

Abstract
The GxxxG motif is frequently found at the dimerization interface of a transmembrane structural motif called GASright, which is characterized by a short interhelical distance and a right-handed crossing angle between the helices. In GASright dimers, such as glycophorin A (GpA), BNIP3, and members of the ErbB family, the backbones of the helices are in contact, and they invariably display networks of 4 to 8 weak hydrogen bonds between Cα–H carbon donors and carbonyl acceptors on opposing helices (Cα–H···O=C hydrogen bonds). These networks of weak hydrogen bonds at the helix–helix interface are presumably stabilizing, but their energetic contribution to dimerization has yet to be determined experimentally. Here, we present a computational and experimental structure-based analysis of GASright dimers of different predicted stabilities, which show that a combination of van der Waals packing and Cα–H hydrogen bonding predicts the experimental trend of dimerization propensities. This finding provides experimental support for the hypothesis that the networks of Cα–H hydrogen bonds are major contributors to the free energy of association of GxxxG-mediated dimers. The structural comparison between groups of GASright dimers of different stabilities reveals distinct sequence as well as conformational preferences. Stability correlates with shorter interhelical distances, narrower crossing angles, better packing, and the formation of larger networks of Cα–H hydrogen bonds. The identification of these structural rules provides insight on how nature could modulate stability in GASright and finely tune dimerization to support biological function.
Introduction
Oligomerization is critical for the biological function of many membrane proteins. In particular, oligomerization is important for the bitopic or “single-pass” proteins [i.e., those that span the membrane bilayer with a single transmembrane (TM) helix], which are the largest class of integral membrane proteins.1−3 Over 2300 single-pass proteins are predicted to exist in the human proteome alone, including oligomerizing systems such as receptor tyrosine kinases,4−8 cytokine receptors,9,10 integrins,11,12 cadherins,13 apoptotic regulators,14−16 enzymes,17 immunological complexes,18 and many more.19 The TM helices often have a critical role in driving and modulating the oligomerization of these systems, frequently acting in cooperation with the proteins’ soluble domains. Deciphering the rules that govern TM helix oligomerization in these systems is critical to understanding function and mechanisms of disease in a broad array of biological events.
The oligomerization of TM helices is often mediated by structural motifs that are evolutionarily optimized for protein–protein interactions.20,21 One of the most prevalent dimerization motifs for single-pass proteins is the fold of the glycophorin A dimer (GpA), which is named GASright from the right-handed crossing angle between the helices (near −40°), and the presence of small amino acids (Gly, Ala, Ser: GAS).20 These small residues are arranged to form GxxxG and GxxxG-like sequence motifs (GxxxG, GxxxA, SxxxG, etc.)22−24 typically found at the GASright dimerization interface (Figure 1a). As extensively reviewed by Teese and Langosch, GxxxG sequence motifs are prevalent in biology, and they are frequently associated with parallel, right-handed GASright structures (although GxxxG can also be found in antiparallel or left-handed dimers and even at lipid-binding sites).19 The sequence context surrounding the GxxxG motif can modulate stability,25,26 and thus, the versatile GASright motif can be found both in proteins that form very stable “structural” dimers (such as GpA27 and BNIP316), as well as in weaker and dynamic systems in which changes in conformation or oligomerization state are necessary for supporting function (such as signaling in members of the ErbB receptor tyrosine kinase4,5,7,28−30 and integrin families31−33). Despite its common occurrence and importance, however, the fundamental physical rules that determine the strength of GASright dimerization are yet not well understood.
Figure 1.
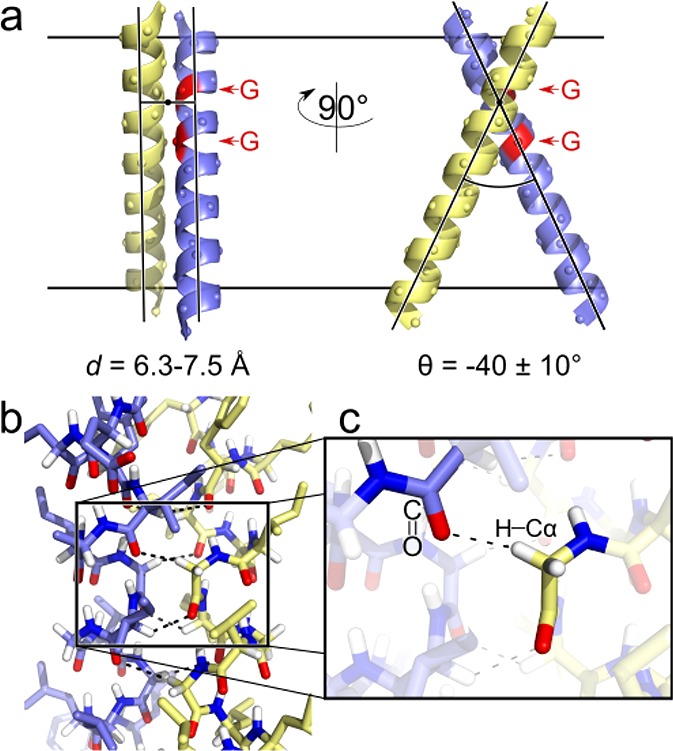
The GASright dimerization motif. (a) The GASright motif is a right-handed helical dimer with a short interhelical distance (6.3–7.5 Å) and a right-handed crossing angle of approximately −40°. The GxxxG sequence pattern at the crossing point (red) allows the backbones to come into contact. (b) The contact enables formation of networks of weak interhelical H bonds between Cα–H donors and carbonyl oxygen acceptors (shown in detail in c).
The major unknown is the contribution of weak hydrogen bonds that occur at the interface of GASright dimers to the free energy of dimerization. GASright invariably displays networks of hydrogen bonds formed by Cα–H carbon donors and carbonyl acceptors (Cα–H···O=C), occurring in four to eight instances between atoms on opposing helices at the association interface (Figure 1, panels b and c).34 In general, hydrogen bonding can be a stabilizing force in membrane proteins, and it has been shown that “canonical” hydrogen bonds (i.e., those formed by oxygen or nitrogen donors) can drive the interaction of TM helices.35−39 Carbon is a weaker donor than oxygen or nitrogen, but Cα–H groups are activated by the flanking electron-withdrawing amide groups in the peptide backbone, and thus the strength of Cα–H hydrogen bonds has been estimated to be as much as one-half of the N–H donors in vacuum.40,41 Therefore, it is plausible that multiple Cα–H hydrogen bonds occurring at the dimerization interface would contribute significantly to the free energy of association in GASright dimers.34,42 Nevertheless, experimental demonstration of this hypothesis has, so far, remained elusive.
A major technical challenge in measuring the contribution of Cα–H hydrogen bonds to TM helix association in GASright dimers is the fact that both the donor and acceptor groups are part of the backbone, making a rational mutation strategy difficult to implement. To date, there have been only two experimental studies that have probed the contribution of Cα–H hydrogen bonds in membrane proteins. One of these studies was not performed on a GASright dimer but rather on the 7-TM helix membrane protein bacteriorhodopsin.43 The study focused on the interaction between a Cα–H hydrogen bond donor and a threonine hydroxyl group acceptor and found that the removal of the side-chain acceptor group by mutation did not destabilize folding. However, it should be noted that the study targeted one isolated Cα–H hydrogen bond that occurs in the context of a large, multispan membrane protein. A second study investigated the energy of interaction of a Cα–H hydrogen bond in a GASright dimer by IR spectroscopy, estimating a favorable interaction energy of −0.88 kcal/mol between the Cα–H donor of Gly 79 and the carbonyl of Ile 76 of GpA.44 This result supports the notion that Cα–H hydrogen bonds are likely significantly stabilizing. However, it is understood that geometry can play a significant role in determining the strength of Cα–H hydrogen bonds,45 and this study is limited to a single specific bond among the many found in GpA. Moreover, the study measured hydrogen bonding strength but not its contribution to the free energy of dimerization, which has not been yet directly assessed.
The hypothesis that Cα–H hydrogen bonds are major contributors to the free energy of GASright dimerization remains compelling, particularly given by the unique ability of the structural motif to form this unusual feature. In fact, among all possible symmetric homodimeric configurations, GASright is the only one that promotes the formation of a large number of concurrent Cα–H hydrogen bonds.42 This ability arises from three unique aspects of the geometry of GASright: (1) a crossing angle that precisely aligns Cα–H donors and carbonyl acceptors across two helices, (2) the presence of Gly at certain specific positions (producing the GxxxG pattern), where they are necessary to prevent clashing between the close helices, and (3) the ability of those same Gly residues to increment the number of Cα–H bonds by donating their second Hα. Therefore, GASright appears to be a structural motif optimized for the formation of Cα–H hydrogen bond networks.
We found that an algorithm (CATM) based on the simultaneous optimization of van der Waals forces and Cα–H hydrogen bonding was able to predict the small database of known three-dimensional structures of GASright homodimers to near atomic precision,42 another finding that indirectly reinforces the importance of these forces in dimerization. The CATM algorithm was later successfully applied to predict the interface of a previously uncharacterized GxxxG-containing dimer, ADCK3, a mitochondrial protein that plays an essential role in the biosynthesis of coenzyme Q.17 CATM can capture, with remarkable accuracy, the structural features of a variety of GASright dimers. The success in predicting structure raises the question of whether the underlying energetic model can also capture, at least in part, the energetics of GASright dimerization.
To address this question, here we have combined CATM with a high-throughput biological assay to examine the relationship between structure and stability of GASright dimers of various geometries. We have applied CATM to the over 2300 sequences of TM domains of single-pass proteins present in the human genome, predicting the structure of hundreds of potential GASright dimers. We then selected candidates that represent a range of predicted dimerization stabilities and assessed their association propensity with TOXCAT, a widely used in vivo reporter assay that is sensitive to the relative association of TM dimers in a biological membrane.46 After several steps of experimental validation, we obtained computational and experimental measurements for 26 well-behaved candidate GASright homodimers. We observe a significant correlation in the overall trend of energies predicted computationally and the dimerization propensities measured experimentally. These data provide the first experimental evidence for a model in which a combination of van der Waals forces and Cα–H hydrogen bonding acts as a primary source of stability, modulating the strength of GASright association.
Results and Discussion
Structural Prediction of GASright Homodimers
The CATM algorithm is designed to predict the structure of potential GASright homodimers from the amino acid sequence of a TM domain by docking the two helices and simultaneously optimizing van der Waals interactions, weak and canonical hydrogen bondings, and an implicit membrane solvation model. The algorithm only considers potential GASright conformations, and it does not explore the entire conformational range of a generic TM helix dimer, which makes it efficient and capable of searching for potential GASright dimers in high-throughput in large databases of TM sequences.
To create a diverse set of predicted GASright dimer structures to be tested experimentally, we drew sequences from the human proteome. The Uniprot database of annotated protein sequences currently identifies 2383 human proteins containing a single TM domain.47 When these TM domain sequences were run through CATM, they produced 1141 potential GASright dimers with a negative (i.e., favorable) energy score (dimer energy–monomer energy). The CATM scores assume a broad range of association energies, from −70 to 0 kcal/mol, with a skewed bell distribution (Figure 2a). The left tail of the distribution contains sequences enriched in well-packed structures with extensive Cα–H hydrogen bonding networks that are predicted to be very stable (Figure 2, panels b and c). The top 10% of the predicted structures form an average of 6.0 ± 1.7 Cα–H bonds. The predicted structures represent a rich repertoire of potential GASright dimers covering a wide range of predicted stabilities for follow-up experimental analysis.
Figure 2.

Energy distribution of CATM predicted GASright dimers in human single-pass sequences. (a) Histogram of calculated energies of human GASright dimers. CATM identified 1141 sequences that produced a model with a negative (favorable) energy of association. (b) Extensive complementary packing, as well as (c) the characteristic networks of Cα–H hydrogen bonds displayed by the lowest energy structures, chondroitin sulfate glucuronyltransferase (Uniprot accession Q9P2E5).
For the subsequent experimental phase, we did not consider any sequence whose dimer interface contained strongly polar residues (Asp, Glu, Arg, Lys, His, Asn, and Gln) or Pro. We chose to exclude these residues because proline has a tendency to form kinks in helices that are difficult to predict,48,49 while strongly polar residues have a propensity to drive TM association through the formation of interhelical hydrogen bonds.37−39,50 Their inclusion would have increased the probability of dimers mediated by nonspecific interfaces, breaking the desired structural correspondence between the model predicted by CATM and the constructs in experimental conditions. These exclusions reduced the number of available sequences to 668. We also excluded sequences with predicted marginal stability (a score higher than −5 kcal/mol). From the remaining 604 sequences, we randomly selected 65 diverse candidates for experimental analysis (Tables S1–S4).
Experimental Strategy: TOXCAT Assay Using Standardized Sequences
To experimentally assess the dimerization of the 65 predicted GASright dimers and their mutants, we used TOXCAT, a widely adopted assay that measures TM homo-oligomerization in biological membranes.46 This system is based on the in vivo expression of a chimeric protein in the inner membrane of Escherichia coli in which the TM domain of interest is fused to the ToxR transcriptional activator. Dimerization of the TM helices brings together two ToxR subunits, which bind to a specific promoter, activating the expression of the reporter gene chloramphenicol acetyltransferase (CAT). Quantification of CAT thus provides an indication of the extent of TM helix–helix association in a biological membrane (Figure 3a).
Figure 3.
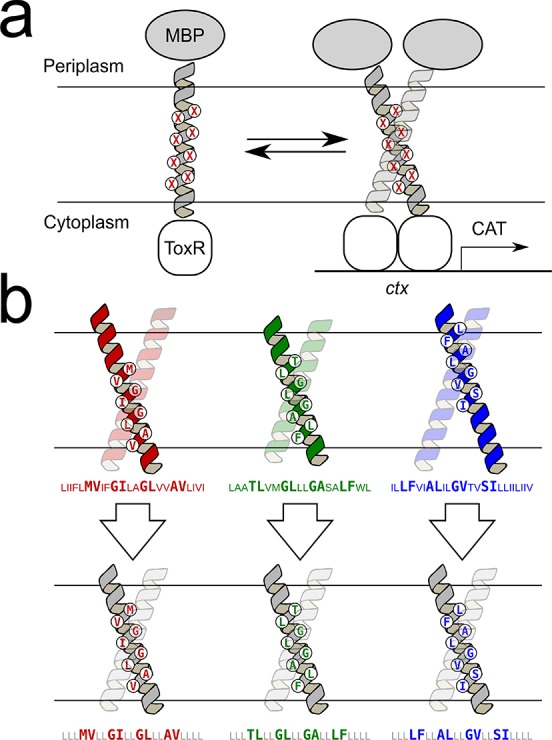
Experimental design. (a) TOXCAT is an in vivo assay based on a construct in which the TM domain under investigation is fused to the ToxR transcriptional activator. TM association results in the expression of a reporter gene in E. coli cells, which can be quantified. (b) To reduce variability in TOXCAT, the eight interfacial amino acids identified by CATM in the wild-type sequences (top) were “stitched” into a standardized poly-Leu sequence (bottom). Standardization of the predicted constructs retains the geometry of the interface while controlling the length of the TM helix, the position of the crossing point, and the hydrophobicity of the TM segment.
The general relationship between reporter gene expression in TOXCAT and thermodynamic stability of any given dimer is likely complex, but reasonable correlation has been found for collections of point mutants of GpA and their energy of dimerization in detergents.51,52 In these studies, the constructs are homogeneous, having identical length of the TM region, nearly identical sequence, and comparable hydrophobicity. Because TOXCAT’s response may be dependent on these variables,15,53,54 controlling them is likely to simplify the comparison between constructs. The predicted lengths of the TM domains of the 2383 human single-pass sequences in Uniprot range widely,22 and their estimated ΔG of membrane insertion ranges from −6.7 to +11.9 kcal/mol (using the biological ΔGapp predictor55). To reduce heterogeneity as much as possible, we adopted a strategy of “stitching” the 8 positions predicted by CATM to be at the helix–helix interface of a standardized TM helix of 21 amino acids consisting of a poly-Leu backbone (LLLxxLLxxLLxxLLxxLILI, where the x represents the variable interfacial positions).
As illustrated in Figure 3b, this stitching strategy ensures that all constructs have the same TM domain length and that the predicted interface is centered in the middle of the membrane. Perhaps most importantly, the standardized sequence reduces the variability in hydrophobicity. Because the noninterfacial residues in all the constructs remain constant, the ΔGapp range for membrane insertion is reduced to −6.6 to −2.9 kcal/mol, likely leading to a more consistent expression of the constructs in the E. coli membrane. Another important reason for standardizing all noninterfacial positions is that the strategy removes potential alternative dimerization interfaces that may be present within the wild-type sequence because only the amino acids involved in the predicted GASright interfaces are carried over into the standardized constructs.
There is an existing precedent for such a strategy with GASright homodimers: it has been shown that the interfacial residues of GpA in a leucine backbone behave similarly to the wild-type sequence.46 In addition, a pure poly-Leu sequence has a relatively low propensity for self-association in TOXCAT,37,39,56 which is important for reducing the risk of alternate interfaces. To ensure that the interfaces of the standardized sequences were consistent with those initially predicted for the wild-type sequences, the standardized sequences were also evaluated with CATM (Tables S1–S4). We found that CATM consistently predicts nearly identical interfaces for wild-type and standardized constructs. The computed energies that we report for our analysis below correspond to those calculated using the standardized poly-Leu construct and not the original wild-type sequences.
Experimental Validation of the Predicted Structures
To partially validate the predicted structural models, we adopted a mutagenesis strategy. Saturation mutagenesis has been commonly used to identify or confirm the interface of TM dimers.15,17,57−59 Because it would be impractical to perform saturation mutagenesis of all 65 candidate constructs, we opted to introduce in each construct a single mutation predicted to be highly detrimental, selecting the most sensitive interfacial position of GASright homodimers, the so-called “C1” position, as defined in our previous work.42 The C1 position is one of the residues near the crossing point of the helical dimer. In GASright homodimers, C1 is required to be occupied by Gly in order to allow contact between the backbones of the two helices.42 Substitution of Gly at C1 with a large hydrophobic amino acid, such as Ile, would push the helices apart and completely eliminate any potential association mediated by the predicted interface. We computationally verified that all models of C1Gly→Ile variants contained significant clashes. Introduction of this control enabled the removal of constructs that retained significant association in TOXCAT after the C1Gly→Ile mutation, since these results suggest that the dimerization observed experimentally was not mediated by the predicted GASright structural model (or, alternatively, that a second possible dimerization interface is also present in the construct, which is not disrupted by the C1Gly→Ile mutation).
To confirm proper membrane insertion, each of the 65 constructs and their C1Gly→Ile variants was tested for its ability to support growth in minimal medium containing maltose as the only carbon source, as standard practice in TOXCAT.46 A total of 15 constructs (wild type or C1 variant) did not fully grow in these conditions (Table S2). These constructs were not further considered in the study. We then eliminated constructs whose TOXCAT signal was below the minimal threshold of a pure poly-Leu construct because we would not be able to differentiate specific GASright-mediated dimerization from background association. A pure poly-Leu construct displays approximately 30% of the CAT expression level of the GpA standard, therefore any construct below the 30% threshold was eliminated (10 constructs, Table S3).
Finally, any constructs whose C1Gly→Ile control variant scored above 30% of relative CAT expression level were also eliminated from the analysis because they did not match our expected model, as explained earlier (14 constructs, Table S4). As an exception to this rule, if a C1Gly→Ile mutation reduced the “wild-type” CAT activity by at least 75% we retained it for analysis, even if it was above the 30% threshold, because of the dramatic reduction in dimerization (3 constructs). The final 26 GASright constructs are listed in Table S1. Their predicted structural models are illustrated in Figure S1. The progression from the 2328 genomic sequence to the final 26 experimental constructs is summarized in Table S5. We verified the expression of the ToxR-TM-MBP chimeras of the 26 constructs by Western blots: the constructs displayed rather homogeneous levels of expression, with a standard deviation of 22% (Figure S2).
Cα–H Hydrogen Bonds and vdW Predict Experimental Association Propensities
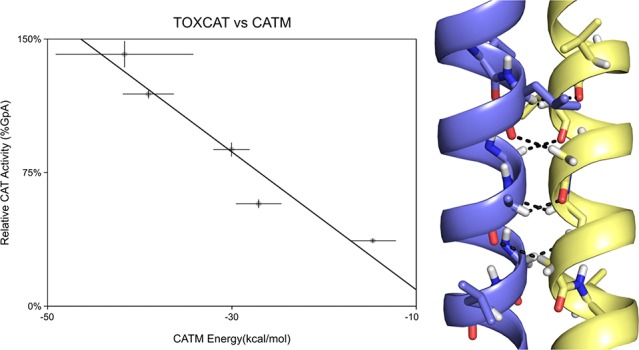
The comparison of association energies calculated with CATM and dimerization propensities assessed by TOXCAT for the 26 selected constructs is shown in Figure 4a. The plot shows a statistically significant correlation (R2 = 0.441, p < 0.0005, t test of linear regression slope). One clear outlier is present in the plot (the TNR12 construct, TOXCAT 119%, CATM −6 kcal/mol, highlighted in gray): if this point is excluded, the R2 increases to 0.647 (p < 0.000005). The correlation is also statistically significant by rank order correlation coefficient analysis, which does not assume a linear model (r = −0.683, p < 0.005, and r = −0.827, p < 0.000001, with and without TNR12, respectively).60 Some of the variance is likely due to the biological nature of the TOXCAT assay, some to imprecision by CATM in predicting the structures, and the remaining variance can be attributed to the limitations of the energy model, which was constructed solely on its ability to predict structure. However, the energetic model is clearly able to capture the trend of dimerization propensities observed experimentally.
Figure 4.

Comparison of CATM energies with apparent TOXCAT dimerization. (a) Comparison of CATM energy score of 26 sequences and their TOXCAT signal (measured as the enzymatic activity of the reporter gene CAT). The points are color-coded according to the grouping in (b). The error bars represent the standard deviation among replicates. The dashed line represents the linear regression fit of the data, with the exclusion of the outlier point highlighted in gray (R2 = 0.647, p < 0.000005). (b) Same data as in (a), grouped and averaged in five bins based on CAT activity from weak (>25%, magenta) to very strong (>125%, blue), in 25% intervals. The error bars represent the standard error of the average. The dashed line is the linear regression of the data (R2 = 0.931, p < 0.01). The groups are the base of the analysis reported in Figure 5.
Structural and Sequence Analysis of Groups with Distinct Stability
Interesting differences in structural and sequence features are observed among constructs with different dimerization propensities. To appreciate these structural and energetic properties that distinguish strong from weaker dimers, we grouped the data according to five levels of TOXCAT signal, using five 25%-wide bins, from very weak (25–50% GpA) to very strong apparent dimerization (>125% GpA). We first confirmed that the energy model is sensitive enough to distinguish between the five stability groups. Indeed, proportionality is retained after TOXCAT and CATM values are averaged within each groups (Figure 4b). Linear regression of these averaged values produces a significant fit (p < 0.01), with a R2 value of 0.931 if the TNR12 outlier is excluded, and a R2 value of 0.883 when TNR12 is included (p < 0.05, Figure S3). The regression analyses of Figure 4 (panels a and b) produce two distinct equations of the line, which is an expected mathematical outcome of averaging. However, it should be noted that a linear relationship is likely not the correct physical model and is not necessarily expected.61 What is important is that there is proportionality between TOXCAT and CATM outcomes, and that the energetic model is able to clearly differentiate among the five sets of constructs. Therefore, the grouping is suitable for a comparative analysis of sequence and structural features that characterize constructs with increasing apparent stability. Statistical analysis of the trends independent of grouping is also provided.
Stability Correlates with Sequence Biases
The results of the sequence and structural features of the five groups are summarized in Table 1 and Figure 5. Some sequence biases at the interface of the predicted dimers were already present in the initial pool of 604 sequences, as expected for a selection of GASright dimers (Figure S4). Most notably, the sequences are enriched with GxxxG and GxxxG-like motifs, and Gly is nearly absolutely preserved at position C1, where this amino acid is required for interhelical backbone contact (the nomenclature of the positions is defined in Figure 5b).42 However, on top of these biases, a number of interesting trends emerged within our experimental pool that correlate statistically with their stability.
Table 1. Energetic and Geometric Properties of Groups of Constructs of Different Apparent Dimerization and Statistical Significance of the Distributionsa.
| TOXCAT range (% GpA) | 25–50% | 50–75% | 75–100% | 100–125% | 125+% | correlation with TOXCAT |
|---|---|---|---|---|---|---|
| number of constructs | 7 | 7 | 4 | 4 | 3 | |
| average TOXCAT (% GpA) | 37 ± 1 | 58 ± 2 | 88 ± 4 | 118 ± 2 | 141 ± 7 | |
| CATM energy score (kcal/mol)b | –14.7 ± 2.5 | –27.1 ± 2.5 | –30.0 ± 2.0 | –39.1 ± 2.8 | –41.7 ± 7.5 | p < 0.000001d |
| van der Waals (kcal/mol) | –26.2 ± 5.3 | –33.7 ± 4.5 | –33.6 ± 2.1 | –39.3 ± 2.4 | –39.0 ± 11.1 | p < 0.005d |
| Cα-H hydrogen bonding (kcal/mol) | –5.2 ± 1.1 | –8.0 ± 1.9 | –9.7 ± 0.5 | –12.0 ± 2.3 | –13.0 ± 0.8 | p < 0.000001d |
| solvation (kcal/mol) | 16.7 ± 1.9 | 14.2 ± 1.9 | 13.3 ± 2.0 | 11.7 ± 2.4 | 10.6 ± 2.7 | p < 0.00005d |
| crossing angle (deg) | –51 ± 4 | –47 ± 6 | –49 ± 2 | –41 ± 7 | –39 ± 7 | p < 0.01d |
| number of Cα-H hydrogen bonds | 4.6 ± 1.0 | 5.1 ± 1.1 | 6.0 ± 0.0 | 7.5 ± 1.0 | 8.0 ± 0.0 | N/Ae |
| interface surface area (Å2) | 4810 ± 490 | 4660 ± 500 | 4630 ± 190 | 4770 ± 540 | 4510 ± 280 | – |
| interhelical distance (Å) | 7.1 ± 0.2 | 6.7 ± 0.3 | 6.5 ± 0.1 | 6.4 ± 0.1 | 6.5 ± 0.0 | p < 0.00005d |
| van der Waals/interface surface area [kcal/(mol Å2)] | –0.0054 ± 0.0012 | –0.0073 ± 0.0015 | –0.0073 ± 0.0005 | –0.0083 ± 0.0008 | –0.0086 ± 0.0021 | p < 0.001d |
| sequences with GxxxG | 2/7 | 4/7 | 3/4 | 4/4 | 3/3 | p < 0.01f |
| sequences with Sm-xxx-Smc | 6/7 | 7/7 | 4/4 | 4/4 | 3/3 | – |
| sequences with Gly at N1 | 0/7 | 3/7 | 3/4 | 4/4 | 3/3 | p < 0.0001f |
| sequences with Gly at C1 | 7/7 | 7/7 | 4/4 | 4/4 | 3/3 | – |
| sequences with Gly at C5 | 2/7 | 1/7 | 0/4 | 2/4 | 1/3 | – |
All values are reported as averages ± standard deviation unless noted. The outlier TNR12 was excluded from the 100–125% group.
Sm-xxx-Sm are defined by any combinations of Gly, Ala, Ser, and Cys at the first and last position.
Rank order (Spearman) correlation analysis.60
Rank correlation statistics not applicable to noncontinuous variable.
Point Biserial Correlation analysis.62
Figure 5.

Sequence and structural bias occur in groups with different stabilities. (a) Sequences of the 26 constructs ranked by TOXCAT signal showing the groups, as defined in Figure 4. GxxxG motifs are underlined with a solid line, GxxxG-like motifs with a dotted line. Color coding as in Figure 4b. (b) Nomenclature of the interfacial positions, as defined previously.42 The sequence and structural biases of the groups of experimental constructs (orange symbols) are illustrated for (c) the number of Cα–H hydrogen bonds, which increases with stability, (d) the interhelical distance, and (e) crossing-angle, which decrease with stability, and the fraction of sequences containing (f) GxxxG and (g) Gly at the N1 position, which also increase. Data also reported in Table 1. The same trends are observed in groups of different stabilities computed from the entire data set of 670 structures predicted from the human proteome (blue symbols).
The first trend is the frequency of the GxxxG motif, which increases from the least to the most stable groups (Figure 5c, orange symbols, and Table 1). In particular, the three more stable groups (>75%, >100%, >125%) contain GxxxG motifs in all but one construct, formed by the C1 Gly and a second Gly either at N1 (the position at i-4 from C1) or at C5 (the position at i+4); conversely, in the two less stable groups (>50% and >25%) GxxxG is found in just 43% of the sequences. The biased distribution of GxxxG containing sequences is confirmed, independently from the grouping scheme, using Point Biserial correlation statistics, which measures correlation between a continuous variable (TOXCAT signal) and a binary variable (occurrence of GxxxG) (correlation coefficient r = +0.63, p < 0.001).62 The fact that GxxxG is present in the most stable constructs is not surprising. However, it should be noted that some low-stability sequences also contain GxxxG, further demonstrating that the presence of the sequence motif is not the sole determinant of stability.25,26 Notably, all sequences that do not contain a GxxxG motif contain a Small-xxx-Small motif (i.e., GxxxA, SxxxG, etc.), with the exception of one low affinity construct (1A32–2).
The GxxxG motifs in the sequence can be formed by the invariable Gly at C1 together with a second Gly at either N1 or C5. However, the marked increase of GxxxG in the most stable constructs is primarily due to the presence of a Gly at position N1 (Figure 5d). In the three most stable groups 10 out of 11 of the sequences have a Gly at N1, whereas Gly occurs rarely at the same position in the lower stability groups (p < 0.0001). Conversely, Gly at C1 is rarer and its presence does not correlate with apparent stability.
Stability Correlates with Structural Features
These trends suggest that distinct sequence biases occur among GASright dimers of different stabilities. To understand their physical basis, we looked at how structural parameters varied as a function of apparent stability. We observed numerous structure-related differences, which are summarized in Table 1 and Figure 5. The analysis indicates that as stability increases, (i) the distance between the helices becomes increasingly shorter, (ii) the crossing angle becomes smaller, (iii) the structural models become increasingly better packed, and (iv) they display larger networks of Cα–H hydrogen bonds.
The interhelical distance (measured between the helical axes) progressively decreases from an average of 7.1 Å for the lowest stability set, down to 6.5 Å for the most stable group, which is near the closest two helices can approach before their backbones would sterically clash (Figure 5e). The correlation between TOXCAT and interhelical distance is statistically significant (rank order spearman correlation coefficient r = +0.74, p < 0.0005, Table 1). We also observe a reduction of the interhelical angle, which progressively decreases toward −40° (p < 0.01, Figure 5f). These geometric changes are favored by the presence of Gly at N1, as discussed in the previous section.
The tighter interhelical contact in the most stable constructs leads to an increase of favorable van der Waals interactions between the helices (p < 0.005), which improve by 49% from the lowest to the highest dimerizing groups from −26.2 to −39.0 kcal/mol. These improved van der Waals interactions do not originate from a larger dimer interface (which remains relatively constant across the sets), therefore they are attributable to better packing. The more intimate interhelical contact also favors a very significant change in hydrogen bonding: the number of interhelical Cα–H hydrogen bonds increases from 4.6 to 8.0 on average (Figure 5g). Correspondingly, the average contribution of hydrogen bonding to the binding energy more than doubles from −5.2 to −13.0 kcal/mol (p < 0.000001). Finally, we observe a reduction of the cost of desolvating the helices (from +17 kcal/mol to +11 kcal/mol) that also contributes to the better energy score computed in CATM for the more stable dimers (p < 0.00005).
To further investigate these sequence and geometry biases, we performed a similar analysis on the entire set of 604 poly-Leu predicted structures, grouping the results by decreasing CATM energy (blue symbols in Figure 5 and Table S7). We observe very similar trends across all examined variables. A progressive reduction of the crossing angle and interhelical distance is observed as the energy score decreases, along with an increased number of Cα–H hydrogen bonds and improvement of the packing. Similarly, presence of a GxxxG motif increases, reaching 100% in the lowest energy groups. The trend mirrors the presence of Gly at position N1, whereas the presence of Gly at C5 (the second position that can form a GxxxG motif with C1) also increases but not as dramatically, topping at 50% in the lowest energy group.
In summary, the model suggests that the stronger interactions tend to be formed by helices that have a closer distance and a smaller crossing angle. These geometries tend to be favored by the presence of a second Gly at N1, forming a GxxxG motif with the Gly at C1, although the precise stability and conformation of each dimer is influenced by its entire sequence context. It is possible that some of the observed results may be influenced by the current experimental conditions. For example, the optimal crossing angle could be sensitive to the thickness of the membrane and the length of the TM helices, which were not varied in either the computational or the TOXCAT experiments. Nevertheless, these biases provide important insight into how the GASright sequence is able to modulate stability, a feature that is likely important for a structural motif that is found in both stable constitutive dimers, as well as in weaker “dynamic” or transient dimers, where dissociation or conformational change is required for the function of the protein.
Conclusions
An unusual interaction is at the core of one of the most common transmembrane motifs, and yet the contribution of these Cα–H hydrogen bond networks to the free energy of dimerization has remained uncertain. This is in part due to scarce availability of structures, which poses a serious hurdle to understanding the structural basis of TM helix oligomerization. Structure-based analysis has been possible for a few structurally characterized dimeric systems, such as GpA25,63 and BNIP3.16 Conversely, large-scale comparative analyses, based either on combinatorial libraries,23,64−68 comprehensive protein families,69 or homology-based clusters of human proteins53 have been performed primarily on sequences of unknown structure. Computational modeling has often been applied in coordination to these approaches to aid in the interpretation of experimental data.11,17,18,57,58,70−76 An advance of the present work is the availability of a reliable structural prediction method, which has enabled the design of an experimental analysis of dimers of diverse stabilities to test pre-existing structural and energetic models.
This analysis addressed the question of whether Cα–H hydrogen bonding and van der Waals forces are predictive of the dimerization propensity of GASright dimers. The results provide the first experimental support for the hypothesis that Cα–H hydrogen bonds are indeed major determinants of dimerization in GxxxG-mediated dimers. Our data complement the only other experimental report in the literature that has shown that Cα–H hydrogen bonds have the potential to stabilize GASright, a study by Arbely & Arkin that measured the strength of a single hydrogen bond interaction in a well-characterized GASright model system;44 here, we addressed the role of Cα–H hydrogen bonds as contributor to the free energy of dimerization, examined at the level of the entire structural motif.
We found that a simple energy model combining Cα–H hydrogen bonding and van der Waals already forms a good base when tested in biological membranes, albeit in standardized sequence conditions. The present analysis also provides initial insight on how change in the sequence and geometry may modulate these terms and therefore overall stability in GASright. The results also suggest that, with more data, a similar strategy would likely support the development of a more sophisticated energy function, which would provide further insight into the forces involved in GASright association as well as improve our ability to accurately predict structure and stability of these dimers from primary sequence data alone.
Methods
Software
All calculations were implemented and performed using MSL v. 1.1,77 an open source C++ library that is freely available at http://msl-libraries.org.
Prediction of GASright Structure and Dimerization Energy
The structure of GASright dimers was predicted from a database of 2383 human sequences annotated as single-pass membrane proteins in Uniprot (as of November 2, 2016).47 Structural prediction was performed with the program CATM.42 Side chain mobility was modeled using the energy-based conformer library applied at the 95% level.78 Energies were determined using the CHARMM 22 van der Waals function,79 the IMM1 membrane implicit solvation model,80 and the hydrogen bonding function of SCWRL 4,81 as implemented in MSL,77 with the following parameters for Cα donors, as reported previously: B = 60.278, D0 = 2.3 Å, σd = 1.202 Å, αmax = 74.0°, and βmax = 98.0°.42
The CATM algorithm was described in detail previously.42 Briefly, the sequence of interest is threaded into a set of different registers at each of 463 representative geometries. If sequence-based filtering rules are met, the sequence is built on the backbone in all atoms and the helices are docked by reducing the interhelical distance in steps. At each step, the side chains are optimized and the interaction energy is evaluated until a minimum energy is found. To further optimize the dimer, the geometry is then subjected to Monte Carlo backbone perturbation cycles in which all interhelical parameters (distance, Z shift, axial rotation, and crossing angle) are locally varied. If the final interaction energy (calculated as the energy of the dimer minus the energy of two monomers separated at long distance) is negative, the solution is accepted. The solutions are then clustered using an RMSD criterion to produce a series of distinct models. The computation produced 1141 structures of predicted GASright homodimers. These structures are available at http://seneslab.org/CATM.
Cloning and Expression of Chimeric Proteins in MM39 Cells and MalE Complementation Assay
DNA sequences containing the transmembrane region of interest were cut with NheI and DpnII restriction enzymes and cloned into the NheI-BamHI restriction sites of the pccKAN vector as previously described.17,57 The TOXCAT constructs were transformed into MM39 cells. A freshly streaked colony was inoculated into 3 mL of LB broth containing 100 μg/mL ampicillin and grown overnight at 37 °C. 50 μL of overnight cultures were inoculated into 3 mL of LB broth and grown to an OD600 of 0.8–1.0 at 37 °C. After recording the optical density, 1 mL of cells was spun down for 15 min at 17000g and resuspended in 500 mL of sonication buffer (25 mM Tris-HCl, 2 mM EDTA, pH 8.0). Cells were lysed by probe sonication at medium power for 10 s over ice. An aliquot was removed from each sample and stored in SDS–PAGE loading buffer for immunoblotting. The lysates were then cleared by centrifugation at 17000g, and the supernatant was kept on ice for chloramphenicol acetyltransferase (CAT) activity assay.
To confirm proper membrane insertion and orientation of the TOXCAT constructs, overnight cultures were plated on M9 minimal medium plates containing 0.4% maltose as the only carbon source and grown at 37 °C for 48–72 h. The variants that did not grow in these conditions were not considered for this study.
Chloramphenicol Acetyltransferase (CAT) Spectrophotometric Assay
CAT activity was measured as described.57,82 Briefly, 750 μL of buffer containing 0.4 mg/mL 5,5′- dithiobis(2-nitrobenzoic acid) or Ellman’s reagent and 0.1 M Tris-HCl, pH 7.8, were mixed with 250 μL of 0.4 mM acetyl CoA and 40 μL of cleared cell lysates, and the absorbance at 412 nm was measured for 2 min to establish basal enzyme activity rate. After addition of 40 μL of 2.5 mM chloramphenicol in 10% ethanol, the absorbance was measured for an additional 2 min to determine CAT activity. The basal CAT activity was subtracted, and the value was normalized by the cell density measured as OD600. All measurements were determined by at least four independently cultured biological replicates, each of which was measured with two technical replicates.
Quantification of Expression by Immunoblotting
Protein expression was confirmed by immunoblotting. The cell lysates were normalized by cell density and loaded onto a NuPAGE 4–12% bis-tris SDS–PAGE gel (Invitrogen) and then transferred to PVDF membranes (VWR) for 1 h at 100 millivolts. Blots were blocked using 5% bovine serum albumin (US Biologicals) in TBS-Tween buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween 20) for overnight at 4 °C, incubated with goat biotinylated anti-maltose binding protein antibodies (Vector laboratories) for 2 h at room temperature, followed by peroxidase-conjugated streptavidin antigoat secondary antibodies (Jackson ImmunoResearch) for 2 h at 4 °C. Blots were developed with the Pierce ECL Western Blotting Substrate Kit; 1 mL of ECL solution was added to the blot and incubated for 90 s. Chemiluminescence was measured using an ImageQuant LAS 4000 (GE Healthsciences). Individual bands were quantified by ImageJ.
Acknowledgments
The work was supported by National Science Foundation Grants CHE-1415910 and CHE-1710182. B.K.M. and S.M.A. acknowledge the support of the NLM training grant 5T15LM007359 to the CIBM Training Program. S.M.A. also acknowledges the support of the Dr. James Chieh-Hsia Mai Wisconsin Distinguished Graduate Fellowship. B.K.M. also acknowledges support by a fellowship in Informatics from the PhRMA Foundation. E.J.L. acknowledges the support of a Hilldale Undergraduate Research Fellowship. We are grateful to Dr. Sabareesh Subramaniam for contributions to the development of CATM, to Samson Condon and Claire Armstrong for helpful suggestions and discussion, and to Elizabeth Caselle for critical reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07505.
Predicted structure of the experimental constructs; quantification of constructs expression by immunoblotting; Fig. 4b including the outlier; average amino acid composition at interfacial positions; summary of constructs; energetic and geometric summary of all predicted constructs. (PDF)
Author Present Address
§ Vanderbilt University, Department of Chemistry, 7330 Stevenson Center, Station B 351822, Nashville, TN 37235.
Author Contributions
‡ S.M.A. and B.K.M. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Arkin I. T.; Brunger A. T. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1998, 1429 (1), 113–128. 10.1016/S0167-4838(98)00225-8. [DOI] [PubMed] [Google Scholar]
- Hubert P.; Sawma P.; Duneau J.-P.; Khao J.; Hénin J.; Bagnard D.; Sturgis J. Cell Adh Migr 2010, 4 (2), 313–324. 10.4161/cam.4.2.12430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin E.; von Heijne G. Protein Sci. 1998, 7 (4), 1029–1038. 10.1002/pro.5560070420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharov E. V.; Mineev K. S.; Goncharuk M. V.; Arseniev A. S. Biochim. Biophys. Acta, Biomembr. 2012, 1818 (9), 2158–2170. 10.1016/j.bbamem.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Mineev K. S.; Volynsky P. E.; Ermolyuk Y. S.; Tkach E. N.; Sobol A. G.; Chupin V. V.; Kirpichnikov M. P.; Efremov R. G.; Arseniev A. S. J. Biol. Chem. 2008, 283 (11), 6950–6956. 10.1074/jbc.M709202200. [DOI] [PubMed] [Google Scholar]
- Chung I.; Akita R.; Vandlen R.; Toomre D.; Schlessinger J.; Mellman I. Nature 2010, 464 (7289), 783–787. 10.1038/nature08827. [DOI] [PubMed] [Google Scholar]
- Mineev K. S.; Bocharov E. V.; Pustovalova Y. E.; Bocharova O. V.; Chupin V. V.; Arseniev A. S. J. Mol. Biol. 2010, 400 (2), 231–243. 10.1016/j.jmb.2010.05.016. [DOI] [PubMed] [Google Scholar]
- Anbazhagan V.; Munz C.; Tome L.; Schneider D. J. Mol. Biol. 2010, 404 (5), 773–777. 10.1016/j.jmb.2010.10.026. [DOI] [PubMed] [Google Scholar]
- Matthews E. E.; Thévenin D.; Rogers J. M.; Gotow L.; Lira P. D.; Reiter L. A.; Brissette W. H.; Engelman D. M. FASEB J. 2011, 25 (7), 2234–2244. 10.1096/fj.10-178673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilar M.; Charalampopoulos I.; Kenchappa R. S.; Simi A.; Karaca E.; Reversi A.; Choi S.; Bothwell M.; Mingarro I.; Friedman W. J.; Schiavo G.; Bastiaens P. I. H.; Verveer P. J.; Carter B. D.; Ibáñez C. F. Neuron 2009, 62 (1), 72–83. 10.1016/j.neuron.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W.; Metcalf D. G.; Gorelik R.; Li R.; Mitra N.; Nanda V.; Law P. B.; Lear J. D.; Degrado W. F.; Bennett J. S. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (5), 1424–1429. 10.1073/pnas.0409334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Litvinov R. I.; Vilaire G.; Zhu H.; Li W.; Caputo G. A.; Moore D. T.; Lear J. D.; Weisel J. W.; Degrado W. F.; Bennett J. S. J. Biol. Chem. 2006, 281 (48), 36732–36741. 10.1074/jbc.M605877200. [DOI] [PubMed] [Google Scholar]
- Lai M.-D.; Jing X. Curr. Genomics 2007, 8 (1), 43–49. 10.2174/138920207780076938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharov E. V.; Pustovalova Y. E.; Pavlov K. V.; Volynsky P. E.; Goncharuk M. V.; Ermolyuk Y. S.; Karpunin D. V.; Schulga A. A.; Kirpichnikov M. P.; Efremov R. G.; Maslennikov I. V.; Arseniev A. S. J. Biol. Chem. 2007, 282 (22), 16256–16266. 10.1074/jbc.M701745200. [DOI] [PubMed] [Google Scholar]
- Lawrie C. M.; Sulistijo E. S.; MacKenzie K. R. J. Mol. Biol. 2010, 396 (4), 924–936. 10.1016/j.jmb.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulistijo E. S.; MacKenzie K. R. J. Mol. Biol. 2006, 364 (5), 974–990. 10.1016/j.jmb.2006.09.065. [DOI] [PubMed] [Google Scholar]
- Khadria A. S.; Mueller B. K.; Stefely J. A.; Tan C. H.; Pagliarini D. J.; Senes A. J. Am. Chem. Soc. 2014, 136 (40), 14068–14077. 10.1021/ja505017f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A. M.; Stanley B. J.; Matthews E. E.; Dawson J. P.; Engelman D. M. Biochemistry 2006, 45 (16), 5228–5234. 10.1021/bi052112e. [DOI] [PubMed] [Google Scholar]
- Teese M. G.; Langosch D. Biochemistry 2015, 54 (33), 5125–5135. 10.1021/acs.biochem.5b00495. [DOI] [PubMed] [Google Scholar]
- Walters R. F. S.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (37), 13658–13663. 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.-Q.; Kulp D. W.; Schramm C. A.; Mravic M.; Samish I.; DeGrado W. F. Structure 2015, 23 (3), 527–541. 10.1016/j.str.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A.; Gerstein M.; Engelman D. M. J. Mol. Biol. 2000, 296 (3), 921–936. 10.1006/jmbi.1999.3488. [DOI] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. J. Mol. Biol. 2000, 296 (3), 911–919. 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- Brosig B.; Langosch D. Protein Sci. 1998, 7 (4), 1052–1056. 10.1002/pro.5560070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doura A. K.; Fleming K. G. J. Mol. Biol. 2004, 343 (5), 1487–1497. 10.1016/j.jmb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Li E.; Wimley W. C.; Hristova K. Biochim. Biophys. Acta, Biomembr. 2012, 1818 (2), 183–193. 10.1016/j.bbamem.2011.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. Science 1997, 276 (5309), 131–133. 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- Endres N. F.; Das R.; Smith A. W.; Arkhipov A.; Kovacs E.; Huang Y.; Pelton J. G.; Shan Y.; Shaw D. E.; Wemmer D. E.; Groves J. T.; Kuriyan J. Cell 2013, 152 (3), 543–556. 10.1016/j.cell.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragin P. E.; Mineev K. S.; Bocharova O. V.; Volynsky P. E.; Bocharov E. V.; Arseniev A. S. J. Mol. Biol. 2016, 428 (1), 52–61. 10.1016/j.jmb.2015.11.007. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Mayzel M. L.; Volynsky P. E.; Goncharuk M. V.; Ermolyuk Y. S.; Schulga A. A.; Artemenko E. O.; Efremov R. G.; Arseniev A. S. J. Biol. Chem. 2008, 283 (43), 29385–29395. 10.1074/jbc.M803089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Mitra N.; Gratkowski H.; Vilaire G.; Litvinov R.; Nagasami C.; Weisel J. W.; Lear J. D.; DeGrado W. F.; Bennett J. S. Science 2003, 300 (5620), 795–798. 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- Li R.; Gorelik R.; Nanda V.; Law P. B.; Lear J. D.; DeGrado W. F.; Bennett J. S. J. Biol. Chem. 2004, 279 (25), 26666–26673. 10.1074/jbc.M314168200. [DOI] [PubMed] [Google Scholar]
- Lau T.-L.; Kim C.; Ginsberg M. H.; Ulmer T. S. EMBO J. 2009, 28 (9), 1351–1361. 10.1038/emboj.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A.; Ubarretxena-Belandia I.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (16), 9056–9061. 10.1073/pnas.161280798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie J. U. Curr. Opin. Struct. Biol. 2011, 21 (1), 42–49. 10.1016/j.sbi.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choma C.; Gratkowski H.; Lear J. D.; DeGrado W. F. Nat. Struct. Biol. 2000, 7 (2), 161–166. 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- Zhou F. X.; Merianos H. J.; Brunger A. T.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (5), 2250–2255. 10.1073/pnas.041593698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratkowski H.; Lear J. D.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (3), 880–885. 10.1073/pnas.98.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F. X.; Cocco M. J.; Russ W. P.; Brunger A. T.; Engelman D. M. Nat. Struct. Biol. 2000, 7 (2), 154–160. [DOI] [PubMed] [Google Scholar]
- Vargas R.; Garza J.; Dixon D. A.; Hay B. P. J. Am. Chem. Soc. 2000, 122 (19), 4750–4755. 10.1021/ja993600a. [DOI] [Google Scholar]
- Scheiner S.; Kar T.; Gu Y. J. Biol. Chem. 2001, 276 (13), 9832–9837. 10.1074/jbc.M010770200. [DOI] [PubMed] [Google Scholar]
- Mueller B. K.; Subramaniam S.; Senes A. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (10), E888–895. 10.1073/pnas.1319944111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohannan S.; Faham S.; Yang D.; Grosfeld D.; Chamberlain A. K.; Bowie J. U. J. Am. Chem. Soc. 2004, 126 (8), 2284–2285. 10.1021/ja0317574. [DOI] [PubMed] [Google Scholar]
- Arbely E.; Arkin I. T. J. Am. Chem. Soc. 2004, 126 (17), 5362–5363. 10.1021/ja049826h. [DOI] [PubMed] [Google Scholar]
- Park H.; Yoon J.; Seok C. J. Phys. Chem. B 2008, 112 (3), 1041–1048. 10.1021/jp077285n. [DOI] [PubMed] [Google Scholar]
- Russ W. P.; Engelman D. M. Proc. Natl. Acad. Sci. U. S. A. 1999, 96 (3), 863–868. 10.1073/pnas.96.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2017, 45 (D1), D158–D169. 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senes A.; Engel D. E.; DeGrado W. F. Curr. Opin. Struct. Biol. 2004, 14 (4), 465–479. 10.1016/j.sbi.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Yohannan S.; Yang D.; Faham S.; Boulting G.; Whitelegge J.; Bowie J. U. J. Mol. Biol. 2004, 341 (1), 1–6. 10.1016/j.jmb.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Hong H.; Chang Y.-C.; Bowie J. U. Methods Mol. Biol. 2013, 1063, 37–56. 10.1007/978-1-62703-583-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong M. T.; Jaszewski T. M.; Fleming K. G.; MacKenzie K. R. J. Mol. Biol. 2007, 371 (2), 422–434. 10.1016/j.jmb.2007.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar A.; Weinstein J.; Biran I.; Fridman Y.; Bibi E.; Fleishman S. J. Elife 2016, 5, e12125 10.7554/eLife.12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirrbach J.; Krugliak M.; Ried C. L.; Pagel P.; Arkin I. T.; Langosch D. Bioinformatics 2013, 29 (13), 1623–1630. 10.1093/bioinformatics/btt247. [DOI] [PubMed] [Google Scholar]
- Johnson R. M.; Rath A.; Deber C. M. Biochem. Cell Biol. 2006, 84 (6), 1006–1012. 10.1139/o06-192. [DOI] [PubMed] [Google Scholar]
- Hessa T.; Meindl-Beinker N. M.; Bernsel A.; Kim H.; Sato Y.; Lerch-Bader M.; Nilsson I.; White S. H.; von Heijne G. Nature 2007, 450 (7172), 1026–1030. 10.1038/nature06387. [DOI] [PubMed] [Google Scholar]
- Ruan W.; Lindner E.; Langosch D. Protein Sci. 2004, 13 (2), 555–559. 10.1110/ps.03357404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe L. M.; Taylor K. C.; Subramaniam S.; Khadria A.; Rayment I.; Senes A. Biochemistry 2013, 52 (15), 2574–2585. 10.1021/bi400222r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P.; Zheng B.-K.; Guo P.-R.; Kawakami T.; Luo S.-Z. Biophys. J. 2013, 104 (7), 1435–1444. 10.1016/j.bpj.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei P.; Liu X.; Hu M.-H.; Zuo L.-M.; Kai M.; Wang R.; Luo S.-Z. Protein Sci. 2011, 20 (11), 1814–1823. 10.1002/pro.713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman C. Am. J. Psychol. 1904, 15 (1), 72–101. 10.2307/1412159. [DOI] [Google Scholar]
- MacKenzie K. R.; Fleming K. G. Curr. Opin. Struct. Biol. 2008, 18 (4), 412–419. 10.1016/j.sbi.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate R. F. Ann. Math. Stat. 1954, 25 (3), 603–607. 10.1214/aoms/1177728730. [DOI] [Google Scholar]
- Fleming K. G.; Ren C.-C.; Doura A. K.; Eisley M. E.; Kobus F. J.; Stanley A. M. Biophys. Chem. 2004, 108 (1–3), 43–49. 10.1016/j.bpc.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Dawson J. P.; Melnyk R. A.; Deber C. M.; Engelman D. M. J. Mol. Biol. 2003, 331 (1), 255–262. 10.1016/S0022-2836(03)00714-9. [DOI] [PubMed] [Google Scholar]
- Ridder A.; Skupjen P.; Unterreitmeier S.; Langosch D. J. Mol. Biol. 2005, 354 (4), 894–902. 10.1016/j.jmb.2005.09.084. [DOI] [PubMed] [Google Scholar]
- Unterreitmeier S.; Fuchs A.; Schäffler T.; Heym R. G.; Frishman D.; Langosch D. J. Mol. Biol. 2007, 374 (3), 705–718. 10.1016/j.jmb.2007.09.056. [DOI] [PubMed] [Google Scholar]
- Herrmann J. R.; Panitz J. C.; Unterreitmeier S.; Fuchs A.; Frishman D.; Langosch D. J. Mol. Biol. 2009, 385 (3), 912–923. 10.1016/j.jmb.2008.10.058. [DOI] [PubMed] [Google Scholar]
- Schanzenbach C.; Schmidt F. C.; Breckner P.; Teese M. G.; Langosch D. Sci. Rep. 2017, 7, 43476. 10.1038/srep43476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendrola J. M.; Berger M. B.; King M. C.; Lemmon M. A. J. Biol. Chem. 2002, 277 (7), 4704–4712. 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Kulp D. W.; Lear J. D.; DeGrado W. F. J. Am. Chem. Soc. 2009, 131 (32), 11341–11343. 10.1021/ja904625b. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Luo B.-H.; Barth P.; Schonbrun J.; Baker D.; Springer T. A. Mol. Cell 2009, 34 (2), 234–249. 10.1016/j.molcel.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman D. M.; Adair B. D.; Brünger A.; Flanagan J. M.; Hunt J. F.; Lemmon M. A.; Treutlein H.; Zhang J. Soc. Gen. Physiol. Ser. 1993, 48, 11–21. [PubMed] [Google Scholar]
- Adams P. D.; Arkin I. T.; Engelman D. M.; Brünger A. T. Nat. Struct. Mol. Biol. 1995, 2 (2), 154–162. 10.1038/nsb0295-154. [DOI] [PubMed] [Google Scholar]
- Stouffer A. L.; Nanda V.; Lear J. D.; DeGrado W. F. J. Mol. Biol. 2005, 347 (1), 169–179. 10.1016/j.jmb.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Dixon A. M.; Drake L.; Hughes K. T.; Sargent E.; Hunt D.; Harton J. A.; Drake J. R. J. Biol. Chem. 2014, 289 (17), 11695–11703. 10.1074/jbc.M113.516997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard K. P.; Lear J. D.; DeGrado W. F. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (13), 8568–8572. 10.1073/pnas.132266099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulp D. W.; Subramaniam S.; Donald J. E.; Hannigan B. T.; Mueller B. K.; Grigoryan G.; Senes A. J. Comput. Chem. 2012, 33 (20), 1645–1661. 10.1002/jcc.22968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramaniam S.; Senes A. Proteins: Struct., Funct., Genet. 2012, 80 (9), 2218–2234. 10.1002/prot.24111. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiórkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. B 1998, 102 (18), 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Lazaridis T. Proteins: Struct., Funct., Genet. 2003, 52 (2), 176–192. 10.1002/prot.10410. [DOI] [PubMed] [Google Scholar]
- Krivov G. G.; Shapovalov M. V.; Dunbrack R. L. Proteins: Struct., Funct., Genet. 2009, 77 (4), 778–795. 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw W. V. Methods Enzymol. 1975, 43, 737–755. 10.1016/0076-6879(75)43141-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.