

ABSTRACT
Tumor cells, which undergo Epithelial-mesenchymal transition (EMT) acquire increased capacities of proliferation, invasion and have the ability to generate metastases by escaping the immune system during their systemic migration. To escape the immune system, cancer cells may induce tolerance or resist elimination by immune effectors via multiple mechanisms and we hypothesized that EMT may control the expression of immune checkpoint inhibitors, then promoting immune evasion. PD-L1 (programmed cell death ligand 1) but not PD-L2 nor Galectin 9 or Death receptor (DR4, DR5 and Fas) and ligands (FasL and TRAIL) expression was up-regulated during cytokine-driven EMT in a reversible manner. Moreover PD-L1 is overexpressed in VIMENTIN positive NSCLC tissues. We also demonstrated that the expression of PD-L1 required both TNFα and TGFβ1. Indeed, TGFβ1 decreased DNMT1 content and that resulted in PD-L1 promoter demethylation whereas TNFα induced the NF-κB pathway that promoted expression of demethylated PD-L1 promoter.
KEYWORDS: death receptors; DNA methylation; epithelial-mesenchymal transition; lung cancer, cancer; NF-kB; PD-L1
Introduction
EMT is a highly conserved cellular process which allows epithelial cells to convert to mesenchymal cells.1 The loss of E-CADHERIN expression is a crucial step in EMT and a hallmark of mesenchymal phenotype, whereas VIMENTIN is a canonical marker used to detect fully transitioned epithelial cells and the acquisition of fibroblastic-like phenotype. EMT is involved in different physio-pathological contexts including: embryonic and organ development (type 1 EMT), tissue regeneration and fibrosis (type 2 EMT), or carcinoma progression (type 3 EMT). Oncogenesis, hypoxia or exposure to chronic inflammation are the main environmental conditions promoting the ability of tumor cells to undergo EMT.2 TGF-β1, TNFα, HGF, EGF, PDGF, Wnt or NOTCH ligands are soluble factors which have been described to control mesenchymal phenotype acquisition during cancer progression.3 Mesenchymal program relies on the expression of specific transcription factors such as ZEB, SNAIL and TWIST families, which repress E-CADHERIN, induce the expression of molecules involved in cytoskeleton regulation and promote the secretion of metalloproteinases involved in invasion.4 Tumor cells, which undergo EMT, show increase proliferation, migration and resistance to apoptosis. By co-opting a program involved in embryonic development, carcinoma cells are able to invade normal tissues and induce metastases. EMT also confers to carcinoma cells, stem cell like properties indispensable to clonal expansion and initiation of metastases.5 Altogether, EMT is one of the major molecular mechanisms carried out during oncogenesis to promote cancer progression. One other crucial step of oncogenesis is the acquisition by cancer cells of the ability to evade the immune system.6 Recent clinical advances in immunotherapy demonstrated that some cancers, with established lymphocyte infiltrates, express immune checkpoint inhibitory molecules, such as PD-L1. Several studies suggested that PD-1/PD-L1 status may be a new indicator of poor prognosis for patients with lung cancers, head and neck or melanoma.7-10 In these patients, treatment with anti-PD-L1/PD-1 restored effective antitumor immunity and induced long lasting objective responses, demonstrating the pivotal role of such immune inhibitory signaling in oncogenesis. However, the understanding of the mechanisms governing PD-L1 expression in cancer may improve the medical approaches to restore immunosurveillance. We therefore hypothesized that the expression of immune checkpoint inhibitors such as PD-L1, might be specifically regulated during EMT to allow invasion and immune escape. Indeed, the role of ZEB-1 and miR200 on PD-L1 in EMT was recently showed in breast cancer models.11 To investigate this issue, we selected a model of reversible EMT based on a TNF-α/TGF-β1 treatment of the NSCLC cell line A549. Expression of death receptors or ligands, NK cell activating or inhibitory molecules, as well as immune checkpoint inhibitors were analyzed together with the EMT status and we observed that induced EMT in NSCLC does not modify the expression of NK cell receptor ligands or inhibitory molecules as well as death receptors or ligands. By contrast, we demonstrated that PD-L1 was strongly up-regulated in in vitro EMT cell models and in NSCLC EMT tissues. Nevertheless, PD-L1 expression was not correlated to EMT specific transcription factors but we evidenced for the first time that the control of PD-L1 expression is coordinated by two processes during EMT. Indeed, PD-L1 expression required both the demethylation of its promoter that was induced by TGFβ treatment and the activation of the NF-κB/IKKε pathway and the recruitment of NF-κB on PD-L1 promoter following TNFα treatment. In summary, our results clearly reported that tumoral microenvironment and EMT that control PD-L1 expression are key points that have to be appreciated in the future for anti-cancer therapies and to limit risks of metastasis.
Results
Effects of cytokine-induced EMT in immune checkpoint inhibitor expression
In order to assess how EMT influences the recognition of tumor cells by lymphocytes during invasion, we developed a reversible model of EMT (cytokines (TNF-α /TGF-β1)-induced EMT).12-14 For this purpose, A549 cell line was exposed for 5 days to TNF-α and TGF-β1 (Fig. 1A). This treatment allowed this epithelial cell line to acquire mesenchymal tumor cells features (Fig. 1B and Supp. Fig. 1). Removal of cytokines led to the restoration of an epithelial phenotype within 5 days (Fig. 1A and 1B). EMT status was confirmed by western-blotting showing that TNF-α and TGF-β1 treatment repressed epithelial marker E-CADHERIN expression but induced overexpression of mesenchymal marker VIMENTIN (Fig. 1C). Similarly, TNF-α and TGF-β1 induced a reversible loss of EPCAM (Fig. 1D). EMT phenotype conferred by exposition to cytokine combination was also associated with a lengthening of cells as observed by F-ACTIN staining (Fig. 1E) and an enhanced invasive capacity of A549 cells (Fig. 1F).
Figure 1.
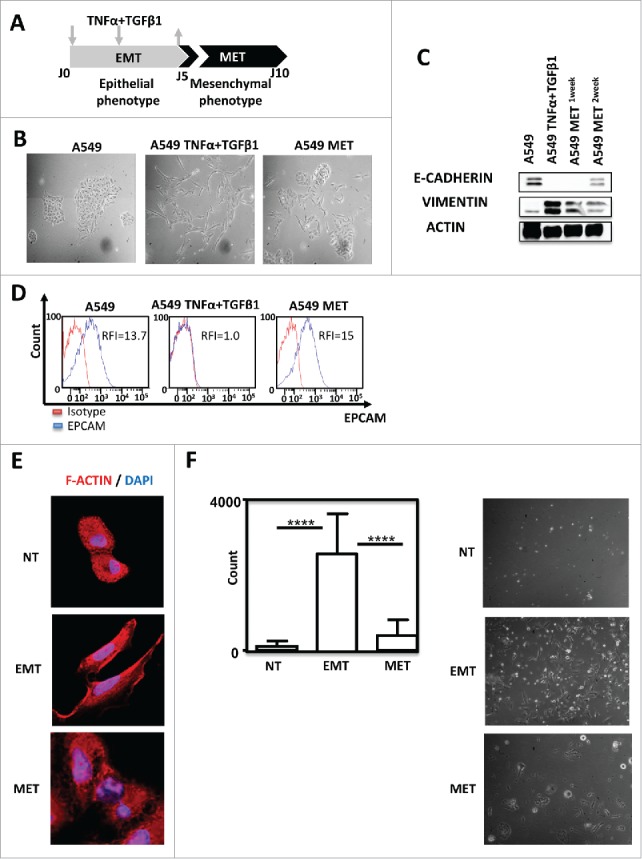
TNF-α and TGF-β1 combination induced A549 EMT in a reversible manner. (A) Pipeline of EMT induction and reversion (MET) in A549 cells. Cells were treated for 5 days with TNFα/TGF-β1 and then the cytokines were removed during the 5 next days (arrows) (B) Lung cancer cells A549 were observed on the fifth day of adherence with or without TNFα/TGF-β1 treatment under the microscope (40X magnification). To observe the cells undergoing MET (mesenchymal epithelial transition), the cytokines TNF-α and TGF-β1 were removed during the next 5 days. (C) Expression of epithelial marker E-CADHERIN and mesenchymal marker VIMENTIN in A549 after treatment with TNF-α and TGF-β1 were measured by Western-Blotting. One or two weeks after the removal of cytokines, expressions of E- CADHERIN and VIMENTIN were tested again. (D) EPCAM staining measured by FACS after treatment with TNF-α and TGF-β1, as well as, after the removal of cytokines during the next five days. The experiments in panels B, C and D were realized 4 times with similar results. (E) Representative staining of F-Actin using Rhodamine Phalloidin in A549 treated or not with TNF-α/TGF-β1. Nuclei were stained with DAPI. (F) The change in the invasive capacity of A549 with or without TNFα/TGF-β1 treatment was measured using Matrigel system. This experiment was performed in duplicate, and repeated 3 times (left: quantification; right: representative pictures).
Effector immune cells can eliminate carcinoma cells throughout different mechanisms: one of which is the production of pro-apoptotic ligands, such as TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL). These compounds can trigger extrinsic apoptosis through the activation of death receptors, Death Receptor 4 (DR4) / Death Receptor (DR5) and Fas. We then first assessed the expression of DR4, DR5 and Fas in A549 cells exposed or not to TNF-α and TGF-β1. We observed that death receptor expressions were not significantly modified during the acquisition of mesenchymal phenotype by A549 (Supp. Fig. 2A). Moreover, EMT did not induce the production of TRAIL or FasL (data not shown) nor HLA-G expression (supp. Fig. 2B). Another mechanism involved in the eradication of tumor cells during immunosurveillance is the recognition of NKG2D ligands (MICA, MICB, ULBP1–3) by CD8 T lymphocytes or NK cells.15-17 TNF-α/TGF-β1 treatment did not influence the expression of the NKG2D ligands (Supp. Fig. 2C). Similarly, EMT did not regulate NKp30-Fc, NKp46-Fc and DNAM-Fc expression. Of note, we did not observe in these experiments any significant modification in MHC class I (Supp. Fig. 2C). Altogether, these experiments showed that the modulation of molecules regulating the recognition of tumor cells by NK was unlikely. Indeed, a 5 days TNF-α and TGF-β1 treatment of A549 did not hamper the capacity of activated NK to recognize and induce A549 cell death nor to produce IFN-γ following co-cultures with this cell line (Supp. Fig. 3A and 3B).
The next set of experiments was performed to investigate the modulation of immune-checkpoint ligands according to EMT status. Using flow cytometry experiments we showed that TNF-α/ TGF-β1 co-treatment did not influence the expression of the TIM3 ligands Galectine 9 or CEACAM1 (CD66). PD-L2, Vista were not expressed in this model (Fig. 2A). On the opposite, EMT induction, (monitored by the loss of EPCAM), led to the up-regulation of PD-L1 (Fig. 2A). Interestingly, PD-L1 expression disappeared following removal of TNF-α and TGF-β1 and induction of MET (Fig. 2B). Finally, we used a specific inhibitor of TGF-β receptor to block TNF-α/TGF-β1-induced EMT (Supp. Fig. 4A and 4B). TGF-β receptor inhibition blocked PD-L1 expression (Supp. Fig. 4C). These results indicated that cytokine-mediated EMT promoted the expression of PD-L1. We next sought to determine which cytokine, TNF-α or TGF-β1 was responsible of the induction of PD-L1. For this purpose, A549 cells were treated with TNF-α, TGF-β1 or with the combination during 5 days. As previously described,13,14,18 TNF-α or TGF-β1 treatment alone failed to drive an efficient mesenchymal transformation of A549 cells in short term cultures (data not shown). Nonetheless a decrease of EPCAM expression was observed following treatment with TGF-β1 and at a lower level with TNF-α alone and was more important with the combined TNF-α/TGF-β1 treatment (Fig. 2C). In addition, treatment with each cytokine alone, was not able to induce PD-L1 expression but cytokine combination promoted significant up-regulation of PD-L1 (Fig. 2C). The up-regulation of PD-L1 by exposition to TNF-α and TGF-β1 was also confirmed using ELISA (Fig. 2D) and qRT-PCR (Fig. 2E).
Figure 2.
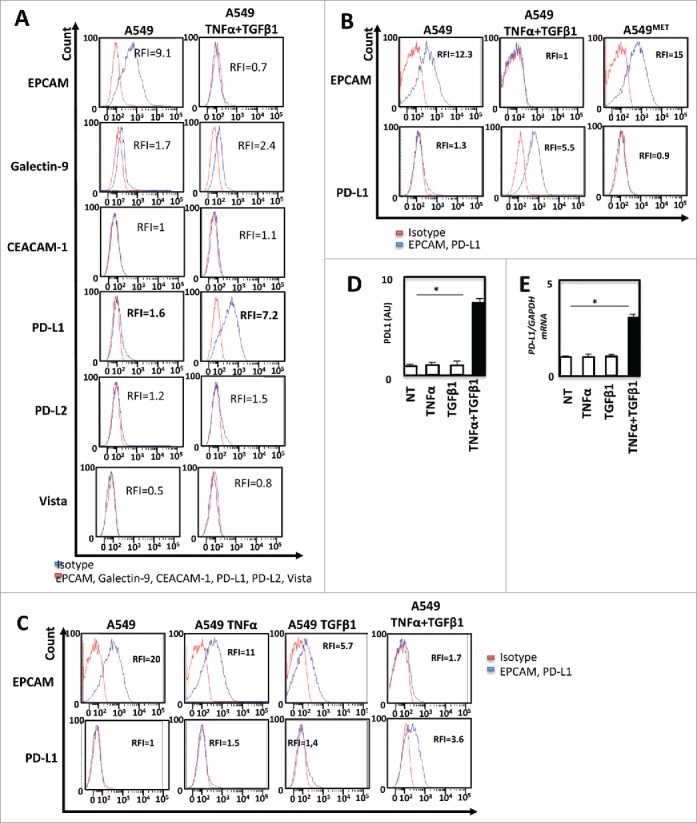
TNF-α and TGF-β1 cooperate to promote PD-L1 expression during EMT (A) The expression of immune-checkpoint inhibitors as Galectin-9, CEACAM1, PD-L1, PD-L2, Vista before and after induction of EMT on A549 cells was measured by FACS and measurements were independently repeated at least three times. (B) By FACS the changes in the expression of PD-L1 and EPCAM were also confirmed with 5 days of treatment and after the removal of cytokines during the next five days. (C) At last, the changes in the expression of PD-L1 with TNF-α or TGF-β1 alone, as well as with the combination of these cytokines were measured by FACS. (D and E) Expression of PD-L1 protein and mRNA were measured following TNFα/TGF-β1 treatment by ELISA or qRT-PCR. These experiments were repeated three times with similar results.
Expression of PDL1 is dependent of EMT
The increased transcription of PD-L1 during cytokine-induced EMT, prompted us to characterize the expression of EMT-related transcriptional regulators in A549 cells. Western blotting experiments showed an increased expression of ZEB-1 and SNAIL1 in cells treated with TNF-α/TGF-β1, while GLI, SNAIL2/SLUG, TWIST levels were not modified by the treatment (Fig. 3A). qRT-PCR analyses confirmed the induced expression of SNAI1 (coding SNAIL1) and ZEB-1 mRNA during EMT (Fig. 3B). Of note, TNF-α alone increased ZEB-1 while TGF-β1 exposition enhanced SNAIL1 expression ruling out a direct role of these transcriptional modulators in PD-L1 regulation. Moreover, SNAI1 stable transfection in A549 cells (Fig. 3C), did not increase the expression of PD-L1 expression, even when this cell line was exposed to TNF-α and TGF-β1 compared to control cells (Fig. 3D). Finally, we inhibited the SMAD pathway using a chemical inhibitor (SIS3) and once again this treatment did not block PD-L1 induction (Supp. Fig. 4B). These data strongly suggested that PD-L1 regulation is, at least in part, independent of classical EMT transcriptional factors.
Figure 3.
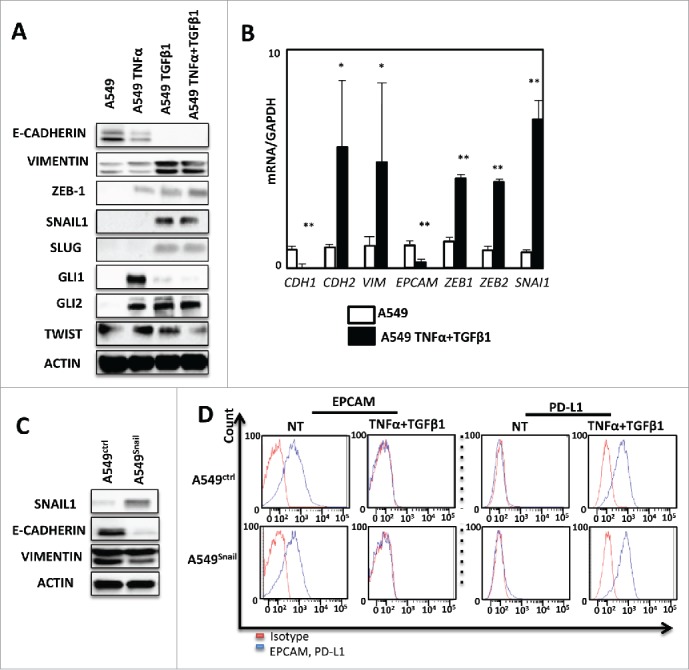
PD-L1 expression is not regulated by conventional EMT transcriptional factors in A549. (A) Expression of several transcription factors of EMT as: ZEB-1, SNAIL-1, SLUG, GLI1, GLI2, TWIST in protein levels was measured by Western Blotting after 5 days of treatment of A549 cells with TNF-α and TGFβ. This experiment was realized 3 times. (B) qRT-PCR analysis was realized to test the expression of different transcription factors of EMT mRNA levels after treatment with cytokines. GAPDH was used as a housekeeping gene. (C) The expression of VIMENTIN and E-CADHERIN were measured using Western-blotting in A549 cells after transfection with SNAIL-1 or control vector. (D) expression of EPCAM and PD-L1 was measured by FACS analysis during treatment with TNF-α and TGFβ on A549 cancer cells transfected with SNAIL-1 or control vector. Results from Fig. 3A and 3 C and 3D are representative examples from at least 3 independent experiments.
In order to understand whether PD-L1 induction is really associated to EMT, we stably expressed E-CADHERIN in A549 cells (Fig. 4A). As expected, both β-CATENIN and E-CADHERIN stainings were increased in E-CADHERIN expressing cells compared to control cells (Fig. 4B). We next quantified PD-L1 expression in these cells and we observed a non-significant decrease of PD-L1 mRNA in E-CADHERIN expressing cells but as mentioned above, basal PD-L1 expression is already low in A549 cells (Fig. 4C). Moreover, the overexpression of E-CADHERIN strongly blocked the TNF-α/TGF-β1-mediated PD-L1 induction (Fig. 4D). These data clearly showed that PD-L1 expression in A549 cells is dependent of TNF-α, TGF-β1 and EMT.
Figure 4.
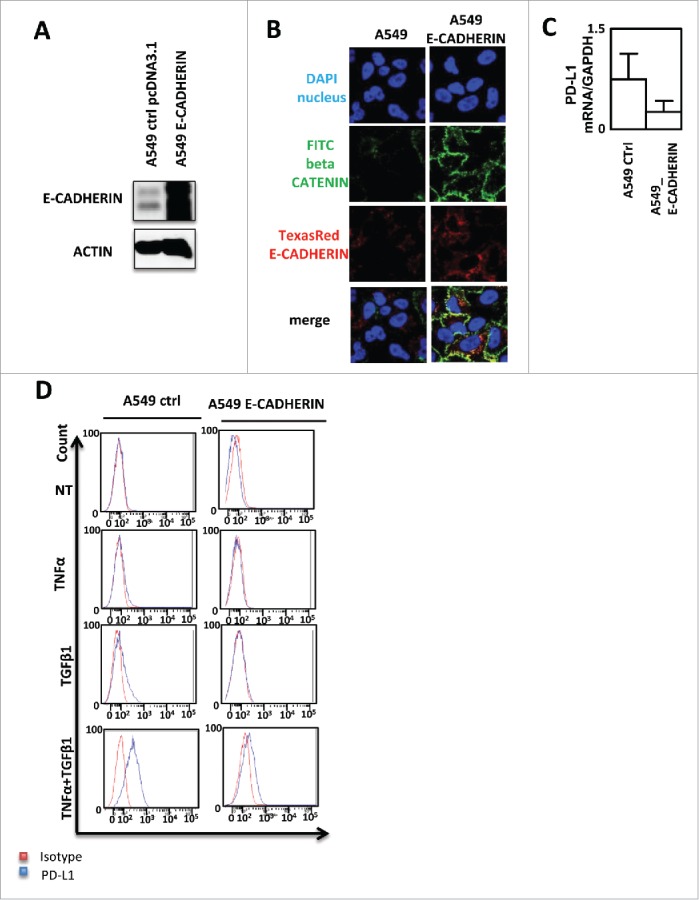
PD-L1 expression blockade by E-CADHERIN overexpression during EMT in A549 cells. (A) Expression of E-CADHERIN was controlled by western blotting in A549 transfected by a pcDNA3.1 void vector or expressing E-CADHERIN. (B) reversion of mesenchymal to epithelial phenotype was measured using immunofluorescence in A549 transfected by a pcDNA3.1 void vector or expressing E-CADHERIN. Nuclei were stained by DAPI (blue), beta CATENIN by FITC (green) and E-CADHERIN by TexasRed (Red). (C) expression of PD-L1 mRNA was measured by qRT-PCR in A549 control or A549 cells transfected with a plasmid expressing E-CADHERIN (D) Expression of PD-L1 was measured by cytometry in A549 transfected by a pcDNA3.1 void vector or expressing E-CADHERIN treated or not with TNFα/TGFβ1.
TNF-α is required for PD-L1 expression via NF-κB signaling during EMT
In order to decipher the mechanisms implicated in the control of PD-L1 expression during cytokine-induced EMT, we first analyzed the transcription factor binding sites within the PD-L1 promoter. NF-κB was the main transcription factor, previously described as promoting EMT.19-22 and presenting a binding site in the PD-L1 promoter. A strong increase of NF-κB activity was observed following treatment with TNF-α alone or TNF-α combined with TGF-β1 but not with TGF-β1 alone (Fig. 5A). Consequently, we next deiced to investigate the role of NF-κB subunits in the control of PD-L1 expression during EMT. JSH23, a pharmacological inhibitor of NF-κB, was used to prevent NF-κB expression and nuclear translocation of NF-κB subunits in A549 cells treated with both TNF-α/TGF-β1. JSH-23 selectively blocked nuclear translocation of active NF-кB without affecting the process of IkB degradation and suppress its binding to its target DNA consensus sequence.23,24 As previously described, JSH23 inhibited the induction of NF-κB subunit in A549 following cytokine-induced EMT (Fig. 5B). Indeed, JSH23 only slightly prevented PD-L1 expression in these cells (Fig. 5C). Interestingly, any effect of JSH23 on PD-L1 expression could also be observed in the epithelial HT29 cells whose basal (without EMT induction) PD-L1 expression is very low and comparable to A549 cells, whereas JSH23 strongly blocked PD-L1 expression in the mesenchymal MDA-MB-231 cells expressing high levels of VIMENTIN and PD-L1 (Supp Fig. 5A–C) suggesting the implication of NF-κB in PD-L1 expression. As expected p52 NF-κB subunit overexpression alone could not restore PD-L1 expression in A549 cells (Supp Fig. 6).
Figure 5.
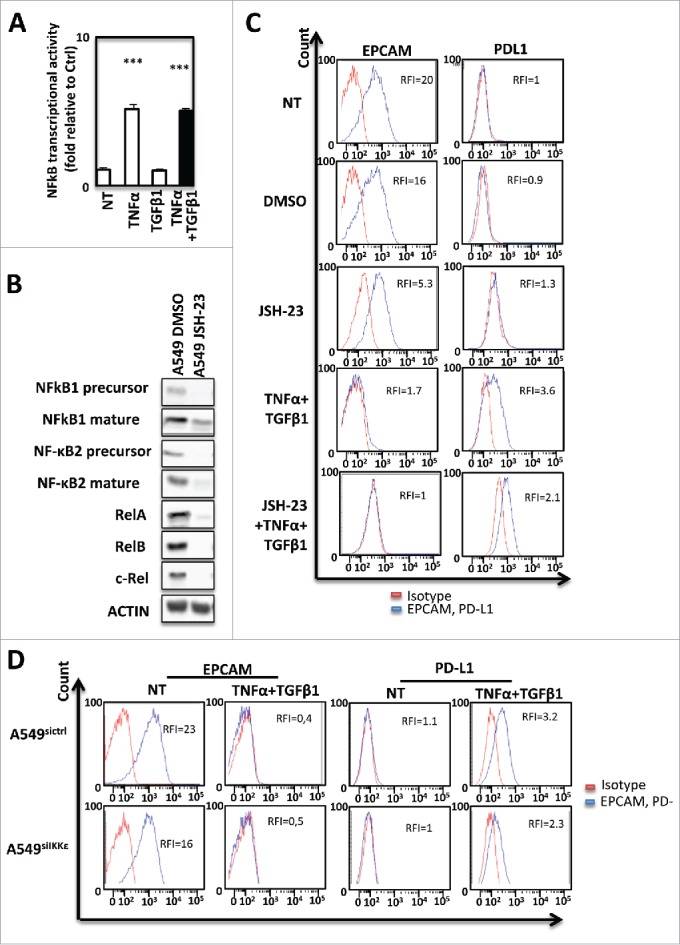
PD-L1 expression during EMT is dependent from NF-kB in signaling in A549 cells. (A) Activity of NF-kB was measured in A549 cells treated or not with TNF-α and TGFβ1. (B) The expression of NF-kB subunits was measured by Western Blotting after treatment with NF-kB inhibitor. (C) The expression of PD-L1 was measured using cytometry following NF-kB inhibition with JSH-23. (D) Expression of EPCAM and PD-L1 was measured using cytometry in A549 treated or not with TNF-α and TGFβ1 and transfected or not with si IKKε. These experiments were repeated tree times with similar results.
Figure 6.
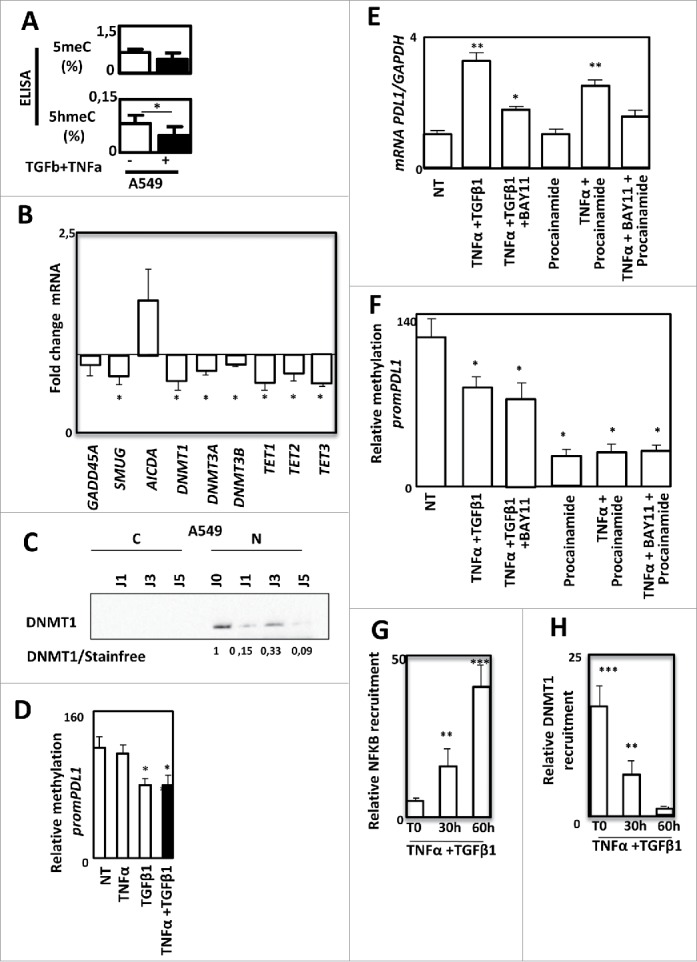
PD-L1 expression is regulated by DNA methylation during EMT in A549 cells. Quantification of 5meC and 5hmeC content using ELISA in A549 cells treated or not with TNF-α and TGFβ1. (B) Quantification of GADD45 A, SMUG, AICA, DNMT1, DNMT3 A, DNMT3B, TET1, TET2 and TET3 gene expression using qRT-PCR in A549 treated or not with TNF-α and TGFβ1. (C) Expression of DNMT1 using Western blotting in cytosolic or nuclear fractions of A549 cells treated or not with TNF-α and TGFβ1. (D) Methylation of PD-L1 promoter was quantified in A549 cells treated or not with TNF-α and/or TGFβ1. (E and F) Expression of PD-L1 mRNA using qRT-PCR and methylation of PD-L1 promoter were quantified in A549 cells treated or not with TNF-α and TGFβ1 in presence or not of BAY11, or cells treated with procalamide or treated with procalamide, TNF-α and BAY11. (G and H) Recruitment of NF-kB and recruitment of DNMT1 were quantified by ChIP in in A549 cells treated or not with TNF-α and TGFβ1.
We next analyzed the direct role of the non canonical kinase IKKε on PD-L1 regulation since IKKε expression was strongly induced by TNF-α/TGF-β1 treatment (Supp. Fig. 7A). To determine whether IKKε was also involved in the NF-κB-dependent PD-L1 induction, IKKε was genetically expressed in A549 cells (Supp Fig. 7B). According to our previous results obtained with NF-κBp52, IKKε ovexpression alone was not able to significantly promote PD-L1 expression. (Supp Fig. 7C and 7D). However, pharmacological inhibition using three different inhibitors (Bx795, IKK3 and Amlexanox) (Supp. Fig. 8A–8C) and siRNA mediated transcription blockade (Fig. 5D) were used to prevent IKKε activity during cytokine-induced EMT and significantly decreased PD-L1 induction. Altogether, these results highlight the involvement of NF-κB/ IKKε signaling in EMT-induced PD-L1 expression on tumor cells but suggest that the NF-κB pathway is only one parameter.
Figure 7.
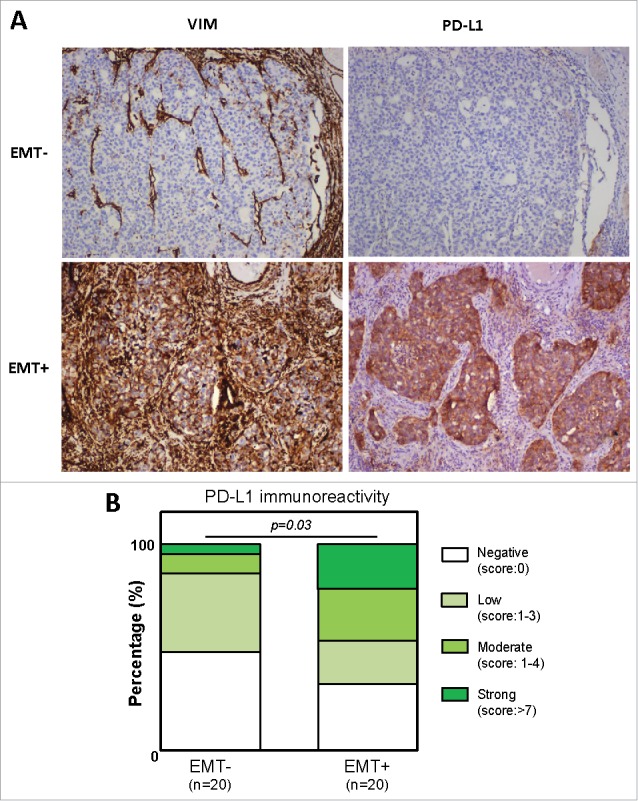
PD-L1 expression is correlated to EMT-like phenotype. PD-L1 and VIMENTIN expression was quantified using IHC in a cohort of 40 non small cell lung carcinoma. (A) Examples of anti-VIMENTIN and anti-PD-L1 immunostainings in EMT- and EMT+ tumors. (B) Quantification of PD-L1 staining in EMT- and EMT+ tumors.
DNA methylation control of the PD-L1 expression by TGF-β1 during EMT
As EMT is a highly plastic process, we next analyzed whether DNA methylation, could be involved in the control of PD-L1 expression. Indeed, a modification of global DNA methylation has already been reported during EMT.25 Moreover, DNMTs have also been directly involved in EMT process.26 We indeed observed a small decrease of DNA methylation and a significant decrease of the 5hmeC following TNF-α/TGF-β1 treatment in A549 cells (Fig. 6A). These modifications were also confirmed in another model, the ACHN cells in which EMT was also induced by TNF-α/TGF-β1 and associated with a moderate increase of PD-L1 expression (Supp. Figure 9A and 9B). We next quantified the expression of genes coding enzymes involved in DNA methylation (Fig. 6B). We found that all DNMTs (DNA methyl transferase) and TET (ten eleven translocation) genes were downregulated by TNF-α/TGF-β1 treatment suggesting that global DNA methylation modification is due to a decline of DNA methylation related proteins. These data were confirmed at the protein level as we showed a decrease of DNMT1 content, the main protein known to process the maintenance of DNA methylation following TNF-α/TGF-β1 treatment in A549 cells (Fig. 6C). A similar decrease of both DNMT1 mRNA and protein were also observed in the ACHN model (Supp. Figures 9C and D). In light of these elements, we wondered whether PD-L1 expression could be regulated by DNA methylation. As shown in Fig. 6D, TNF-α/TGF-β1 effectively reduced the methylation status of PD-L1 promoter. A similar demethylation was also observed using TGF-β1 alone but not with TNF-α alone suggesting that PD-L1 promoter methylation is rather controlled by TGF-β1 than TNF-α (Fig. 6D). Since earlier showed that NF-κB was partially involved in PD-L1 regulation (Fig. 5), and that a synergistic expression of PD-L1 was observed with the combined TNF-α/TGF-β1 treatment (Fig. 2), we asked whether PD-L1 expression could be controlled simultaneously by DNA methylation and NF-κB signaling. Indeed, inhibition of NF-κB, using a specific inhibitor (BAY11), strongly reduced the PD-L1 expression of cells treated with TNF-α/TGF-β1 (Fig. 6E). Inhibition of DNA methylation using procainamide, a specific inhibitor of DNMT1 had no effect suggesting that inhibition of DNA methylation alone could not restore PD-L1 expression. Single treatment of A549 cells with 5-aza-deoxycytidine, another DNA demethylation agent, also failed to restore PD-L1 expression (data not shown). Moreover, inhibition of DNA methylation in presence of TNF-α is sufficient to induce PD-L1 expression while addition of the NF-κB inhibitor blocked this induction (Fig. 6E). We next confirmed that procainamide induced the demethylation of PD-L1 promoter but that the inhibitor of NF-κB had not effect on this methylation (Fig. 6F). Moreover, the mesenchymal MDA-MB-231 cells expressing a high level of PD-L1 in basal conditions (Supp. Fig. 5), also presented an unmethylated PD-L1 promoter (Supp. Figure 10). To confirm that both DNA methylation and NF-κB signaling are required for PD-L1 regulation, we finally analyzed the recruitment of both NF-κB /p65 and DNMT1 on the PD-L1 promoter of A549 cells. We showed that TNF-α/TGF-β1 treatment induced an increase of NF-κB/p65 recruitment concomitant with a loss of DNMT1 recruitment on the PD-L1 promoter (Figu. 6G and 6H).
Finally, to confirm that PD-L1 expression is associated with EMT in vivo, we quantified PD-L1 expression in tumor cells by IHC staining in a cohort of 40 NSCLC patients. These latter were subclassified into two groups based on VIMENTIN expression [EMT-negative (n = 20) versus EMT-positive tumors (n = 20)] (Fig. 7). According to our in vitro experiments, the reported results strongly indicate that PD-L1 expression is correlated to EMT-like phenotype (p = 0.03). Moreover, 6 out of 7 tumors displaying a strong PD-L1 immunoreactivity are in the EMT-positive category. In summary, all our data contribute to show for the first time that PD-L1 expression is regulated by the tumoral microenvironment and is coordinated particularly by the cytokines TNF-α and TGF-β1 involved in EMT induction.
Discussion
EMT is a complex reprogramming of epithelial cells playing a pivotal role in tumor invasion, acquisition of stemness properties and metastasis development. In the present study, we questioned the influence of EMT in immunosurveillance through the study of PD-L1 expression regulation.
We selected a model where EMT can be induced in a reversible manner in order to mimic the invasion phase leading to metastasis formation. As previously described, we observed that TNF-α and TGF-β1 synergized to promote EMT of A549. In this model,22, SNAIL was overexpressed in A549 following TGF-β1 used alone or in combination with TNF-α (Fig. 3A). In the present study, we showed that EMT phenotype conferred by TGF-β1 and TNF-α to A549 was associated with the upregulation of PD-L1, in line with the requirement for an invading tumor cell to evade the immune system. Of note, exposition to TNF-α or TGF-β1 alone did not induce a significant expression of PD-L1. Moreover, SNAIL transfection did not promote PD-L1 on A549 (Fig. 3D), suggesting for the first time that a synergism between TGF-β1 and TNF-α signaling is required to control PD-L1 in this setting.
Hypoxia as well as interactions with stromal cells such as cancer associated fibroblasts and mesenchymal stem cells, or inflammation are elements of the tumor microenvironment fostering EMT.3 We reported that the PD-L1 expression was particularly expressed in VIMENTIN positive NSCL tumors (Fig. 7). TGF-β1 was shown to be of particular importance, among growth-factors and cytokines involved in this dedifferentiation process,27,28 Indeed, TGF-β1 controls the expression of EMT-related transcription factors such as SNAIL, ZEB and TWIST1.27 TNF-α is another cytokine produced within tumor microenvironment by hematopoietic cells such as M2 macrophages and exerting a promoting role on EMT.29,30 TNF-α stimulates NF-κB in cancer cells and increases the ability of TGF-β1 to induce EMT.30,31
5 subunits forms the NF-κB transcription factor family: RelA, RelB, c-Rel, NF-κB1 (p105/p50), NF-κB2 (p100/p52). NF-κB signaling is an important component of the molecular network generated by inflammation in cancer cells.32 Huber MA et al. showed that NF-κB signaling is required for initiation and maintenance of TGF-β1 driven EMT in breast carcinoma.19 It was also previously reported in the A549 model, that mesenchymal NSCLC displays an actively constitutive NF-κB signaling.22 Thus, NF-κB's role in PD-L1 regulation has been addressed. The inhibition of the NF-κB signaling and of the non canonical subunit IKKε blocked PD-L1 expression during EMT induction in A549 cells (Fig. 5). Strikingly, inhibition of the NF-κB pathway, in the MDA-MB-231 cells (associated with a high basal expression of PD-L1), resulted in the abolition of PD-L1 expression (Supp. Fig. 7). On the opposite, ectopic expression of IKKε or NF-κBp52 had minor effects on PD-L1 expression in the A549 cells (Supp. Fig. 6–8). These data revealed the essential role played by the NF-κB/ IKKε signaling in PD-L1 expression. It also highlighted that PD-L1 upregulation during EMT is not only due to this one parameter.
Since epigenetics, has already been involved in EMT, we next wondered whether DNA methylation could also regulate PD-L1 gene expression. We showed that TNF-α/TGF-β1 induced a global DNA demethylation associated with a loss of DNMT1 content and a demethylation of PD-L1 promoter (Fig. 6). Moreover, inhibition of DNA methylation combined with TNF-α treatment restored PD-L1 expression in A549 cells. This observation was explained by a loss of DNMT1 recruitment on the PD-L1 promoter in benefits to NF-κB (Fig. 6G and 6H). According to these observations, we showed that the mesenchymal MDA-MB-231 cells associated with a high expression of PD-L1 presented an unmethylated PD-L1 promoter (Supp. Figure 10). Finally, all these data demonstrate that PD-L1 expression is regulated by a double process: i) it requires the demethylation of its promoter following a loss of DNMT1 in a TGF-β1 dependent manner, ii) it requires the activation of the IKKε/ NF-κB signaling pathway in a TNF-α dependent manner (Supp. Figure 11). It has also been recently reported that the expression of PD-L1 in NSLCC or breast cancers was controlled by ZEB-1 which blocked the expression of the miR200, an inhibitor of PD-L1 expression. These observations confirmed that PD-L1 expression is thinly orchestrated during EMT.11,33 Moreover, the stability of PD-L1 on membranes is also regulated by CMTM6.34 All these data suggest that PD-L1 is an important immune checkpoint and that its expression and stability are thinly controlled by several processes
The regulation of PD-L1 expression became an issue of growing importance regarding the clinical results achieved with anti-PD-1/PD-L1 immunotherapy.35,36 Although PD-L2 expression was observed in cancer cells, This protein is mostly expressed on dendritic cells and macrophages, while PD-L1 expression was mainly reported on tumor cells.37-39 but also on myeloid cells infiltrating cancer microenvironment in particular in tumor associated macrophages.40,41
Medical Strategies to block the action of TGFβ and TNFα in the tumoral microenvironment could help to limit EMT and PD-L1 expression. However, the dual role of TGFβ, depending of cell context, which balances between tumor suppressor gene functions (decrease of proliferation, increase of differentiation and apoptosis) and oncogenic functions (increase of invasion and metastasis) makes this approach difficult (for a review.42).
Recently, using array-based analyses, an increase expression of PD1 was also associated with a demethylation of its promoter in NSCLC tumors compared to tumor-free samples.43 According to previous reports showing that anti-cancer therapies using DNA demethylating agents (DDA) (for a review.44) favor expression of pro-invasiveness genes and metastasis.45,46 our present data suggest that DDA could promote PD1/PD-L1 expression and may contribute to immune escape. On the opposite, an alternative strategy using remethylating agents (e.g. donor of methyl such as folate enriched diets.47,48) might be useful to limit immune escape.
Mesenchymal cells are endowed with properties of proliferation and invasion. Few studies.49-51 have already suggested that EMT foster cancer progression also by acting on mechanisms allowing tumors to evade immune surveillance. A previous work has demonstrated that EMT is involved in the induction of immunosuppression.50 Mesenchymal cells were shown to be able to induce regulatory T cells, to impair dendritic cells in vitro and in vivo partly through TSP1 production and to escape from T-cell mediated lysis.52 Inversely, elements of the immune system can act as inducers of EMT.51 CD8 T cells can stimulate mammary epithelial tumor cells to undergo EMT. In the present study, we established another link between EMT and immune escape through the regulation of PD-L1 and our results strongly suggest that epidrugs could be used in the future to modulate PD-L1 expression and improve immune checkpoints in NSCLC patients.
Materiels and methods
Cell culture
The lung cancer cell line A549 was obtained from the American Type Cell Culture Collection (ATCC) and was cultured in DMEM (Dulbecco's Modified Eagle Medium, Lonza, Paris, France) supplemented with 10% inactivated fetal calf serum (FCS) (Invitrogen, Cergy-Pontoise, France) and 1% Penicillin-Streptomycin (Invitrogen, Cergy-Pontoise, France). Absence of mycoplasma was tested periodically. Cell culture was incubated in 5% CO2 at 37°C. Identification of A549 after transfections is controlled by analyzing the short tandem repeat following to recommendations of ATCC. The cytokines TNF-α and TGFβ were purchased from Peprotech (Neuilly-sur-Seine, France) and were used at the concentration of 20 ng/ml and 4 ng/ml, respectively. JSH-23 (catalog #481408) was obtained from Calbiochem (Lezennes, France) and was used at 25μM. All experiments with cytokines TNF-α and TGFβ were realized at fifth day of treatment if not specified. The IKKε inhibitors Bx795 (catalog #702675-74-9) were used at 1 μM, which was obtained from Sigma-Aldrich (Saint-Quentin-Fallavier, France). Procainamide (Euromedex) was used at 0.5 mM and BAY11 (Calbiochem) was used at 5 μM.
Plasmids and transfections
A549 cells were transfected by plasmids encoding SNAIL-1-pBabe puro (plasmid 23347, Addgene, Cambridge, USA), NFkB2-p52-cFlag-pcDNA3 (plasmid 20019, Addgene, Cambridge, USA), pBabe-Puro-Flag-IKBKE (plasmid 15292, Addgene, Cambridge, USA), psiRNA-h7SKGFPzeo (plasmid ksirn4-gz21, Invivogen, San Diego, USA), to obtain stable cells A549Snail-pBabe, A549NFkBp52-pcDNA3, A549IKKε-pBabe, A549siIKKε-h7SK respectively. In parallel transfections with the empty vectors were realized as a control. All transfections were performed using the Lipofectamine® 3000 Reagent (Life Technologies, Saint Aubin, France) following the manufacturer's instructions and four days after transfection the cells undergo selection by geneticin at 1 mg/mL (# 10131-027, Life Technologies, New York, USA), by puromycin (#ant-pr-1, Invivogen, San Diego, USA) at 2μg/ml or by Zeocyne (#P/N46–0509, Invitrogen, Cergy-Pontoise, France) at 500 μg/ml according to vector.
Flow cytometer
For membrane staining, cells were incubated with corresponding antibody for 30 min at 4°C then washed with PBS (phosphate buffered saline) and centrifuged for 10 minutes at 300 g. Ten thousands cells from each sample were evaluated for fluorescence detection using BD FACSCanto cytometer. (Becton Dickinson, Le Pont de Claix, France) and analysed in FACS Diva software. The overlay graphs are made with the Flow Jo software. Mouse IgG1, κ Isotype Control-APC, EpCAM-APC, MHC-I (HLA A,B,C) –APC, Mouse IgG1, κ Isotype Control-PE, PD-L1 (CD274)–PE, PD-L2 (CD273)–PE (BD Biosciences, Le Pont de Claix, France), Mouse IgG1, Isotype Control-PE, DR4-PE, DR5-PE, MICB–PE, Mouse IgG1, Isotype Control–PE, TRAIL (B-S23)–PE, Mouse IgG2 a, Isotype Control-PE, Fas-PE, Negativ control IgG1 (Diaclone Gen-Probe, Besançon, France), Mouse IgG1, κ Isotype Control-PE, Fas-L (CD178)-PE, Galectin-9-PE (BioLegend, San Diego, USA), Mouse IgG2B, Isotype Control-AF488, MICA -AF488, Mouse IgG2 a, Isotype Control-AF488, ULBP1-AF488, Mouse IgG2 a, Isotype Control-PE, ULBP3–PE (RD Systems, Lille, France), Mouse IgG2 a, κ Isotype Control-PE, HLA-G–PE (eBiosciences, Paris, France) and CD66 (produced in our Lab) were used for flow cytometer analysis (Sigma Aldrich, Saint Quentin, France).
Antibody used for Flow Cytometer: EpCAM, PD-L1, PD-L2 were obtained from BD Biosciences (Le Pont de Claix, France), DR4, DR5, MICB, TRAIL, Fas from Dicalone (Gen-Probe, Besançon, France), Fas-L, Galectin-9 from Biolegend (San Diego, USA), MICA, ULBP1, ULBP3 from RD Systems (Lille, France), HLA-G from eBiosciences, (Paris, France) and CD66 was produced in our laboratory.
Western blotting
Cellular proteins were separated by electrophoresis (SDS-PAGE) in a polyacrylamide gel in presence of sodium dodecylsulfate (SDS) according to their molecular weight, then transferred to a polyvinylidene difluoride film (PVDF) for staining by specific antibody and then detection by secondary antibody coupled to peroxidase. Antibodies used are listed in Supp. Table 1.
Confocal microscopy
Cells were cultured in Labteck slides (Sigma-Aldrich, France). Cells were fixed with 4% paraformaldehyde and then permeabilized with 0.1% Triton X100. Cells were stained with appropriate antibodies (Beta-CATENIN and E-CADHERIN (OZYME, Montigny-le-Brretoneux, France) or ACTIN was stained with Phalloidin-Rhodamin (P1951, Sigma-Aldrich, France). Nuclei were stained with DAPI. Images were collected with an Olympus FV1000 laser scanning confocal microscope.
qRT-PCR
Total RNA was isolated using RNeasy Mini Kit (Qiagen, Hilden, Germany). Reverse transcription were performed using M-MLV (Sigma-Aldrich, M-1302 according to manufacturer's instructions (Sigma-Aldrich). Quantitative PCR (qPCR) were done in duplicate using the StepOne Real-Time PCR system (Applied Biosystems), Power SYBR Green PCR Master Mix (Applied Biosystems, 4367659), according to manufacturer's instructions. Expression level was normalized to that of glyceraldehyde-3- phosphate dehydrogenase (GAPDH). Primers for CDH1, CDH2, VIMENTIN, EPCAM, ZEB-1, ZEB-2 and SNAI1 were obtained from53 GADD45 A, SMUG, AICA, DNMT1, DNMT3 A, DNMT3B, TET1, TET2 and TET3 primers were obtained from QuantiTect Primer Assays.
Invasion test
Cell invasion assay was performed using cell culture inserts with 8 μm pore size coated with Matrigel. A549 cells (180.000 cells/well) suspended with 100μl DMEM were seeded in the upper side of the chamber. In the lower side of the chamber, 500μl DMEM was added. TGFβ and/or TNF-α were added into both sides of the chamber. After 24 h, cells on the lower surface of the chamber were trypsinized, suspended in DMEM and counted with a trypan blue. The experiments were performed with duplicate, and repeated 3 times.
Epigenetics
The ChIP were performed using ChIP-IT kit (Active Motif) as preconized by the manufacturer and chromatin was obtained from cells sonicated with the Bioruptor (Diagenode). For the DNA methylation experiments, gDNA was extracted using the Nucleospin tissue kit (Macherey Nagel) or using the QiaCube (Qiagen) and global DNA methylation or global 5hmeC content were measured by ELISA on 100 ng gDNA using the MethylFlash methylated DNA quantification kit (Epigentek) or the MethylFlash Global DNA Hydroxymethylation (5-hmC) ELISA Easy Kit (Epigentek). PD-L1 promoter methylation was quantified by qMSRE with the OneStep qMethyl (ZYMOresearch) on the region -173/-68 with the primers 5′-TCCTAGGACACCAACACTAGATACCTAAAC-3′ and 5′-TCTGCCCAAGGCAGCAA-3′. Antibodies used are listed in Table 1.
Tissue samples, immunohistochemistry and immunostaining assessment
Paraffin-embedded tissue specimens of non-small cell lung cancer were collected [in collaboration with the Tissue Biobank of the University of Liege (Liege, Belgium)] and stained with both anti-vimentin (clone V9, Ventana Medical Systems, Tucson, AZ, USA) and anti-PD-L1 (knockout validated clone 28–8, Abcam, Cambridge, MA, USA) antibodies. The original diagnosis of each case was confirmed by experienced histopathologists. The protocol was approved by the Ethics Committee of the University Hospital of Liege. Immunohistochemical analysis was performed with a standard protocol detailed previously.54 Based on epithelial vimentin expression, the tissue samples were classified into two groups: EMT-positive versus EMT-negative. Regarding PD-L1 immunoreactivity in epithelial (tumor) cells, a semi-quantitative score of the intensity (0: negative; 1: low; 2: moderate; 3: strong) and extent (0: <5% of positive cells; 1: 6–33%; 2: 34–66%; 3: >67%) of the staining according to an arbitrary scale was used. As previously described,55 the results obtained with the two scales were multiplied and a global score ranged between 0 and 9 was obtained. All immunolabelled tissues were evaluated by two experienced histopathologists.
ELISA assay and NFKB activity
Cells were cultured in 25 cm2 dishes and treated with TNFα, TGFβ or combination of these two cytokines. IFNy production was measured by using IFNy ELISA kit (Diaclone, France) with 100 µl of supernatant of treated cells in according to manufacturer's instructions. PD-L1 was measured using the Human PD-L1 ELISa kit (Abcam ab214565). NKkB activity was measured using the TransAM® NFKB kit (Active Motif).
Supplementary Material
Funding Statement
This work was supported by the Ligue Contre le Cancer (001AC.2015), LabeX IGO, University of Franche-Comté, Région Franche-Comté and the ANR(ANR-11-LABX-0016-01).
Conflict of interest
“The authors declare no potential conflicts of interest.”
Acknowledgments
“The PFC/AN's work was realized in the context of the LabEX IGO program supported by the National Research Agency via the investment of the future program ANR-11-LABX-0016-01”. This work was also supported by the university of Franche-Comté and grants of “La Ligue contre le cancer #001AC.2015” and “la Région Franche-Comté”.
Author contributions
AA, AK, GJ, GD, MV, PJR, BJ, TC, HM, HE, PP, HE, NA, CPF performed experiments; BC, HE, CPF analyzed results; AA, BC, HE wrote the manuscript; AO, GY, HE, GJ, CB, CPF, BGM, PP revised the manuscript; all authors provided final approval.
References
- 1.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1(6–7):303–314. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7(2):131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 4.Garg M: Epithelial-mesenchymal transition – activating transcription factors – multifunctional regulators in cancer. World J Stem Cells. 2013;5(4):188–195. doi: 10.4252/wjsc.v5.i4.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: Concepts and molecular links. Semin Cancer Biol. 2012;22(5–6):396–403. doi: 10.1016/j.semcancer.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Chen YB, Mu CY, Huang JA. Clinical significance of programmed death-1 ligand-1 expression in patients with non-small cell lung cancer: A 5-year-follow-up study. Tumori. 2012;98(6):751–755. [DOI] [PubMed] [Google Scholar]
- 8.Shi MH, Xing YF, Zhang ZL, Huang JA, Chen YJ. [Effect of soluble PD-L1 released by lung cancer cells in regulating the function of T lymphocytes]. Zhonghua Zhong Liu Za Zhi. 2013;35(2):85–88. [DOI] [PubMed] [Google Scholar]
- 9.Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, Martinez A, Nuciforo P, Comerma L, Alos L, et al.. Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res. 2017;77(13):3540–3550. doi: 10.1158/0008-5472.CAN-16-3556. [DOI] [PubMed] [Google Scholar]
- 10.Ascierto PA, McArthur GA. Checkpoint inhibitors in melanoma and early phase development in solid tumors: what's the future? J Transl Med. 2017;15(1):173. doi: 10.1186/s12967-017-1278-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noman MZ, Janji B, Abdou A, Hasmim M, Terry S, Tan TZ, Mami-Chouaib F, Thiery JP, Chouaib S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology. 2017;6(1):e1263412. doi: 10.1080/2162402X.2016.1263412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005;6:56. doi: 10.1186/1465-9921-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamauchi Y, Kohyama T, Takizawa H, Kamitani S, Desaki M, Takami K, Kawasaki S, Kato J, Nagase T. Tumor necrosis factor-alpha enhances both epithelial-mesenchymal transition and cell contraction induced in A549 human alveolar epithelial cells by transforming growth factor-beta1. Exp Lung Res. 2010;36(1):12–24. doi: 10.3109/01902140903042589. [DOI] [PubMed] [Google Scholar]
- 14.Saito A, Suzuki HI, Horie M, Ohshima M, Morishita Y, Abiko Y, Nagase T. An integrated expression profiling reveals target genes of TGF-beta and TNF-alpha possibly mediated by microRNAs in lung cancer cells. PLoS One. 2013;8(2):e56587. doi: 10.1371/journal.pone.0056587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348(6230):136–139. doi: 10.1126/science.1258867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 2001;413(6852):165–171. doi: 10.1038/35093109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–738. doi: 10.1038/nature01112. [DOI] [PubMed] [Google Scholar]
- 18.Liu X. Inflammatory cytokines augments TGF-beta1-induced epithelial-mesenchymal transition in A549 cells by up-regulating TbetaR-I. Cell Motil Cytoskeleton. 2008;65(12):935–944. doi: 10.1002/cm.20315. [DOI] [PubMed] [Google Scholar]
- 19.Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114(4):569–581. doi: 10.1172/JCI200421358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Radisky DC, Bissell MJ. NF-kappaB links oestrogen receptor signalling and EMT. Nat Cell Biol. 2007;9(4):361–363. doi: 10.1038/ncb0407-361. [DOI] [PubMed] [Google Scholar]
- 21.Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, et al.. Epithelial-mesenchymal transition induced by TNF-alpha requires NF-kappaB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012;72(5):1290–1300. doi: 10.1158/0008-5472.CAN-11-3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar M, Allison DF, Baranova NN, Wamsley JJ, Katz AJ, Bekiranov S, Jones DR, Mayo MW. NF-kappaB regulates mesenchymal transition for the induction of non-small cell lung cancer initiating cells. PLoS One. 2013;8(7):e68597. doi: 10.1371/journal.pone.0068597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, Kim Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett. 2004;571(1–3):50–54. doi: 10.1016/j.febslet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Negi G, Sharma SS. JSH-23 targets nuclear factor-kappa B and reverses various deficits in experimental diabetic neuropathy: effect on neuroinflammation and antioxidant defence. Diabetes Obes Metab. 2011;13(8):750–758. doi: 10.1111/j.1463-1326.2011.01402.x. [DOI] [PubMed] [Google Scholar]
- 25.Cardenas H, Vieth E, Lee J, Segar M, Liu Y, Nephew KP, Matei D. TGF-beta induces global changes in DNA methylation during the epithelial-to-mesenchymal transition in ovarian cancer cells. Epigenetics. 2014;9(11):1461–1472. doi: 10.4161/15592294.2014.971608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LH, Hsu WL, Tseng YJ, Liu DW, Weng CF. Involvement of DNMT 3B promotes epithelial-mesenchymal transition and gene expression profile of invasive head and neck squamous cell carcinomas cell lines. BMC Cancer. 2016;16:431. doi: 10.1186/s12885-016-2468-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19(2):156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121(10):3804–3809. doi: 10.1172/JCI57099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012;72(19):4883–4889. doi: 10.1158/0008-5472.CAN-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, et al.. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145(6):926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawata M, Koinuma D, Ogami T, Umezawa K, Iwata C, Watabe T, Miyazono K. TGF-beta-induced epithelial-mesenchymal transition of A549 lung adenocarcinoma cells is enhanced by pro-inflammatory cytokines derived from RAW 264.7 macrophage cells. J Biochem. 2012;151(2):205–216. doi: 10.1093/jb/mvr136. [DOI] [PubMed] [Google Scholar]
- 32.Karin M: Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441(7092):431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 33.Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al.. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;5:5241. doi: 10.1038/ncomms6241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al.. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549(7670):101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]
- 37.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 38.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, et al.. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 39.Noguchi T, Ward JP, Gubin MM, Arthur CD, Lee SH, Hundal J, Selby MJ, Graziano RF, Mardis ER, Korman AJ, et al.. Temporally Distinct PD-L1 Expression by Tumor and Host Cells Contributes to Immune Escape. Cancer Immunol Res. 2017;5(2):106–117. doi: 10.1158/2326-6066.CIR-16-0391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211(5):781–790. doi: 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al.. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280 A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-beta pathway for cancer therapy. Clin Cancer Res. 2012, 18(17):4514–4521. doi: 10.1158/1078-0432.CCR-11-3224. [DOI] [PubMed] [Google Scholar]
- 43.Marwitz S, Scheufele S, Perner S, Reck M, Ammerpohl O, Goldmann T. Epigenetic modifications of the immune-checkpoint genes CTLA4 and PDCD1 in non-small cell lung cancer results in increased expression. Clin Epigenetics. 2017;9:51. doi: 10.1186/s13148-017-0354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pechalrieu D, Etievant C, Arimondo PB. DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem Pharmacol. 2017, 129:1–13. doi: 10.1016/j.bcp.2016.12.004. [DOI] [PubMed] [Google Scholar]
- 45.Sato N, Maehara N, Su GH, Goggins M. Effects of 5-aza-2′-deoxycytidine on matrix metalloproteinase expression and pancreatic cancer cell invasiveness. J Natl Cancer Inst. 2003;95(4):327–330. doi: 10.1093/jnci/95.4.327. [DOI] [PubMed] [Google Scholar]
- 46.Ateeq B, Unterberger A, Szyf M, Rabbani SA. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes in vitro and in vivo. Neoplasia. 2008;10(3):266–278. doi: 10.1593/neo.07947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pufulete M, Al-Ghnaniem R, Khushal A, Appleby P, Harris N, Gout S, Emery PW, Sanders TA. Effect of folic acid supplementation on genomic DNA methylation in patients with colorectal adenoma. Gut. 2005;54(5):648–653. doi: 10.1136/gut.2004.054718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hervouet E, Debien E, Campion L, Charbord J, Menanteau J, Vallette FM, Cartron PF. Folate supplementation limits the aggressiveness of glioma via the remethylation of DNA repeats element and genes governing apoptosis and proliferation. Clin Cancer Res. 2009;15(10):3519–3529. doi: 10.1158/1078-0432.CCR-08-2062. [DOI] [PubMed] [Google Scholar]
- 49.Knutson KL, Lu H, Stone B, Reiman JM, Behrens MD, Prosperi CM, Gad EA, Smorlesi A, Disis ML. Immunoediting of cancers may lead to epithelial to mesenchymal transition. J Immunol. 2006;177(3):1526–1533. doi: 10.4049/jimmunol.177.3.1526. [DOI] [PubMed] [Google Scholar]
- 50.Kudo-Saito C, Shirako H, Takeuchi T, Kawakami Y. Cancer metastasis is accelerated through immunosuppression during Snail-induced EMT of cancer cells. Cancer Cell. 2009, 15(3):195–206. doi: 10.1016/j.ccr.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 51.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, et al.. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69(7):2887–2895. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akalay I, Janji B, Hasmim M, Noman MZ, Andre F, De Cremoux P Bertheau P, Badoual C, Vielh P, Larsen AK, et al.. Epithelial-to-Mesenchymal Transition and Autophagy Induction in Breast Carcinoma Promote Escape from T-cell-Mediated Lysis. Cancer Res. 2013;73(8):2418–2427. doi: 10.1158/0008-5472.CAN-12-2432. [DOI] [PubMed] [Google Scholar]
- 53.Gemmill RM, Roche J, Potiron VA, Nasarre P, Mitas M, Coldren CD, Helfrich BA, Garrett-Mayer E, Bunn PA, Drabkin HA. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011;300(1):66–78. doi: 10.1016/j.canlet.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Herfs M, Longuespee R, Quick CM, Roncarati P, Suarez-Carmona M, Hubert P, Lebeau A, Bruyere D, Mazzucchelli G, Smargiasso N, et al.. Proteomic signatures reveal a dualistic and clinically relevant classification of anal canal carcinoma. The Journal of pathology. 2017;241(4):522–533. doi: 10.1002/path.4858. [DOI] [PubMed] [Google Scholar]
- 55.Hubert P, Herman L, Roncarati P, Maillard C, Renoux V, Demoulin S, Erpicum C, Foidart JM, Boniver J, Noel A, et al.. Altered alpha-defensin 5 expression in cervical squamocolumnar junction: Implication in the formation of a viral/tumour-permissive microenvironment. The Journal of pathology. 2014;234(4):464–477. doi: 10.1002/path.4435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.