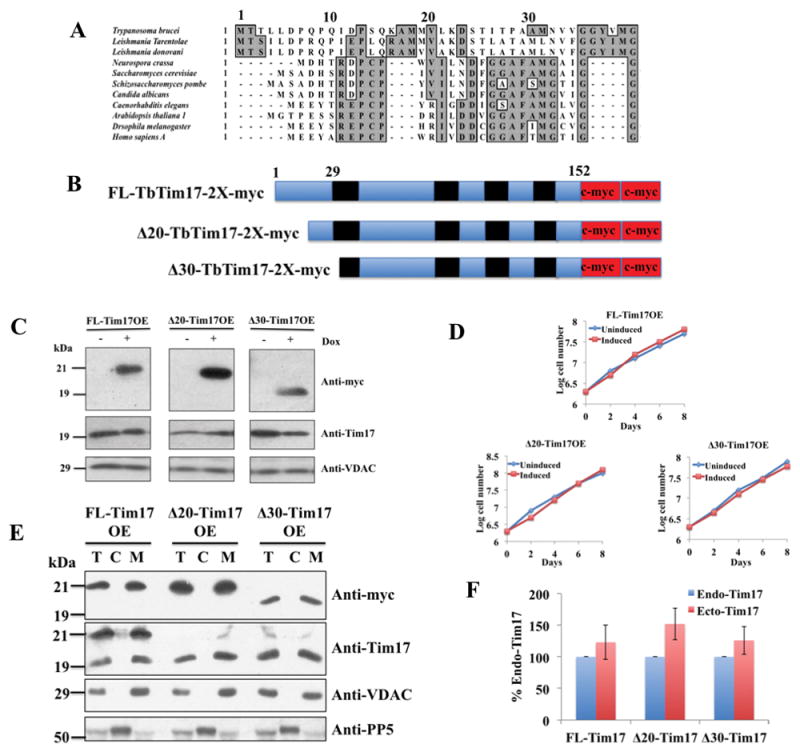
Sequence alignment of the N-terminal region of Tim17 in different organisms, schematics, expression, and sub-cellular localization of the N-terminal deletion mutants of TbTim17. (A) Primary sequence alignment of Tim17 N-terminal region in different organisms. Conserved residues are in grey boxes. (B) Schematics of the FL-, Δ20- and Δ30-TbTim17-2X-myc proteins. The predicted transmembrane domains are in black and myc epitope tags are in red. Numbers indicate the position of the amino acid residues. (C) Inducible expression of FL-, Δ20-, and Δ30-Tim17-2X-myc in T. brucei. Stable transfectants for FL-Tim17OE (over expressed), Δ20-Tim17OE, and Δ30-Tim17OE were grown in the absence (−) and presence (+) of doxycycline (Dox) for 48 hours. Equal numbers of cells were harvested and total cellular proteins were analyzed by SDS-PAGE and immunoblot analysis using anti-myc, -TbTim17, and –VDAC antibodies. 5 × 106 cells were loaded per lane (D) Growth kinetics of FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE in the absence (uninduced) and presence (induced) of doxycycline. Cells were counted up to 8 days and the log of cumulative cell numbers was plotted against time. (E) The FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE cells were grown in the presence of doxycycline for 48 h. Cells were harvested and sub-cellular fractionation were performed. Total (lane T), Cytosolic (lane C), and mitochondrial (lane M) fractions were analyzed by SDS-PAGE and immunoblotting using anti-myc, -Tim17, -VDAC, and -PP5 antibodies. (F) The intensity of protein bands for the endogenous (Endo) Tim17 (recognized by anti-Tim17 antibody) and the ectopically (Ecto) expressed FL and N-terminal deletion mutants (recognized by anti-myc antibody) in the mitochondrial fractions were quantitated by densitometry, normalized with the levels of VDAC in the corresponding samples. Levels of ectopically expressed proteins are presented as percent of the endogenous TbTim17 levels in the corresponding cell lines. Standard errors were calculated from 3 independent experiments.
Fig. 2.

Constitutive expression (CE) of the FL and N-terminal deletion mutants of TbTim17 in Tim17 knockdown (KD) T. brucei. (A) The schematic diagram of the pHD1344 constitutive expression vector for FL and N-terminal deletion mutants and the tetracycline-inducible p2T7-177/Tim17(3′UTR) RNAi construct. SAS, splice acceptor site; Puro R and Phleo R, puromycin and phleomycin resistance genes, respectively; Tet R, tetracyclin repressor. Arrows indicate the direction of transcription. (B) Growth kinetics of Tim17KD, Tim17KD/Δ20-Tim17CE, Tim17KD/Δ30-Tim17CE, and Tim17KD/FL-Tim17CE in the absence (uninduced) and presence (induced) of doxycycline. Cell numbers were counted up to 10 days and the log of cumulative cell numbers was plotted against time. Results represent three independent experiments. (C) Immunoblot analysis of total cellular proteins harvested at different time points (0–4 days) after induction with doxycycline. TbTim17KD cells, and the double-transfected T. brucei cells Tim17KD/Δ20-Tim17CE, Tim17KD/Δ30-Tim17CE, and Tim17KD/FL-Tim17CE were grown in the presence of doxycycline for induction of Tim17 RNAi targeted to the 3′UTR of the TbTim17 transcript. Proteins were analyzed by SDS-PAGE and immunoblot analysis using antibodies for T. brucei Tim17, VDAC and also for the myc epitope tag. Equal numbers of cell (5 × 106/ml) were loaded per lane. (D) The intensity of protein bands for the endogenous (Endo) Tim17 (recognized by anti-Tim17 antibody) and the ectopically (Ecto) expressed FL and N-terminal deletion mutants (recognized by anti-myc antibody) were quantitated by densitometry, normalized with the levels of VDAC in the corresponding samples and were presented as percent of the endogenous TbTim17 levels at 0 time point in the corresponding cell line. Standard errors were calculated from 3 independent experiments. (E) The Tim17KD/FL-Tim17CE, Tim17KD/Δ20-Tim17CE, and Tim17KD/Δ30-Tim17CE cells were grown in the presence of doxycycline for 96 h. Cells were harvested and sub-cellular fractionation were performed. Total (lane T), Cytosolic (lane C), and mitochondrial (lane M) fractions were analyzed by SDS-PAGE and immunoblotting using anti-myc, -Tim17, -VDAC, and -PP5 antibodies.
To verify the localization of FL-TbTim17-2X-myc, Δ20-TbTim17-2X-myc, and Δ30-TbTim17-2X-myc in T. brucei, sub-cellular fractionation was performed. Analysis of proteins from the total, cytosol and mitochondrial fractions by SDS-PAGE and western blotting using antibodies for myc epitope revealed that FL-, Δ20-, and Δ30-TbTim17-2X-myc were localized in the mitochondria of T. brucei (Fig. 1E). Myc-tagged proteins were detected by the anti-myc antibody, as expected. Anti-TbTim17 antibody recognized both the ectopically expressed (21 kDa) and the endogenous TbTim17 (19 kDa) in FL-TbTim17-OE cells similar to our previous report [29]. The ectopically expressed Δ20- and Δ30-TbTim17-2X-myc were not detected by anti-TbTim17 antibody as this antibody was developed against the first 18 amino acid residues of this protein. VDAC and PP5 were used as the mitochondrial and cytosolic markers, respectively. Quantitation of the levels of the endogenous and ectopically expressed TbTim17 in different cell lines revealed that the FL-, Δ20-, and Δ30-TbTim17-2X-myc were overexpressed about 123%, 152%, and 126% of the endogenous TbTim17 levels in the respective cell line (Fig. 1F).
3.2. The N-terminal deletion mutants of TbTim17 couldn’t complement the growth phenotype caused by TbTim17 KD
TbTim17 is essential for T. brucei cell growth. To investigate the effect of truncation of TbTim17 N-terminus on its function, we generated a TbTim17 RNAi cell line by targeting the 3′UTR of the TbTim17 transcript as described [29]. Then we further transfected this cell with either FL-TbTim17-2X-myc or the N-terminal deletion mutant constructs for expression of these proteins constitutively (Fig. 2A). The double-transfected cell lines Tim17KD/Δ20-Tim17CE and Tim17KD/Δ30-Tim17CE were grown in the presence and absence of doxycycline for induction of RNAi. Tim17KD/FL-TbTim17CE, and TbTim17KD cells were also grown in parallel as controls. Upon induction of TbTim17 RNAi, the cell growth of the single-transfected TbTim17KD T. brucei declined over the time, as shown previously (Fig. 2B). Both Tim17KD/Δ20-Tim17CE and Tim17KD/Δ30-Tim17CE cell lines showed growth phenotype similar to the TbTim17KD (Fig. 2B), showing that none of these mutants were able to rescue the inhibition of cell growth caused by TbTim17 RNAi. However, as shown previously [29], expression of the FL-TbTim17-2X-myc completely restored the inhibition of cell growth due to TbTim17KD (Fig. 2B). These results indicate that the N-terminal deletion mutants are non-functional thus unable to complement the loss of function of the endogenous TbTim17.
To verify that the mutant proteins are expressed in TbTim17 RNAi cells, immunoblot analysis of the total cellular proteins harvested at different time periods during cell growth was performed using anti-myc and anti-TbTim17 antibodies (Fig. 2C). Both of the Δ20- and Δ30-TbTim17-2X-myc mutants were expressed constitutively (0, 2, and 4 days) similar to the FL-TbTim17-2X-myc, as detected by the anti-myc antibodies. This antibody did not recognize any proteins from TbTim17KD cells, as expected. Anti-myc antibodies recognized a doublet for FL-TbTim17-2X-myc, which could be due to spontaneous degradation of the expressed protein. A similar banding pattern was observed previously when we expressed FL-TbTim17-2X-myc either in wild type or in TbTim62KD cells [16]. We didn’t see a doublet in Fig. 1C and E, which could be due to incomplete separation or oversaturation of the FL-TbTim17-2X-myc band. Probing the blots with anti-TbTim17 antibody revealed that the levels of the endogenous TbTim17 gradually decreased in all cells after induction of RNAi. VDAC was used as a loading control. We noticed that the level of reduction of the endogenous TbTim17 varies in different cell lines. For that reason we quantitate the levels of endogenous and constitutively expressed TbTim17 and its mutants for each cell line (Fig. 2D). Endogenous TbTim17 levels were reduced ~72%, ~46%, and ~42% in Tim17KD/FL-Tim17CE, Tim17KD/Δ20-Tim17CE, and Tim17KD/Δ30-Tim17CE, respectively within day 4 post-induction. In Tim17KD/FL-Tim17CE cells, 126% over expression of the FL protein completely restored the growth phenotype. Whereas, 85% to 99% over expression of the N-terminal mutants failed to complement the loss (42 to 46%) of the endogenous protein. Therefore we confirmed that the N-terminal mutants are non-functional.
In order to verify that the FL-, Δ20-, and Δ30-TbTim17-2X-myc were targeted to mitochondria in TbTim17KD cells, total, cytosolic and mitochondrial fractions isolated after 4 days of induction of RNAi were analyzed by western blot using anti-myc and anti-TbTim17 antibodies (Fig. 2E). Results showed that FL- and the N-terminal mutants are localized in mitochondria similar to the residual endogenous TbTim17. The doublet bands for FL-TbTim17 were also enriched in the mitochondrial fraction, showing that these bands are not generated due to improper targeting. PP5 was used as the cytosolic marker.
3.3. The N-terminal deletion mutants of TbTim17 failed to restore the defect of mitochondrial protein import caused by TbTim17 KD
Next, we used an in vivo assay system to compare import efficiency of MRP2 into mitochondria of Tim17KD, Tim17KD/Δ20-TbTim17CE, Tim17KD/Δ30-TbTim17CE, and Tim17KD/FL-TbTim17CE cells. For these assays MRP2-2X-myc was expressed in an inducible manner in the above cell lines (Fig. 3A) and mitochondrial proteins were analyzed at different time points after induction. In TbTim17KD cells, MRP2-2X-myc was localized in mitochondria at earlier time points after induction with doxycycline, however the levels dropped sharply as the endogenous TbTim17 levels gradually reduced by RNAi, simultaneously (Fig. 3B), showing that the import of MRP2-2X-myc into mitochondria depends on TbTim17. We also observed a sharp decrease of MRP2-2X-myc in Tim17KD/Δ20-TbTim17CE and Tim17KD/Δ30-TbTim17CE mitochondria after day 2 post-induction (Fig. 3B and C). However, MRP2-2X-myc accumulated into mitochondria and its levels were consistent from day 2 to 6 post-induction of Tim17KD/FL-TbTim17CE cells (Fig. 3B and C). These results showed that the protein import defect in TbTim17 KD mitochondria couldn’t be complemented by the N-terminal truncation mutants but was fully restored by the FL-TbTim17. Probing the blots with anti-TbTim17 antibody revealed that the endogenous TbTim17 levels decreased in mitochondria harvested from all cell types after induction of TbTim17 RNAi. Anti-myc antibody also recognized the Δ20-TbTim17-2X-myc, Δ30-TbTim17-2X-myc, and FL-TbTim17-2X-myc in mitochondria at all time points tested although the expression levels of these mutants varied with post-induction days (Fig. 3B). VDAC was used as the loading control. Similar to our previous experiment we quantitate the endogenous and ectopically expressed TbTim17 and its mutants in respective cell lines at different time points (Fig. 3D). Results showed that the endogenous TbTim17 levels were decreased after 4 days of induction of RNAi and reached below 50% at Day 6 in all cell lines. The levels of ectopically expressed FL-, Δ20-, and Δ30-Tim17 were within the range of 100–142%, 104–118%, and 125–166%, respectively. Therefore, in each case ectopically expressed protein levels were more than 100% of the endogenous day 1 TbTim17 levels. Together, these results showed that constitutive expression of FL-Tim17 does but the N-terminal mutants do not support MRP2 import into TbTim17 KD mitochondria. Thus, N-terminal region of TbTim17 is critical for functioning of this protein.
Fig. 3.

In vivo import assays for MRP2 into mitochondria in Tim17KD and Tim17KD complemented by either FL-Tim17CE or Δ20-Tim17CE or Δ30-Tim17CE T. brucei. (A) The schematic diagram of the pHD1344 constitutive expression vector for FL and N-terminal deletion mutants, the tetracycline-inducible p2T7-177/Tim17(3′UTR) RNAi construct, and MRP2-2X-myc/pLew-Bsd inducible expression construct. SAS, splice acceptor site; Puro R, Phleo R, and BsdS R, puromycin, phleomycin, and blasticidin resistance genes, respectively; Tet R, tetracyclin repressor. Arrows indicate the direction of transcription. Chromosomal integration sites of each of these constructs are shown. (B) The cell lines Tim17KD/MRP2, Tim17KD/Δ20-Tim17CE/MRP2, Tim17KD/Δ30-Tim17CE/MRP2, and Tim17KD/FL-Tim17CE/MRP2, were induced with doxycycline. Cells were harvested at different time points (0–6 days) after induction. Mitochondrial fractions were analyzed by SDS-PAGE and immunoblot analysis using anti-myc, -VDAC, and -Tim17 antibodies. (C) Intensities of the MRP2-2X-myc protein bands were quantitated, normalized with the intensities for the corresponding VDAC protein bands and plotted against time. Results represent 3 independent experiments. (D) The intensity of protein bands for the endogenous (Endo) Tim17 (recognized by anti-Tim17 antibody) and the ectopically (Ecto) expressed FL and N-terminal deletion mutants (recognized by anti-myc antibody) were quantitated by densitometry, normalized with the levels of VDAC in the corresponding samples and were presented as percent of the endogenous TbTim17 levels at day 1 post induction in the corresponding cell lines. Standard errors were calculated from 3 independent experiments.
3.4. The N-terminal deletion mutants of TbTim17 are not inserted into mitochondrial inner membrane in T. brucei
Next we investigated if the N-terminal deletion mutants of TbTim17 were properly inserted into the IM and assembled into the functional complex because failure to do so could make these mutants non-functional. For this purpose we first assessed the integration of the FL-, Δ20-, and Δ30-TbTim17-2X-myc into the mitochondrial membrane of T. brucei. Mitochondria from these cells grown in the presence of doxycycline for 48 h were isolated and proteins were extracted with Na2CO3. The soluble and pellet fractions were separated by centrifugation and proteins from each fraction were analyzed by SDS-PAGE and immunoblotting (Fig. 4A). Immunoblot analysis with the anti-myc antibody revealed that FL-, and Δ30-TbTim17-2X-myc were present in the pellet fraction, indicating these proteins are membrane integrated thus resistant to alkali. However, Δ20-TbTim17-2X-myc was alkali-sensitive and completely extracted into the supernatant fraction, showing that this mutant protein is not membrane integrated into T. brucei mitochondria. Antibodies against TAO and mHsp70 were used as controls. TAO, an integral membrane protein [39] was found in the pellet, whereas mHSP70 (38), a matrix protein was present in the soluble fraction, as expected.
Fig. 4.
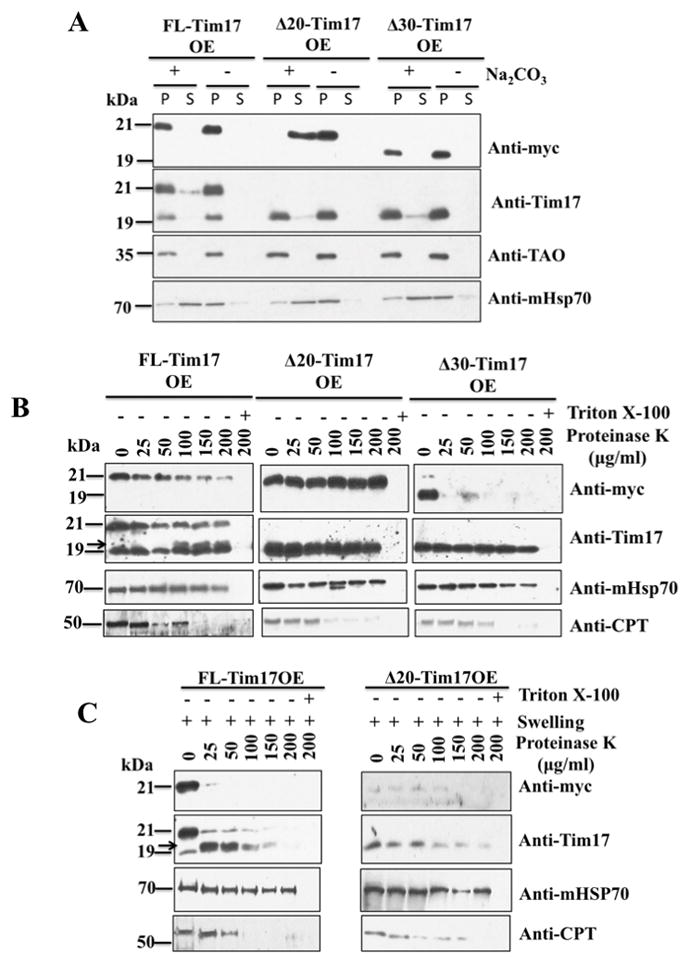
Sub-mitochondrial localization of the N-terminal deletion mutants of TbTim17. (A) Alkali extraction of the mitochondria isolated from FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE after growing in the presence of doxycycline for 48 h. Proteins from equal volume of the supernatant (lane S) and pelleted (lane P) fractions were analyzed by immunoblotting using anti-myc, -Tim17, -TAO, and –mHSP70 antibodies. (B) Mitochondria isolated from the FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE T. brucei grown for 48 h in the presence of doxycycline were treated with various concentration (0–200 μg/ml) of proteinase K as described in the materials and Methods. Proteins were analyzed by immunoblotting using anti-myc, -Tim17, mHSP70, and –CPT antibodies. (C) Mitochondria from the FL-Tim17OE and Δ20-Tim17OE T. brucei were swelled in hypotonic buffer to rupture the mitochondrial outer membrane. Mitoplasts were recovered by centrifugation and treated with various concentrations (0–200 μg/ml) of proteinase K as described above. Proteins were analyzed by immunoblot analysis using anti-myc, -Tim17, -mHSP70, and -CPT antibodies. Arrow shows a ~19 kDa cleaved product of FL-Tim17-2X-myc.
In order to determine the sub-mitochondrial localization of the mutant proteins, isolated mitochondria from the respective cells were treated with increasing concentrations (0–200 μg/ml) of proteinase K. We found that a majority of FL-TbTim17-2X-myc was protected from protease digestion (Fig. 4B). However, in comparison to the endogenous TbTim17, FL-TbTim17-2X-myc was slightly more sensitive to Proteinase K, this could be due to over-expression of the FL-TbTim17-2X-myc. Interestingly, Δ20-TbTim17-2X-myc was completely protected from protease digestion (Fig. 4C), revealing that although this mutant is not membrane integrated, at least it crossed the mitochondrial outer membrane (OM). Disruption of the mitochondrial membrane by Triton X-100 along with proteinase K treatment completely digests both the FL- and Δ20-TbTim17-2X-myc, showing that these proteins are not protease resistant. In contrast to Δ20-TbTim17-2X-myc, Δ30-TbTim17-2X-myc was mostly digested even at 25 μg/ml proteinase K (Fig. 4B), indicating that this mutant protein was improperly associated with the OM. The endogenous TbTim17, an IM protein, and mHsp70, a matrix protein were protected from protease digestion as expected. However, an outer membrane protein, carnitine palmitoyl transferase (CPT) [30] was digested by proteinase K concentrations above 50–100 μg/ml.
We further examine the location of the Δ20-TbTim17-2X-myc by proteinase K digestion of mitochondria after swelling. The Δ20-TbTim17-2X-myc, was mostly undetected after swelling of the mitochondria (Fig. 4C), only a residual amounts were present in samples treated with proteinase K (0–100 μg/ml), showing that this mutant protein is primarily accumulated in the IMS region and could be peripherally associated with the IM or IM proteins, thus after rupture of the mitochondrial outer membrane most of this mutant protein is lost. The FL-TbTim17-2X-myc was detected by both anti-myc and anti-TbTim17 antibody after swelling of mitochondria, but mostly digested after proteinase K digestion (Fig. 4C). A cleaved product of ~19 kDa was generated after digestion with 25 or 50 μg/ml proteinase K, indicating that the C-terminal of TbTim17 is exposed in the IMS. The endogenous TbTim17 was gradually degraded in both samples with increasing concentrations of proteinase K. The matrix-localized mHsp70 was protected and CPT mostly degraded, as expected. All together our results indicate that the N-terminal region of TbTim17 is critical for proper sub-mitochondrial sorting of this protein.
To investigate if these N-terminal mutants of TbTim17 are also missorted when constitutively expressed in TbTim17KD cells, proteinase K digestion assays were performed using isolated mitochondria from Tim17KD/FL-Tim17CE, Tim17KD/Δ20-Tim17CE, and Tim17KD/Δ30-Tim17CE T. brucei after growing for 4 days in the presence of doxycycline (Fig. S1). We found that Fl-, Δ20-, and Δ30-Tim17-2X-myc primarily were protected from protease digestion similar to the endogenous TbTim17. However, when mitochondria were swelled (mitoplast) before digestion only FL-Tim17-2X-myc was partially protected. We noticed some differences in the result from OE and CE mitochondria during proteinase K digestion assays particularly Δ30-mutant was protected in CE but not in OE mitochondria, which could be due to differences in the expression levels of the mutant or some technical variability of this sensitive assay. We also couldn’t exclude the possibilities that the levels of endogenous TbTim17 and a constitutive versus inducible expression of the ectopic proteins had some effect on sub-mitochondrial localization of TbTim17 mutants. However, in both cases (OE and CE) mutant proteins appear more sensitive to proteinase K after swelling of mitochondria. Furthermore, alkali-extraction of Tim17KD/FL-Tim17CE, Tim17KD/Δ20-Tim17CE, and Tim17KD/Δ30-Tim17CE mitochondria further revealed that Δ20-Tim17-2X-myc was not integrated into mitochondrial membrane but FL- and Δ30-Tim17-2X-myc behave like membrane integral proteins (Fig. S2), as we found in OE mitochondria.
Together, our results show that similar in the parental cell, the N-terminal mutants were not targeted to proper sub-mitochondrial location in Tim17 KD cells. However, FL-Tim17 was sorted properly and thus remains functional.
3.5. The N-terminal deletion mutants of TbTim17 are not assembled into the TIM complex
Analysis of mitochondria samples isolated from FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE cells on BN-PAGE followed by immunoblot using anti-myc antibody revealed that FL-TbTim17-2X-myc was able to assemble into the TbTim17 complexes, as shown previously [16,29] (Fig. 5A). However, Δ20-TbTim17-2X-myc, and Δ30-TbTim17-2X-myc were not properly assembled into the TbTim17 protein complex (Fig. 5A). Moreover, Δ30-TbTim17-2X-myc was detected in two protein complexes at ~100 and ~180 kDa, which could be the aggregated forms of the ectopically expressed Δ30-TbTim17-2X-myc. A single band of ~800 kDa was detected in Δ20-Tim17OE sample, however, that could be a non-specific interaction. Anti-TbTim17 antibody recognized the endogenous TbTim17 complex within the region 300 to >800 kDa in all samples. Similarly, Cytochrome c1 antibody recognized the Cytochrome b-c1 reductase complex at comparable levels in each sample. Together, these results revealed that the first 30 amino acids of the N-terminal domain are critical for the assembly of TbTim17 into the TbTIM complex.
Fig. 5.
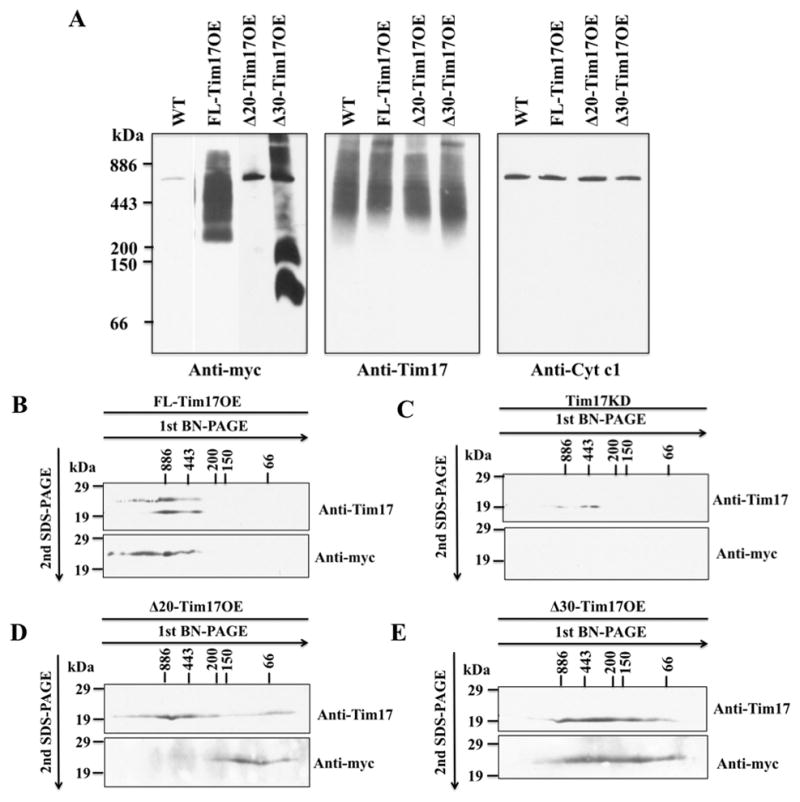
Analysis of the TbTim17 protein complex in mitochondria. (A) BN-PAGE analysis of the mitochondrial membrane protein complexes. Mitochondrial proteins isolated from wild type (WT), FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE T. brucei grown for 48 h in the presemce of doxycycline were solubilized with digitonin (1%). The solubilized supernatant were clarified by centrifugation at 100,000 × g and analyzed by BN-PAGE. Protein complexes were detected by immunoblot analysis using anti-myc, -Tim17, -cytochrome c1 (Cyt c1) antibodies. Molecular size markers are indicated. (B-E) Gel strips representing the individual lanes for FL-Tim17OE (B), Tim17KD (C), Δ20-Tim17OE (D), and Δ30-Tim17OE (E) samples were excised from the first dimension BN-PAGE and subjected to 12% Tricine-SDS-PAGE. Proteins were transferred to a nitrocellulose membrane, and blots were probed with anti-myc and anti-Tim17 antibodies.
We also performed 2D-BN-SDS-PAGE to further analyze the TbTim17 complexes in these cells (Fig. 5B–E). FL-Tim17-2X-myc protein complex as detected by anti-myc antibody was found at a similar size range of the endogenous TbTim17 (Fig. 5B). Anti-TbTim17 recognized both the endogenous and the ectopically expressed TbTim17, as expected. TbTim17 complex was hardly detected in Tim17 KD cells by anti-TbTim17 antibody and anti-myc antibody did not recognize any proteins in this sample (Fig. 5C). In contrast to FL-Tim17-2X-myc, Δ20-TbTim17-2X-myc was detected by anti-myc antibody only at lower molecular mass (~66 to ~150 kDa). Anti-TbTim17 antibody recognized the endogenous TbTim17 complex primarily in the ~300 to ~800 kDa region in Δ20-Tim17OE sample (Fig. 5D). These results indicated that Δ20-TbTim17-2X-myc was not assembled into the matured complex as shown in 1D-gel, but only accumulated in the lower molecular mass complexes or in the aggregated form of this protein. Interestingly, these low-mass complexes were not detected in the 1D-gel, which could be due to greater sensitivity of the anti-myc antibody for denatured than native protein. A small portion of the endogenous TbTim17 was also detected in the range below ~66 kDa, indicating that expression of Δ20-TbTim17-2X-myc may reduce the stability of the matured complex. We found Δ30-TbTim17-2X-myc in a broad region (~800 to <66 kDa) as detected by anti-myc antibody (Fig. 6E), showing that this mutant protein was not properly assembled into TbTim17 complex thus separated from the native complex during solubilization. Interestingly, we found that the endogenous TbTim17 complex was also destabilized in Δ30-TbTim17OE mitochondria as recognized by anti-TbTim17 antibody (Fig. 5E). Instead of a larger complex of ~300 to >800 kDa in WT mitochondria, TbTim17 is present in a broader range protein complexes (~800 to <66 kDa) on 2D-BN-SDS-PAGE of Δ30-TbTim17OE mitochondria sample. Although the reason for destabilization of TbTim17 complex is not clear at this moment we speculate that accumulation of the mutant proteins hampers association of the endogenous Tim17 with other proteins in the IM, IMS, or OM and that causes instability of the TbTim17 complex. However, further experiments are needed to elucidate the actual mechanism. All together these results revealed that the N-terminal hydrophilic region of TbTim17 is necessary for assembly of TbTim17 into the TbTIM complex.
Fig. 6.
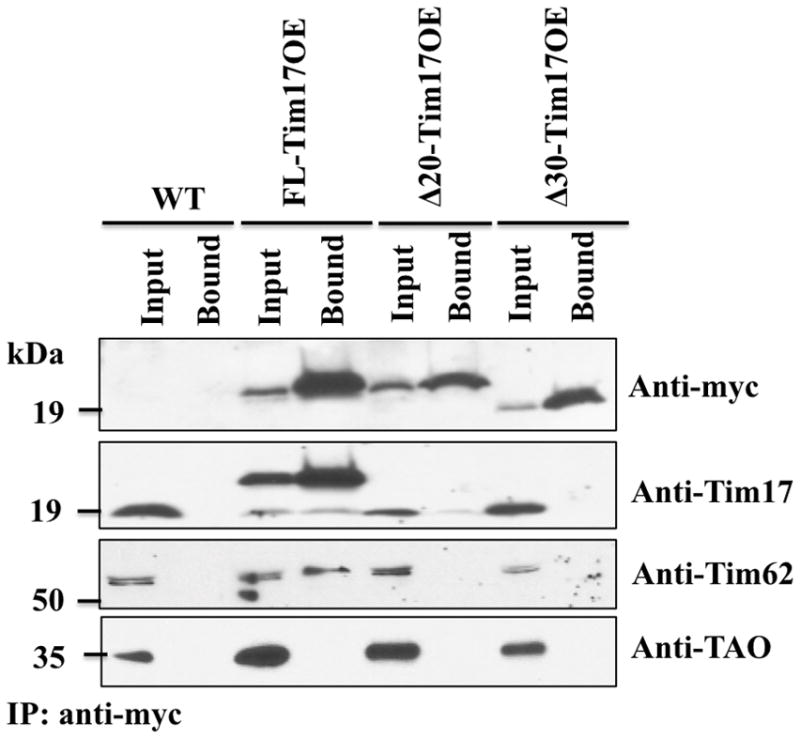
Co-immunoprecipitation analysis of the N-terminal deletion mutants of TbTim17 with other TbTims. Mitochondrial proteins isolated from the wild type (WT), FL-Tim17OE, Δ20-Tim17OE, and Δ30-Tim17OE T. brucei grown for 48 h in the presence of doxycycline were solubilized with digitonin (1%). The soluble proteins were immunoprecipitated (IP) with anti-myc-coupled agarose beads. Proteins from the Input (10%) and precipitated (Bound, 50%) fractions were analyzed by immunoblotting using anti-myc, -Tim17, -Tim62, and TAO antibodies.
3.6. The N-terminal deletion mutants of TbTim17 are not associated with its interacting partners
To further analyze the association of FL-TbTim17-2X-myc, Δ20-TbTim17-2X-myc, and Δ30-TbTim17-2X-myc with TbTim17 and other known interaction partners of this protein in T. brucei, co-IP experiments were performed. Anti-myc antibody pulled down FL-TbTim17-2X-myc, Δ20-TbTim17-2X-myc, and Δ30-TbTim17-2X-myc from the respective mitochondrial extracts (Fig. 6). This antibody did not detect any protein in the immunoprecipitate from wild-type mitochondrial extracts. Probing of the same blot with anti-TbTim17 antibody showed that TbTim17 was co-immunoprecipitated with FL-TbTim17-2X-myc but not with Δ20-TbTim17-2X-myc, and Δ30-TbTim17-2X-myc from the respective mitochondrial extract, suggesting that the first 30 amino acids are critical for the homotypic association of TbTim17 in the endogenous complex. TbTim17 appears to be an abundant protein in the TbTIM complex. Although a definite molar ratio has not been determined yet, it is anticipated that more than one molecule of TbTim17 is present in each complex. In fungi, Tim23, Tim17, and Tim22 are present in dimeric forms and Tim23 dimer associates with two molecules of Tim17 to form the TIM23-17 import channel [9,10, 24]. Therefore, it can be anticipated that TbTim17 is present in multimeric form to form the TbTIM complex.
Previously, we identified two trypanosome-specific components of the TbTIM17 protein complex [15,16]. One of those proteins in particular TbTim62 was shown to play a role in the assembly and stability of this complex. Immunodetection with an anti-TbTim62 antibody demonstrated that the FL-TbTim17 protein was able to associate with TbTim62. When the first 20 and 30 amino acids were deleted from the N-terminal domain of TbTim17 the association with TbTim62 was disrupted. These results further indicate that the N-terminal deletion mutants of TbTim17 were not assembled into the TbTIM complex, thus were not associated with other components of this complex.
All together, we showed that deletion of the first 20 or 30 amino acid residues of TbTim17 didn’t hamper this protein to be targeted to mitochondria, however this region is critical for proper sorting and assembly of TbTim17 into the matured TbTIM complex. A schematic model for sub-mitochondrial sorting of these mutants is shown in Figure 7. We speculated that like other polytopic IM proteins in fungi, TbTim17 crosses the OM in a partially folded conformation. The N-terminal region is released from the ATOM channel and is recognized by other proteins in the IMS or IM for further assembly into the TbTIM complex. The N-terminal deletion mutants of TbTim17 are unable to interact with these assembly factor(s) thus are not properly sorted to the IM. The Δ20-TbTim17 mutant most likely translocated through the OM but destined to the IMS region and could be peripherally associated with either the OM or IM. On the other hand Δ30-TbTim17 either partially or fully translocated through OM but is improperly integrated into the membrane.
Fig. 7.

A working model showing the effect of N-terminal truncation on TbTim17 assembly. The N-terminal 30 amino acid residues of TbTim17 are in red and the rest of the protein are in black lines. ATOM; archaic translocase of mitochondrial outer membrane, TIM; translocase of mitochondrial inner membrane, MOM; mitochondrial outer membrane, MIM; mitochondrial inner membrane; IMS; intermembrane space. FL-TbTim17 is translocated through ATOM and assembled into the TIM complex. However, Δ20-, and Δ30-Tim17 are targeted to mitochondria but failed to be assembled in the TIM.
4. Discussion
Here we demonstrated a unique role of the N-terminal domain of TbTim17 for sorting and membrane insertion of this protein into T. brucei mitochondria. We found that the N-terminal truncation mutants of TbTim17 are non-functional because these mutant proteins are neither assembled into the TbTIM nor associated with either Tim62 or Tim17. In contrast, ectopically expressed FL-TbTim17 was properly sorted and assembled into the matured complex thus fully capable to complement the deficiency of the endogenous TbTim17 during mitochondrial protein import. It is likely that the N-terminal region of TbTim17 interacts with other proteins and that is required for its insertion and assembly into the TIM complex. This is the first report for identification of an essential structural domain of TbTim17.
We found that the N-terminal truncation mutants of TbTim17 were expressed and properly targeted to mitochondria in T. brucei. We did not observe any mutant proteins to be accumulated in the cytosolic fractions. Therefore mitochondrial-targeting signal of TbTim17 is not located in this region, as expected. Previous studies showed that Saccharomyces cerevisiae (Sc) Tim17 and Tim23, each possesses an internal targeting and membrane insertion signal, which is located within the 3rd and 4th transmembrane domains of these proteins (40). However, the N-terminal regions of either of these ScTims do not have any membrane insertion signal. Particularly, an N-terminal deletion mutant of ScTim17 was properly assembled into the TIM23-17 complex [41]. This is in contrast to our finding, which showed that the N-terminal hydrophilic region of TbTim17 is critical for proper insertion of this protein into the MIM, suggesting that the import and sorting process of TbTim17 is probably different from its fungal counterpart.
On the other hand, we showed previously that while expressed in T. brucei, ScTim23, ScTim17, and ScTim22 were targeted to mitochondria but only ScTim17 at least partially assembled into the TbTim17 protein complex, thus ScTim17 could partly complement the function of TbTim17 [29]. This indicates that mitochondrial protein import and sorting machinery in T. brucei is capable of importing heterologous ScTim17 with different sorting and insertion signal. This further suggests that TbTim17 and ScTim17 could be inserted into the IM in T. brucei by different pathways. Alternatively, TbTim17 may possess additional internal targeting and insertion signal similar to ScTim17. However, this internal signal of TbTim17 is not sufficient for proper membrane insertion of this protein. Furthermore, while expressed in S. cerevisiae, TbTim17 destined to mitochondria but persisted as a non-functional protein, possibly because it was not assembled to the ScTim23-17 complex [29]. Together, it suggests that the protein import apparatus in T. brucei is relatively more flexible to accommodate heterologous protein than in higher eukaryotes.
It has also been shown that the N-terminal region of ScTim17 possesses other critical function. The ΔN-ScTim17 is non-functional in S. cerevisiae as it failed to interact with the matrix targeted substrate proteins [41]. Therefore, interaction of the N-terminal domain of ScTim17 with the presequence is necessary for import of the precursor protein through the TIM23-17 channel. Recent data also showed that cysteine residue at position 10 is involved in disulfide bond formation with cysteine at position 77 [42,43]. Mutation of any of these two cysteines is detrimental for Tim17 function, particularly the gating of the TIM23 channel. However, none of these cysteine residues is conserved in TbTim17. Using our truncation mutants we also couldn’t determine if the N-terminal domain of TbTim17 is involved in the substrate binding and translocation processes. Further analysis by site-directed mutagenesis followed by cross-linking with the translocation intermediate may provide answer to this question.
We identified TbTim62, a novel subunit of the TbTIM17 complex. TbTim62 interacts with TbTim17 and is present in a ~150 kDa protein complex on BN-PAGE [16]. A smaller fraction of TbTim17 also found in a ~150 kDa complex, which is not formed in TbTim62 KD mitochondria and TbTim17 is not assembled into the larger matured complex, instead the protein is degraded and TbTim17 levels reduced [16]. Therefore, interaction with TbTim62 is found to be crucial for the assembly and stability of the TbTIM17 complex. Here we showed that unlike the FL-TbTim17 the N-terminal truncation mutants of TbTim17 are not stably associated with TbTim62 that could explain why these mutants are not assembled into the matured complex. It is possible that the N-terminal region of TbTim17 interacts with TbTim62 to form a stable assembly intermediate. However, it has to be determined first if these two proteins directly interact with each other. We also found that the ectopically expressed FL-TbTim17 associates with the endogenous TbTim17, indicating that TbTim17 could be in a dimeric or multimeric form in the TbTIM complex. In contrast to the FL-TbTim17, the N-terminal truncation mutants were not associated with the endogenous TbTim17, which further show that these mutant proteins were not assembled in the TbTIM complex. Further investigation is on-going to find if the N-terminal region of TbTim17 is directly involved for its dimerization and also for its interaction with TbTim62.
Overall we show that N-terminal hydrophilic domain of TbTim17 possesses a unique signal for membrane insertion and assembly of this protein that is not found in the homologous Tims in other eukaryotes.
Supplementary Material
Acknowledgments
We thank George Cross for the pLew100 vectors and the T. brucei 427 29-13 cell line, Jason Carnes for the pHD1344 vector, Larry Simpson for CPT, and Paul Englund for mHSP70 antibodies. Work was supported by grants 1RO1AI125662 and 2SC1GM081146 from the National Institute of Health to MC. EW and JTS were supported by training grants T32HL007737, T32AI007281, and 2R25GM059994. The Molecular Biology Core Facility at Meharry Medical College is supported by NIH grant U54RR026140/U54MD007593.
References
- 1.Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, Roger AJ. Phylogenetic analyses support the monophyly of Excavata and resolve relatioships among eukaryotic supergroups. Proc Natl Acad Sci USA. 2009;106:3859–64. doi: 10.1073/pnas.0807880106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yaro M, Munyard KA, Stear MJ, Groth DM. Combatting African Trypanosomiasis (AAT) in livestock: The potential role of trypanotolerance. Vet Parasitol. 2016;225:43–52. doi: 10.1016/j.vetpar.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Morrison LJ, Vezza L, Rowan T, Hope JC. Animal African Trypanosomiasis: Time to increase focus on clinically relevant parasite and host species. 2016;32:599–607. doi: 10.1016/j.pt.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 4.Lukes J, Hashimi H, Zikova A. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr Genet. 2005;48:277–299. doi: 10.1007/s00294-005-0027-0. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri M, Ott RD, Hill GC. Trypanosome alternative oxidase: from molecule to function. Trends Parasitol. 2006;22:484–491. doi: 10.1016/j.pt.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Acestor N, Panigrahi AK, Ogata Y, Anupama A, Stuart KD. Protein composition of Trypanosoma brucei mitochondrial membrane. Proteomics. 2009;9:5497–5508. doi: 10.1002/pmic.200900354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Cui J, Nilsson D, Gunasekhara K, Chanfon A, Song X, Wang H, Xu Y, Ochsenreiter T. The Trypanosoma brucei MitoCarta and its regulation and splicing pattern during development. Nucl Acid Res. 2010;38:7378–7387. doi: 10.1093/nar/gkq618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Annu Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: from proteomics to functional mechanisms. Nat Rev Mol Cell Biol. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- 10.Schulz C, Schendzielorz A, Rehling P. Unlocking the presequence import pathway. Trends Cell Biol. 2015;25:265–75. doi: 10.1016/j.tcb.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Paschen SA, Neupert W, Rapaport D. Biogenesis of beta-barrel membrane proteins of mitochondria. Trends Biochem Sci. 2005;30:575–82. doi: 10.1016/j.tibs.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 12.Endo T, Yamano K. Transport of proteins across or into the mitochondrial outer membrane. Biochim Biophys Acta. 2010;1803:706–14. doi: 10.1016/j.bbamcr.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Ceh-Pavia E, Spiller MP, Lu H. Folding and biogenesis of mitochondrial small Tim proteins. Int J Mol Sci. 2013;14:16685–705. doi: 10.3390/ijms140816685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chatzi A, Sideris DP, Katrakili N, Pozidis C, Tokatlidis K. Biogenesis of yeast Mia40-uncoupling folding from import and atypical recognition features. FEBS J. 2013;280:4960–69. doi: 10.1111/febs.12482. [DOI] [PubMed] [Google Scholar]
- 15.Singha UK, Hamilton V, Duncan MR, Weems E, Tripathi MK, Chaudhuri M. The Translocase of Mitochondrial Inner Membrane in Trypanosoma brucei. J Biol Chem. 2012;287:14480–93. doi: 10.1074/jbc.M111.322925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singha UK, Hamilton V, Chaudhuri M. Tim62: a novel mitochondrial protein in Trypanosoma brucei, is essential for assembly and stability of the Tim17 protein complex. J Biol Chem. 2015;290:23226–39. doi: 10.1074/jbc.M115.663492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pusnik M, Schmidt O, Perry AJ, Oelijeklaus S, Niemann M, Warcheid B, Lithgow T, Meisinger C, Schneider A. Mitochondrial preprotein translocase of trypanosomatids has a bacterial origin. Curr Biol. 2011;21:1738–43. doi: 10.1016/j.cub.2011.08.060. [DOI] [PubMed] [Google Scholar]
- 18.Mani J, Desy S, Niemann M, Chanfon A, Oeljeklaus S, Pusnik M, Schmidt O, Gerbeth C, Meisinger C, Warscheid B, Schneider A. Mitochondrial protein import receptors in kinetoplastids reveal convergent evolution over large phylogenetic distances. Nat Commun. 2015;6:6646. doi: 10.1038/ncomms7646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harsman A, Oeljeklaus S, Wenger C, Huot JL, Warscheid B, Schneider A. The non-canonical mitochondrial inner membrane presequence translocase of trypanosomatids contains two essential rhomboid-like proteins. Nat Commun. 2016;7:13707. doi: 10.1038/ncomms13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerscher O, Holder J, Srinivasan M, Leung RS, Jensen RE. The Tim54p-Tim22p complex mediates insertion of proteins into the mitochondrial inner membrane. J Cell Biol. 1997;139:1663–75. doi: 10.1083/jcb.139.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferramosca A, Zara V. Biogenesis of mitochondrial carrier proteins: molecular mechanisms of import into mitochondria. Biochim Biophys Acta. 2013;1833:494–502. doi: 10.1016/j.bbamcr.2012.11.014. [DOI] [PubMed] [Google Scholar]
- 22.Bomer U, Rassow J, Zufall N, Pfanner N, Meijer M, Maarse AC. The preprotein translocase of inner mitochondrial membrane: evolutionary conservation of targeting and assembly of Tim17. J Mol Biol. 1996;262:389–395. doi: 10.1006/jmbi.1996.0522. [DOI] [PubMed] [Google Scholar]
- 23.van der Laan M, Hutu DP, Rehling P. On the mechanism of preprotein import by the mitochondrial preprotein translocase. Biochim Biophys Acta. 2010;1803:732–39. doi: 10.1016/j.bbamcr.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Truscott KN, Kovermann P, Geissler A, Merlin A, Meijer M, Driessen AJ, Rassow J, Pfanner N, Wagner R. A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat Struct Biol. 2001;8:1074–1082. doi: 10.1038/nsb726. [DOI] [PubMed] [Google Scholar]
- 25.Mokaranjac D, Paschen SA, Kozany C, Prokisch H, Hoppins SC, Nargang FE, Neupert W, Hell K. Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 2003;22:816–825. doi: 10.1093/emboj/cdg090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gevorkyan-Airapetov L, Zohary K, Popov-Celeketic D, Mapa K, Hell K, Neupert W, Azem A, Mokranjac D. Interaction of Tim23 with Tim50 is essential for protein translocation by the mitochondrial TIM23 complex. J Biol Chem. 2009;284:4865–72. doi: 10.1074/jbc.M807041200. [DOI] [PubMed] [Google Scholar]
- 27.Rassow J, Dekker PJT, Wilpe SV, Meijer M, Soll J. The preprotein translocase of the mitochondrial inner membrane: function and evolution. J Mol Biol. 1998;286:105–120. doi: 10.1006/jmbi.1998.2455. [DOI] [PubMed] [Google Scholar]
- 28.Murcha MW, Wang Y, Narsai R, Whelan J. The plant mitochondrial protein import apparatus – The differences make it interesting. Biochim Biophys Acta. 2014;1840:1233–45. doi: 10.1016/j.bbagen.2013.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Weems E, Singha UK, Hamilton V, Smith JT, Waegemann K, Mokranjac D, Chaudhuri M. Functional complementation analyses reveal that the single PRAT-family protein of Trypanosoma brucei is a divergent homolog of Tim17 in Saccharomyces cerevisiae. Eukaryot Cell. 2015;14:286–96. doi: 10.1128/EC.00203-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singha UK, Peprah E, Williams S, Walker R, Saha L, Chaudhuri M. Characterization of the mitochondrial inner membrane translocator Tim17 from Trypanosoma brucei. Mol Biochem Parasitol. 2008;159:30–43. doi: 10.1016/j.molbiopara.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tschopp F, Charrière F, Schneider A. In vivo study in Trypanosoma brucei links mitochondrial transfer RNA import to mitochondrial protein import. EMBO Rep. 2011;12:825–32. doi: 10.1038/embor.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biebinger S, Wirtz LE, Lorenz P, Clayton C. Vectors for inducible expression of toxic gene products in bloodstream and procyclic Trypanosoma brucei. Mol Biochem Parasitol. 1997;85:99–112. doi: 10.1016/s0166-6851(96)02815-0. [DOI] [PubMed] [Google Scholar]
- 33.Wirtz E, Leal S, Ochatt C, Cross GA. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol. 1999;99:89–101. doi: 10.1016/s0166-6851(99)00002-x. [DOI] [PubMed] [Google Scholar]
- 34.Miller MM, Halbig K, Cruz-Reyes J, Read LK. RBP16 stimulates trypanosome RNA editing in vitro at an early step in the editing reaction. RNA. 2006;12:1292–303. doi: 10.1261/rna.2331506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singha UK, Sharma S, Chaudhuri M. Down regulation of mitochondrial porin inhibits cell growth and alters respiratory phenotype in Trypanosoma brucei. Euk Cell. 2009;8:1418–28. doi: 10.1128/EC.00132-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaudhuri M. Cloning and characterization of a novel serine/threonine protein phosphatase type 5 from Trypanosoma brucei. Gene. 2001;266:1–13. doi: 10.1016/s0378-1119(01)00367-5. [DOI] [PubMed] [Google Scholar]
- 37.Priest JW, Hajduk SL. Developmental regulation of Trypanosoma brucei cytochrome c reductase during bloodstream to procyclic differentiation. Mol. Biochem. Parasitol. 1994;65:291–304. doi: 10.1016/0166-6851(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 38.Effron PN, Torri AF, Engman DM, Donelson JE, Englund PT. A mitochondrial heat shock protein from Crithidia fasiculata. Mol. Biochem. Parasitol. 1993;59:191–200. doi: 10.1016/0166-6851(93)90217-l. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhuri M, Ajayi W, Hill GC. Biochemical and molecular properties of the Trypanosoma brucei alternative oxidase. Mol. Biochem. Parasitol. 1998;95:53–68. doi: 10.1016/s0166-6851(98)00091-7. [DOI] [PubMed] [Google Scholar]
- 40.Káldi K, Bauer MF, Sirrenberg C, Neupert W, Brunner M. Biogenesis of Tim23 and Tim17, integral components of the TIM machinery for matrix-targeted preproteins. EMBO J. 1998;17:1569–76. doi: 10.1093/emboj/17.6.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meier S, Neupert W, Herrmann JM. Conserved N-terminal negative charges in the Tim17 subunit of the TIM23 translocase play a critical role in the import of preproteins into mitochondria. J Biol Chem. 2005;280:7777–85. doi: 10.1074/jbc.M412158200. [DOI] [PubMed] [Google Scholar]
- 42.Wrobel L, Sokol AM, Chojnacka M, Chacinska A. The presence of disulfide bonds reveals an evolutionarily conserved mechanism involved in mitochondrial protein translocase assembly. Sci Rep. 2016;6:27484. doi: 10.1038/srep27484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramesh A, Peleh V, Martinez-Caballero S, Wollweber F, Sommer F, van der Laan M, Schroda M, Alexander RT, Campo ML, Herrmann JM. A disulfide bond in the Tim23 complex is crucial for voltage gating and mitochondrial protein import. J Cell Biol. 2016;214:417–31. doi: 10.1083/jcb.201602074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.