

Abstract
Krüppel-like factor 4 (KLF4, GKLF) is a zinc-finger transcription factor involved in a large variety of cellular processes, including apoptosis, cell cycle progression, as well as stem cell renewal. KLF4 is critical for cell fate decision and has an ambivalent role in tumorigenesis. Emerging data keep reminding us that KLF4 dysregulation either facilitates or impedes tumor progression, making it important to clarify the regulating network of KLF4. Like most transcription factors, KLF4 has a rather short half-life within the cell and its turnover must be carefully orchestrated by ubiquitination and ubiquitin–proteasome system. To better understand the mechanism of KLF4 ubiquitination, we performed a genome-wide screen of E3 ligase small interfering RNA library based on western blot and identified SCF-FBXO32 to be a new E3 ligase, which is responsible for KLF4 ubiquitination and degradation. The F-box domain is critical for FBXO32-dependent KLF4 ubiquitination and degradation. Furthermore, we demonstrated that FBXO32 physically interacts with the N-terminus (1–60 aa) of KLF4 via its C-terminus (228–355 aa) and directly targets KLF4 for ubiquitination and degradation. We also found out that p38 mitogen-activated protein kinase pathway may be implicated in FBXO32-mediated ubiquitination of KLF4, as p38 kinase inhibitor coincidently abrogates endogenous KLF4 ubiquitination and degradation, as well as FBXO32-dependent exogenous KLF4 ubiquitination and degradation. Finally, FBXO32 inhibits colony formation in vitro and primary tumor initiation and growth in vivo through targeting KLF4 into degradation. Our findings thus further elucidate the tumor-suppressive function of FBXO32 in breast cancer. These results expand our understanding of the posttranslational modification of KLF4 and of its role in breast cancer development and provide a potential target for diagnosis and therapeutic treatment of breast cancer.
INTRODUCTION
Zinc-finger containing transcription factor Krüppel-like factor 4 (KLF4) is an epithelium-enriched regulator, which has an essential role in cell cycle regulation, programmed cell death and stem cell reprogramming.1–5 KLF4 is considered as a suppressor of cell proliferation6 for its transactivation of cell cycle inhibitor p21Cip1/Waf1 7 and suppression of several cell cycle promoting genes.8–10 Nevertheless, KLF4 also acts as an anti-apoptotic gene by directly binding to the promoter of p53 and inhibiting its expression.11 Furthermore, KLF4 is important in cell fate decision undergoing certain intracellular stresses.12 Thus, KLF4 is associated with several human diseases, especially cancer. Conflicting evidences surprisingly uncovered the fact that KLF4 may have an ambivalent role in tumorigenesis in a tissue-specific manner. Actually, KLF4 acts as a tumor suppressor in colon,13 gastric,14 esophageal,15,16 bladder17 and lung cancer,18 whereas in pancreatic19 and breast cancer,20,21 KLF4 is believed to function as an oncogene. For example, KLF4 is a key regulator in the initiation of premalignant intraepithelial neoplasia by mutant Kras in pancreatic cancer.19 Owing to its importance in physiological and pathological processes, KLF4 is critically regulated on different levels. Studies in recent years gradually unveiled the underlying mechanism of KLF4 regulation. Given the short half-life of KLF4, we focused on its posttranslational regulation, especially the ubiquitination regulation. The E3 ligase Cdh1-anaphase-promoting complex,22 β-TrCP23 and pVHL24,25 are reported to mediate KLF4 ubiquitination and degradation. Anaphase-promoting complex governs transforming growth factor-β-induced KLF4 proteolysis in colon cancer cells.22 β-TrCP ubiquitinates KLF4 in response to phosphorylation by extracellular signal-regulated kinases 1 and 2 and mediates mouse embryonic stem cell renewal.23 In addition, tumor-suppressor pVHL inhibits KLF4-dependent proliferation via mediating KLF4 ubiquitination and degradation.24 These studies partially explained the post-translational regulation mechanism of KLF4 protein, and more importantly, they also implied that different E3 ligases may be involved in specific molecular context. In fact the detailed mechanism of KLF4 function and regulation in breast cancer still remains largely unknown. In order to get a comprehensive understanding about the ubiquitination regulation of KLF4 in breast cancer, we performed a genome-wide screening using an ubiquitin-conjugated small interfering RNA (siRNA) library and identified FBXO32 as a novel E3 ligase, which is responsible for KLF4 ubiquitination and degradation in breast cancer cell lines.
FBXO32 belongs to the F-box protein super family that functions as substrate recognition subunit in SCF E3 ligase complex.26–29 FBXO32 is originally identified to be specifically expressed in muscle where it has a pivotal role in muscle atrophy.26,28,30 However, emerging evidences show that FBXO32 is also involved in the process of tumorigenesis. The observation that FBXO32 is generally downregulated in ovarian and gastric cancer via promotor hypermethylation suggest that it may function as a tumor suppressor.31,32 In addition, a recent study identified the classic oncogene c-Myc as an ubiquitination substrate of FBXO32, supporting the speculation that FBXO32 acts as a tumor suppressor in cancer development.33 Currently, the role of FBXO32 in tumorigenesis still remains unclear, and the underlying mechanism is poorly understood.
In this study, we report that FBXO32 interacts with KLF4 and mediates KLF4 degradation through the ubiquitin–proteasome pathway in the presence of active p38/mitogen-activated protein kinase (MAPK) phosphorylation cascade. FBXO32 deficiency in breast cancer cells leads to KLF4 accumulation and facilitates tumorigenesis both in vitro and in vivo. Our study identified a novel E3 ligase, which is responsible for KLF4 protein turnover and provided a possible mechanism of KLF4 regulation and function in breast cancer tumorigenesis and development.
RESULTS
KLF4 has a rather short half-life
KLF4 is a transcription factor regulating a network of genes involved in various cellular functions, and it is a protein with a very short half-life.34 We examined the protein stability of KLF4 and found that KLF4 protein rapidly declined after cycloheximide treatment in MCF7 cells (Figure 1a), and MG132 treatment led to an obvious accumulation of KLF4 protein (Figure 1b). Furthermore, rapid degradation of exogenous KLF4 protein was observed in HEK293T cells and additional MG132 inhibited KLF4 degradation (Figure 1c). These results strongly suggest that KLF4 has a short half-life and is regulated through ubiquitin–proteasome pathway.
Figure 1.
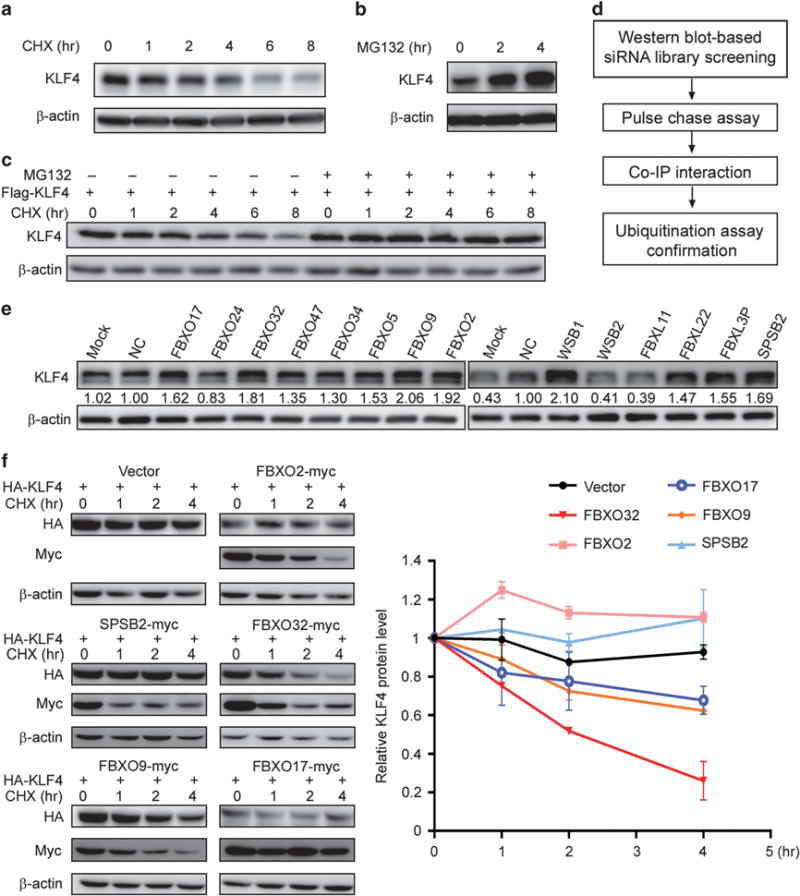
A genome-wide functional screen for E3 ubiquitin ligase(s) targeting KLF4 protein for degradation. (a, b) KLF4 protein has a short half-life and is degraded via ubiquitin–proteasome pathway. MCF7 cells were plated in six-well plates. Twenty-four hours later, the cells were treated with 50 μM CHX (a) or 20 μM MG132 (b) for the indicated time. KLF4 level was analyzed by western blot. (c) Exogenously expressed KLF4 is degraded through proteasomal pathway. HEK293T cells were transfected with Flag-tagged KLF4, treated with 50 μM CHX for indicated time with or without 20 μM MG132. The exogenous KLF4 was probed by immunoblot with Flag antibody. (d) Experimental procedure flow chart for identification of the E3 ubiquitin ligase(s) targeting KLF4 protein. (e) A second round of siRNA screening was carried out to confirm the candidates. KLF4 level was probed by immunoblot after siRNA knockdown of 14 candidates. Quantification was made by Image J (NIH, Bethesda, MD, USA). (f) FBXO32 facilitates KLF4 degradation. HA-tagged KLF4 is co-transfected with five E3 ligase candidates individually. Twenty-four hours after transfection, the cells were treated with CHX for indicated time. KLF4 and E3 ligase candidates were analyzed by western blot with antibodies against HA and myc. Quantification was made by Image J (NIH) and the statistics were done with GraphPad Prism.
FBXO32 promotes KLF4 degradation
To investigate the E3 ligases affecting KLF4 protein degradation, we designed a western blot-based siRNA screening strategy and performed a genome-wide screening with an E3 ligase siRNA library in MCF7 cells. The overall screening strategy was described by Figure 1d. We examined KLF4 protein level after individual knockdown of the genes in the library, and genes whose knockdown resulted in a remarkable increase in KLF4 protein level were chosen as potential candidates that may be responsible for KLF4 ubiquitination and proteasomal degradation.
A total of 14 genes were identified as positive candidates after the first round screening. To exclude false-positive candidates, it was necessary to make a further confirmation. Hence, we redid the siRNA knockdown of the 14 genes and finally narrowed the candidates down to eight genes whose knockdown led to a >1.5-fold increase in KLF4 protein level: FBXO17, FBXO32, FBXO9, FBXO2, WSB1, FBXL22, FBXL3P and SPSB2 (Figure 1e). We successfully cloned five of these candidates (FBXO2, FBXO9, FBXO17, FBXO32 and SPSB2) and performed a CHX pulse-chase assay to test the impact of these candidates on KLF4 protein stability. We co-transfected the KLF4 gene with individual E3 ligase candidates and examined the stability of the exogenous KLF4 protein. The result revealed that FBXO32 greatly accelerated KLF4 degradation, whereas other candidates had only moderate effect on KLF4 protein degradation (Figure 1f). We then chose FBXO32 for the following study.
FBXO32 interacts with and ubiquitinates KLF4
We then performed a co-immunoprecipitation (IP) assay to test the interaction between FBXO32 and KLF4. Flag-tagged KLF4 was co-transfected with indicated plasmids into HEK293T cells. Tagged KLF4 and its interacting partners were immunoprecipitated with anti-FLAG M2 affinity gel. Subsequent western blot analysis showed that FBXO32 physically interacted with KLF4 (Figure 2a). We performed ubiquitination assay to test whether FBXO32 ubiquitinates KLF4 in vivo. We co-transfected either empty vector or Myc-tagged FBXO32 with Flag-tagged KLF4 and HA-tagged ubiquitin in HEK293T cells. We found that KLF4 ubiquitination was remarkably increased under FBXO32 expression (Figure 2b). In order to further confirm that FBXO32 facilitates KLF4 degradation through ubiquitin–proteasome pathway, we co-transfected FBXO32 and KLF4 in HEK293T cells, and found that FBXO32 expression strongly reduced KLF4 protein expression, whereas MG132 treatment restored KLF4 protein level (Figure 2c). These results suggest that FBXO32 facilitates KLF4 degradation through ubiquitin–proteasome pathway.
Figure 2.
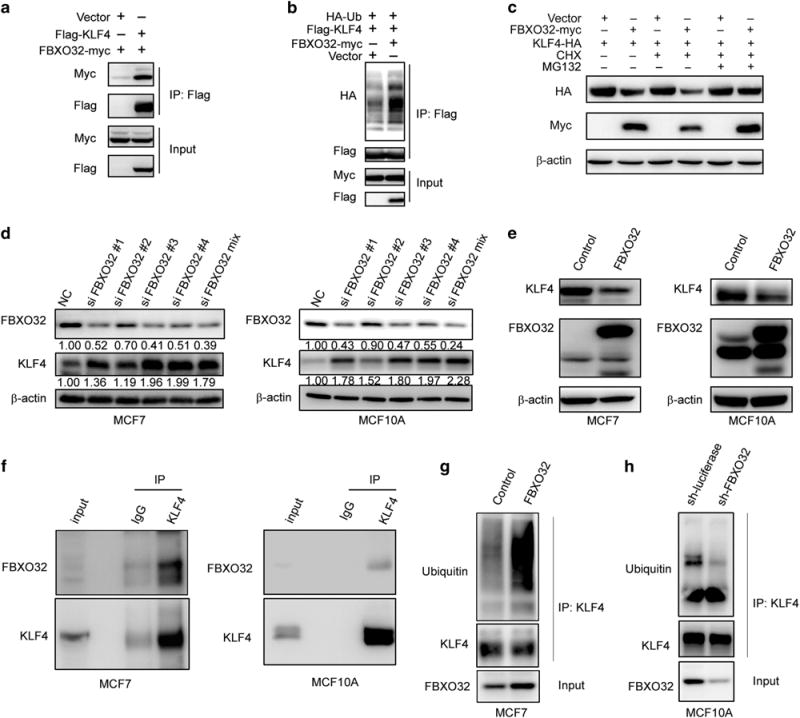
FBXO32 interacts with KLF4 and mediates KLF4 ubiquitination and degradation. (a) Exogenous FBXO32 interacts with KLF4. HEK293T cells were transfected with Flag-tagged KLF4 and either empty vector or the Myc-tagged FBXO32. Twenty-four hours after transfection, the cells were treated with 20 μM MG132 for 4 h. The co-IP experiment was performed using anti-Flag M2 affinity gel. The immunoprecipitates were analyzed by western blot with anti-Myc antibody. (b) FBXO32 increases exogenous KLF4 ubiquitination. Indicated plasmids were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with 20 μM MG132 for 6 h to accumulate the ubiquitinated KLF4. Anti-Flag M2 affinity gel was used to immunoprecipitate tagged KLF4. The ubiquitin was probed by HA-specific antibody to reflect the ubiquitination of KLF4. (c) MG132 restores FBXO32-mediated KLF4 decreasing. HA-tagged KLF4 is transfected with either the empty vector or the Myc-tagged FBXO32 in HEK293T cells. Twenty-four hours after transfection, CHX and MG132 were added as indicated for 4 h. The cell lysates were subjected to western blot to evaluate the protein level of KLF4. (d) FBXO32 knockdown leads to KLF4 protein accumulation. siRNAs specifically targeting FBXO32 are transfected into MCF7 and MCF10A cells, the expression of KLF4 and FBXO32 was tested by western blot. (e) FBXO32 overexpression decreases the protein level of KLF4. MCF7 and MCF10A cells stably expressed FBXO32 were established. The cell lysates with equal amount of total protein were used to test the protein levels of FBXO32 and KLF4 via immunoblotting. (f) Endogenous FBXO32 interacts with KLF4. MCF7 or MCF10A cells cultured in 10 cm dish were treated with MG132 for 4 h and harvested in 1 ml of cell lysis buffer. IP was performed with anti-KLF4 primary antibody. The immunoprecipitates were analyzed by western blot and detected with anti-FBXO32 antibody. (g, h) Effect of FBXO32 on the ubiquitination of endogenous KLF4. Control and FBXO32 overexpressed MCF7 cells (g) or control and FBXO32 knockdown MCF10A cells (h) were treated with MG132 for 6 h and subjected to ubiquitination assay. Immunoprecipitated KLF4 proteins were analyzed with western blot and detected with ubiquitin antibody.
To test the effect of FBXO32 on endogenous KLF4, we transiently knocked down FBXO32 in MCF7 and MCF10A cells and observed elevated KLF4 protein expression (Figure 2d). We also generated MCF7 and MCF10A cells that stably over-express FBXO32, and both cells displayed a remarkable decrease in KLF4 protein level (Figure 2e). Furthermore, co-IP analysis with endogenous KLF4 and FBXO32 revealed their interaction under physiological conditions (Figure 2f). Overexpression of FBXO32 could increase endogenous KLF4 ubiquitination (Figure 2g), whereas knockdown of FBXO32 led to a decrease in KLF4 ubiquitinantion (Figure 2h). These results further confirm that FBXO32 is a potential E3 ligase responsible for KLF4 ubiquitination and proteasomal degradation.
FBXO32 recognizes and interacts with KLF4 via its C-terminal and F-box domain is required for FBXO32 function
FBXO32 functions as substrate recognition component of SCF E3 ligase.27,29 In order to further investigate the mechanism of FBXO32 and KLF4 interaction, we developed a series of FBXO32 deletion mutants (Figure 3a). Previous studies showed that the F-box domain is responsible for binding with Cullin1 to maintain the integrity of the whole complex,35 suggesting that F-box is critical for the activity of the E3 ligase function. We used pulse-chase assay to test the role of F-box in KLF4 degradation, and observed that wild-type FBXO32 dramatically reduced the KLF4 half-life, however, the FBXO32-mediated KLF4 degradation was completely blocked by F-box deletion (Figure 3b). An ubiquitination assay was performed to test the effect of F-box on KLF4 ubiquitination. We found that deletion of F-box abrogated the FBXO32-dependent KFL4 ubiquitination (Figure 3c). These results indicate that F-box domain is critical for FBXO32 function.
Figure 3.
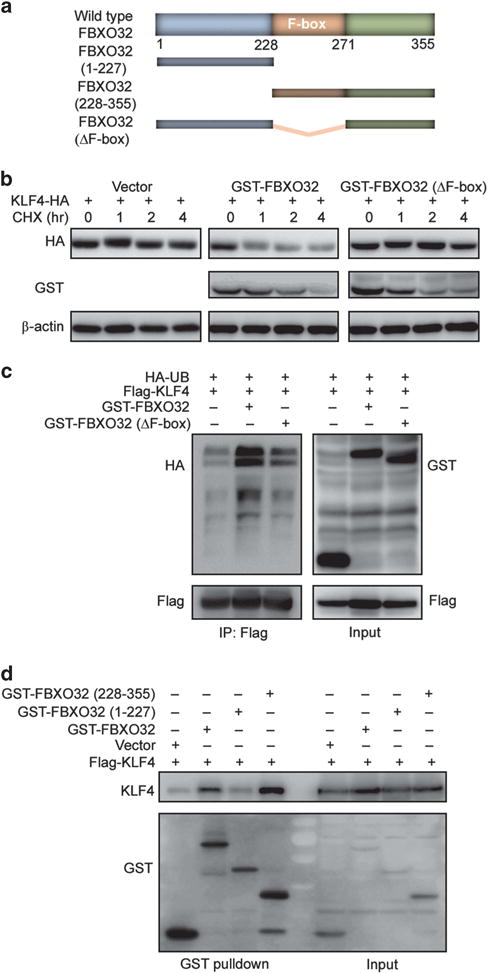
The C-terminus of FBXO32 is responsible for recognizing and interacting with KLF4 and the F-box domain is critical for FBXO32 E3 ligase function. (a) Schematic of human FBXO32 with previously identified domains as well as deletion mutants generated in the present work. (b) The F-box domain is critical for the E3 ligase activity of FBXO32. HA-tagged KLF4 and GST-tagged FBXO32 were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with CHX for indicated time. The cell lysates were analyzed by western blot for KLF4 protein level. (c) Deletion of F-box abrogated FBXO32-mediated KLF4 ubiquitination. Flag-tagged KLF4 and GST-tagged wild-type or F-box deletion mutant FBXO32 were co-transfected into HEK293T cells. The cells were treated with MG132 and subjected to ubiquitination assay as described. (d) FBXO32 interacts with KLF4 via its C-terminus. Indicated plasmids were transfected into HEK293T cells. Twenty-four hours later, the cells were treated with MG132 and subjected to GST pulldown. The precipitates were analyzed by western blot.
To understand which domain of FBXO32 is responsible for KLF4 recognition and interaction, we used a co-IP assay to test the binding ability of these deletion mutants. We found that deletion of the C-terminus (228-355aa) abolished FBXO32 and KLF4 interaction, whereas deletion of the N-terminus (1-227aa) had little effect on FBXO32 and KLF4 interaction (Figure 3d). This result indicates that it is the C-terminus of FBXO32, which recognizes and interacts with KLF4.
The N-terminus of KLF4 is critical for interaction with FBXO32
To identify the regions of KLF4 responsible for degradation, we generated several Flag-tagged KLF4 deletion mutants (Figure 4a). We first tested the stability of these deletion mutants. The mutants were individually co-transfected with either empty vector or wild-type FBXO32 in HEK293T cells, and 24 h after transfection the cell lysates were subjected to western blot. The result exhibited that deletion of the 1–60 amino acids eliminated FBXO32-mediated KLF4 degradation (Figure 4b), suggesting this region was critical for KLF4 degradation. The 1–60 aa were reported to be responsible for KLF4 interaction with pVHL,24 thus we tested the binding ability of N-terminal deletion mutants to explore the detail of KLF4 interaction with FBXO32. Co-IP assay showed that deletion of the 1–60 amino acids abrogated KLF4 and FBXO32 interaction (Figure 4c). Therefore, we proposed that amino acids 1–60 of KLF4 were necessary for FBXO32 binding. This result, together with the fact that KLF4 truncation (61–470) becomes much more stable than wild-type KLF4, supports the hypothesis that FBXO32 mediates KLF4 degradation through physical interaction with its 1–60 aa.
Figure 4.
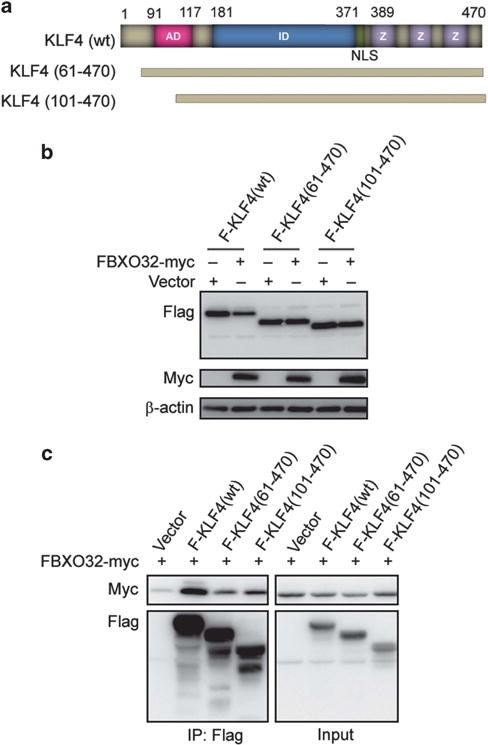
The KLF4 N-terminus is critical for interaction with FBXO32. (a) Schematic of human KLF4 with functional domain, as well as deletion mutants generated in this study. AD, transactivation domain; ID, transrepression domain; NLS, nuclear localization signal; Z, zinc-finger domain. (b) Deletion of amino acids 1–60 stabilizes KLF4. Flag-tagged wild-type or deletion mutants of KLF4 were transfected into HEK293T cells together with empty vector or Myc-tagged FBXO32. Twenty-four hours after transfection, the protein level of wild-type KLF4 or deletion mutants were tested by immunoblotting with antibody against Flag. (c) Deletion of amino acids 1–60 eliminates KLF4 and FBXO32 interaction. Indicated plasmids were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with MG132 for 4 h. Co-IP was performed with anti-Flag M2 affinity gel.
Active p38/MAPK pathway is necessary for FBXO32-mediated KLF4 degradation
Conventionally, F-box proteins recognize and interact with substrates as the substrates are phosphorylated.36 In mouse embryonic stem cells, KLF4 is targeted to proteasomal degradation by β-TrCP while phosphorylated by extracellular signal-regulated kinases 1 and 2.23 However, FBXO32 is reported to destabilize c-Myc independent of phosphorylation.33 Thus, we wondered whether phosphorylation is required for initiating FBXO32-mediated KLF4 ubiquitination and degradation. Hence, we tested KLF4 protein level after inhibition of several phosphorylation pathways, and found that treatment with p38/MAPK inhibitor led to a marked increase of the KLF4 protein level in MCF7 cells (Figure 5a).
Figure 5.
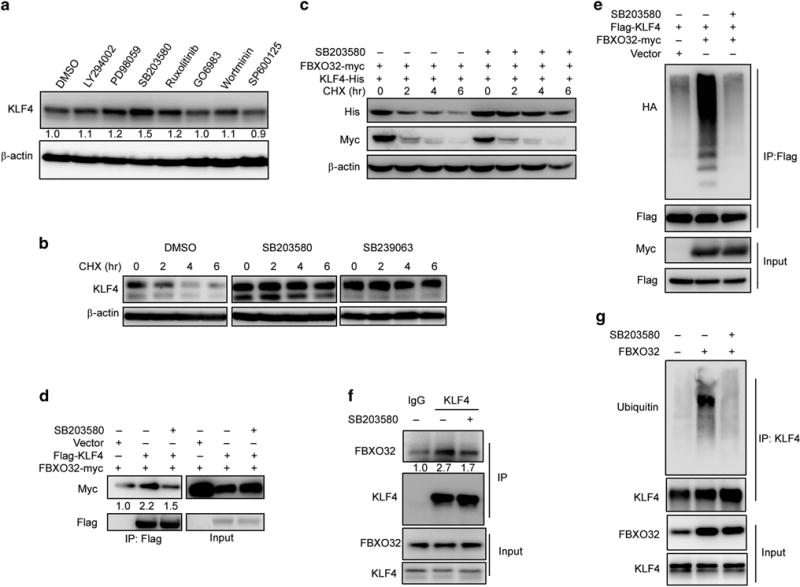
Active p38/MAPK pathway is necessary for FBXO32-mediated KLF4 degradation. (a) p38 inhibitor leads to KLF4 accumulation. MCF7 cells plated in six-well plate were treated with DMSO or indicated inhibitors for 6 h. The protein level of KLF4 was detected by immunoblot. (b) KLF4 protein half-life is elongated under p38 inhibition. MCF7 cells plated in six-well plate were treated with DMSO or p38 inhibitors (SB203580 and SB239063) for 6 h. Then CHX was added 0, 2, 4 or 6 h before harvest. KLF4 protein level was tested by immunoblotting. (c) p38 inhibitor abolishes FBXO32-mediated KLF4 degradation. HEK293T cells were transfected with His-tagged KLF4 and Myc-tagged FBXO32. Twenty-four hours after transfection, the cells were treated with DMSO or p38 inhibitor for 6 h followed by treatment with CHX for indicated time. Western blot was performed to examine the protein level of KLF4. (d, f) p38 inhibitor disrupts KLF4 and FBXO32 interaction. HEK293T cells were transfected with Flag-tagged KLF4 and Myc-tagged FBXO32 or empty vector. Twenty-four hours after transfection, the cells were treated with DMSO or SB203580 for 6 h followed by 4-h treatment with MG132. The cell lysates were precipitated with anti-Flag M2 affinity gel and the precipitates were analyzed by western blot (d). MCF7 cells cultured in 10 cm dishes were treated with either DMSO or SB203580 for 6 h. The cell lysates were subjected to co-IP analysis by KLF4 antibody. The immunoprecipitates were blotted by FBXO32 antibody (f). (e, g) p38 inhibitor decreased FBXO32-mediated KLF4 ubiquitination. Indicated plasmids were co-transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with DMSO or SB203580 for 6 h followed by 6- h treatment with MG132. The cells were subjected to ubiquitination assay and the ubiquitinated KLF4 were detected by immunoblotting with anti-HA antibody (e). FBXO32 overexpressed MCF7 cells cultured in 10 cm dishes were treated with either DMSO or SB203580 for 6 h. The cell lysates were subjected to ubiquitination analysis. Ubiquitinated KLF4 proteins were revealed by immunoblotting with ubiquitin antibody (g).
To further confirm whether KLF4 accumulation under p38/MAPK inhibition is via posttranslational regulation, we tested the protein stability of KLF4 after p38/MAPK inhibition. Pulse-chase analysis revealed that p38 inhibitor SB203580 and SB239063 greatly prolonged KLF4 half-life (Figure 5b). We subsequently examined the relationship between p38/MAPK pathway and FBXO32-mediated KLF4 degradation. FBXO32 and KLF4 were co-transfected in HEK293T cells with or without p38 inhibitor treatment. Then cycloheximide was added for indicated time. We found that p38 inhibition almost abolished the FBXO32-mediated KLF4 degradation (Figure 5c). Consistently, our results displayed that p38/MAPK inhibition greatly weakened FBXO32 and KLF4 interaction (Figure 5d), as well as FBXO32-mediated KLF4 ubiquitination (Figure 5e). We observed the same effect of p38/MAPK inhibition on endogenous KLF4 and FBXO32 interaction (Figure 5f) and FBXO32-dependent KLF4 ubiquitination (Figure 5g). Taken together, these results suggest that FBXO32-mediated KLF4 ubiquitination and degradation is dependent on an active p38/MAPK pathway.
Loss of FBXO32 expression facilitates tumorigenesis
Previous reports suggested that FBXO32 may be a tumor suppressor in ovarian and gastric cancer.31,32 In multiple breast cancer cell lines, FBXO32 is downregulated by polycomb protein EZH2, and knockdown or pharmacologically depletion of EZH2 leads to an increase in FBXO32 expression and facilitates breast cancer cell apoptosis.37,38 We used premalignant human breast epithelial cell line MCFCA1a to investigate the impact of FBXO32 and the concomitant change in KLF4 on tumorigenesis. We developed FBXO32 knockdown MCFCA1a cells and FBXO32/KLF4 double knockdown MCFCA1a cells (Figure 6a), and tested tumorigenesis by soft agar colony-forming assay. As shown in Figure 6b, FBXO32 knockdown greatly increased the colony-forming ability of MCFCA1a cells and simultaneously knockdown of KLF4 could largely reverse the tumorigenic effect of FBXO32 knockdown.
Figure 6.
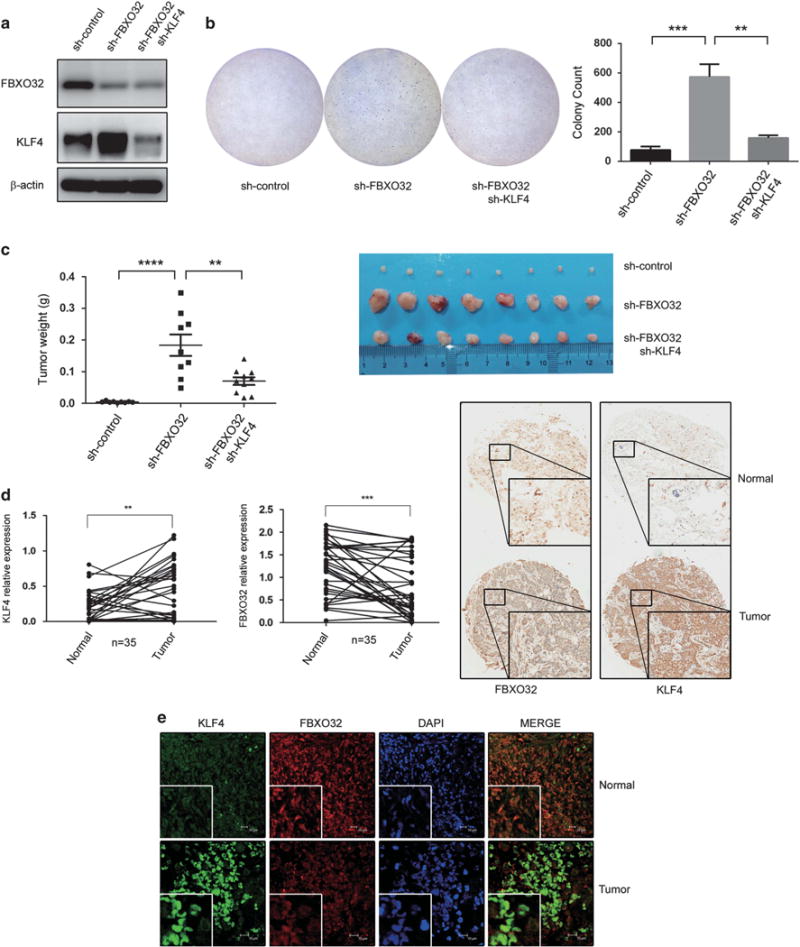
FBXO32 facilitates tumorigenesis both in vitro and in vivo. (a) FBXO32/KLF4 double knockdown MCFCA1a cells. MCFCA1a cells with FBXO32 stable knockdown and FBXO32/KLF4 double knockdown were developed using a lentivirus system. Expression of FBXO32 and KLF4 were analyzed by western blot. (b) FBXO32 inhibits tumorigenesis through mediating proteasomal degradation of KLF4 in MCFCA1a cells. In all, 30 000 MCFCA1a cells were plated in 60-mm dish to grow in soft agar. Three weeks later, the cells were dyed with crystal violet and the colony numbers were counted. (c) FBXO32 inhibits tumorigenesis through mediating proteasomal degradation of KLF4 in a mouse model. In total, 4 × 105 MCFCA1a cells were injected orthotopically into BALB/c nude mice (n = 8). The mice were killed 3 weeks later. The xenografts were weighed and image of primary tumors was presented. (d) Tissue microarray sections were stained by KLF4 and FBXO32 antibodies. Representative staining of KLF4 and FBXO32 (20 ×) was shown and the tissue staining was quantified and analyzed. (e) Immunofluorescent double staining of human breast cancer tissues. Tissue microarray section was immunoblotted by KLF4 and FBXO32 antibodies and labeled with second antibody conjugated with FITC and CY3. Immunofluorescence labeled tissue samples were then scanned by laser confocal microscopy. Representative staining were presented. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To test the functional impact of FBXO32 on breast cancer tumorigenesis in vivo, we injected MCFCA1a cells orthotopically in the mammary fat pad to evaluate the tumorigenesis and tumor growth in a mouse model. The MCFCA1a cells with FBXO32 stable knockdown showed a remarkably increased primary tumor growth. In addition, the oncogenic effect caused by FBXO32 knockdown could be markedly impaired by KLF4 knockdown (Figure 6c). These results suggest that FBXO32 knockdown greatly improves breast cancer tumorigenesis both in vitro and in vivo, which is at least partially dependent on KLF4 accumulation.
To validate the roles of FBXO32 and KLF4 in tumorigenesis, we performed immunohistochemistry staining to examine the expression of FBXO32 and KLF4 in 35 paired breast cancer and adjacent normal tissue samples. Consistent with previous report,25 KLF4 showed a significant increase in breast cancer tissues compared with adjacent normal tissues. And FBXO32 was remarkably down-regulated in the same samples (Figure 6d). A representative image was shown in Figure 6d. In addition, expression of FBXO32 and KLF4 in human breast tumor tissues were detected by immunofluorescent double staining (Figure 6e). This expression pattern suggests that dysregulation of KLF4 by FBXO32 may contribute to the process of tumorigenesis of breast cancer.
DISCUSSION
KLF4 is originally identified as a zinc-finger transcription factor, which is highly expressed in colon, especially in the well-differentiated cells.1 KLF4 is responsible for differentiation of esophageal, skin and colonic cells,16,39,40 and also functions as a negative regulator of cell cycle.6–10 Another striking potential of KLF4 is to induce pluripotent stem cells and to mediate stem cell renewal.5,23 Like other transcription factors of great importance, KLF4 level is critically manipulated on different levels and its dysregulation is associated with several human diseases including cancer.4 Although initially defined as a tumor suppressor, the oncogenic role of KLF4 is becoming more clearly in recent years. KLF4 is just proved to be a critical initiator of early pancreatic cancer.19 In addition, in breast cancer, higher KLF4 level usually implies high risk of tumorigenesis and poor prognosis,20 but the underlying mechanism still remains obscure. Therefore, better understanding of the regulatory mechanisms of the KLF4 protein level will provide additional insights in inhibiting KLF4-associated tumorigenesis in breast cancer.
Previous results showed that KLF4 is a labile protein with a high turnover rate.34 Kim et al.23 discovered that E3 ligase β-TrCP targets KLF4 to polyubiquitination and degradation in mouse embryonic cells in response to extracellular signal-regulated kinases 1 and 2 phosphorylation. Gamper et al.24 and Hu et al.25 identified pVHL as a protein that governs KLF4 turnover and illustrated that estrogen-induced downregulation of pVHL facilitates accumulation of KLF4 in breast cancer cells. Suppression of pVHL in response to estrogen signaling pathway results in elevation of KLF4 protein and subsequent mitotic effect. These studies just revealed a tip of the iceberg in the regulation mechanism of KLF4 in breast cancer. Consider the complexity and diversity of ubiquitination regulation, we suspect that there should be additional E3 ligases regulating KLF4 degradation in different circumstances or in response to different stimuli.
Searching for E3 ligases that target a specific protein substrate of interest seems to be a time-consuming and serendipitous task, whereas the application of an ubiquitin-conjugated siRNA library makes it possible. A successful case is the study of identifying novel E3 ligase targets Snail for ubiquitination and degradation through a luciferase-based screening method by Zheng et al.41 By the genome-wide screening with the ubiquitin-conjugated siRNA library, we finally identified F-box protein FBXO32 to be a novel E3 ligase, which specifically recognizes KLF4 and mediates its ubiquitination and proteasomal degradation. The pulse-chase assay revealed the degradation effect of FBXO32 on KLF4. The co-IP test and the ubiquitination assay confirmed the interaction between these two proteins and the increased ubiquitination of KLF4 in the presence of FBXO32. Then, we stably overexpressed FBXO32 in various breast cancer cell lines and found out a predominant decrease in KLF4 protein level, which is in agreement with the observation that KLF4 protein significantly increased when FBXO32 was specifically knocked down. Thus, we conclude that FBXO32 is a liable E3 ligase that mediates KLF4 turnover in breast cancer cells. Interestingly, a recent study reported that FBXO22 degrades KLF4 in hepatocellular carcinoma,42 whereas in our situation knockdown of FBXO22 had negligible effect on KLF4 protein level.
FBXO32 is originally defined as a muscle-specific gene involved in muscle atrophy. As an F-box protein, FBXO32 assembles with Cul1 to form a functional E3 ligase complex via its F-box domain.26–28 Intriguingly, in certain circumstances F-box domain is even dispensable for the E3 ligase function of FBXO32,38 thus we tested the role of F-box domain in FBXO32-mediated KLF4 degradation. As shown in Figure 3, deletion of F-box eliminated FBXO32-mediated KLF4 ubiquitination and degradation, which is consistent with the initial observation that F-box is necessary for the E3 ligase function of FBXO32. Therefore, F-box domain is required, at least in this situation, for FBXO32 function as an E3 ligase.
The substrate recognition domain of FBXO32 is also undefined. Unlike the FBXWs and the FBXLs, the FBXOs do not have any defined substrate-recognizing domain.29 However, the region C-terminal to the F-box domain is thought to be responsible for FBXO32 to interact with their substrates.26 Consistent with this, our result revealed that it is the C-terminal residues but not the N-terminal residues of FBXO32 that interact with KLF4. Notably, there is a class II PDZ domain at the extreme C-terminus of FBXO32 protein.26 As a protein-interacting motif, the PDZ could possibly be the exact substrate-interacting domain of FBXO32 whereas further validation is necessary.
In this study, we observed that inhibition of p38/MAPK signaling pathway delayed KLF4 degradation and abolished FBXO32-mediated KLF4 ubiquitination. Considering that F-box proteins usually recognize and interact with substrates in response to substrate phosphorylation,29 we speculated that KLF4 could be phosphorylated through p38/MAPK pathway before degraded by FBXO32. Coincidently, it is reported that FBXO32 itself is probably under regulation of p38/MAPK pathway, which means activation of p38/MAPK pathway leads to activated transcription of FBXO32.43,44 Although the detailed mechanism and the related signaling pathway still remain to be further explored. P38 MAPK pathway is involved in cellular response to a large variety of stress or stimuli.45 By inducing cell cycle arrest, differentiation and apoptosis, p38 can function as a tumor suppressor especially at the initiation of cellular transformation.46,47 Notably, the ability of p38 to inhibit stem cell renewal may also account for its tumor-suppressive effect.48 Consider that KLF4 is an important regulator of stem cell reprogramming,5 it is possible that KLF4 is a functional downstream target in p38-mediated inhibition of stem cell self-renewal. In addition, inactivation of p38 MAPK pathway may contribute to KLF4 accumulation and subsequent tumor initiation.
KLF4 is usually believed to act as an oncogene in breast cancer progression. The oncogenic role of KLF4 in breast cancer is probably related to its potential of inhibiting apoptosis and mediating cancer cell stemness.21 On the other hand, although enriched in muscle, several studies in recent years have unveiled the emerging role of FBXO32 in tumorigenesis.31–33,37,38 The fact that FBXO32 is reported to be downregulated in ovarian and esophageal cancer suggests that FBXO32 may function as a tumor suppressor.31,32 In addition, the fact that oncoproteins like c-Myc and p21 are ubiquitination substrates of FBXO32 further supported the plausible tumor-suppressive effect of FBXO32.33,38 As we identified FBXO32 as a novel E3 ligase responsible for KLF4 turnover, we further investigated the role of FBXO32-mediated KLF4 degradation in breast tumorigenesis. Our results revealed that stable knockdown of FBXO32 and the subsequent KLF4 accumulation in MCFCA1a cells led to a considerable increase in malignant transformation both in soft agar colony-forming assay and in a mouse model. The immunostaining results in paired breast cancer and adjacent normal tissues further validated the tumor-suppressive role of FBXO32 and the oncogenic role of KLF4 in breast cancer progression.
Collectively, our study provided a new insight into the mechanism of tumorigenesis in breast cancer. We found that FBXO32 is a novel E3 ligase for KLF4 turnover and further illustrated the molecular mechanism of FBXO32-mediated KLF4 ubiquitination and degradation. Functional study revealed that loss of FBXO32 expression facilitated breast cancer tumorigenesis both in vitro and in vivo. Moreover, this observation further verified the oncogenic role of KLF4 in breast cancer. In conclusion, our results made a regulatory connection between the tumor-suppressor FBXO32 and the oncogene KLF4 and provided a comprehensive understanding about the underlying mechanism of tumorigenesis in breast cancer.
MATERIALS AND METHODS
Cell culture
HEK293T, MCF7 and MCF10A cells were derived from American Type Culture Collection (Rockville, MD, USA). HEK293T and MCF7 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (Hyclone, South Logan, UT, USA). MCF10A and MCFCA1a cells49 were cultured in DMEM/F12 (1:1) (Gibco) with 5% horse serum (Hyclone).
Expression plasmids
The plasmids expressing KLF4, FBXO32 and other E3 ligase candidates were cloned from complementary DNA derived from MCF7 cells. The amino terminal KLF4 deletion mutants were constructed as described previously.22 Deletion mutants of FBXO32 were generated using the PCR directed mutagenesis method.
Antibodies and reagents
The following antibodies were used in western blot: anti-FBXO32 (NBP1-36955, Novus, Littleton, CO, USA); anti-FBXO32 (ab168372, Abcam, Cambridge, MA, USA); anti-KLF4 (sc-20691, Santa-Cruz, Dallas, TX, USA); anti-KLF4 (ab75486, Abcam); anti-ubiquitin (3936, CST, Billerica, MA, USA); anti-HA (sc-805, Santa-Cruz); anti-GST (G7781, Sigma, St Louis, MO, USA); anti-Flag (F7425, Sigma), anti-Myc (562, MBL, Woburn, MA, USA). The following antibodies were used in immunohistochemistry and immunofluorescent double staining: anti-FBXO32 (LS-B9748, Lifespan Biosciences, Seattle, WA, USA) and anti-KLF4 (NBP2-24749, Novus). The anti-KLF4 antibody (12173, CST) and anti-FLAG M2 Affinity Gel (A2220, Sigma) were used for IP. The Glutathione Sepharose 4B (17-0756-01, GE Healthcare, Amersham, UK) was used in GST pulldown. Cycloheximide (C7698) and MG132 (C2211) were purchased from Sigma. SB203580 was obtained from Promega (Madison, WI, USA) and SB239063 was purchased from Selleckchem (Houston, TX, USA).
siRNA library screening
The human ubiquitin conjugation ON-TARGET plus siRNA library against 195 ubiquitin ligases were purchased from Dharmacon (Dharmacon siRNA Technologies, Chicago, IL, USA). The screening was carried out using MCF7 cells. The protein level of KLF4 was detected with western blot 48 h after transfection.
The co-IP assay
To assess the interaction of exogenously expressed proteins, indicated plasmids were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with MG132 for 4 h before harvested. The whole-cell lysates were incubated with anti-Flag M2 affinity gel at 4 °C overnight. The beads were washed with cell lysis buffer and the immunoprecipitates were analyzed with western blot. To assess the interaction of endogenously expressed proteins, MCF7 cells or MCF10A cells were harvested after indicated treatment or not. The whole-cell lysates were incubated with 2 μg of KLF4 antibody or normal rabbit IgG at 4 °C overnight, and two additional hours with 20 μl of 50% protein A agarose. The immunoprecipitates were analyzed by western blot.
GST pulldown
Indicated plasmids were transfected into HEK293T cells. Twenty-four hours after transfection, the cells were treated with MG132 for 4 h, then lysed with cell lysis buffer. The cell lysates were incubated with glutathione sepharose at 4 °C overnight. The sepharose was collected by centrifugation and washed with phosphate-buffered saline. The concentrated proteins were eluted and subjected to western blot.
The in vivo ubiquitination assay
Cells with indicated treatment were incubated with MG132 for 6 h and lysed with denaturing lysis buffer. The cell lysates were boiled for 10 min, renatured with bovine serum albumin washing buffer and subjected to IP with anti-Flag M2 affinity gel or KLF4 antibody. The immunoprecipitates were analyzed by western blot.
Soft agar colony formation assay
In all, 30 000 MCFCA1a cells in 0.36% agarose was put on top of the bottom layer, which is composed of 0.75% agarose in DMEM plus 10% fetal bovine serum in a 60 mm dish. The dishes were kept in 37 °C incubator for 3 weeks and the colonies were stained with crystal violet.
Animal studies
Female BALB/c nude mice 6 weeks of age were used for orthotopic primary tumor formation. The mice were randomly divided into three groups (eight for each group). In all, 4 × 105 MCFCA1a cells suspended in 0.1 ml of saline were injected directly into the mammary fat pad. Three weeks later, the mice were killed and the primary tumors were weighted. The mice were annotated by number and the investigator was blinded to the groups. All procedures and experimental protocols were approved by Institutional Animal Care and Use Committee of Chinese Academy of Medical Sciences Cancer Hospital.
Immunohistochemistry
Tissue microarrays containing 35 paired tumor and adjacent normal tissues collected at Chinese Academy of Medical Sciences Cancer Hospital were stained by an immunoperioxidase method as described previously.50 Stained sections were scanned and analyzed with Aperio ImageScope system (Leica Biosystems, Buffalo Grove, IL, USA). Representative regions were scored according to the intensity (value of 0 for negative, 1 for weak positive, 2 for positive and 3 for strong positive) and extent (as a percentage of total cells) of staining. The investigator was blinded to the sample when grading. This study was approved by the ethical committee of the Chinese Academy of Medical Sciences Cancer Hospital, and informed consent was obtained from each patient.
Immunofluorescent double staining
Paraffin-embedded tissue array was blotted with KLF4 antibody and then labeled with FITC-conjugated goat-anti-rabbit antibody. Then the sample was incubated with FBXO32 antibody and labeled with CY3-conjugated goat-anti-rabbit antibody. The sample was then incubated with 4,6-diamidino-2-phenylindole for 5 min. The section was mounted and scanned with laser confocal microscopy.
Statistical analysis
Each experiment was repeated at least three times. For the protein degradation curves, all data are representative of the mean value of three independent experiments. The ± s.d were represented as error bars. For the soft agar colony-forming assay, the data from three independent experiments were subjected to one-way analysis of variance (two-tailed) by GraphPad Prism Version 5.01 (GraphPad Software, LaJolla, CA, USA). The ± s.e.m were represented as error bars. In the animal model, primary tumor weights were analyzed with unpaired t-test (two-tailed). For immunohistochemistry, statistical analyses were performed using paired t-test (two-tailed). For unpaired t-test and one-way analysis of variance, the variances between groups were compared by F-test and they are not significantly different, so we assumed equal variances; no sample was excluded from the analysis and all data meet normal distribution. P-values <0.05 were considered as significant.
Acknowledgments
This project was funded by National Natural Science Foundation of China (81130043 and 81420108025) and grants from the Ministry of Science and Technology (2016YFC1302100 and 2013CB911004).
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. A gene for a novel zinc-finger protein expressed in differentiated epithelial cells and transiently in certain mesenchymal cells. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 2.Rowland BD, Peeper DS. KLF4, p21 and context-dependent opposing forces in cancer. Nat Rev Cancer. 2006;6:11–23. doi: 10.1038/nrc1780. [DOI] [PubMed] [Google Scholar]
- 3.Ghaleb AM, Katz JP, Kaestner KH, Du JX, Yang VW. Kruppel-like factor 4 exhibits antiapoptotic activity following gamma-radiation-induced DNA damage. Oncogene. 2007;26:2365–2373. doi: 10.1038/sj.onc.1210022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiol Rev. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Shields JM, Christy RJ, Yang VW. Identification and characterization of a gene encoding a gut-enriched Kruppel-like factor expressed during growth arrest. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang W, Geiman DE, Shields JM, Dang DT, Mahatan CS, Kaestner KH, et al. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoon HS, Ghaleb AM, Nandan MO, Hisamuddin IM, Dalton WB, Yang VW. Kruppel-like factor 4 prevents centrosome amplification following gamma-irradiation-induced DNA damage. Oncogene. 2005;24:4017–4025. doi: 10.1038/sj.onc.1208576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shie JL, Chen ZY, Fu M, Pestell RG, Tseng CC. Gut-enriched Kruppel-like factor represses cyclin D1 promoter activity through Sp1 motif. Nucleic Acids Res. 2000;28:2969–2976. doi: 10.1093/nar/28.15.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon HS, Yang VW. Requirement of Kruppel-like factor 4 in preventing entry into mitosis following DNA damage. J Biol Chem. 2004;279:5035–5041. doi: 10.1074/jbc.M307631200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Q, Hong Y, Zhan Q, Shen Y, Liu Z. Role for Kruppel-like factor 4 in determining the outcome of p53 response to DNA damage. Cancer Res. 2009;69:8284–8292. doi: 10.1158/0008-5472.CAN-09-1345. [DOI] [PubMed] [Google Scholar]
- 13.Zhao W, Hisamuddin IM, Nandan MO, Babbin BA, Lamb NE, Yang VW. Identification of Kruppel-like factor 4 as a potential tumor suppressor gene in colorectal cancer. Oncogene. 2004;23:395–402. doi: 10.1038/sj.onc.1207067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei D, Kanai M, Huang S, Xie K. Emerging role of KLF4 in human gastrointestinal cancer. Carcinogenesis. 2006;27:23–31. doi: 10.1093/carcin/bgi243. [DOI] [PubMed] [Google Scholar]
- 15.Wang N, Liu ZH, Ding F, Wang XQ, Zhou CN, Wu M. Down-regulation of gut-enriched Kruppel-like factor expression in esophageal cancer. World J Gastroenterol. 2002;8:966–970. doi: 10.3748/wjg.v8.i6.966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tetreault MP, Yang Y, Travis J, Yu QC, Klein-Szanto A, Tobias JW, et al. Esophageal squamous cell dysplasia and delayed differentiation with deletion of kruppel-like factor 4 in murine esophagus. Gastroenterology. 2010;139:e179. doi: 10.1053/j.gastro.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohnishi S, Ohnami S, Laub F, Aoki K, Suzuki K, Kanai Y, et al. Downregulation and growth inhibitory effect of epithelial-type Kruppel-like transcription factor KLF4, but not KLF5, in bladder cancer. Biochem Biophys Res Commun. 2003;308:251–256. doi: 10.1016/s0006-291x(03)01356-1. [DOI] [PubMed] [Google Scholar]
- 18.Hu W, Hofstetter WL, Li H, Zhou Y, He Y, Pataer A, et al. Putative tumor-suppressive function of Kruppel-like factor 4 in primary lung carcinoma. Clin Cancer Res. 2009;15:5688–5695. doi: 10.1158/1078-0432.CCR-09-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei D, Wang L, Yan Y, Jia Z, Gagea M, Li Z, et al. KLF4 is essential for induction of cellular identity change and acinar-to-ductal reprogramming during early pancreatic carcinogenesis. Cancer Cell. 2016;29:324–338. doi: 10.1016/j.ccell.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster KW, Frost AR, McKie-Bell P, Lin CY, Engler JA, Grizzle WE, et al. Increase of GKLF messenger RNA and protein expression during progression of breast cancer. Cancer Res. 2000;60:6488–6495. [PubMed] [Google Scholar]
- 21.Yu F, Li J, Chen H, Fu J, Ray S, Huang S, et al. Kruppel-like factor 4 (KLF4) is required for maintenance of breast cancer stem cells and for cell migration and invasion. Oncogene. 2011;30:2161–2172. doi: 10.1038/onc.2010.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu D, Wan Y. Regulation of Kruppel-like factor 4 by the anaphase promoting complex pathway is involved in TGF-beta signaling. J Biol Chem. 2011;286:6890–6901. doi: 10.1074/jbc.M110.179952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim MO, Kim SH, Cho YY, Nadas J, Jeong CH, Yao K, et al. ERK1 and ERK2 regulate embryonic stem cell self-renewal through phosphorylation of Klf4. Nat Struct Mol Biol. 2012;19:283–290. doi: 10.1038/nsmb.2217. [DOI] [PubMed] [Google Scholar]
- 24.Gamper AM, Qiao X, Kim J, Zhang L, DeSimone MC, Rathmell WK, et al. Regulation of KLF4 turnover reveals an unexpected tissue-specific role of pVHL in tumorigenesis. Mol Cell. 2012;45:233–243. doi: 10.1016/j.molcel.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu D, Zhou Z, Davidson NE, Huang Y, Wan Y. Novel insight into KLF4 proteolytic regulation in estrogen receptor signaling and breast carcinogenesis. J Biol Chem. 2012;287:13584–13597. doi: 10.1074/jbc.M112.343566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kipreos ET, Pagano M. The F-box protein family. Genome Biol. 2000;1:REVIEWS3002.1–REVIEWS3002.7. doi: 10.1186/gb-2000-1-5-reviews3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 29.Skaar JR, Pagan JK, Pagano M. SnapShot: F box proteins I. Cell. 2009;137:1160–1160 e1161. doi: 10.1016/j.cell.2009.05.039. [DOI] [PubMed] [Google Scholar]
- 30.Jogo M, Shiraishi S, Tamura TA. Identification of MAFbx as a myogenin-engaged F-box protein in SCF ubiquitin ligase. FEBS Lett. 2009;583:2715–2719. doi: 10.1016/j.febslet.2009.07.033. [DOI] [PubMed] [Google Scholar]
- 31.Chou JL, Su HY, Chen LY, Liao YP, Hartman-Frey C, Lai YH, et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest. 2010;90:414–425. doi: 10.1038/labinvest.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo W, Zhang M, Guo Y, Shen S, Guo X, Dong Z. FBXO32, a new TGF-beta/Smad signaling pathway target gene, is epigenetically inactivated in gastric cardia adenocarcinoma. Neoplasma. 2015;62:646–657. doi: 10.4149/neo_2015_078. [DOI] [PubMed] [Google Scholar]
- 33.Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W. FBXO32 targets c-Myc for proteasomal degradation and inhibits c-Myc activity. J Biol Chem. 2015;290:16202–16214. doi: 10.1074/jbc.M115.645978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen ZY, Wang X, Zhou Y, Offner G, Tseng CC. Destabilization of Kruppel-like factor 4 protein in response to serum stimulation involves the ubiquitin-proteasome pathway. Cancer Res. 2005;65:10394–10400. doi: 10.1158/0008-5472.CAN-05-2059. [DOI] [PubMed] [Google Scholar]
- 35.Sarikas A, Hartmann T, Pan ZQ. The cullin protein family. Genome Biol. 2011;12:220. doi: 10.1186/gb-2011-12-4-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cardozo T, Pagano M. The SCF ubiquitin ligase: insights into a molecular machine. Nat Rev Mol Cell Biol. 2004;5:739–751. doi: 10.1038/nrm1471. [DOI] [PubMed] [Google Scholar]
- 37.Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–1063. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18:1771–1779. doi: 10.1038/cdd.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster KW, Liu Z, Nail CD, Li X, Fitzgerald TJ, Bailey SK, et al. Induction of KLF4 in basal keratinocytes blocks the proliferation-differentiation switch and initiates squamous epithelial dysplasia. Oncogene. 2005;24:1491–1500. doi: 10.1038/sj.onc.1208307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shie JL, Chen ZY, O’Brien MJ, Pestell RG, Lee ME, Tseng CC. Role of gut-enriched Kruppel-like factor in colonic cell growth and differentiation. Am J Physiol Gastrointest Liver Physiol. 2000;279:G806–G814. doi: 10.1152/ajpgi.2000.279.4.G806. [DOI] [PubMed] [Google Scholar]
- 41.Zheng H, Shen M, Zha YL, Li W, Wei Y, Blanco MA, et al. PKD1 phosphorylation-dependent degradation of SNAIL by SCF-FBXO11 regulates epithelial-mesenchymal transition and metastasis. Cancer Cell. 2014;26:358–373. doi: 10.1016/j.ccr.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tian X, Dai S, Sun J, Jin G, Jiang S, Meng F, et al. F-box protein FBXO22 mediates polyubiquitination and degradation of KLF4 to promote hepatocellular carcinoma progression. Oncotarget. 2015;6:22767–22775. doi: 10.18632/oncotarget.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derbre F, Ferrando B, Gomez-Cabrera MC, Sanchis-Gomar F, Martinez-Bello VE, Olaso-Gonzalez G, et al. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: role of p38 MAPKinase and E3 ubiquitin ligases. PloS One. 2012;7:e46668. doi: 10.1371/journal.pone.0046668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner EF, Nebreda AR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. 2009;9:537–549. doi: 10.1038/nrc2694. [DOI] [PubMed] [Google Scholar]
- 46.Dolado I, Swat A, Ajenjo N, De Vita G, Cuadrado A, Nebreda AR. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11:191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 47.Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, Kenner L, et al. p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet. 2007;39:741–749. doi: 10.1038/ng2033. [DOI] [PubMed] [Google Scholar]
- 48.Ventura JJ, Tenbaum S, Perdiguero E, Huth M, Guerra C, Barbacid M, et al. p38alpha MAP kinase is essential in lung stem and progenitor cell proliferation and differentiation. Nat Genet. 2007;39:750–758. doi: 10.1038/ng2037. [DOI] [PubMed] [Google Scholar]
- 49.Santner SJ, Dawson PJ, Tait L, Soule HD, Eliason J, Mohamed AN, et al. Malignant MCF10CA1 cell lines derived from premalignant human breast epithelial MCF10AT cells. Breast Cancer Res Treat. 2001;65:101–110. doi: 10.1023/a:1006461422273. [DOI] [PubMed] [Google Scholar]
- 50.Luo A, Yu X, Li G, Ma G, Chen H, Ding F, et al. Differentiation-associated genes regulated by c-Jun and decreased in the progression of esophageal squamous cell carcinoma. PloS One. 2014;9:e96610. doi: 10.1371/journal.pone.0096610. [DOI] [PMC free article] [PubMed] [Google Scholar]