

ABSTRACT
Human γδT cell immunotherapy is well tolerated and has shown promising results in clinical trials; however, its antitumor efficacy is limited, including results in solid tumors. Ex-vivo expanded γδT cell stimulated by zoledronic acid (ZOL) activates the γδT cell subpopulation of so called Vγ9Vδ2 T cells. To improve the clinical outcomes of Vγ9Vδ2 T cell (abbreviated as γδT cell here) immunotherapy, we aimed to increase the cytotoxicity of γδT cells by focusing on two issues: recognition of tumor cells by γδT cells and the effector (γδT cell)-to-target (tumor cell) (E/T) ratio. Ex vivo-expanded γδT cells showed potent cytotoxicity against urinary bladder cancer (UBC) cells in in vitro assays. Combination treatment with standard anticancer agents showed that low dose gemcitabine pretreatment significantly enhanced the cytotoxicity of γδT cells by upregulating the expression of MICA and MICB (MICA/B), which are tumor-associated antigens recognized by γδT cells. These effects were abrogated by small interfering RNA-mediated knockdown of MICA/B in UBC cells, suggesting that pre-exposing cancer cells to anticancer agents could be a promising strategy. A bladder instillation approach was used to increase the E/T ratio. The efficacy of ex vivo-expanded γδT cell immunotherapy was examined in an orthotopic xenograft model. In Vivo Imaging System analysis revealed the potent cytotoxicity of weekly intravesical administration of γδT cells, and weekly gemcitabine pretreatment enhanced the cytotoxicity of γδT cells in vivo. In conclusion, intravesical γδT cell immunotherapy and combination therapy with low dose gemcitabine may be a promising strategy in UBC.
KEYWORDS: γδT cell, bladder instillation, chemo-immunotherapy, gemcitabine, MICA/B, NKG2D, orthotopic xenograft model, Vγ9Vδ2 T cell
Introduction
Urinary bladder cancer (UBC) is a common malignancy worldwide. Approximately 70% of UBC patients are diagnosed with non-muscle invasive UBC (NMIUC) by transurethral resection of bladder tumors (TUR-Bt).1,2 Morales et al. reported the first clinical use of intravesical Bacillus Calmette-Guerin (BCG) for the treatment of NMIUC in 1976. Intravesical BCG treatment reduces the risk of recurrence and progression to a greater extent than other treatments and therefore continues to play a central role in the treatment of NMIUC.3 There is currently no gold standard for salvage intravesical therapy after BCG treatment failure. In many cases, these tumors have the potential to progress to muscle invasive cancer, leading to systemic metastasis.4-6 Radical cystectomy remains the treatment of choice after BCG failure, which inevitably impairs the quality of life of patients.3,4
Human Vγ9Vδ2 T cell immunotherapy may be a unique potential therapeutic option. The activation of Vγ9Vδ2 T cells by aminobisphosphonates in vitro and in vivo has been described first by Wilheim and Kunzmann.7-9 γδT cells are T cells that express TCRγδ and consist of a small proportion of T cells in the peripheral blood.10-12 γδT cells recognize and are activated by non-peptide phosphorylated antigens. Zoledronic acid (ZOL) is one of the best studied nitrogen-containing aminobisphosphonates in the field of γδT cell research. ZOL inhibits farnesyl pyrophosphonate (FPP) synthase in the mevalonate pathway in target cells, leading to the accumulation of isopentenyl pyrophosphate (IPP), which stimulates and activates TCRγδ.7-9,13,14
Ex-vivo expanded Vγ9Vδ2 T cells show potent cytotoxicity against various types of cancer cells in a major histocompatibility complex (MHC)-unrestricted manner.7-9,15-18 Although several clinical trials of systemic cancer immunotherapy using ex vivo-expanded Vγ9Vδ2 T cells have been conducted, the results are unsatisfactory. The effector (γδT cell)-to-target (tumor cell) (E/T) ratio is an important factor determining cytotoxicity. We hypothesized that the E/T ratio may be insufficient in the systemic administration of ex vivo-expanded γδT cells, and that effector cell administration into a localized body cavity such as the urinary bladder may improve the E/T ratio to achieve a maximum cytotoxic effect.
Anticancer agents have long been believed to suppress immune functions in cancer patients because of bone marrow suppression.19 However, recent studies show that some agents integrate the cytotoxicity of immune cells against cancer cells.20,21
Therefore, we asked whether pretreatment of cancer cells with standard chemotherapeutic agents would increase the cytotoxicity of γδT cells.
Pretreatment with gemcitabine at a low dose, which did not induce cell death, increased the cytotoxicity of γδT cells by upregulating the expression of MHC class I-related chain protein A/B (MICA/B), which γδT cells use to recognize cancer cells. The safety and manageable toxicity of gemcitabine in the clinical setting have been demonstrated, supporting that the combination of gemcitabine and γδT cell therapy is a promising treatment strategy. The aim of the present study was to investigate the cytotoxicity of ex vivo-expanded γδT cell-based chemo-immunotherapy in vitro using cytotoxicity assays and in vivo in an orthotopic xenograft model.
Results
Cell expansion using rhIL2 and ZOL gave rise to ex vivo-expanded human Vγ9Vδ2 T cells with a well-differentiated TCRγδ positive phenotype
ZOL and recombinant human IL2 (rhIL2) play crucial roles in the expansion of human γδT cells.22,23 ZOL at a dose of 5 μM was added to AlyS505N medium supplemented with 10% human AB serum on day 0 based on previous results showing that the optimal ZOL concentration for stimulating human γδT cells was 0.5–5 μM.24 In addition, cells were serially stimulated with rhIL2 (100 U/ml) daily with a change to fresh medium until the culture endpoint. After the expansion of γδT cells, clusters and aggregates were observed starting on day 3. On day 11, mature expanded γδT cells were harvested and frozen under liquid nitrogen until use.
γδT cells were defined as CD3+/TCRγδ+, and this population was achieved in >80% of cultured cells on day 11 as shown by flow cytometry (Fig. 1A). The absolute number of γδT cells reached a maximum at 1600-fold expansion and the proportion for Vγ9Vδ2 T cells among total T cells (CD3 positive cells) and total γδT cells (TCRγδ positive cells) was 73.6% and 92.0% respectively on day 11 (Fig. 1B). The surface expression of NKG2D, TCRVγ9, and TCRVδ2 in cultured γδT cells and the intracellular levels of perforin and Granzyme B were determined on days 0 and 11. The results showed that ex vivo cultured γδT cells expressed the NKG2D receptor on the cell surface (Fig. 1C), and both the TCRVγ9-positive lineage and the TCRVδ2-positive lineage were expanded by stimulation with ZOL and rhIL2 (Fig. 1D). Activation of ex vivo cultured γδT cells was confirmed by the intracellular staining of granules containing perforin and Granzyme B, which are important for cancer cell apoptosis and could be detected at the culture endpoint compared with the beginning point. PBMCs on day 0 stimulated with the Cell Stimulation cocktail at 500 × for 4 h were also used as positive controls (Fig. 1E).
Figure 1.
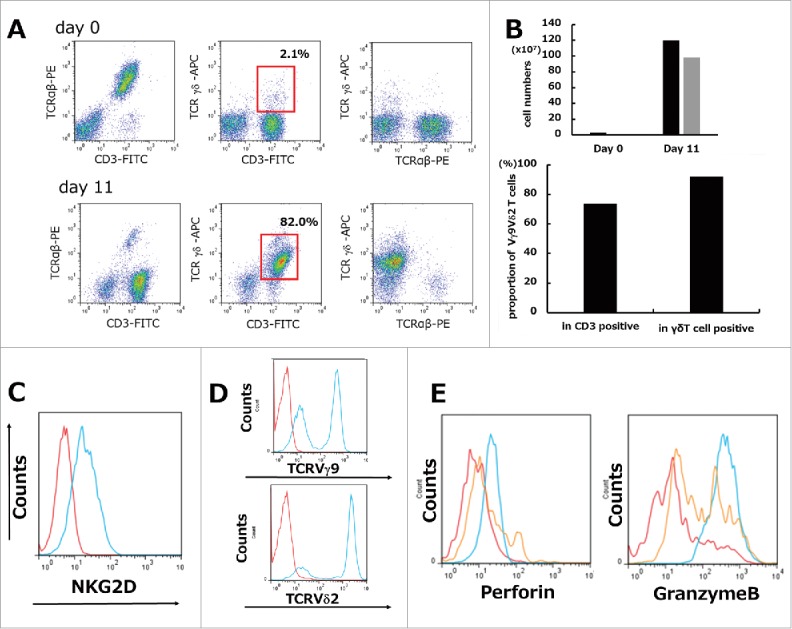
Ex vivo mature human γδT cells were expanded from PBMCs stimulated by ZOL and rhIL2. (A) Representative data from healthy volunteer-derived γδT cells. Isolated PBMCs showed a minor subset of TCRγδ-positive (gated on lymphocytes by FSC/SSC) cells on day 0. Representative flow cytometric analysis showing the profiles of γδT cells in which >80% achieved a CD3+/TCRγδ+ population (red squares) on day 11. (B) The absolute cell number of γδT cells during 11 days of culture showed a maximum 1600-fold increase. Black bars: total cells; gray bars: γδT cells (upper graph). Proportion for Vγ9Vδ2 T cells among total T cells (CD3 positive cells) and total γδT cells (TCRγδ positive cells) was 73.6% and 92.0% respectively on day 11 (lower graph). (C) Expression of NKG2D on γδT cells. Representative flow cytometric profiles are shown as histograms. Blue line: expression of NKG2D on γδT cells (gated on TCRγδ+) on day 11; red line: background control. (D) Phenotypic analysis of γδT cells. Representative flow cytometric profiles are shown as histograms (gated on TCRγδ+). Lineage expressing Vγ9Vδ2 can be expanded by ZOL. Blue line: expression of TCRVγ9 or TCRVδ2 on γδT cells (gated on TCRγδ+) on day 11; red line: background control. (E) Intracellular granule (perforin, Granzyme B) staining was performed. Red line: day 0. Blue line: day 11. Orange line: PBMCs stimulated with Cell Stimulation cocktail 500X (2 μl/ml) were used as positive controls. Representative images of histograms are shown.
Human Vγ9Vδ2 T cells showed strong cytotoxicity against UBC cells in in vitro cytotoxicity assays
UBC cells pretreated with 5 μM ZOL for 24 h showed markedly increased sensitivity to human γδT cell-mediated lysis after a 4 h co-culture (Fig. 2 and S1), consistent with previous reports showing that pretreatment of cancer cells with ZOL is a promising approach to enhance the cytotoxicity of γδT cells. The cytotoxicity of γδT cells was assessed by flow cytometry with CFSE/propidium iodide (PI) double staining. First, UBC cells and UB cells were labeled with CFSE and seeded in 6-well plates. The next day, γδT cells were added to the plate at the indicated E/T ratio. After 4 h of co-culture, PI was used to distinguish living cells from those that had undergone cell death. The percentage of PI-positive UBC cells markedly increased according to the E/T ratio and ZOL pretreatment of the target UBC cells (Fig. 2A, Fig. 2B, and S1). By contrast, γδT cells showed little cytotoxicity against normal UB cells, consistent with previous reports.19,25,26 Confocal microscopy scanning revealed that CFSE-labeled γδT cells attached to PKH26-labeled UBC cells, leading to apoptosis within 4 h of co-culture. Representative images are shown (Fig. 2C), which indicate that γδT cells may kill UBC cells in a contact-dependent manner. Taken together, these results indicated that γδT cells exerted potent cytotoxic effects against UBC cells.
Figure 2.
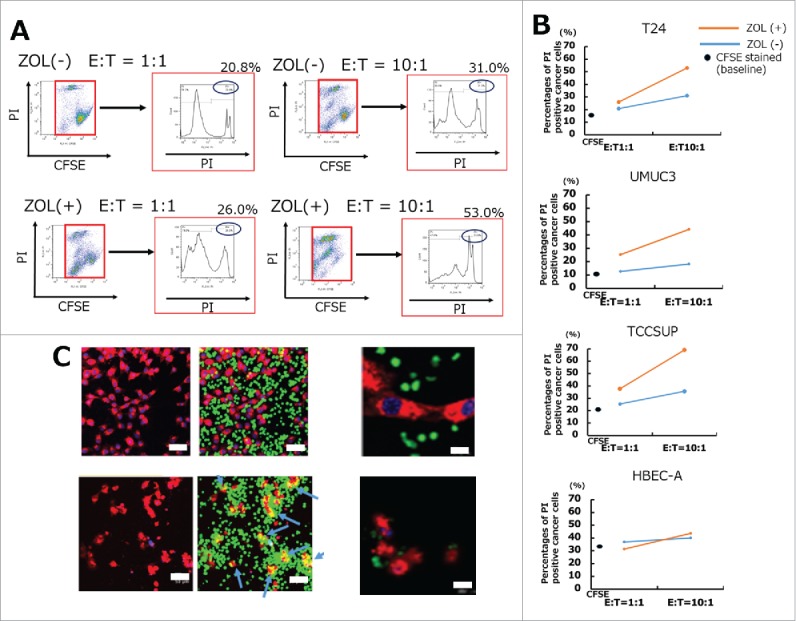
In vitro cytotoxicity assays demonstrated that UBC cells were lysed by human Vγ9Vδ2 T cells. (A) UBC and UB cells were stained with 0.5 μM CFSE and stimulated with or without 5 μM ZOL overnight. Results of typical flow cytometric analysis of γδT cell cytotoxicity in the T24 cell line are shown. (B) A summary of γδT cell cytotoxicity against several UBC cells and UB cells is shown. UBC and UB cells stained with CFSE 0.5 μM that were not co-cultured with γδT cells were used as the negative control. Cytotoxicity assays were performed in the absence (blue bars) or presence (orange bars) of 5 μM ZOL pretreatment. Black spots indicate the apoptotic percentages of CFSE stained baseline cancer cells. Mean value of triplicate wells is shown and representative data of three independent experiments are shown. (C) Imaging of cancer cells lysed by human γδ T cells using LSM 510. After 4 h of co-culture, γδT cells attached to cancer cells and lysed them by direct contact (blue arrows) (lower panels). Scale bars, 50 μm and 200 μm.
Pretreatment with anticancer agents enhanced the cytotoxicity of human Vγ9Vδ2 T cells against UBC cells
Pretreatment with 5 μM ZOL for 24 h increased the cytotoxicity of γδ T cells compared with that in the absence of ZOL pretreatment (Fig. 2A, Fig. 2B, and S1). We hypothesized that pretreatment of cells with anticancer agents in combination with ZOL may increase the cytotoxicity of γδT cells via an immunomodulatory mechanism. To rule out potential bias introduced by the cytotoxicity of the anticancer agents themselves, UBC cells were treated with gemcitabine (GEM) and cisplatin (CDDP) for 24 and 72 h, and the growth inhibitory effects were examined using the WST-8 assay. The cytotoxic effect of each agent against UBC cells is shown in Fig. 3A and C. Based on the results of the WST-8 assay, cells were pretreated with 5 μM GEM or 5 μM CDDP for 24 h, a dose at which these agents showed little cytotoxicity against UBC cells. Pretreatment with GEM at 5 μM for 24h increased human γδT cell-induced cytotoxicity; CDDP pretreatment had no effect (Fig. 3B). As shown in Fig. 3A, these results suggested that the increase in cytotoxicity was not a consequence of growth inhibition by GEM pretreatment. Next, we investigated whether pretreatment with other anticancer agents against UBC could enhance the cytotoxicity of human γδT cells. Pretreatment of cells with several agents at a low dose that did not inhibit cell growth at 24 h followed by the WST-8 assay demonstrated their synergistic effect on increasing the cytotoxicity of γδT cells in combination with ZOL pretreatment with the following exceptions: mitomycin C (MMC) pretreatment in T24, adriamycin (ADR) in TCCSUP, methotrexate (MTX) in TCCSUP, and vinblastine (VBL) in TCCSUP (Fig. 3D). In all cases, in the absence of γδT cells, anticancer agents combined with ZOL had little effects on cancer cell death (Fig. S3). Based on these results, the next experiments aimed to elucidate the underlying mechanisms.
Figure 3.

Pretreatment with anticancer agents synergistically enhanced human Vγ9Vδ2 T cell cytotoxicity against UBC cells along with ZOL pretreatment. (A and C) Growth inhibitory effects on UBC cells. Cells were pretreated with various concentrations of anticancer agents for 24 and 72 h. Solid line: 24 h; dashed line: 72 h. Data represent the mean ± SD of triplicate cultures. (B) Pretreatment with 5 μM gemcitabine for 24 h synergistically enhanced γδT cell cytotoxicity against T24 UBC cells along with ZOL pretreatment, whereas 5 μM cisplatin had no effect. Data represent the mean ± SD of triplicate wells. (D) Pretreatment with anticancer agents (MTX, VBL, ADR, and MMC) enhanced γδT cell cytotoxicity in combination with ZOL except in some cases. Although agents had minimal cytotoxicity at lower concentrations (MTX, VBL, ADR, and MMC at 100 nM) at 24 h, as determined by the WST-8 assay, pretreatment with these agents enhanced γδT cell cytotoxicity except in some cases (MMC pretreatment in T24, ADR in TCCSUP, MTX in TCCSUP, and VBL in TCCSUP). Statistical significance is displayed as ** for P < 0.01. N.S.: not significant.
Gemcitabine pretreatment upregulated MICA/B and ULBP family expressions in UBC cells
To examine whether pretreatment with anticancer agents had an effect on MICA/B expression, T24 and TCCSUP cells were treated with several anticancer agents at sub-lethal concentrations, including GEM, CDDP, MTX, VBL, ADR, and MMC. The results showed that GEM and other agents upregulated MICA/B expression at a low dose that did not affect the growth of UBC cells, whereas CDDP and ZOL had no effect (Fig. 4A, Fig. 4B, and S2). Of these agents, we focused on GEM because it is a common drug in the clinical setting, and the effects of GEM on MICA and MICB mRNA expression were investigated in T24 and TCCSUP cells. Quantitative RT-PCR analysis showed that treatment with 5 μM GEM for 24 h upregulated both MICA and MICB in T24 cells and MICB in TCCSUP cells (Fig. 4C). We also examined whether pretreatment with GEM had an effect on other NKG2DL than MICA/B such as ULBP1 and ULBP2/5/6 in UBC cells. The results showed that GEM significantly upregulated ULBP1 and ULBP2/5/6 as well as MICA/B expression, at a low dose that did not affect the growth of UBC cells, whereas ZOL had no effect (Fig. S4). By contrast, it was of note that pretreatment with GEM didn't change MICA/B expressions on normal bladder cell line, HBEC-A (Fig. S5).
Figure 4.

Changes of MICA/B expression in UBC cells following treatment with gemcitabine and in vitro cytotoxicity assay in MICA/B-knockdown UBC cells. (A) MICA/B expression in T24 and TCCSUP cells before and after ZOL, cisplatin, gemcitabine, and both ZOL and gemcitabine treatment. All agents were used at a concentration of 5 μM for 24 h. MICA/B expression was examined by FACS. A representative histogram is shown. Red histogram: background control; blue: no treatment; orange: agent treatment. (B) Median fluorescence intensity (MFI) of MICA/B expression in each agent-treated sample was quantified and normalized to that of the untreated control sample. Data represent the mean ± SD of triplicate wells. (C) T24 and TCCSUP cells were treated with 5 μM gemcitabine for 24 h. MICA and MICB mRNA transcripts were examined by quantitative RT-PCR. (D) Effects of MICA/B small interfering RNA (siMICA/B). (E) In vitro cytotoxicity assay was performed on MICA/B-knockdown UBC cells. (F) In vitro cytotoxicity assay was performed on UBC cells in the presence of anti-MICA/B blocking mAb or isotype control. (G) In vitro cytotoxicity assay was performed on UBC cells in the presence of anti-NKG2D blocking mAb or isotype control. Data represent the mean ± SD of triplicate wells. Statistical significance is displayed as ** for P < 0.01.
MICA/B-knockdown (MICA/B-KD) decreased the cytotoxicity of human Vγ9Vδ2 T cells against UBC cells
To determine the effect of MICA/B-knockdown (MICA/B-KD) on the cytotoxicity of γδT cells, MICA/B were knocked down by siRNA transfection (siMICA/B). siMICA/B efficiently downregulated MICA/B mRNA expression in control cells and in 5 μM GEM-pretreated cells (Fig. 4D). To determine whether the antitumor effect of γδT cells was mediated by the NKG2D receptor, the cytotoxic effects of γδT cells were investigated in MICA/B-KD UBC cells. The results showed that human γδ T cell-mediated cell lysis was significantly decreased in GEM-treated MICA/B-KD UBC cells compared with that in siControl cells (Fig. 4E). However, the cytotoxicity of γδT cells was higher in GEM-treated MICA/B-KD T24 cells than in untreated MICA/B-KD T24 cells. One possible explanation was that siMICA/B didn't completely abrogate the basal activity of MICA/B in GEM-treated T24 cells compared with untreated T24 cells shown in Fig. 4D. This additive MICA/B activity might contribute to the GEM induced cytotoxicity of γδT cells. Another possible explanation might be that there are other mechanisms contributing to the GEM-induced increase in cytotoxicity (Fig. 4E). Next, we performed these experiments using anti MICA/B blocking mAb instead of siRNA. The results showed that human γδT cell-mediated cell lysis was significantly decreased in GEM-treated MICA/B-blocking UBC cells compared with that in GEM-treated isotype control cells (Fig. 4F). Collectively, these data support the concept that downregulation of MICA/B plays a critical role in the cytotoxicity of γδT cells induced by GEM.
NKG2D receptor modulated the additional cytotoxicity of human Vγ9Vδ2 T cells against UBC cells
To elucidate the role of NKG2D receptor in Vγ9Vδ2 T cells, we conducted the NKG2D blocking experiments in T24 and TCCSUP cells since other NKG2D ligands than MICA/B could be blocked. The experiment protocol was the same with the cytotoxicity assay performed in MICA/B knockdown cells (Fig. 4E). We used the blocking mAb specific for NKG2D instead of MICA/B. As expected, blocking of NKG2D receptor resulted in a markedly inhibition of target GEM-treated UBC cell lysis by γδT cells (Fig. 4G). These data support our concept that GEM increases the cytotoxicity of γδT cells via upregulation of NKG2D ligands.
Successful antitumor activity of human Vγ9Vδ2 T cells in in vivo orthotopic UBC xenograft models
To determine whether γδT cells could exert strong antitumor effects in vivo, we established an in vivo orthotopic UBC xenograft model. For this purpose, 5–6-week-old female SCID mice were intravesically administrated with 1.0 × 106 UBC-luc cells through a 24 gauge catheter on day 0. Of the luciferase transfected UBC cell lines tested (T24, TCCSUP, and UMUC-3), only UMUC-3 cell line could dwell and grow in the urinary bladder of mice successfully. Therefore, we conducted in vivo experiments focusing on UMUC3-luc cells. Anti-asialo GM1 antibody (100 μg/body, i.p.) was injected immediately before UMUC3-luc cell transplantation on day 0 to deplete the NK activities of these SCID mice as previously described.27 In vivo orthotopic UBC xenografts were established and used to examine the antitumor activity of γδT cells (Fig. 5A). Mice were randomized into two groups as follows: (i) γδT cell-treated (n = 6; days 7, 14, 21, and 28; 1 × 107 γδT cells along with 5 μM ZOL dissolved in 150 μl PBS by intravesical injection) and (ii) PBS treated (n = 7; days 7, 14, 21, and 28; 150 μl PBS by intravesical injection). Mice were analyzed using an In Vivo Imaging System (IVIS) (Fig. 5B). γδT cell-treated mice showed markedly lower UMUC3-luc expressing bioluminescense intensity than PBS-treated mice (Fig. 6A and D). In the experiments shown in Fig. 6A, one mouse treated by γδT cell died at the 2-week. This was a consequence of anesthesia because this mouse died immediately after induction of anesthesia. Also, weekly, repeated urethral ligation for 4h should be highly stressful for 5-6-week-old very young mice, sometimes mice unexpectedly died irrelevant to cancer growth. By contrast, PBS treated mice obviously became weaken and moved slowly at 3-week and 4-week. These mice undoubtfully died related to cancer growth. In addition, we observed differences in the laparotomy findings, with large intravesical tumors detected in PBS-treated mice, whereas the bladders of γδT cell-treated mice were almost intact on day 30 (Fig. 6B). In the histopathological analysis, H&E-stained images of bladders showed gross tumor invasion and mitotic cells in PBS-treated mouse specimens. By contrast, inhibition of tumor growth was obvious and tissues were almost intact in γδT cell plus ZOL-treated mice, and the normal structures were preserved, similar to control mice (Fig. 6C). Collectively, these data demonstrated that γδT cells have strong antitumor activity in orthotopic xenograft models without prominent adverse effects on the normal urinary bladder.
Figure 5.

Experimental design of the in vivo orthotopic mouse model. (A) Example of the 5–6 week orthotopic xenograft model. On the initial day, mice were intravesically treated with UMUC3-luc expressing cells (1.0 × 106 cells) following anti-asialo GM (100 μg/body i.p.) antibody treatment. (B) Flowchart of the experimental design. UMUC3-luc cells (1 × 106) were orthotopically transplanted into 5–6-week-old female SCID mice by intravesical administration on day 0. The transplanted tumors were allowed to establish for 7 days. The mice were then randomized and divided into two groups followed by treatment by intravesical injections of 1 × 107 γδT cells along with 5 μM ZOL or PBS once a week for a total of four injections. (C) Flowchart of the experimental design in combination with gemcitabine. Same as in (B), with intraperitoneal gemcitabine pretreatment once a week for a total of four injections (days 6, 13, 20, and 27).
Figure 6.
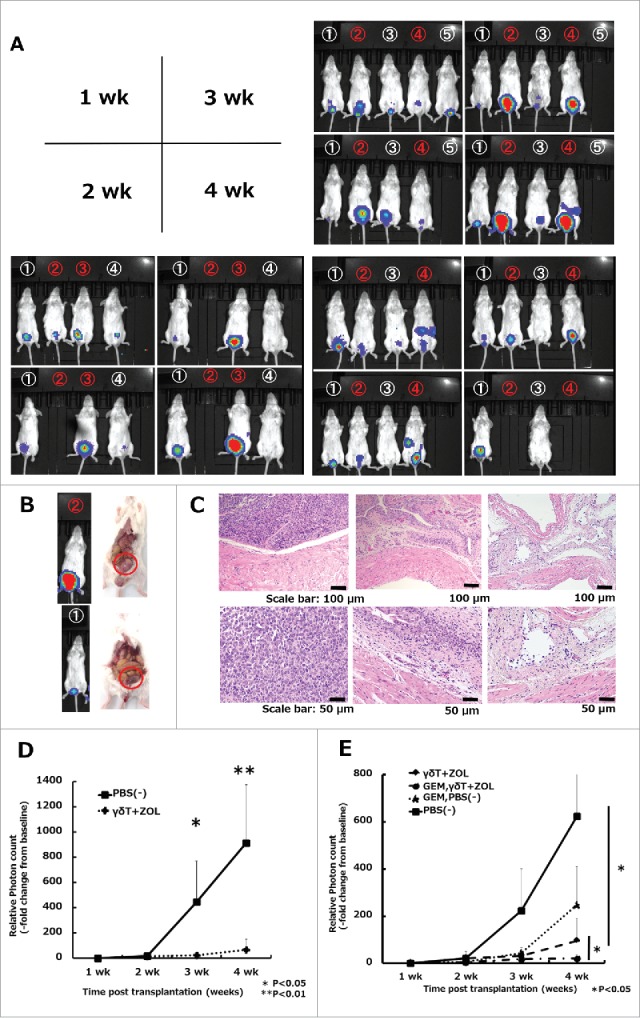
In vivo effects of Vγ9Vδ2 T cell weekly intravesical repeat treatment in combination with gemcitabine in an orthotopic mouse model. (A) Effects of weekly repeat treatment with γδT cells combined with 5 μM ZOL. Numbers in white denote γδT cell plus ZOL-treated mice, whereas numbers in red denote PBS-treated mice (vehicle control). The bioluminescense intensity of γδT cell plus ZOL-treated mice was markedly inhibited when compared with that in the vehicle-control arm, especially at 3 and 4 weeks. A blank space under the number indicates that the mice died during the analysis period. Representative images of three independent experiments are shown. (B) Representative laparotomy findings on day 30. Upper: PBS treatment; lower: γδT cell with 5 μM ZOL treatment. (C) Representative images of H&E-stained bladder sections on day 30. Left: PBS-treated mouse; middle: γδT cell-treated mouse; right: non-tumor transplanted mouse (control). Original magnification: × 10 (upper panels), × 20 (lower panels); scale bars: 100 μm (upper panels), 50 μm (lower panels). (D) Results of IVIS measurements. Experimental design is shown in Fig. 5B. Tumor growth was detected by cancer cell expressing luciferase signal (exposure: 3 min; binning: large) and bioluminescence intensity was quantified using the region of interest (ROI) tool. Results are expressed as the mean ± SEM (n = 6–7 per group). (E) Results of IVIS measurements in the four groups. The experimental design is shown in Fig. 5C. Tumor burden and bioluminescence intensity were most reduced in the ZOL and γδT cell in combination with the gemcitabine group when compared with the other groups. Weekly, repeated pretreatment with GEM (5 μM; dissolved in 200 μL PBS; four i.p. injections) synergistically enhanced the cytotoxicity of γδT cells when compared with the GEM untreated group in vivo with a statistically significant difference. Results are expressed as the mean ± SEM (n = 4–6 per group). Statistical significance is displayed as ** for P < 0.01 and * for P < 0.05.
GEM increased the cytotoxicity of human Vγ9Vδ2 T cells in combination with ZOL in an orthotopic xenograft model
The in vivo antitumor effects of γδT cells and ZOL in combination with GEM were examined in our orthotopic xenograft model. At 6 days after tumor establishment, mice were randomized into four groups, with two groups added to the two groups described above: (iii) GEM + PBS-treated (5 μM GEM/200 μl i.p. on days 6, 13, 20, and 27 + 150 μl PBS on days 7, 14, 21, and 28 by intravesical injection) and (iv) GEM + γδT cells-treated (5 μM GEM/200 μl i.p. on days 6, 13, 20, and 27 + 1 × 107 γδT cells along with 5 μM ZOL dissolved in 150 μl PBS on days 7, 14, 21, and 28 by intravesical injection). Mice were analyzed by IVIS weekly (Fig. 5C). Mice treated with γδT cells plus ZOL in combination with GEM showed greater decreases in tumor burden and bioluminescence intensity than the other groups. Moreover, GEM pretreatment synergistically enhanced the cytotoxicity of γδT cells in vivo when compared with the group without GEM pretreatment and the difference was statistically significant (P = 0.029) (Fig. 6E and S6). These results indicated that γδT cell adoptive immunotherapy in combination with GEM is an effective strategy against tumor growth in vivo and may be a promising approach to the treatment of UBC.
Discussion
Increasing evidence indicates that cancer immunotherapy with ex vivo-expanded human Vγ9Vδ2 T cells and ZOL has cytotoxic effects on malignant tumors.28,29 Human Vγ9Vδ2 T cells can recognize small nonpeptidic phosphorylated compounds such as IPP through the TCRγδ receptor, and ZOL pretreatment is widely used to induce IPP accumulation in cancer cells.14,30 TCRγδ-mediated signaling constitutes the basis of current γδT cell-based immunotherapeutic strategies against cancer.31 Moreover, human Vγ9Vδ2 T cells recognize and kill target cancer cells through a number of mechanisms, including interactions between NKG2D and NKG2DL.
In 1999, Bauers et al. reported that MICA is a functional ligand that stimulates the NKG2D receptor.32 NKG2D is a C-type lectin receptor shared by NK cells, γδT cells, and CTLs, which recognize NKG2DL such as MICA, MICB, and UL16-binding proteins (ULBP1-6). MICA/B molecules are not expressed in most normal tissues, whereas they are abundant in many cancer tissues and virus or bacteria-infected transformed cells. TCRγδ stimulation alone is insufficient to fully achieve γδT cell cytotoxicity, and an additional co-stimulatory signal via receptors such as NKG2D is required.33 Rincon-Orozco et al. and Wrobel et al. reported the function of NKG2D as costimulatory receptor in tumor killing.34,35 Previous studies highlighted the importance of NKG2D/NKG2DL interactions for cancer cell recognition and γδT cell-mediated cytotoxicity.34-37 In the present study, in vitro cytotoxicity assays demonstrated that the cytotoxicity of γδT cells against UBC cells was markedly increased by ZOL pretreatment (Fig. 2A, Fig. 2B, and S1). Consistent with previous reports, human γδT cells showed little cytotoxicity against normal UB cells (Fig. 2B and S1).32,37 Next, we determined the effect of anticancer agents on the cell surface expression of MICA/B because low dose DNA damaging anticancer agents upregulate NKG2DLs in various cancer cell lines.38,39 Todaro et al. reported the effect of chemotherapeutic agents on the stress-inducible upregulation in cancer initiating cells.40,41
Several anticancer agents affect immune reactions and upregulate cancer antigens in target cells, thereby activating immune cells.42-44 Skov et al. reported that HDAC inhibitors, which have anticancer activity, upregulate NKD2DLs on the surface of several cancer cells.45 In this study, the GEM and other agents increased the cytotoxic effects of γδT cells despite showing minimal cytotoxicity at the indicated concentrations for 24 h (Fig. 3). GEM was mainly used in this study to increase the sensitivity of γδT cell mediated cytotoxicity since GEM remains the first line anticancer agent against UBC in clinical setting. Thus, combinatory GEM and adoptive transfer of γδT cell may be more clinically applicable than other agents. Next, to determine whether the NKG2D-MICA/B signaling pathway was involved in the increased cytotoxicity, we investigated the effect of MICA/B-KD on the cytotoxicity of γδT cells against UBC cells. Our results showed that siRNA against MICA/B significantly abrogated the increased cytotoxicity (Fig. 4E). Also, these findings were confirmed by MICA/B blocking experiments (Fig. 4F). Moreover, other ligands than MICA/B such as ULBP1-6 expression were also upregulated by GEM treatment (Fig. S4) and blocking NKG2D receptor led to decreased UBC cell lysis by γδT cells. These findings support the role of NKG2D as a key receptor for the recognition of target cancer cells and suggest that anticancer agents promote an “eat me-like” signal by upregulating MICA/B in target cells. Combination γδT cell therapy incorporating low dose GEM to promote the activation of γδT cells via NKG2D could be a promising anticancer strategy.
Weekly, repeated intravesical γδT cell infusion therapy showed a strong cytotoxic effect in our orthotopic xenograft model (Fig. 6). In previous work, we reported that human γδT cell intravesical treatment showed potent cytotoxicity in an in vivo orthotopic xenograft model.46 In that study, mice were divided into four groups as follows: (i) PBS infusion; (ii) γδT cell infusion; (iii) ZOL infusion; and (iv) γδT cell plus ZOL simultaneous infusion. Our results showed that γδT cells combined with ZOL infusion treatment significantly decreased bioluminescense intensity in mice compared with other groups. Based on these results, we tested the effect of simultaneous infusion of γδT cells and ZOL, though it was different from in vitro cytotoxicity assays, where ZOL was treated 24h prior to γδT cell addition. In our previous study, intravesical treatment was performed for five sequential days starting on day 4 after tumor transplantation, when faint bioluminescense intensity was detected by IVIS. Unlike our previous protocol, here we used a weekly bladder instillation treatment protocol as it is a clinically acceptable schedule. Moreover, we showed that low dose GEM pretreatment synergistically enhanced the cytotoxicity of γδT cells in our in vivo orthotopic xenograft model.
Kobayashi et al. reported that adoptive transfer of γδT cells stimulated with 2M3B1PP combined with ZOL and rhIL2 against advanced renal cell carcinoma resulted in a good response.47 Bennouna et al. reported the infusion of BrHHP-stimulated γδT cells in non-small lung cell carcinoma patients and showed that these autologous γδT cells were safe and well tolerated.48 Although many clinical studies and trials of γδT cell-based immunotherapy have been reported, the outcomes were not necessarily satisfactory.49-51 One possible explanation is that most of these clinical trials were implemented through a systemic infusion approach. This limitation could be overcome if γδT cells can reach target cancer tissues locally. The bladder instillation approach is therefore of interest because the urinary bladder in pooled urine phase is an obstructive space, and γδT cells can directly migrate into cancer tissues. Clinical trials of intravesical γδT cell immunotherapy remain to be performed, and the bladder instillation strategy may open up new avenues in γδT cell research. The results of our in vivo mouse xenograft experiments indicate that the bladder infusion approach may be clinically applicable and could have beneficial effects in patients with refractory UBC.
Currently, intravesivcal BCG therapy is the gold standard of superficial bladder tumor treatment following TUR-Bt. However, we could not discuss about the possibility of combining γδT cell adoptive transfer with BCG therapy using our orthotopic xenograft model since SCID mice lack the innate immune system so as not to reject the transplanted human tumor cells, one limitation might be that we could not examine the efficacy of BCG, which depends on an intact immune system for adequate response.
In conclusion, we showed that GEM pretreatment increased the cytotoxicity of human γδT cells partially by upregulating the NKG2D ligands MICA/B in target cancer cells. These results indicate that γδT cell-based chemo-immunotherapy is a promising approach in combination with pretreatment with chemotherapy agents. Our results strongly suggest that the bladder infusion strategy could make a breakthrough in γδT cell-based immunotherapy. Further research is needed to uncover the mechanisms underlying the increased cytotoxicity induced by chemotherapeutic agents. We would like to plan an early phase clinical trial in the near future to verify the efficacy and safety of intravesical administration of γδT cells in human subjects.
Materials and methods
Cell lines and reagents
The UBC cell lines UMUC3, T24, and TCCSUP were purchased from American Type Culture Collection. Luciferase stably-expressing cancer cell lines were generated by transfection with Red Firefly vector (#16157 Thermo Fisher) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions, and all cancer cell lines were cultured in RPMI1640 (Wako Pure Chemical Industries) supplemented with 10% heat-inactivated fetal-bovine serum (FBS; Sigma Aldrich) and 1% penicillin and streptomycin (PC/SM; Wako Pure Chemical Industries). Cells were kept in a 37°C incubator with 5% CO2, and all experiments were performed within 30 passages. HBEC-A (human normal urinary bladder epithelium) was purchased (#KP-4109, Kurabo Corporation) and cultured in UrolifeA medium using a proliferation kit (#LUC-LL0063, Kurabo Corporation). ZOL was a gift from Dr. Yoshimasa Tanaka, Nagasaki University, and recombinant human IL2 (rhIL2) (#093-03953) and anticancer agents (gemcitabine, cisplatin, methotrexate, vinblastine, adriamycin, and mitomycin C) were all purchased from Wako Pure Chemical. Anti-human MICA/B antibody was purchased from eBiosciences (#12-5788, clone6D4). siMICA/B and siControl were purchased from R&D Systems (#sc-43931 and sc-37007).
Anti-human ULBP-1 antibody and anti-human ULBP-2/5/6 antibody were purchased from R&D Systems (#170818 and #165903). CFSE was from Dojindo Corporation (#341-06443).
Expansion of human Vγ9Vδ2 T cells
Healthy donors provided written informed consent for the use of peripheral blood for research purposes in accordance with the Declaration of Helsinki and under approval by Kyoto Pharmaceutical University Review Board. Peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque (GE Healthcare). PBMCs were cultured in AlyS505N medium (Funakoshi) supplemented with 10% heat-inactivated AB human serum, and rhIL2 (100 U/ml) and ZOL (5 μM) were added on day 0, with additional rhIL2 (100 U/ml) added every day during the culture period. The medium was appropriately scaled up dependent on cell expansion. These cells were used as effector cells (E) in the cytotoxicity assay. Cells were analyzed by flow cytometry on day 11 and CD3+/TCRγδ+ cells were determined as γδT cells.
Flow cytometric analysis
Cells were suspended in 100 μl PBS at a density of 1 × 107 cells/ml and stained with fluorochrome-conjugated monoclonal antibodies (mAbs) specific for cell surface antigens corresponding to CD3-FITC (#555332), TCRαβ-PE (#564728), TCRγδ-APC (#555718), CD45-FITC (#555482), CD45-PE (#555483), CD45-APC (#555485), TCRVγ9-PE (#331307, cloneB3), TCRVδ2-PE (#555717) and NKG2D-FITC (#11-5878, clone1D11). After 20 min incubation on ice in the dark, cells were washed twice, and the pellet was resuspended in 500 μl PBS and examined by flow cytometry on a FACSCalibur system. Data were analyzed using Flowjo software (Flowjo LLC).
For intracellular staining, 5 × 107/ml cells in 100 μl were stained by TCRγδ mAb, fixed, and permeabilized for 20 min on ice with 4% paraformaldehyde (PFA) and 0.2% Tween 20. Cells were then washed twice, resuspended in 100 μl PBS, stained with anti-Perforin-Alexafluor 488 (#563764, cloneδG9) or anti-Granzyme B-PE mAb (#561142, cloneGB11), incubated for an additional 20 min at room temperature in the dark, and washed twice. Finally, cells were resuspended in 500 μl PBS and examined by flow cytometry. To retain the granules inside cells, the GolgiStop reagent (#554724, eBioscience) was added during the final 4 h of culture. The Cell Stimulation cocktail 500 × (#00-4970, eBioscience) was used to activate PBMCs as a positive control in the presence of GolgiStop.
In vitro cytotoxicity assay
In vitro cytotoxicity was determined by flow cytometry using CFSE and PI staining. Cancer cells were labeled with CFSE at a final concentration of 0.5 μM and incubated for 30 min in a 5% CO2 atmosphere. Then, CFSE-labeled cancer cells were centrifuged, washed twice, resuspended in complete medium, and seeded at a density of 1.0 × 106 cells per 2 ml medium per well in 6-well plates. If indicated, immediately after UBC cells were seeded to the 6-well plates, ZOL and/or anticancer agents were added to the medium at the indicated dilution concentration. After 24 h of incubation, γδT cells (E/T ratio: 1/1, 5/1, and 10/1) were added to each well after removing harmful anticancer agents and replacing the medium with fresh medium. After co-culture for 4 h, samples were collected, washed twice, the pellet was resuspended in 500 μl PBS, and 2.5 μl PI (1 mg/ml) was added to each sample before flow cytometric analysis. Samples were examined using a FACSCalibur system, and the CFSE+/PI+ population was measured as apoptotic cancer cells. In MICA/B blocking experiments, blocking mAb specific for MICA/B (#558032, clone6D4, BD) or isotype control mAb (#555571, cloneG155-178, BD) were added to the medium in the 6-well plate at the final concentration of 2.5 μg/ml for 30 min, prior to co-incubation with γδT cells. In NKG2D blocking experiments, blocking mAb specific for NKG2D (#149810, R&D) or isotype control mAb (#11711, R&D) were added to the medium in the 6-well plate at the final concentration of 2.5 μg/ml for 30 min, prior to co-incubation with γδT cells.
WST-8 assay
The antiproliferative activities of various anticancer agents against UBC cell lines were examined using the WST-8 assay according to the manufacturer's protocol (Nacalai Tesque). Briefly, 3.0 × 104 cells per well were seeded in 96-well plates in the presence of different concentrations of anticancer agents in a final volume of 100 μl and incubated for 24 or 72 h at 37°C. The Cell Count Reagent SF (Nacalai Tesque) was added at 10 μl per well. After incubation for 3 h, the absorbance at 450 nm (reference wavelength: 630 nm) was read using a microplate reader (Model 680 Microplate Reader; Bio-Rad). Values were normalized to the untreated (control) sample and expressed as the relative fold increase.
SiRNA transfection of cancer cells
A predesigned double-stranded siRNA (MICA/B siRNA, sc-43931, Santa Cruz Biotechnology Inc.) was used to inhibit MICA/B expression. The siControl nontargeting siRNA (control siRNA-A, sc-37007, Santa Cruz Biotechnology Inc.) was used as the reference control. Transfection was performed in UBC cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. Transfected cells were seeded into 6-well plates at a density of 1 × 106 cells per well and incubated for 24 h. The expression of MICA and MICB was analyzed by RT-PCR.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from UBC cells pretreated with or without the indicated anticancer agents using the Illustra RNAspin Mini RNA Isolation kit (GE Healthcare) following the manufacturer's protocol. RNA was quantified by NanoDrop 2000 (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized using the High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative RT-PCR was performed using the Thermal Cycler Dice Real Time System II (Takarabio). PCR amplification was performed as follows: denaturation at 95°C for 10 sec, annealing at 55–57°C for 15 sec, and extension at 72°C for 1 sec. The PCR reaction was terminated after 40–50 cycles. The primer sequences and probes were designed using the Universal Probe Library (UPL). Primers used in this study were as follows:
MICA (Hu) gene: forward, 5′-GGCATCTTCCCTTTTGCAC-3′,
reverse, 5′-GGACAGCACCGTGAGGTTAT3′;
MICB (Hu) gene: forward, 5′CTGAGAAGGTGGGGACGTA-3′,
reverse, 5′-CGAAGACTGTGGGGCTCA-3′;
18S-rRNA gene: forward, 5′-GCAATTATTCCCATGAACG-3′,
reverse, 5′-GGGACTTAATCAACGCAAGC-3′.
The relative expression of MICA and MICB in each sample was normalized to that of 18S-rRNA (internal control).
Immunofluorescence image analysis
T24 cancer cells were labeled with PKH26 (red) (Sigma Aldrich) and nuclei were labeled with Hoechst33342 (blue) (Thermo Fisher Scientific) according to the manufacturer's instructions.
γδT cells were labeled with CFSE (green) at a final concentration of 0.5 μM for 30 min in the dark. Images were captured using a confocal laser scanning microscope (LSM510). Data were analyzed by ImageJ software.
In vivo orthotopic UBC xenograft model
Female SCID mice (5–6-weeks old) were purchased from Japan SLC and injected with 1 × 106 UMUC3-luc cells dissolved in 150 μl PBS on day 0 by transurethral injection. On day 6 or 7, mice were monitored by IVIS, and luciferase expressing cell signals were measured and quantified. In all intravesical procedures, mice urethra were ligated immediately after γδT cells/PBS infusions and ligated suture were released 4h later and spontaneous voiding restarted.
If bioluminescence was not detected or extra urinary bladder bioluminescence was suspected, mice were excluded from the study. Mice were divided into two groups: group one received intravesical administration of γδT cells (1 × 107 cells) plus 5 μM ZOL dissolved in 150 μl PBS, followed by treatment once a week for a total of four injections (days 7, 14, 21, and 28); group 2 received intravesical administration of 150 μl PBS in the same way (days 7, 14, 21, and 28). Bioluminescense intensity was monitored by IVIS weekly as cancer cell growth can be visualized noninvasively. In the analysis using four groups, the same experimental design was used except that both groups were treated with 5 μM GEM dissolved in 200 μl PBS by intraperitoneal injection (days 6, 13, 20, and 27) before intravesical treatment once a week for a total of four injections.
The relative fold change of each week's value from the baseline value (1 week) was individually calculated. All mouse experiments were performed under pathogen-free conditions in line with Institutional Animal Care protocols approved by Kyoto Pharmaceutical University.
Histopathology
Following IVIS measurements, the urinary bladder and kidney were removed, excised, fixed in 4% PFA, paraffin embedded, sectioned (3 μm), and stained with hematoxylin and eosin (H&E). Images were captured using a microscope (Olympus BX50).
Statistical analysis
Continuous variables were analyzed using the Student's t-test. Multiple comparisons between groups were made using the Bonferroni/Dunn test. P-values of <0.05 were considered statistically significant.
Supplementary Material
Funding Statement
This work was supported by a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology in Japan (MEXT, 15K10604 to HN and 26461436 to EA) and MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2017–2021 (S1511024L to EA). The authors have no conflicts of interest to declare.
Abbreviations
- CFSE
carboxy-fluorescein succinimidyl ester
- GEM
gemcitabine
- MHC
Major Histocompatibility Complex
- MICA/B
MHC class I-related chain protein A/B
- NKG2D
Natural Killer Group 2 member D
- NKG2DL
NKG2D ligand.
- PBS
phosphate buffered saline
- TCR
T cell receptor
- UBC
urinary bladder cancer
- ZOL
zoledronic acid
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Tetsuya Takada and Syohei Kawanishi (Department of Clinical and Translational Physiology, Kyoto Pharmaceutical University) for technical advice and assistance in biological experiments, and Dr. Kiyoshi Sato (Department of Thoracic Surgery, Osaka Medical University) for special advice regarding the method for generating the UBC orthotopic transplantation mouse model. We also thank Yoko Nakagawa (Department of CCMT, Kyoto University) for technical advice on how to appropriately culture human γδT cells.
References
- 1.Ghasemzadeh A, Bivalacqua TJ, Hahn NM, Drake CG. New Strategies in Bladder Cancer: A Second Coming for Immunotherapy. Clin Cancer Res. 2016;22:793–801. doi: 10.1158/1078-0432.CCR-15-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barocas DA, Globe DR, Colayco DC, Onyenwenyi A, Bruno AS, Bramley TJ, Spear RJ. Surveillance and treatment of non-muscle-invasive bladder cancer in the USA. Advances in urology. 2012;2012:421709. doi: 10.1155/2012/421709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morales A, Eidinger D, Bruce AW. Intracavitary Bacillus Calmette-Guerin in the Treatment of Superficial Bladder Tumors. The Journal of urology. 2017;197:S142–s5. doi: 10.1016/j.juro.2016.10.101. [DOI] [PubMed] [Google Scholar]
- 4.Sylvester RJ. Bacillus Calmette-Guerin treatment of non-muscle invasive bladder cancer. International journal of urology: official journal of the Japanese Urological Association. 2011;18:113–20. doi: 10.1111/j.1442-2042.2010.02678.x. [DOI] [PubMed] [Google Scholar]
- 5.Zlotta AR, Fleshner NE, Jewett MA. The management of BCG failure in non-muscle-invasive bladder cancer: an update. Canadian Urological Association journal = Journal de l'Association des urologues du Canada. 2009;3:S199–205. doi: 10.5489/cuaj.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herr HW, Dalbagni G. Defining bacillus Calmette-Guerin refractory superficial bladder tumors. The Journal of urology. 2003;169:1706–8. doi: 10.1097/01.ju.0000062605.92268.c6. [DOI] [PubMed] [Google Scholar]
- 7.Kunzmann V, Bauer E, Wilhelm M. Gamma/delta T-cell stimulation by pamidronate. The New England journal of medicine. 1999;340:737–8. doi: 10.1056/NEJM199903043400914. [DOI] [PubMed] [Google Scholar]
- 8.Kunzmann V, Bauer E, Feurle J, Weissinger F, Tony HP, Wilhelm M. Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood. 2000;96:384–92. [PubMed] [Google Scholar]
- 9.Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, Tony HP. Gammadelta T cells for immune therapy of patients with lymphoid malignancies. Blood. 2003;102:200–6. doi: 10.1182/blood-2002-12-3665. [DOI] [PubMed] [Google Scholar]
- 10.Bonneville M, O'Brien RL, Born WK. Gammadelta T cell effector functions: A blend of innate programming and acquired plasticity. Nature reviews Immunology. 2010;10:467–78. doi: 10.1038/nri2781. [DOI] [PubMed] [Google Scholar]
- 11.Vantourout P, Hayday A. Six-of-the-best: unique contributions of gammadelta T cells to immunology. Nature reviews Immunology. 2013;13:88–100. doi: 10.1038/nri3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Acker HH Anguille S, Van Tendeloo VF Lion E. Empowering gamma delta T cells with antitumor immunity by dendritic cell-based immunotherapy. Oncoimmunology. 2015;4:e1021538. doi: 10.1080/2162402X.2015.1021538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rei M, Pennington DJ, Silva-Santos B. The emerging Protumor role of gammadelta T lymphocytes: implications for cancer immunotherapy. Cancer Res. 2015;75:798–802. doi: 10.1158/0008-5472.CAN-14-3228. [DOI] [PubMed] [Google Scholar]
- 14.Sandstrom A, Peigne CM, Leger A, Crooks JE, Konczak F, Gesnel MC, Breathnach R, Bonneville M, Scotet E, Adams EJ. The intracellular B30.2 domain of butyrophilin 3A1 binds phosphoantigens to mediate activation of human Vgamma9Vdelta2 T cells. Immunity. 2014;40:490–500. doi: 10.1016/j.immuni.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayday AC. Gammadelta T cells and the lymphoid stress-surveillance response. Immunity. 2009;31:184–96. doi: 10.1016/j.immuni.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Fisher JP, Flutter B, Wesemann F, Frosch J, Rossig C, Gustafsson K, Anderson J. Effective combination treatment of GD2-expressing neuroblastoma and Ewing's sarcoma using anti-GD2 ch14.18/CHO antibody with Vgamma9Vdelta2+ gammadeltaT cells. Oncoimmunology. 2016;5:e1025194. doi: 10.1080/2162402X.2015.1025194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchida R, Ashihara E, Sato K, Kimura S, Kuroda J, Takeuchi M, Kawata E, Taniguchi K, Okamoto M, Shimura K, et al.. Gamma delta T cells kill myeloma cells by sensing mevalonate metabolites and ICAM-1 molecules on cell surface. Biochem Biophys Res Commun. 2007;354:613–8. doi: 10.1016/j.bbrc.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 18.Ashihara E, Munaka T, Kimura S, Nakagawa S, Nakagawa Y, Kanai M, Hirai H, Abe H, Miida T, Yamato S, et al.. Isopentenyl pyrophosphate secreted from Zoledronate-stimulated myeloma cells, activates the chemotaxis of gammadeltaT cells. Biochem Biophys Res Commun. 2015;463:650–5. doi: 10.1016/j.bbrc.2015.05.118. [DOI] [PubMed] [Google Scholar]
- 19.Chen G, Emens LA. Chemoimmunotherapy: Reengineering tumor immunity. Cancer Immunol Immunother. 2013;62:203–16 doi: 10.1007/s00262-012-1388-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nature reviews Immunology. 2009;9:353–63 doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mattarollo SR, Kenna T, Nieda M, Nicol AJ. Chemotherapy and zoledronate sensitize solid tumour cells to Vgamma9Vdelta2 T cell cytotoxicity. Cancer Immunol Immunother. 2007;56:1285–97. doi: 10.1007/s00262-007-0279-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kabelitz D, Wesch D, He W. Perspectives of gammadelta T cells in tumor immunology. Cancer Res. 2007;67:5–8. doi: 10.1158/0008-5472.CAN-06-3069. [DOI] [PubMed] [Google Scholar]
- 23.Stresing V, Daubine F, Benzaid I, Monkkonen H, Clezardin P. Bisphosphonates in cancer therapy. Cancer Lett. 2007;257:16–35. doi: 10.1016/j.canlet.2007.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Sato K, Kimura S, Segawa H, Yokota A, Matsumoto S, Kuroda J, Nogawa M, Yuasa T, Kiyono Y, Wada H, et al.. Cytotoxic effects of gammadelta T cells expanded ex vivo by a third generation bisphosphonate for cancer immunotherapy. Int J Cancer. 2005;116:94–9. doi: 10.1002/ijc.20987. [DOI] [PubMed] [Google Scholar]
- 25.Chitadze G, Lettau M, Luecke S, Wang T, Janssen O, Furst D, Mytilineos J, Wesch D, Oberg HH, Held-Feindt J, et al.. NKG2D- and T-cell receptor-dependent lysis of malignant glioma cell lines by human gammadelta T cells: Modulation by temozolomide and A disintegrin and metalloproteases 10 and 17 inhibitors. Oncoimmunology. 2016;5:e1093276. doi: 10.1080/2162402X.2015.1093276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruenbacher G, Nussbaumer O, Gander H, Steiner B, Leonhartsberger N, Thurnher M. Stress-related and homeostatic cytokines regulate Vgamma9Vdelta2 T-cell surveillance of mevalonate metabolism. Oncoimmunology. 2014;3:e953410. doi: 10.4161/21624011.2014.953410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habu S, Fukui H, Shimamura K, Kasai M, Nagai Y, Okumura K, Tamaoki N. In vivo effects of anti-asialo GM1. I. Reduction of NK activity and enhancement of transplanted tumor growth in nude mice. Journal of immunology (Baltimore, Md: 1950). 1981;127:34–8. [PubMed] [Google Scholar]
- 28.Deniger DC, Moyes JS, Cooper LJ. Clinical applications of gamma delta T cells with multivalent immunity. Frontiers in immunology. 2014;5:636. doi: 10.3389/fimmu.2014.00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buccheri S, Guggino G, Caccamo N, Li Donni P, Dieli F. Efficacy and safety of gammadeltaT cell-based tumor immunotherapy: A meta-analysis. Journal of biological regulators and homeostatic agents. 2014;28:81–90. [PubMed] [Google Scholar]
- 30.Harly C, Guillaume Y, Nedellec S, Peigne CM, Monkkonen H, Monkkonen J, Li J, Kuball J, Adams EJ, Netzer S, et al.. Key implication of CD277/butyrophilin-3 (BTN3A) in cellular stress sensing by a major human gammadelta T-cell subset. Blood. 2012;120:2269–79. doi: 10.1182/blood-2012-05-430470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Henry O, Distefano MD, Wang YC, Raikkonen J, Monkkonen J, Tanaka Y, Morita CT. Butyrophilin 3A1 plays an essential role in prenyl pyrophosphate stimulation of human Vgamma2Vdelta2 T cells. Journal of immunology (Baltimore, Md: 1950). 2013;191:1029–42. doi: 10.4049/jimmunol.1300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science (New York, NY). 1999;285:727–9. doi: 10.1126/science.285.5428.727. [DOI] [PubMed] [Google Scholar]
- 33.Nedellec S, Sabourin C, Bonneville M, Scotet E. NKG2D costimulates human V gamma 9V delta 2 T cell antitumor cytotoxicity through protein kinase C theta-dependent modulation of early TCR-induced calcium and transduction signals. Journal of immunology (Baltimore, Md: 1950). 2010;185:55–63. doi: 10.4049/jimmunol.1000373. [DOI] [PubMed] [Google Scholar]
- 34.Rincon-Orozco B, Kunzmann V, Wrobel P, Kabelitz D, Steinle A, Herrmann T. Activation of V 9V 2 T Cells by NKG2D. The Journal of Immunology. 2005;175:2144–51. doi: 10.4049/jimmunol.175.4.2144. [DOI] [PubMed] [Google Scholar]
- 35.Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, Kabelitz D, Wesch D. Lysis of a broad range of epithelial tumour cells by human gamma delta T cells: involvement of NKG2D ligands and T-cell receptor- versus NKG2D-dependent recognition. Scand J Immunol. 2007;66:320–8. doi: 10.1111/j.1365-3083.2007.01963.x. [DOI] [PubMed] [Google Scholar]
- 36.Cerwenka A, Baron JL, Lanier LL. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:11521–6. doi: 10.1073/pnas.201238598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waldhauer I, Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–43. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 38.Gasser S, Raulet D. The DNA damage response, immunity and cancer. Seminars in cancer biology. 2006;16:344–7. doi: 10.1016/j.semcancer.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Krieg S, Ullrich E. Novel immune modulators used in hematology: Impact on NK cells. Frontiers in immunology. 2012;3:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Todaro M, Orlando V, Cicero G, Caccamo N, Meraviglia S, Stassi G, Dieli F. Chemotherapy sensitizes colon cancer initiating cells to Vgamma9Vdelta2 T cell-mediated cytotoxicity. PloS one. 2013;8:e65145. doi: 10.1371/journal.pone.0065145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Todaro M, Meraviglia S, Caccamo N, Stassi G, Dieli F. Combining conventional chemotherapy and gammadelta T cell-based immunotherapy to target cancer-initiating cells. Oncoimmunology. 2013; 2:e25821. doi: 10.4161/onci.25821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Raulet DH. Roles of the NKG2D immunoreceptor and its ligands. Nature reviews Immunology. 2003;3:781–90. doi: 10.1038/nri1199. [DOI] [PubMed] [Google Scholar]
- 43.Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016;76:1683–9. doi: 10.1158/0008-5472.CAN-15-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang B, Sikorski R, Sampath P, Thorne SH. Modulation of NKG2D-ligand cell surface expression enhances immune cell therapy of cancer. J Immunother. 2011;34:289–96. doi: 10.1097/CJI.0b013e31820e1b0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skov S, Pedersen MT, Andresen L, Straten PT, Woetmann A, Odum N. Cancer cells become susceptible to natural killer cell killing after exposure to histone deacetylase inhibitors due to glycogen synthase kinase-3-dependent expression of MHC class I-related chain A and B. Cancer Res. 2005;65:11136–45. doi: 10.1158/0008-5472.CAN-05-0599. [DOI] [PubMed] [Google Scholar]
- 46.Yuasa T, Sato K, Ashihara E, Takeuchi M, Maita S, Tsuchiya N, Habuchi T, Maekawa T, Kimura S. Intravesical administration of gammadelta T cells successfully prevents the growth of bladder cancer in the murine model. Cancer Immunol Immunother. 2009;58:493–502. doi: 10.1007/s00262-008-0571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobayashi H, Tanaka Y, Yagi J, Minato N, Tanabe K. Phase I/II study of adoptive transfer of gammadelta T cells in combination with zoledronic acid and IL-2 to patients with advanced renal cell carcinoma. Cancer Immunol Immunother. 2011;60:1075–84. doi: 10.1007/s00262-011-1021-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bennouna J, Levy V, Sicard H, Senellart H, Audrain M, Hiret S, Rolland F, Bruzzoni-Giovanelli H, Rimbert M, Galéa C, et al.. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vgamma9Vdelta2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol Immunother. 2010;59:1521–30. doi: 10.1007/s00262-010-0879-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abe Y, Muto M, Nieda M, Nakagawa Y, Nicol A, Kaneko T, Goto S, Yokokawa K, Suzuki K. Clinical and immunological evaluation of zoledronate-activated Vgamma9gammadelta T-cell-based immunotherapy for patients with multiple myeloma. Experimental hematology. 2009;37:956–68. doi: 10.1016/j.exphem.2009.04.008. [DOI] [PubMed] [Google Scholar]
- 50.Nakajima J, Murakawa T, Fukami T, Goto S, Kaneko T, Yoshida Y, Takamoto S, Kakimi K. A phase I study of adoptive immunotherapy for recurrent non-small-cell lung cancer patients with autologous gammadelta T cells. European journal of cardio-thoracic surgery: official journal of the European Association for Cardio-thoracic Surgery. 2010;37:1191–7. doi: 10.1016/j.ejcts.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 51.Nicol AJ, Tokuyama H, Mattarollo SR, Hagi T, Suzuki K, Yokokawa K, Nieda M. Clinical evaluation of autologous gamma delta T cell-based immunotherapy for metastatic solid tumours. British journal of cancer. 2011;105:778–86. doi: 10.1038/bjc.2011.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.