

Abstract
Transition-metal-catalyzed chlorosulfonylation of 5-ethynylpyrimidine nucleosides provided (E)-5-(β-chlorovinyl)sulfones A, which undergo nucleophilic substitution with amines or thiols affording B. The treatment of vinyl sulfones A with ammonia followed by acid-catalyzed hydrolysis of the intermediary β-sulfonylvinylamines gave 5-(β-keto)sulfones C. The latter reacts with electrophiles, yielding α-carbon-alkylated or -sulfanylated analogues D. The 5′-triphosphates of A and C were incorporated into double-stranded DNA, using open and one-nucleotide gap substrates, by human or Escherichia coli DNA-polymerase-catalyzed reactions.
Introduction
The 5-modified pyrimidine nucleosides have been extensively studied, and many analogues exhibit a wide range of biological activity.1,2 For example, (E)-5-(2-bromovinyl)-2′-deoxyuridine3 and bicyclic furannopyrimidine-2-one analogues display antiviral activity against varicella zoster virus (VZV)4 and 5-(trifluoromethyl)-2′-deoxyuridine is used for the treatment of colorectal cancer.5 The introduction of reactive groups at the C5 position of pyrimidine nucleobases such as alkyne6−9 or azide10−13 for Cu-catalyzed or strain-promoted click chemistry, aldehyde14 for reductive aminations,15 ene/diene for cycloaddition reactions,16−19 vinyl sulfonamide for Michael additions,20,21 or chloroacetamide for nucleophilic substitutions22 has been explored23 for imaging cellular DNA, bioconjugation with DNA-bound proteins, and applications in the fluorescent bioanalysis of DNA and RNA.24 The 5′ triphosphates8,10,20,25 or 3′ phosphoroamidite26,27 of these probes were incorporated into oligonucleotides by DNA/RNA polymerases or solid-phase synthesis.
The vinyl sulfones are widely used intermediates in organic and medicinal chemistry.28−30 The (β-halo)vinyl sulfones are gaining attention as a new class of reactive sulfones.31 Recent synthetic developments include metal-free stereoselective E-iodosulfonylation of internal alkynes with sodium phenyl sulfinate and iodine to provide tetrasubstituted olefins,32 di-t-butyl peroxide/I2-promoted difunctionalization of alkynes with sodium benzenesulfinates to give (E)-β-iodovinyl sulfones,33 and multicomponent reactions with insertion of sulfur dioxide.34 The application of (α-halo)vinyl sulfones to organic synthesis as well as nucleoside and medicinal chemistry is also documented.35−37 The β-keto sulfones are also important synthetic synthons.38,39 They have been synthesized from terminal alkynes via pyridine-catalyzed dioxygen-triggered radical reactions40 or in reactions of aryl/heteroaryl acetylenes with sulfonyl chloride in the presence of catalytic amount of p-toluenesulfonic acid.41 Both (β-halovinyl)sulfones and (β-keto)sulfones exhibit important biological activities. For example, vinyl sulfones act as inhibitors of cysteine proteases28 and human cathepsin L,42 whereas (β-keto)sulfones are selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1).43
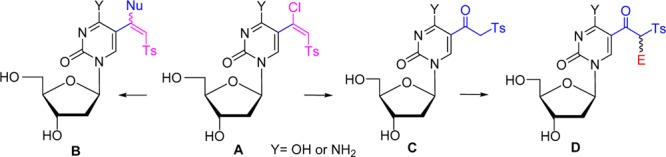
We have recently reported a stereoselective synthesis of (E)-5-(β-halovinyl)sulfone derivatives of uracil nucleosides (e.g., II, Figure 1) by transition-metal-catalyzed or radical-mediated halovinylsulfonylation of 5-ethynyluracils I with TsNa or TsNHNH2 in the presence of NXS (X = Br, I) or FeX3 (X = Cl, Br), respectively, as halogen sources.44 5-(β-Halovinyl)sulfones II underwent efficient stereoselective addition–elimination with nucleophiles (NuH) such as thiols or amines to provide E-β-thiovinyl or Z-β-aminovinyl sulfones III and IV. The 3′,5′-di-O-acetyl-5-(E)-(1-chloro-2-tosylvinyl)-2′-deoxyuridine (II; X = Cl, R = Ac; compound 9a in Table 1) inhibited the growth of L1210 (IC50 5.6 μM), CEM, and HeLa cancer cells in the lower micrometer range.45 It also displayed micromolar activity against varicella zoster virus (VZV; EC50 4 μM).45 Moreover, the lack of activity of 9a with the thymidine kinase-deficient VZV strain implies the importance of phosphorylation in the metabolism of these compounds.
Figure 1.

Synthesis of uridine 5-(β-halovinyl)sulfone analogues and their reactions with nucleophiles.
Table 1. Conversion of 5-(β-Chlorovinyl)sulfones to 5-(β-Keto)sulfonesa.

| entry | substrate | product | yield (%)b |
|---|---|---|---|
| 1 | 5 | 12 | 72 |
| 2 | 9a | 12 | 65 |
| 3 | 9b | 13 | 74 |
| 4 | 9c | 13 | 70 |
| 5 | 4 | 14 | 59 |
| 6 | 3 | 15 | 64 |
(i) 5-(β-Chlorovinyl)sulfones 3–5, 9a–c (0.2 mmol) in NH3/MeOH, room temperature (rt), 3–12 h; (ii) 0.1 M HCl in MeCN, rt, 2 h.
Isolated yields.
In this article, we extend the halosulfonylation reaction to cytosine nucleosides and describe the conversion of the uridine and cytidine 5-(β-halovinyl)sulfone derivatives to 5-(β-keto)sulfone probes. We report, herein, two classes of 5-modified uracil and cytosine nucleosides, their selective bioconjugation with nucleophiles or electrophiles, and the incorporation of their 5′-phosphates into double-stranded DNA by human or Escherichia coli DNA-polymerase-catalyzed reactions. One class contains a (β-chlorovinyl)sulfone probe that efficiently reacts with nucleophiles, including amino acid thiols, via the addition–elimination pathway, whereas the second class bears a β-keto sulfone probe at the C5 position that reacts with electrophiles and disulfides.
Results and Discussion
Synthesis and Reactivity of (β-Chlorovinyl)sulfone or (β-Keto)sulfone Probes
The treatment of 2′,3′,5′-tri-O-acetyl-5-ethynylcytosine 1 with tosyl hydrazide (3 equiv) in the presence of FeCl3 (2 equiv) and t-butyl hydroperoxide (TBHP; 4 equiv) gave (E)-5-(β-chlorovinyl)sulfone 3 (Scheme 1). Analogous chlorosulfonylation of the protected 5-ethynyl-2′-deoxycytidine 2 provided β-chlorovinyl sulfone 4, which also demonstrated the stability of the glycosylic bond in the labile 2′-deoxy substrates.
Scheme 1. Synthesis of 5-(β-Chlorovinyl)sulfone Derivatives of Cytidine and 2′-Deoxycytidine.

The 5-(β-halovinyl)sulfones underwent an efficient addition–elimination reaction with nucleophiles such as amines or thiols,44 as demonstrated by the reaction of 4 with n-PrSH, which provided 7 (80%, Scheme 2). The treatment of 5-(1-chloro-2-tosylvinyl)-2′-deoxyuridine 5 (56 mM; H2O/MeOH, 1:4) with tripeptide l-glutathione (1.5 equiv) in the presence of trimethylamine (TEA, 3 equiv) at room temperature (rt) for 4 h led to nucleophilic substitution of chloride to give 8a (55%) after high-performance liquid chromatography (HPLC) purification. 5′-O-phosphate 6 (47 mM; H2O/MeOH, 4:1; see Scheme 4 for the synthesis of 6) coupled (rt, 4 h) with l-glutathione in aqueous solution in the presence of TEA (3 equiv) affording 8b (54%) after purification on a Sephadex column. Phosphate 6 (34 mM) also reacted with glutathione (1.2 equiv) in triethylammonium acetate (TEAA) buffer, under the conditions similar to those for the bioconjugation of glutathione with a chloroacetamide probe attached to the C5 position of dCMP (87 mM),22 providing 8b (58%). Because bioconjugation of a chloroacetamide probe attached to short oligodeoxynucleotides with peptides or proteins also occurred efficiently at a lower concentration (0.1 μM),22 it can be expected that 5-(β-chlorovinyl)sulfone-modified DNA might also serve as a probe for bioconjugation with peptides or proteins.
Scheme 2. Nucleophilic Substitution of 5-(β-Chlorovinyl)sulfones with Thiols.
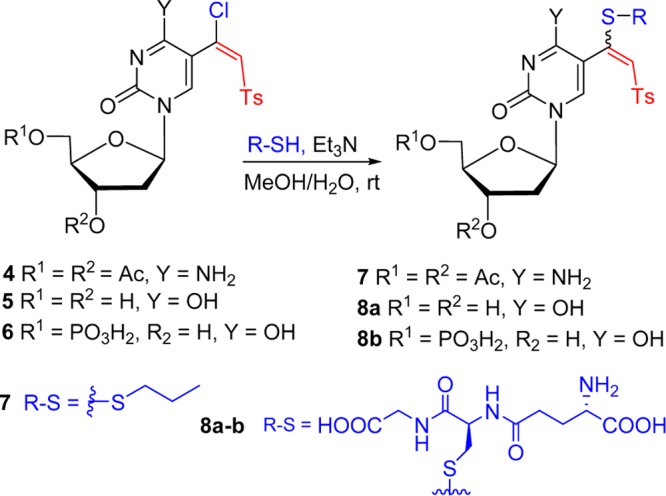
Scheme 4. Synthesis of 5′-Phosphates of 5-(β-Chlorovinyl) and 5-(β-Keto)sulfones of 2′-Deoxyuridine and 2′-Deoxycytidine.

The treatment of 2′,3′,5′-tri-O-acetyluridine 5-(β-chlorovinyl)sulfone 9b with methanolic ammonia at rt resulted in a concomitant deacetylation and vinylic substitution to give intermediary (Z)-β-sulfonylvinylamine (enamine) 10 as a single isomer.44,46 We now found that careful acid hydrolysis of 10 with HCl (pH ∼ 3–4) in MeCN afforded 5-(β-keto)sulfone 13 (74% from 9b; Table 1, entry 3). Subjection of the unprotected or protected 5-(β-chlorovinyl)sulfones of 2′-deoxyuridine (5 or 9a) and unprotected uridine (9c) to this amination/hydrolysis sequence afforded 5-(β-keto)sulfones (12 or 13; entries 1–2 and 4). Cytidine and 2′-deoxycytidine sulfones (3 or 4) were converted via a similar two-step one-pot protocol into 5-(β-keto)sulfones 14 and 15, illustrating its general character (entries 5 and 6). It is noteworthy that 2′-deoxycytidine substrate 4 provided β-sulfonylvinylamine 11 as a mixture of E/Z isomers (3:7), which was confirmed by the correlation between H6 (7.84 ppm) and the vinylic proton (4.95 ppm) in the nuclear Overhauser enhancement spectra for the major isomer. The β-keto sulfones attached to the C5 position of pyrimidine nucleobases can be envisioned as mechanistically different probes than 5-(β-chlorovinyl)sulfones as they can trap electrophiles rather than nucleophiles because they possess a methylene acidic proton (pKa = 10–11).47
As anticipated, the treatment of 5-(β-keto)sulfone 12 with BnBr in the presence of aqueous NaOH at rt afforded α-monobenzylated product 16a as a diastereotopic mixture with no α-dialkylated product observed (Table 2, entry 1). However, the benzylation was also observed at the N3 position and byproduct 16b (17%) was isolated. The alkylation of 12 with MeI or allyl bromide and of 13 with allyl bromide also provided α-alkylated products 17–19 (entries 2–4). As expected, 2′-deoxycytosine 14 and cytidine sulfones 15, which lack an acidic proton at the N3 position, afforded α-alkylated products 20–22 with higher yields (entries 5–7).
Table 2. α-Alkylation of the 5-(β-Keto)sulfones of Uracil and Cytosine Nucleosidesa.

| entry | substrate | E–Z | product | yield (%)b |
|---|---|---|---|---|
| 1 | 12 | BnBr | 16ac | 50 |
| 2 | 12 | MeI | 17 | 49 |
| 3 | 12 | AllBr | 18 | 46 |
| 4 | 13 | AllBr | 19 | 42 |
| 5 | 14 | BnBr | 20 | 68 |
| 6 | 14 | MeI | 21 | 80 |
| 7 | 15 | BnBr | 22 | 72 |
5-(β-Keto)sulfones 12–15 (0.1 mmol), NaOH/H2O (0.2 mmol)/MeOH, rt, 4–12 h.
Isolated yields.
Also isolated was 16b (17%).
The 5-(β-keto)sulfones also react with disulfides in the presence of a TEA-trapping electrophilic phenylsulfenyl48 ion. Thus, the treatment of 2′-deoxyuridine-derived sulfone 12 with phenyl disulfide (2 equiv) in the presence of TEA in MeCN led to the α-sulfanylation, affording 23 (36%) as a 1:1 mixture of diastereomers (Scheme 3, method A). Analogous treatment of 12 with 4-chlorophenyl disulfide yielded 24 (44%). Sulfanylation of 12 was also successful with alkyl disulfides (e.g., MeSSMe), providing alkylsulfanylated product 25 in 40% yield.
Scheme 3. α-Sulfanylation of 5-(β-Keto)sulfones of 2′-Deoxyuridine.
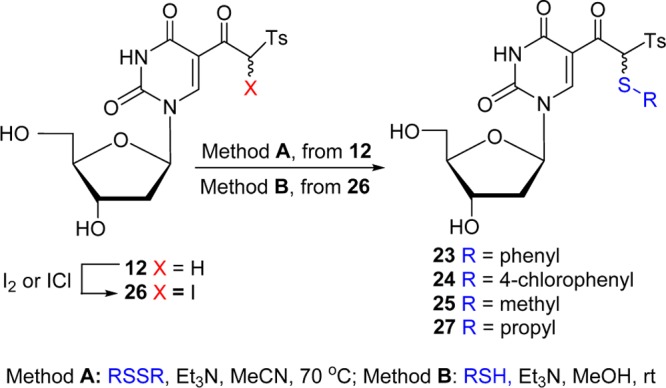
The 5-(β-keto)sulfones can also be α-sulfanylated by a sequence of halogenation and nucleophilic substitution reactions. Thus, the treatment of (β-keto)sulfone 12 with iodine monochloride/NaOH or iodine in the presence of H2O2/AcOH49 afforded 5-(α-iodo-β-keto)sulfone 26 (35 or 41%) as a 1:1 diastereomeric mixture. Subsequent, displacement of iodide from 26 with PrSH/TEA gave 5-(2-propanethio-2-tosylacetyl)-2′-deoxyuridine 27 (Scheme 3, method B).
Phosphorylation and Polymerase-Catalyzed Incorporation into DNA
The uracil and cytosine nucleosides modified at the C5 position with (β-chlorovinyl)sulfone or (β-keto)sulfone probes were incorporated into DNA fragments via polymerase-catalyzed reactions. Phosphorylation of (β-chlorovinyl)sulfone 5 with POCl3 (2.5 equiv; 0 °C, 30 min) in the presence of proton sponge (2.5 equiv)50 followed by quenching of the crude reaction mixture with triethylammonium bicarbonate (TEAB) and purification on a Sephadex column gave 5′-monophosphate 6 (40%; Scheme 4). The reaction of 5 with POCl3 (2.5 equiv)/proton sponge (2.5 equiv) followed by the treatment with tributylammonium pyrophosphate (TBAPP, 4.2 equiv) and tributylamine (TBA, 2.7 equiv) afforded 5′-triphosphate 28 after Sephadex purification. High-resolution mass spectrometry (HRMS) confirmed the presence of a (β-chlorovinyl) unit in 28. Subjection of 2′-deoxyuridine 5-(β-keto)sulfone 12 to the similar phosphorylation sequence afforded triphosphate 29. Analogous phosphorylation of 2′-deoxycytidine 5-(β-keto)sulfone 14 provided triphosphate 30.
The 2′-deoxyuridine- and 2′-deoxycytidine-modified nucleotides 28–30 were incorporated into double-stranded DNA using a bacterial replication DNA polymerase, the Klenow fragment of DNA polymerase I (pol I),51 and a human repair DNA polymerase, DNA polymerase β (pol β).52 The incorporation into double-stranded DNA was examined under the conditions that mimic the situations during both DNA leading and lagging strand syntheses using an open template substrate and a one-nucleotide gap substrate, respectively (Figures 2–4). The results showed that pol β was unable to insert 28 into an open template substrate (Figure 2a, lanes 8–12). On the other hand, 5 U pol I was able to incorporate 28 and dTTP into this template (Figure 2a, compare lane 13 with lane 7). Interestingly, both pol β and pol I inserted 28 into the substrate containing one-nucleotide gap (Figure 2b). It is noteworthy that at a high concentration, 5 U pol I is able to incorporate two nucleotides of 28 by the misincorporation of 28 with a template G. The results showed that 5-(β-chlorovinyl)sulfone 28 can be incorporated into DNA by replication DNA polymerases during leading strand DNA synthesis, whereas it can be incorporated in double-stranded DNA by both replication and repair DNA polymerases during DNA lagging strand synthesis. The sequences of oligonucleotides for constructing the substrates for testing the incorporation of 28 are listed in Table 3. The DNA substrate constructed by the oligonucleotides is illustrated below Table 3.
Figure 2.

Incorporation of 2′-deoxyuridine 5-(β-chlorovinyl)sulfone phosphate 28 into a DNA open template substrate (a) and a one-nucleotide gap substrate (b) by DNA polymerases. Lane 1 represents only substrate in both panels. Substrates were incubated with pol β or pol I at 37 °C for 15 min. In panel a, lanes 2–6 represent the reactions containing 1, 5, 10, 25, and 50 nM pol β along with 50 μM dTTP, respectively. Lanes 8–12 indicate the reactions containing various concentrations of pol β (1, 5, 10, 25, and 50 nM) with 50 μM 28. Lanes 7 and 13 represent the reactions containing 5 U pol I. In panel b, lanes 2–7 represent the reactions containing 0.5, 1, 5, 10, 25, and 50 nM pol β along with 50 μM dTTP, respectively. Lanes 9–14 indicate the reactions containing various concentrations of pol β (0.5, 1, 5, 10, 25, and 50 nM) with 50 μM 28. Lanes 8 and 15 represent the reactions containing 5 U pol I. Substrates were 32P-labeled at the 5′-end of the upstream strand and are illustrated above the gels.
Figure 4.

Incorporation of 2′-deoxycytidine 5-(β-keto)sulfone phosphate 30 into the DNA open template substrate (a) and one-nucleotide gap substrate (b) by DNA polymerases. Substrates were incubated with pol β or pol I at 37 °C for 15 min. In both panels: lanes 1 and 9 represent only substrate; lanes 2–7 represent the reactions containing 0.5, 1, 5, 10, 25, and 50 nM pol β along with 50 μM dCTP, respectively; lanes 10–15 indicate the reactions containing various concentrations of pol β (0.5, 1, 5, 10, 25, and 50 nM, respectively) with 50 μM 30; and lanes 8 and 16 represent the reactions containing 5 U pol I. Substrates were 32P-labeled at the 5′-end of the upstream strand and are illustrated above the gels.
Table 3. Oligonucleotide Sequences for C5-Modified 2′-Deoxyuridine Triphosphates 28 and 29a.
| oligonucleotide | nt | sequence (5′–3′) |
|---|---|---|
| upstream primer | 31 | GCA GTC CTC TAG TCG TAG TAG CAG ATC ATC A |
| downstream primer | 39 | CAA CCG GCA TTA GGT GTA GTA GCT AGA CTT ACT CAT TGC |
| template strand | 71 | GCA ATG AGT AAG TCT AGC TAC TAC ACC TAA TGC CGG TTG ATG ATG ATC TGC TAC TAC GAC TAG AGG ACT GC |

Both pol β and pol I incorporated 29 into DNA open template and one-nucleotide gap substrates (Figure 3). 5 U pol I incorporated same quantity of 29 as dTTP (Figure 3a,b, lane 15). Again, at the high concentrations, 5 U pol I was able to incorporate two nucleotides of 29. However, the incorporation of 29 by pol β into the open template substrate increased along with the increasing concentrations of the enzyme (0.5–50 nM) (Figure 3a, lanes 9–14). For the one-nucleotide gap substrate, 1.0 nM pol β had already inserted 29 into the DNA (Figure 3b, lane 10), indicating pol β incorporated 29 into the one-nucleotide gap substrate with a high efficiency. Our results show that 5-(β-keto)sulfones can be incorporated into double-stranded DNA more efficiently by repair DNA polymerases than β-(chlorovinyl)sulfones. They also suggest that it is possible that both 5′-triphosphates of 5-(β-chlorovinyl)sulfone 28 and 5-(β-keto)sulfone 29, when generated in cells, can be incorporated into genomic DNA by DNA polymerases during DNA replication and repair.
Figure 3.

Incorporation of 2′-deoxyuridine 5-(β-keto)sulfone phosphate 29 into the DNA open template substrate (a) and one-nucleotide gap substrate (b) by DNA polymerases. Substrates were incubated with pol β or pol I at 37 °C for 15 min. In both panels, lane 1 represents only substrate; lanes 2–7 represent the reactions containing 0.5, 1, 5, 10, 25, and 50 nM pol β along with 50 μM dTTP, respectively; lanes 9–14 indicate the reactions containing various concentrations of pol β (0.5, 1, 5, 10, 25, and 50 nM, respectively) with 50 μM 29; lanes 8 and 15 represent the reactions containing 5 U pol I. Substrates were 32P-labeled at the 5′-end of the upstream strand and are illustrated above the gels.
Both pol β and pol I also incorporated 2′-deoxycytidine 5-(β-keto)sulfone phosphate 30 into DNA open template and one-nucleotide gap substrates (Figure 4). 5 U pol I incorporated the same quantity of 30 as dCTP (Figure 4a,b, lane 16). However, the incorporation of 30 by pol β into the open template substrate increased along with the increasing concentrations of the enzyme (0.5–50 nM; Figure 4a, lanes 10–15). For the one-nucleotide gap substrate, 10 nM pol β is required to insert 30 into the DNA (Figure 4b, lane 13). The sequences of oligonucleotides for constructing the substrates for testing the incorporation of 30 are listed in Table 4. The DNA substrate constructed by the oligonucleotides is illustrated below Table 4.
Table 4. Oligonucleotide Sequences for C5-Modified 2′-Deoxycytidine Triphosphate 30a.
| oligonucleotide | nt | sequence (5′–3′) |
|---|---|---|
| upstream primer | 30 | CCT CTT CCG TCT CTT TCC TTT TAC GTC ATC |
| downstream primer | 18 | GGG GGC AGA CTG GGT GGC |
| template strand | 49 | GCC ACC CAG TCT GCC CCC GGA TGA CGT AAA AGG AAA GAG ACG GAA GAG G |

In summary, we have developed two mechanistically different probes attached to the C5 position of uracil and cytosine nucleosides. The first 5-(β-chlorovinyl)sulfone probe efficiently reacts with nucleophiles, such as thiols, at rt in aqueous MeOH solution via the conjugated addition–elimination pathway. The second 5-(β-keto)sulfone probe reacts with electrophiles such as alkyl halides or aryl/alkyl disulfides. These nucleosides with reactive β-chlorovinyl and β-keto sulfone groups were converted into their 5′-triphosphates and were efficiently incorporated into double-stranded DNA using open and one-nucleotide gap substrates by human or E. coli DNA-polymerase-catalyzed reactions. Our results also suggest that if 5-(β-chlorovinyl)sulfone and 5-(β-keto)sulfone 5′-triphosphates are generated in cells, they could be incorporated into genomic DNA by DNA polymerases during DNA replication and repair.
Experimental Section
1H (400 MHz), 13C (100.6 MHz), and 31P (161.9 MHz) NMR spectra were recorded at ambient temperature in solutions of DMSO-d6 unless otherwise noted. Reaction progress was monitored by thin-layer chromatography (TLC) on Merck Kieselgel 60-F254 sheets with product detection by 254 nm light. HRMS were obtained in time-of-flight (electrospray ionization, ESI) or ESI-Fourier transform ion cyclotron resonance mode. Products were purified by column chromatography using Merck Kieselgel 60 (230–400 mesh) or by automated flash chromatography using a CombiFlash system. Reagent-grade chemicals were used, and solvents were dried by reflux and distillation from CaH2 under N2 unless otherwise specified, and an atmosphere of N2 was used for reactions.
(E)-2′,3′,5′-Tri-O-acetyl-5-(1-chloro-2-tosylvinyl)cytidine (3)
p-Toluenesulfonyl hydrazide (90 mg, 0.5 mmol), FeCl3·6H2O (91 mg, 0.33 mmol), and TBHP (70% in water; 84 μL, 0.67 mmol) were added to a stirring solution of 2′,3′,5′-tri-O-acetyl-5-ethynylcytidine53 (1; 66 mg, 0.167 mmol) in CH3CN at rt. The resulting mixture was stirred at 80 °C for 5 h. The volatiles were evaporated, and the residue was purified by column chromatography (MeOH/CHCl3; 0 → 5%) to give 3 (48 mg, 60%): 1H NMR (CDCl3) δ 2.10 (s, 3H), 2.13 (s, 3H), 2.15 (s, 3H), 2.39 (s, 3H), 4.38–4.46 (m, 3H), 5.38 (“t”, J = 5.6 Hz, 1H), 5.49 (dd, J = 5.4, 4.0 Hz, 1H), 6.07 (d, J = 4.0 Hz, 1H), 7.10 (s, 1H), 7.32 (d, J = 8.4 Hz, 2H), 7.66 (d, J = 8.4 Hz, 2H), 7.86 (s, 1H); 13C NMR (CDCl3) δ 20.61, 20.63, 20.9, 21.8, 63.1, 70.1, 74.0, 80.1, 89.4, 100.8, 128.1, 130.4, 136.5, 137.8, 139.3, 144.0, 146.0, 153.2, 160.7, 169.5, 169.6, 170.5. HRMS calcd for C24H2735ClN3O10S [M + H]+ 584.1100, found 584.1132.
(E)-3′,5′-Di-O-acetyl-5-(1-chloro-2-tosylvinyl)-2′-deoxycytidine (4)
The treatment of 3′,5′-di-O-acetyl-5-ethynylcytidine532 (156 mg, 0.465 mmol) with TsNHNH2 (261 mg, 1.39 mmol) in the presence of FeCl3·6H2O (252 mg, 0.93 mmol), as described for 3, gave 4 (168 mg, 68%): 1H NMR (CDCl3) δ 2.10 (s, 3H), 2.12 (s, 3H), 2.20–2.27 (m, 1H), 2.41 (s, 3H), 2.70 (ddd, J = 14.0, 5.2, 2.0 Hz, 1H), 4.29–4.38 (m, 2H), 4.42 (dd, J = 12.8, 5.2 Hz, 1H), 5.21–5.27 (m, 1H), 6.33 (dd, J = 8.0, 5.6 Hz, 1H), 7.01 (s, 1H), 7.32 (d, J = 8.0 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.91 (s, 1H); 13C NMR (CDCl3) δ 21.02, 21.04, 21.8, 38.8, 63.9, 74.5, 82.9, 86.7, 100.0, 127.9, 130.3, 136.6, 136.7, 140.0, 143.9, 145.9, 154.2, 161.4, 170.5, 170.7. HRMS calcd for C22H2535ClN3O8S [M + H]+ for 526.1045, found 526.1046.
5-(1-Chloro-2-tosylvinyl)-2′-deoxyuridine 5′-O-Monophosphate (6)
(MeO)3PO (0.85 mL; dried over 3A molecular sieves) was added to the flame-dried flask containing 5-(β-chloro)vinyl sulfone 5 (55 mg, 0.124 mmol; dried in vacuum (65 °C over P2O5)) and proton sponge (67 mg, 0.31 mmol), and the resulting solution was stirred at 0 °C for 5 min under an Ar atmosphere. Freshly distilled POCl3 (29 μL, 47.5 mg, 0.31 mmol) was then added, and stirring was continued for 30 min at 0 °C. The reaction mixture was quenched by adjusting pH to 7.5–7.8 with triethylammonium bicarbonate (TEAB) buffer (2 M, several drops). The residue was dissolved in water (5 mL) and was extracted with EtOAc (3 × 5 mL). The water layer was evaporated and co-evaporated (three times) with a mixture of EtOH/H2O (1:1, 5 mL). The residue was chromatographed on a DEAE-Sephadex A-25 column (30 × 1 cm2; 3 g of resin) with TEAB (0.05 → 0.25 M), and the appropriate fractions (TLC, Rf 0.45; i-PrOH/H2O/NH4OH, 7:2:1) were evaporated in vacuum and co-evaporated five times with a mixture of EtOH/H2O (1:1, 10 mL) to remove excess of TEAB salt to give 6 (26 mg, 40%) as a triethylammonium salt: 1H NMR (D2O) δ 2.26–2.48 (m, 2H), 2.41 (s, 3H), 3.90–4.01 (m, 2H), 4.12–4.20 (m, 1H), 4.46–4.57 (m, 1H), 6.24 (t, J = 6.0 Hz, 1H), 7.41 (d, J = 8.0 Hz, 2H), 7.42 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.96 (s, 1H); 13C NMR (D2O) δ 20.8, 38.5, 64.0, 71.0, 85.8, 85.9, 108.5, 127.7, 130.1, 134.6, 135.5, 140.8, 142.7, 146.8, 150.8, 161.7; 31P NMR (D2O) δ 2.69; HRMS calcd for C18H1935ClN2O10PS [M – H]− 521.0192, found 521.0199.
3′,5′-Di-O-acetyl-5-(1-propylthio-2-tosylvinyl)-2′-deoxycytidine (7)
n-PrSH (18.1 μL, 14.5 mg, 0.19 mmol) and TEA (56 μL, 39.5 mg, 0.39 mmol) were sequentially added to a stirred solution of 4 (35 mg, 0.0665 mmol) in MeOH (2 mL) at ambient temperature. After 6 h, the volatiles were evaporated and the residue was purified by silica gel column chromatography (MeOH/CHCl3; 0 → 2%) to give 7 (E/Z, 1:1; 30 mg, 80%) as a white solid: 1H NMR δ 0.94 (t, J = 7.2 Hz, 3H), 1.58 (sextet, J = 7.2 Hz, 2H), 1.87 (s, 1.5H), 1.92 (s, 1.5H), 2.07 (s, 1.5H), 2.08 (s, 1.5H), 2.08–2.22 (m, 1H), 2.37 (s, 1.5H), 2.38 (s, 1.5H), 2.30–2.44 (m, 1H), 2.74–2.93 (m, 2H), 4.09 (dd, J = 12.0, 3.6 Hz, 0.5H), 4.14–4.21 (m, 2H), 4.24 (“q”, J = 3.2 Hz, 0.5H), 5.10–5.21 (m, 1H), 6.16 (t, J = 7.2 Hz, 0.5H), 6.23 (t, J = 7.0 Hz, 0.5H), 6.53 (s, 0.5H), 6.59 (s, 0.5H), 5.61–6.75 (m, 1H), 7.31 (d, J = 9.6 Hz, 1H), 7.35 (s, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.54 (s, 1H), 7.56 (d, J = 8.0 Hz, 1H), 7.58 (d, J = 8.0 Hz, 1H), 13C NMR δ 13.3, 13.4, 20.2, 20.41, 20.43, 20.5, 21.1, 34.0, 36.8, 37.4, 63.7, 63.8, 74.0, 74.1, 81.1, 81.4, 84.7, 85.4, 101.1, 101.5, 124.1, 124.6, 127.01, 127.03, 129.7, 129.8, 138.7, 139.0, 143.8, 143.9, 149.0, 153.8, 153.9, 161.8, 162.2, 169.8, 170.0, 170.11, 170.12. HRMS calcd for C25H32N3O8S2 [M + H]+ 566.1625, found 566.1643.
S-[(E)-1-(2′-Deoxyuridin-5-yl)-2-tosylvinyl]-l-glutathione (8a)
5-(β-Chloro)vinyl sulfone 5 (25 mg, 0.056 mmol) was dissolved in the mixture of MeOH/H2O (1 mL, 4:1). l-Glutathione (25.8 mg, 0.084 mmol) and TEA (23.5 μL, 17 mg, 0.17 mmol) were then sequentially added, and the resulting mixture was stirred at ambient temperature for 4 h. The volatiles were evaporated, and the residue was purified by RP-HPLC (Phenomenex Gemini RP-C18 column (250 × 10 mm2, 5 μm particle size) using a gradient of MeCN in water (5 → 10%) for 45 min and a flow rate of 2 mL/min; tR = 22 min) to give 8a (22 mg, 55%) as a triethylammonium salt (white solid): 1H NMR (D2O) δ 2.14 (q, J = 7.2 Hz, 2H), 2.27–2.35 (m, 1H), 2.38–2.46 (m, 1H), 2.43 (s, 3H), 2.51 (t, J = 8.0 Hz, 2H), 3.22–3.42 (m, 2H), 3.72–3.86 (m, 5H), 4.06–4.12 (m, 1H), 4.45 (br, 1H), 4.65–4.69 (m, 1H), 6.24 (t, J = 6.4 Hz, 1H), 6.94 (br, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.94 (s, 1H); 13C NMR (D2O) δ 20.7, 26.0, 31.4, 39.3, 43.4, 51.8, 54.2, 61.0, 70.2, 85.9, 87.0, 99.6, 107.4, 127.3, 130.2, 132.1, 134.0, 136.2, 142.0, 146.4, 150.7, 161.9, 170.7, 173.9, 174.9, 176.1; HRMS calcd for C28H35N5NaO13S2 [M + Na]+ 736.1565, found 736.1513.
S-[(E)-1-(2′-Deoxyuridin-5-yl)-2-tosylvinyl]-l-glutathione 5′-O-Monophosphate (8b)
l-Glutathione (22 mg, 0.07 mmol) and TEA (20 μL, 14.3 mg, 0.14 mmol) were sequentially added into the stirred solution of 5′-monophosphate 6 (25 mg, 0.047 mmol) in MeOH/H2O (1 mL, 1:4) at ambient temperature. The resulting mixture was stirred for 4 h. The volatiles were evaporated, the residue was purified on a DEAE-Sephadex A-25 column (30 × 1 cm2; 3 g of resin) with TEAB (0.1 → 0.3 M), and the appropriate fractions (TLC, Rf 0.25; i-PrOH/H2O/NH4OH, 7:2:1) were evaporated in vacuum and co-evaporated five times with a mixture of EtOH/H2O (1:1, 10 mL) to remove excess of TEAB salt to give 8b (20 mg, 54%) as a triethylammonium salt (off-white solid): 1H NMR (D2O) δ 2.13 (dt, J = 12.0, 7.8 Hz, 1H, 2H), 2.23–2.38 (m, 2H), 2.41 (s, 3H), 2.47–2.55 (m, 2H), 3.26–3.30 (m, 1H), 3.38 (dd, J = 12.0, 7.8 Hz, 1H), 3.70–3.76 (m, 3H), 3.92–4.02 (m, 2H), 4.09–4.19 (m, 1H), 4.48 (br, 1H), 4.60–4.71 (m, 1H), 6.22 (t, J = 6.8 Hz, 1H), 6.85 (s, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 8.0 Hz, 2H), 7.77 (s, 1H); 13C NMR (D2O) δ 20.8, 26.1, 31.4, 33.3, 38.7, 42.2, 48.7, 51.8, 54.2, 58.9, 64.6, 85.8, 85.9, 107.9, 127.3, 129.9, 130.2, 141.9, 146.3, 148.6, 150.7, 161.8, 170.9, 173.9, 174.9, 176.2; 31P NMR (D2O) δ 3.77; HRMS calculated for C28H35N5O16PS2 [M – H]− 792.1264, found 792.1264.
Note: l-Glutathione (6.4 mg, 0.02 mmol) was added to a solution of 5-(β-chloro)vinyl sulfone 6 (9 mg, 0.017 mmol) in TEAA buffer (0.5 mL, 0.3 M, pH = 8.3) at rt, and the mixture was stirred for 12 h. Purification as described above gave 8b (7.8 mg, 58%).
5-(1-Amino-2-tosylvinyl)-2′-deoxycytidine (11)
β-Chlorovinylsulfone 4 (150 mg, 0.285 mmol) was dissolved in NH3/MeOH (5 mL) at 0 °C (ice bath). After stirring for 12 h, the volatiles were evaporated and the residue was column-chromatographed (MeOH/CHCl3; 0 → 10%) to afford 11 (E/Z, 30:70; 77 mg, 64%) as a white solid: 1H NMR δ 1.98–2.07 (m, 1H), 2.10–2.22 (m, 1H), 2.38 (s, 2.1H), 2.40 (s, 0.9H), 3.46–3.57 (m, 1.4H), 3.64–3.70 (m, 0.6H), 3.78 (q, J = 3.6 Hz, 0.7H), 3.84 (q, J = 3.5 Hz, 0.3H), 4.16–4.22 (m, 0.7H), 4.26–4.31 (m, 0.3H), 4.71 (s, 0.3H), 4.95 (s, 0.7H), 4.98 (t, J = 5.4 Hz, 0.7H), 5.19 (d, J = 4.4 Hz, 0.7H), 5.22–5.25 (m, 0.3H), 5.27 (d, J = 4.4 Hz, 0.3H), 6.08 (t, J = 6.5 Hz, 0.7H), 6.13 (t, J = 6.4 Hz, 0.3H), 6.32–6.43 (br s, 1H), 6.84–7.01 (br s, 2H), 7.37 (d, J = 8.4 Hz, 1.4H), 7.45 (d, J = 8.4 Hz, 0.6H), 7.52–7.65 (br s, 1H), 7.72 (d, J = 8.0 Hz, 0.6H), 7.80 (d, J = 8.0 Hz, 1.4H), 7.84 (s, 0.7H), 8.50 (s, 0.3H); 13C NMR δ 20.9, 40.6, 61.1, 70.2, 85.5, 87.6, 90.9, 103.6, 125.6, 129.4, 141.3, 141.9, 142.4, 151.4, 153.8, 162.0; HRMS calcd for C18H23N4O6S [M + H]+ 423.1333, found 423.1348.
Conversion of 5-(β-Chlorovinyl)sulfones into 5-(β-Keto)sulfones: Procedure A
Acetyl-protected (3, 4, 9a, or 9b) or -unprotected (5 or 9c) 5-(1-chloro-2-tosylvinyl)pyrimidine nucleosides were dissolved in methanolic ammonia, and the resulting mixture was stirred at 0 °C → rt for 3–12 h. The volatiles were evaporated, and the residue was dissolved in MeCN (4 mL). The solution was acidified to pH ∼ 3–4 with dil. HCl (aq) and stirred for 2 h and then neutralized with dil. NaOH (aq) to pH ∼ 6–7. The volatiles were evaporated, and the residue was column-chromatographed to give products 12–15.
5-(2-Tosylacetyl)-2′-deoxyuridine (12)
The treatment of 5(44) (300 mg, 0.68 mmol) with methanolic ammonia (10 mL) for 3 h and subsequent acid hydrolysis, as described in procedure A, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 12 (207 mg, 72%) as a white solid: 1H NMR δ 2.04–2.18 (m, 1H), 2.19–2.28 (m, 1H), 2.40 (s, 3H), 3.50–3.66 (m, 2H), 3.87 (dd, J = 6.4, 3.2 Hz, 1H), 4.13–4.28 (m, 1H), 5.10 (t, J = 4.6 Hz, 1H), 5.15 (s, 2H), 5.29 (d, J = 4.4 Hz, 1H), 6.06 (t, J = 6.2 Hz, 1H), 7.42 (d, J = 8.0 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 8.69 (s, 1H), 11.78 (s, 1H); 13C NMR δ 20.9, 40.6, 60.8, 64.3, 70.3, 85.7, 87.9, 111.0, 127.6, 129.5, 136.8, 144.1, 148.0, 148.7, 160.7, 183.5; HRMS calcd for C18H20N2NaO8S [M + Na]+ for 447.0833, found 447.0874.
Analogous treatment of 9a(44) (90 mg, 0.17 mmol) with methanolic ammonia (3 mL) for 12 h, as described in procedure A, also gave 12 (47 mg, 65%).
5-(2-Tosylacetyl)uridine (13)
The treatment of 9c(44) (96 mg, 0.20 mmol) with methanolic ammonia (3 mL) and subsequent acid hydrolysis, as described in procedure A (3 h), followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 13 (64.5 mg, 70%) as a white solid: 1H NMR δ 2.40 (s, 3H), 3.53–3.61 (m, 1H), 3.64–3.73 (m, 1H), 3.88–3.99 (m, 2H), 4.02–4.09 (m, 1H), 5.07–5.24 (m, 4H), 5.50 (d, J = 5.2 Hz, 1H), 5.75 (s, 1H), 7.43 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 8.78 (s, 1H), 11.79 (s, 1H); 13C NMR δ 21.0, 60.1, 64.2, 69.3, 74.3, 84.7, 89.3, 111.2, 127.9, 129.6, 136.8, 144.2, 148.2, 149.4, 161.2, 184.0; HRMS calcd for C18H21N2O9S [M + H]+ for 441.0962, found 441.0954.
Analogous treatment of 9b(44) (95 mg, 0.16 mmol) with methanolic ammonia (3 mL) for 12 h, as described in procedure A, also gave 13 (53 mg, 74%).
5-(2-Tosylacetyl)-2′-deoxycytidine (14)
The treatment of 4 (150 mg, 0.28 mmol) with methanolic ammonia (5 mL) for 12 h and subsequent acid hydrolysis, as described in procedure A, followed by column chromatography (MeOH/CHCl3; 0 → 10%) gave 14 (70 mg, 59%): 1H NMR δ 2.06–2.18 (m, 1H), 2.26–2.35 (m, 1H), 2.41 (s, 3H), 3.59–3.69 (m, 1H), 3.73 (dt, J = 12.0, 4.0 Hz, 1H), 3.90 (q, J = 3.6 Hz, 1H), 4.28 (quint, J = 4.8 Hz, 1H), 4.86 (d, J = 14.4 Hz, 1H), 4.95 (d, J = 14.0 Hz, 1H), 5.29 (d, J = 4.4 Hz, 1H), 5.33 (t, J = 5.2 Hz, 1H), 6.09 (t, J = 6.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 8.0 Hz, 2H), 8.01–8.15 (m, 2H), 9.00 (s, 1H); 13C NMR δ 21.0, 41.1, 60.6, 62.5, 69.2, 86.6, 88.0, 103.0, 128.0, 129.9, 136.0, 144.8, 152.2, 159.0, 162.6, 185.3; HRMS calcd for C18H22N3O7S [M + H]+ 424.1173, found 424.1176.
5-(2-Tosylacetyl)cytidine (15)
The treatment of 3 (146 mg, 0.25 mmol) with methanolic ammonia (4 mL) for 12 h and subsequent acid hydrolysis, as described in procedure A, followed by column chromatography (MeOH/CHCl3; 0 → 8%) gave 15 (70 mg, 64%): 1H NMR δ 2.40 (s, 3H), 3.69 (ddd, J = 12.4, 4.8, 2.0 Hz, 1H), 3.86 (ddd, J = 12.4, 4.8, 2.0 Hz, 1H), 3.92–3.97 (m, 1H), 3.98–4.02 (m, 1H), 4.03–4.09 (m, 1H), 4.81 (d, J = 14.4 Hz, 1H), 4.95 (d, J = 14.0 Hz, 1H), 5.04 (d, J = 6.4 Hz, 1H), 5.45 (t, J = 5.2 Hz, 1H), 5.57 (d, J = 4.8 Hz, 1H), 5.71 (d, J = 2.0 Hz, 1H), 7.44 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.0 Hz, 2H), 8.00–8.22 (m, 2H), 9.05 (s, 1H); 13C NMR δ 21.1, 59.1, 62.4, 67.9, 74.9, 83.9, 91.2, 102.8, 127.9, 129.9, 136.2, 144.6, 152.3, 152.4, 162.6, 185.4; HRMS calcd for C18H22N3O8S [M + H]+ 440.1122, found 440.1133.
Alkylation at the α-Carbon of 5-(β-Keto)sulfones: Procedure B
The NaOH/H2O solution (3 M, 0.2 mmol) was added to a stirred solution of 5-(β-keto)sulfones 12–15 (0.1 mmol) in MeOH (2 mL) at ambient temperature. After 20 min, the electrophile source (R–X, 0.2 mmol) was added and the resulting solution was stirred for 4–12 h. The reaction mixture was then neutralized with dil. HCl to pH ∼ 6.5–7, and the volatiles were evaporated. The residue was column-chromatographed to give products 16–22.
5-(2-Benzyl-2-tosylacetyl)-2′-deoxyuridine (16a) and 3-N-Benzyl-5-(2-benzyl-2-tosylacetyl)-2′-deoxyuridine (16b)
The treatment of 12 (25 mg, 0.058 mmol) with NaOH (aq) (3.0 M, 39 μL) and BnBr (14 μL, 20 mg, 0.117 mmol) for 4 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 16b (6 mg, 17%; 1:1 mixture of diastereomers) followed by 16a (15 mg, 50%; as a 1:1 mixture of diastereomers). Compound 16a had: 1H NMR δ 2.05–2.21 (m, 1.5H), 2.22–2.30 (m, 0.5H), 2.40 (s, 3H), 3.09–3.26 (m, 2H), 3.55–3.65 (m, 2H), 3.87 (q, J = 3.2 Hz, 1H), 4.17–4.23 (m, 0.5H), 4.24–4.29 (m, 0.5H), 5.10–5.18 (m, 1H), 5.27 (d, J = 4.0 Hz, 0.5H), 5.32 (d, J = 4.0 Hz, 0.5H), 5.97 (t, J = 6.2 Hz, 0.5H), 6.06 (t, J = 6.2 Hz, 0.5H), 6.47–6.61 (m, 1H), 6.99–7.08 (m, 2H), 7.10–7.24 (m, 3H), 7.41 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 8.63 (s, 0.5H), 8.69 (s, 0.5H), 11.40 (s, 1H); 13C NMR δ 21.1, 31.3, 31.5, 40.5, 40.9, 60.81, 60.83, 69.9, 70.31, 70.33, 85.9, 86.0, 86.4, 88.0, 88.1, 110.8, 111.3, 126.6, 128.5, 128.8, 128.9, 129.7, 134.6, 136.22, 136.24, 145.1, 148.2, 148.3, 148.9, 161.31, 161.32, 186.4, 186.6; HRMS calcd for C25H27N2O8S [M + H]+ 515.1483, found 515.1457. Compound 16b had: 1H NMR δ 2.04–2.14 (m, 1H), 2.15–2.29 (m, 1H), 2.30 (s, 1.5H), 2.32 (s, 1.5H), 3.13–3.31 (m, 2H), 3.55–3.68 (m, 2H), 3.86–3.92 (m, 1H), 4.18–4.28 (m, 1H), 4.87–4.97 (m, 2H), 5.21 (q, J = 4.0 Hz, 1H), 5.31 (d, J = 4.4 Hz, 0.5H), 5.36 (d, J = 4.4 Hz, 0.5H), 5.97 (t, J = 5.2 Hz, 0.5H), 6.07 (t, J = 6.4 Hz, 0.5H), 6.46 (dd, J = 11.2, 3.6 Hz, 0.5H), 6.51 (dd, J = 12.0, 4.8 Hz, 0.5H), 7.03–7.09 (m, 2H), 7.13–7.23 (m, 4H), 7.29–7.38 (m, 6H), 7.58 (d, J = 8.4 Hz, 1H), 7.59 (d, J = 8.0 Hz, 1H), 8.65 (s, 0.5H), 8.73 (s, 0.5H); 13C NMR (DMSO-d6) δ 21.11, 21.12, 31.2, 31.3, 40.7, 41.0, 43.9, 45.7, 60.5, 60.6, 69.6, 69.7, 70.5, 70.6, 87.0, 87.4, 88.2, 88.3, 110.2, 110.8, 126.7, 127.8, 128.4, 128.7, 129.0, 134.4, 136.2, 136.3, 145.0, 145.2, 146.6, 146.8, 149.1, 149.2, 160.3, 160.4, 186.7, 186.9; HRMS calcd for C32H33N2O8S [M + H]+ 605.1952, found 605.1976.
5-(2-Methyl-2-tosylacetyl)-2′-deoxyuridine (17)
The treatment of 12 (40 mg, 0.094 mmol) with NaOH (aq) (3 M, 63 μL) and MeI (11.7 μL, 26.7 mg, 0.188 mmol) for 4 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 17 (21 mg, 49%) as a 1:1 mixture of diastereomers: 1H NMR δ 1.32 (t, J = 7.6 Hz, 3H), 2.11–2.17 (m, 1H), 2.20–2.30 (m, 1H), 2.39 (s, 1.5H), 2.40 (s, 1.5H), 3.53–3.67 (m, 2H), 3.88 (q, J = 2.8 Hz, 1H), 4.21–4.29 (m, 1H), 5.13 (dd, J = 9.2, 4.4 Hz, 1H), 5.30 (d, J = 4.3 Hz, 0.5H), 5.32 (d, J = 4.3 Hz, 0.5H), 5.97 (q, J = 6.8 Hz, 1H), 6.05 (t, J = 6.4 Hz, 0.5H), 6.11 (t, J = 6.4 Hz, 0.5H), 7.42 (d, J = 8.0 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 8.72 (s, 0.5H), 8.73 (s, 0.5H), 11.78 (s, 1H); 13C NMR δ 11.0, 11.2, 21.1, 40.6, 40.8, 60.8, 60.9, 65.4, 70.0, 86.0, 86.1, 88.0, 88.1, 111.0, 111.1, 128.8, 128.9, 129.6, 129.7, 134.5, 144.8, 144.9, 147.8, 148.0, 149.1, 149.2, 161.3, 161.4, 187.6; HRMS calcd for C19H23N2O8S [M + H]+ 439.1170, found 439.1147.
5-(2-Allyl-2-tosylacetyl)-2′-deoxyuridine (18)
The treatment of 12 (36 mg, 0.084 mmol) with allyl bromide (14.6 μL, 20.3 mg, 0.17 mmol) and NaOH (aq) (3 M, 56 μL) for 4 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 18 (18 mg, 46%) as a ∼1:1 mixture of diastereomers: 1H NMR δ 2.11–2.22 (m, 1.5H), 2.23–2.34 (m, 0.5H), 2.39 (s, 3H), 2.66–2.77 (m, 2H), 3.56–3.67 (m, 2H), 3.89 (q, J = 4.0 Hz, 1H), 4.21–4.29 (m, 1H), 4.92–5.01 (m, 2H), 5.12–5.16 (m, 1H), 5.30 (d, J = 4.4 Hz, 0.5H), 5.34 (d, J = 4.0 Hz, 0.5H), 5.52–5.69 (m, 1H), 6.02 (t, J = 6.4 Hz, 0.5H), 6.09 (t, J = 6.4 Hz, 0.5H), 6.17 (d, J = 2.8 Hz, 0.5H), 6.20 (d, J = 2.8 Hz, 0.5H), 7.42 (d, J = 8.2 Hz, 1H), 7.43 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 7.6 Hz, 2H), 8.70 (s, 0.5H), 8.74 (s, 0.5H), 11.79 (s, 1H); 13C NMR δ 21.1, 30.0, 30.1, 40.6, 40.9, 60.8, 60.9, 68.8, 70.01, 70.03, 86.0, 86.4, 88.1, 88.2, 107.7, 111.0, 113.0, 117.7, 117.8, 128.8, 129.0, 129.8, 132.91, 132.93, 134.5, 145.1, 145.2, 148.3, 149.0, 149.1, 161.4, 161.5, 186.7; HRMS calcd for C21H25N2O8S [M + H]+ 465.1326, found 465.1341.
5-(2-Allyl-2-tosylacetyl)uridine (19)
The treatment of 13 (18 mg, 0.04 mmol) with allyl bromide (7 μL, 9.68 mg, 0.08 mmol) and NaOH (aq) (3 M, 26 μL) for 4 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 19 (8 mg, 42%; ∼1:1 mixture of two diastereomers): 1H NMR δ 2.39 (s, 3H), 2.54–2.57 (m, 1H), 2.68–2.77 (m, 1H), 3.59–3.62 (m, 1H), 3.69–3.77 (m, 1H), 3.92–3.40 (m, 2H), 4.00–4.10 (m, 1H), 4.94–5.00 (m, 2H), 5.13 (d, J = 5.2 Hz, 0.5H), 5.20 (d, J = 5.6 Hz, 0.5H), 5.23–5.29 (m, 1H), 5.49 (d, J = 5.2 Hz, 0.5H), 5.58 (d, J = 5.2 Hz, 0.5H), 5.61–5.66 (m, 1H), 5.71 (d, J = 3.6 Hz, 0.5H), 5.77 (d, J = 4.0 Hz, 0.5H), 6.16 (dd, J = 6.0, 3.6 Hz, 0.5H), 6.18 (dd, J = 6.4, 4.0 Hz, 0.5H), 7.42 (d, J = 8.0 Hz, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.60–7.68 (m, 2H), 8.81 (s, 0.5H), 8.89 (s, 0.5H), 11.81 (s, 1H); 13C NMR δ 21.1, 30.0, 30.1, 59.9, 60.2, 68.8, 68.9, 69.4, 74.4, 74.7, 84.8, 85.0, 89.3, 89.6, 111.3, 111.6, 117.7, 117.8, 128.8, 128.9, 129.7, 132.9, 134.5, 145.22, 145.23, 148.5, 148.6, 149.2, 161.41, 161.43, 186.5, 186.6; HRMS calcd for C21H25N2O9S [M + H]+ 481.1275, found 481.1265.
5-(2-Benzyl-2-tosylacetyl)-2′-deoxycytidine (20)
The treatment of 14 (36 mg, 0.085 mmol) with BnBr (20.4 μL, 29.4 mg, 0.17 mmol) and NaOH (aq) (3 M, 56 μL) for 8 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 20 (30 mg, 68%; ∼1:1 mixture of two diastereomers): 1H NMR δ 1.95–2.01 (m, 1H), 2.23–2.30 (m, 1H), 2.38 (s, 1.5H), 2.40 (s, 1.5H), 3.20–3.31 (m, 2H), 3.64 (ddd, J = 12.0, 5.2, 1.6 Hz, 0.5H), 3.76 (dd, J = 7.2, 4.4 Hz, 1H), 3.83–3.91 (m, 1H), 3.97 (dd, J = 6.8, 3.2 Hz, 0.5H), 4.20 (quint, J = 4.8 Hz, 0.5H), 4.38 (quint, J = 4.6 Hz, 0.5H), 5.29 (d, J = 4.4 Hz, 0.5H), 5.35 (d, J = 4.4 Hz, 0.5H), 5.48 (dd, J = 11.2, 3.2 Hz, 0.5H), 5.57 (dd, J = 10.8, 4.4 Hz, 0.5H), 5.68 (t, J = 5.0 Hz, 0.5H), 5.75 (t, J = 4.6 Hz, 0.5H), 5.96 (t, J = 6.0 Hz, 0.5H), 6.01 (t, J = 5.8 Hz, 0.5H), 7.04–7.24 (m, 5H), 7.42 (d, J = 8.4 Hz, 1H), 7.46 (d, J = 8.4 Hz, 1H), 7.66–7.71 (m, 2H), 7.98–8.15 (m, 2H), 8.92 (s, 0.5H), 9.05 (s, 0.5H); 13C NMR δ 21.0, 21.1, 31.7, 32.1, 41.3, 41.4, 60.4, 60.8, 68.4, 68.9, 69.3, 69.4, 86.1, 86.8, 87.7, 88.0, 104.0, 104.6, 126.7, 126.8, 128.4, 128.5, 128.7, 128.8, 133.1, 133.7, 133.8, 135.7, 145.3, 149.8, 150.9, 151.9, 162.1, 162.4, 187.3, 187.8; HRMS calcd for C25H28N3O7S [M + H]+ 514.1642, found 514.1635.
5-(2-Methyl-2-tosylacetyl)-2′-deoxycytidine (21)
The treatment of 14 (18 mg, 0.043 mmol) with MeI (5.5 μL, 12.2 mg, 0.086 mmol) and NaOH (aq) (3 M, 29 μL) for 12 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 21 (15 mg, 80%; ∼1:1 mixture of two diastereomers): 1H NMR δ 1.34 (d, J = 6.8 Hz, 1.5H), 1.35 (d, J = 6.8 Hz, 1.5H), 2.03–2.18 (m, 1H), 2.25–2.34 (m, 1H), 2.40 (s, 3H), 3.63 (dt, J = 11.6, 4.4 Hz, 0.5H), 3.69–3.75 (m, 1H), 3.77–3.88 (m, 1H), 3.94 (dd, J = 7.6, 3.6 Hz, 0.5H), 4.20 (quint, J = 4.6 Hz, 0.5H), 4.39 (quint, J = 4.8 Hz, 0.5H), 5.19 (q, J = 7.6 Hz, 0.5H), 5.24–5.31 (m, 1H), 5.33–5.48 (m, 1.5H), 6.01 (t, J = 5.6 Hz, 0.5H), 6.15 (t, J = 6.0 Hz, 0.5H), 7.43 (d, J = 7.6 Hz, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.64 (d, J = 8.0 Hz, 1H), 8.04–8.25 (m, 2H), 9.07 (s, 0.5H), 9.16 (s, 0.5H); 13C NMR δ 12.0, 12.02, 21.1, 41.1, 41.2, 60.31, 63.32, 63.3, 68.8, 68.9, 86.0, 86.8, 87.5, 88.0, 103.1, 103.6, 129.0, 129.1, 129.91, 129.93, 133.3, 133.7, 145.1, 145.2, 151.7, 151.9, 152.3, 162.7, 162.9, 188.5, 188.6; HRMS calcd for C19H24N3O7S [M + H]+ 438.1329, found 438.1317.
5-(2-Benzyl-2-tosylacetyl)cytidine (22)
The treatment of 15 (30 mg, 0.068 mmol) with BnBr (16.2 μL, 23.2 mg, 0.135 mmol) and NaOH (aq) (3 M, 46.0 μL) for 8 h, as described in procedure B, followed by column chromatography (MeOH/CHCl3; 0 → 5%) gave 22 (26 mg, 72%; ∼1:1 mixture of diastereomers): 1H NMR δ 2.38 (s, 1.5H), 2.39 (s, 1.5H), 3.19–3.31 (m, 2H), 3.74–3.90 (m, 2.5H), 3.93–4.05 (m, 2H), 4.14 (dd, J = 11.2, 6.0 Hz, 0.5H), 5.04 (d, J = 6.0 Hz, 0.5H), 5.19 (d, J = 6.4 Hz, 0.5H), 5.49 (dd, J = 12.0, 3.6 Hz, 0.5H), 5.52–5.62 (m, 2.5H), 5.76 (t, J = 3.6 Hz, 0.5H), 5.85 (t, J = 4.0 Hz, 0.5H), 7.10–7.24 (m, 5H), 7.42 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 8.0 Hz, 1H), 7.99–8.07 (m, 1.5H), 8.15 (d, J = 3.6 Hz, 0.5H), 8.90 (s, 0.5H), 9.06 (s, 0.5H); 13C NMR δ 21.1, 31.7, 31.8, 59.2, 59.7, 67.5, 68.1, 68.3, 69.1, 74.7, 75.0, 83.9, 84.0, 90.7, 91.2, 104.1, 104.5, 126.7, 126.8, 128.4, 128.5, 128.8, 128.9, 133.7, 134.0, 135.6, 145.2, 145.3, 151.4, 151.9, 162.1, 162.3, 187.3, 187.9; HRMS calcd for C25H28N3O8S [M + H]+ 530.1592, found 530.1577.
Incorporation of the Aryl/Alkyl Sulfanyl Group at the α-Carbon of 5-(β-Keto)sulfone of 2′-Deoxyuridine (23–25): Procedure C
5-(2-Tosylacetyl)-2′-deoxyuridine 12 (0.1 mmol) was dissolved in 3 mL of MeCN. Then, phenyl disulfide (0.2 mmol) and TEA (0.2–0.5 mmol) were sequentially added, and the resulting mixture was placed in an oil bath and stirred at 70 °C for 36–72 h. The volatiles were evaporated, and the residue was column-chromatographed to afford products 23–25.
5-(2-Phenylthio-2-tosylacetyl)-2′-deoxyuridine (23)
The treatment of 12 (50 mg, 0.115 mmol) with phenyl disulfide (51 mg, 0.23 mmol) and TEA (32 μL, 24 mg, 0.23 mmol), as described in procedure C, for 36 h followed by column chromatography (MeOH/CHCl3; 0 → 5%) afforded 23 (22 mg, 36%) as a 1:1 mixture of diastereomers: 1H NMR δ 2.14–2.34 (m, 2H), 2.38 (s, 3H), 3.57–3.66 (m, 2H), 3.84–3.94 (m, 1H), 4.18–4.30 (m, 1H), 5.14 (t, J = 4.4 Hz, 0.5H), 5.16 (t, J = 4.4 Hz, 0.5H), 5.30 (d, J = 4.4 Hz, 0.5H), 5.33 (d, J = 4.4 Hz, 0.5H), 6.04 (d, J = 6.0 Hz, 0.5H), 6.07 (d, J = 6.0 Hz, 0.5H), 7.20–7.48 (m, 8H), 7.69 (d, J = 8.0 Hz, 2H), 8.84 (s, 0.5H), 8.85 (s, 0.5H), 11.91 (s, 0.5H), 11.93 (s, 0.5H); 13C NMR δ 20.1, 39.7, 39.8, 59.7, 68.81, 68.83, 73.0, 73.2, 85.4, 85.5, 87.1, 106.5, 108.2, 127.2, 128.21, 128.22, 128.52, 128.53, 128.6, 130.42, 130.43, 144.31, 144.33, 148.0, 148.1, 148.2, 160.72, 160.73, 183.2, 183.3; HRMS calcd for C24H25N2O8S2 [M + H]+ 533.1047, found 533.1069.
5-(2-(4-Chlorophenylthio)-2-tosylacetyl)-2′-deoxyuridine (24)
The treatment of 12 (50 mg, 0.116 mmol) with 4-chlorophenyl disulfide (67 mg, 0.23 mmol) and TEA (32 μL, 24 mg, 0.23 mmol) for 36 h, as described in procedure C, followed by column chromatography (MeOH/CHCl3; 0 → 5%) afforded 24 (29 mg, 44%) as a 1:1 mixture of diastereomers: 1H NMR δ 2.14–2.29 (m, 2H), 2.39 (s, 3H), 3.55–3.60 (m, 1H), 3.62–3.67 (m, 1H), 3.90 (quint, J = 2.8 Hz, 1H), 4.20–4.28 (m, 1H), 5.12–5.17 (m, 1H), 5.30 (d, J = 4.0 Hz, 0.5H), 5.33 (d, J = 4.4 Hz, 0.5H), 6.05 (t, J = 6.2 Hz, 0.5H), 6.07 (t, J = 6.0 Hz, 0.5H), 7.31 (d, J = 10.2 Hz, 1H), 7.37–7.43 (m, 6H), 7.68 (d, J = 8.4 Hz, 2H), 8.84 (s, 1H), 11.93 (s, 0.5H), 11.95 (s, 0.5H); 13C NMR δ 21.1, 40.3, 40.4, 60.6, 60.7, 69.82, 69.83, 73.9, 74.0, 86.3, 88.1, 109.1, 129.11, 129.12, 129.51, 129.52, 129.53, 129.6, 130.5, 131.0, 133.3, 145.41, 145.43, 149.00, 149.02, 149.2, 161.7, 161.8, 183.9, 184.0; HRMS calcd for C24H24ClN2O8S2 [M + H]+ 567.0657, found 567.0681.
5-(2-Methylthio-2-tosylacetyl)-2′-deoxyuridine (25)
The treatment of 12 (25 mg, 0.058 mmol) with dimethyl disulfide (26 μL, 27 mg, 0.29 mmol) and TEA (40 μL, 29 mg, 0.29 mmol) for 72 h, as described in procedure C, followed by column chromatography (MeOH/CHCl3; 0 → 5%) afforded 25 (11 mg, 40%) as a 1:1 mixture of diastereomers: 1H NMR δ 2.17 (s, 1.5H), 2.18 (s, 1.5H), 2.20–2.33 (m, 2H), 2.42 (s, 3H), 3.53–3.66 (m, 2H), 3.88 (dt, J = 8.4, 3.2 Hz, 1H), 4.20 (dd, J = 9.2, 4.4 Hz, 0.5H), 4.25 (dd, J = 9.4, 4.5 Hz, 0.5H), 5.12 (t, J = 4.4 Hz, 0.5H), 5.13 (t, J = 4.5 Hz, 0.5H), 5.28 (d, J = 4.4 Hz, 0.5H), 5.31 (d, J = 4.4 Hz, 0.5H), 6.01–6.07 (m, 1H), 6.60 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 8.4 Hz, 2H), 8.83 (s, 0.5H), 8.84 (s, 0.5H), 11.88 (s, 1H); 13C NMR δ 14.91, 14.92, 21.1, 41.7, 60.6, 60.7, 69.81, 69.83, 70.6, 70.7, 86.3, 86.5, 88.12, 88.13, 108.8, 108.9, 127.0, 128.0, 129.4, 129.6, 134.6, 145.3, 149.0, 149.2, 161.0, 183.4; HRMS calcd for C19H23N2O8S2 [M + H]+ 471.0890, found 471.0917.
5-(2-Iodo-2-tosylacetyl)-2′-deoxyuridine (26)
NaOH (aq) (3 M, 14 μL) was added into the stirred solution of (β-keto)sulfone 12 (18 mg, 0.042 mmol) in MeOH (1 mL). After 30 min, iodine monochloride (1 M/dichloromethane; 42 μL, 0.042 mmol) was added into the solution and the resulting mixture was continuously stirred for 3 h. The solution was neutralized to pH ∼ 6–7 by adding dil. HCl, and volatiles were evaporated. The residue was column-chromatographed (MeOH/CHCl3; 0 → 5%) to afford 26 (8 mg, 35%) as a 1:1 mixture of diastereomers: 1H NMR δ 2.10–2.25 (m, 2H), 2.40 (s, 3H), 3.53–3.63 (m, 2H), 3.87 (dt, J = 7.6, 4.0 Hz, 1H), 4.17–4.25 (m, 1H), 5.13 (t, J = 4.4 Hz, 0.5H), 5.17 (t, J = 4.4 Hz, 0.5H), 5.28 (d, J = 4.0 Hz, 0.5H), 5.31 (d, J = 4.4 Hz, 0.5H), 5.98–6.07 (m, 1H), 7.30 (s, 0.5H), 7.31 (s, 0.5H), 7.44 (d, J = 8.0 Hz, 2H), 7.75 (d, J = 8.0 Hz, 2H), 8.80 (s, 1H), 11.88 (s, 1H); 13C NMR δ 21.2, 40.7, 60.6, 60.7, 69.8, 69.9, 86.3, 86.5, 88.11, 88.12, 107.7, 107.8, 128.1, 129.62, 129.63, 129.71, 129.72, 133.31, 133.32, 145.3, 145.4, 149.11, 149.12, 161.5, 184.2, 184.3; HRMS calcd for C18H20IN2O8S [M + H]+ 550.9980, found 550.9961.
Alternatively, I2 (24 mg, 0.1 mmol) and 50% H2O2 (aq) (127 μL, 1.88 mmol) were sequentially added into a stirred solution of 12 (80 mg, 0.19 mmol) in AcOH (0.5 mL) at ambient temperature. After 1 h, volatiles were evaporated and the residue was column-chromatographed (CHCl3/MeOH; 100:0 → 95:5) to give 26 (42 mg, 41%).
5-(2-Propanethio-2-tosylacetyl)-2′-deoxyuridine (27)
A 1:1 mixture of PrSH (10 μL, 8.2 mg, 0.11 mmol) and TEA (15 μL, 11 mg, 0.11 mmol) was added into a stirring solution of 26 (40 mg, 0.07 mmol) in MeOH (1.5 mL) at rt. After 8 h, the volatiles were evaporated and the residue was column-chromatographed (MeOH/CHCl3; 0 → 5%) to give 27 (12.5 mg, 34%) as a 1:1 mixture of diastereomers: 1H NMR δ 0.85 (t, J = 7.2 Hz, 3H), 1.48 (sex, J = 7.2 Hz, 2H), 2.14–2.29 (m, 2H), 2.41 (s, 3H), 2.54–2.61 (m, 1H), 2.68–2.76 (m, 1H), 3.53–3.66 (m, 2H), 3.88 (dt, J = 7.2, 3.6 Hz, 1H), 4.19–4.27 (m, 1H), 5.11 (t, J = 4.4 Hz, 0.5H), 5.14 (t, J = 4.4 Hz, 0.5H), 5.29 (d, J = 4.4 Hz, 0.5H), 5.32 (d, J = 4.0 Hz, 0.5H), 6.05 (dd, J = 10.4, 4.4 Hz, 1H), 6.76 (s, 0.5H), 6.78 (s, 0.5H), 7.45 (d, J = 8.0 Hz, 2H), 7.73 (d, J = 8.0 Hz, 2H), 8.82 (s, 1H), 11.91 (s, 1H); 13C NMR δ 13.0, 21.2, 22.0, 34.1, 34.2, 40.7, 40.8, 60.7, 69.8, 69.9, 70.2, 70.3, 86.40, 86.42, 88.11, 88.13, 108.9, 109.0, 129.51, 129.52, 129.7, 134.21, 134.23, 145.1, 145.2, 149.10, 149.12, 149.2, 161.6, 161.7, 184.41, 184.42; HRMS calcd for C21H27N2O8S2 [M + H]+ 499.1203, found 499.1189.
(E)-5-(1-Chloro-2-tosylvinyl)-2′-deoxyuridine 5′-O-Triphosphate (28)
(MeO)3PO (0.5 mL; dried over 3A molecular sieves) was added to the flame-dried flask containing 5-(β-chloro)vinyl sulfone 5 (30 mg, 0.067 mmol; dried in vacuum (65 °C over P2O5)) and proton sponge (37 mg, 0.172 mmol), and the resulting solution was stirred at 0 °C for 5 min under an Ar atmosphere. Freshly distilled POCl3 (16 μL, 26.2 mg, 0.17 mmol) was then added, and stirring was continued for 30 min at 0 °C. Then, tributylammonium pyrophosphate (TBAPP; 0.5 M/dimethylformamide (DMF); 700 μL, 0.35 mmol) and Bu3N (50 μL, 39 mg, 0.21 mmol) were added sequentially and stirred for another 10 min at 0 °C. The reaction mixture was quenched by adjusting pH to 7.5–7.8 with triethylammonium bicarbonate (TEAB) buffer (2 M, several drops). The residue was dissolved in water (5 mL) and was extracted with EtOAc (3 × 5 mL). The water layer was evaporated and co-evaporated (three times) with a mixture of EtOH/H2O (1:1, 5 mL). The residue was chromatographed on a DEAE-Sephadex A-25 column (30 × 1 cm2; 3 g of resin) with TEAB (0.1 → 0.5 M), and the appropriate fractions (TLC, Rf 0.30; i-PrOH/H2O/NH4OH, 5:2:3) were evaporated in vacuum and co-evaporated five times with a mixture of EtOH/H2O (1:1, 10 mL) to remove excess of TEAB salt to give 28 (15 mg, 32%) as a triethylammonium salt: 1H NMR (D2O) δ 2.31–2.38 (m, 1H), 2.41–2.47 (m, 1H), 2.44 (s, 3H), 4.15–4.24 (m, 3H), 4.56–4.67 (m, 1H), 6.27 (t, J = 6.7 Hz, 1H), 7.40–7.47 (m, 3H), 7.65 (d, J = 8.0 Hz, 2H), 8.02 (s, 1H); 13C NMR (D2O) δ 20.9, 38.7, 65.5, 70.9, 85.9, 86.1, 108.6, 127.8, 130.1, 130.2, 134.8, 140.3, 142.8, 146.9, 150.4, 161.4; 31P NMR (D2O) δ −23.15, −11.62, −10.83; HRMS calcd for C18H2135ClN2O16P3S [M – H]− 680.9518, found 680.9517.
5-(2-Tosylacetyl)-2′-deoxyuridine 5′-O-Triphosphate (29)
The treatment of 5-(β-keto)sulfone 12 (40 mg, 0.095 mmol) with POCl3 (22.2 μL, 36.4 mg, 0.24 mmol) and TBAPP (0.5 M/DMF; 800 μL, 110 mg, 0.4 mmol), as described for 28, gave 29 (24 mg, 38%; TLC, Rf 0.30; i-PrOH/H2O/NH4OH, 5:2:3) as a triethylammonium salt: 1H NMR (D2O) δ 2.34–2.40 (m, 1H), 2.45 (s, 3H), 2.46–2.51 (m, 1H), 4.17–4.24 (m, 2H), 4.25–4.31 (m, 1H), 4.59–4.65 (m, 1H), 4.84–4.89 (m, 2H), 6.14 (t, J = 6.4 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.76 (d, J = 8.0 Hz, 2H), 8.47 (s, 1H); 13C NMR (D2O) δ 20.7, 39.3, 58.6, 65.3, 70.9, 86.3, 87.5, 111.0, 128.5, 130.3, 133.9, 147.2, 149.4, 150.0, 161.9, 185.3; 31P NMR (D2O) δ −23.34, −11.61, −10.97; HRMS calcd for C18H22N2O17P3S [M – H]− 662.9857, found 662.9859.
5-(2-Tosylacetyl)-2′-deoxycytidine 5′-O-Triphosphate (30)
The treatment of 5-(β-keto)sulfone 14 (9.3 mg, 0.022 mmol) with POCl3 (5 μL, 8.1 mg, 0.0528 mmol) and TBAPP (0.5 M/DMF; 330 μL, 0.165 mmol), as described for 28, gave 30 (4.5 mg, 31%) as a triethylammonium salt after purification on a DEAE-Sephadex A-25 column (30 × 1 cm2; 5 g of resin) with TEAB (0.2 → 0.7 M) buffer and evaporation and co-evaporation of the appropriate fractions (TLC, Rf 0.25; i-PrOH/H2O/NH4OH, 5:2:3): 1H NMR (D2O) δ 2.23–2.33 (m, 1H), 2.42 (s, 3H), 2.47–2.56 (m, 1H), 4.19–4.35 (m, 3H), 4.54 (dt, J = 10.2, 4.6 Hz, 1H), 5.91–5.00 (m, 2H), 5.97 (t, J = 5.6 Hz, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.75 (d, J = 7.2 Hz, 2H), 8.47 (s, 1H); 13C NMR (D2O) δ 20.8, 40.1, 58.7, 64.6, 69.5, 87.7, 100.0, 104.0, 128.2, 130.5, 133.3, 147.2, 151.1, 154.6, 162.9, 186.5; 31P NMR (D2O) δ −23.24, −11.82, −10.83; HRMS calcd for C18H23N3O16P3S [M – H]− 662.0017, found 662.0043.
Incorporation of 5-Modified Nucleotides into DNA: Materials of Enzymatic Reactions
All DNA oligonucleotides were synthesized by the Integrated DNA Technologies (IDT; Coralville, IA). The radionucleotide [γ-32P]ATP (6000 mCi/mmol) was purchased from MP Biomedicals (Santa Ana, CA). T4 polynucleotide kinase and deoxynucleoside 5′-triphosphates were purchased from Thermo Scientific (Pittsburgh, PA). Micro Bio-Spin TM 6 Columns were purchased from Bio-Rad (Hercules, CA). All other chemicals were purchased from Thermo Scientific (Pittsburgh, PA) and Sigma-Aldrich (St. Louis, MO). Purified human DNA polymerase β (pol β) was purified following the procedures described previously.54,55 The Klenow fragment was obtained from New England Biolabs (Ipswitch, MA).
Oligonucleotide Substrates
Substrates with an upstream primer annealed to the template strand were designated as open template substrates. The substrates were constructed by annealing an upstream primer (31 or 30 nt) with the template strand (71 or 49 nt) at a molar ratio of 1:3. The substrate containing one-nucleotide gap was made by annealing an upstream primer and a downstream primer with the template strand at the molar ratio of 1:3:3. The open template and one-nucleotide gap substrates were employed to mimic the intermediates formed during DNA replication. The sequences of oligonucleotides for constructing the substrates are listed in Tables 3 and 4.
Enzymatic Activity Assay
Nucleotide incorporation by DNA polymerases was performed by incubating different concentrations of pol β or 5 U Klenow fragment with 25 nM 32P-labeled substrates at 37 °C for 15 min according to a method described previously.50,51 The enzymatic reactions were assembled in the presence of triphosphates of 5-(β-chlorovinyl)sulfone of dU 28 (50 μM), 5-(β-keto)sulfone of dU 29 (50 μM), or 5-(β-keto)sulfone of dC 30. This allows us to examine if 28, 29, or 30 can be directly incorporated into a double-stranded DNA during DNA leading and lagging strand maturation. DNA synthesis was separated in a 15% urea denaturing polyacrylamide gel and the synthesized products were detected by a Pharos FX Plus PhosphorImager (Bio-Rad Laboratory, CA).
Acknowledgments
H.H. and Z.W. are recipients of the FIU Presidential and Dissertation Year Fellowships, respectively. This work was partially supported by NIH R01 grant ES023569 (Y.L.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b00584.
1H, 13C, and 31P NMR spectra for all compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Agrofoglio L. A.; Gillaizeau I.; Saito Y. Palladium-Assisted Routes to Nucleosides. Chem. Rev. 2003, 103, 1875–1916. 10.1021/cr010374q. [DOI] [PubMed] [Google Scholar]
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12, 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- De Clercq E.; Descamps J.; De Somer P.; Barr P. J.; Jones A. S.; Walker R. T. (E)-5-(2-Bromovinyl)-2′-deoxyuridine: a potent and selective anti-herpes agent. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 2947–2951. 10.1073/pnas.76.6.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuigan C.; Barucki H.; Carangio A.; Blewett S.; Andrei G.; Snoeck R.; De Clercq E.; Balzarini J.; Erichsen J. T. Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidine nucleosides bearing an aryl side chain. J. Med. Chem. 2000, 43, 4993–4997. 10.1021/jm000210m. [DOI] [PubMed] [Google Scholar]
- Burness C. B.; Duggan S. T. Trifluridine/Tipiracil: A Review in Metastatic Colorectal Cancer. Drugs 2016, 76, 1393–1402. 10.1007/s40265-016-0633-9. [DOI] [PubMed] [Google Scholar]
- Gramlich P. M. E.; Wirges C. T.; Manetto A.; Carell T. Postsynthetic DNA Modification through the Copper-Catalyzed Azide–Alkyne Cycloaddition Reaction. Angew. Chem., Int. Ed. 2008, 47, 8350–8358. 10.1002/anie.200802077. [DOI] [PubMed] [Google Scholar]
- Seela F.; Sirivolu V. R.; Chittepu P. Modification of DNA with Octadiynyl Side Chains: Synthesis, Base Pairing, and Formation of Fluorescent Coumarin Dye Conjugates of Four Nucleobases by the Alkyne-Azide “Click” Reaction. Bioconjugate Chem. 2008, 19, 211–224. 10.1021/bc700300f. [DOI] [PubMed] [Google Scholar]
- Guan L.; van der Heijden G. W.; Bortvin A.; Greenberg M. M. Intracellular Detection of Cytosine Incorporation in Genomic DNA by Using 5-Ethynyl-2′-Deoxycytidine. ChemBioChem 2011, 12, 2184–2190. 10.1002/cbic.201100353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X.; Gerowska M.; El-Sagheer A. H.; Brown T. Enzymatic incorporation and fluorescent labelling of cyclooctyne-modified deoxyuridine triphosphates in DNA. Bioorg. Med. Chem. 2014, 22, 4384–4390. 10.1016/j.bmc.2014.05.050. [DOI] [PubMed] [Google Scholar]
- Ren X.; El-Sagheer A. H.; Brown T. Azide and trans-cyclooctene dUTPs: incorporation into DNA probes and fluorescent click-labelling. Analyst 2015, 140, 2671–2678. 10.1039/C5AN00158G. [DOI] [PubMed] [Google Scholar]
- Neef A. B.; Luedtke N. W. An azide-modified nucleoside for metabolic labeling of DNA. ChemBioChem 2014, 15, 789–793. 10.1002/cbic.201400037. [DOI] [PubMed] [Google Scholar]
- Kumar P.; Hornum M.; Nielsen L. J.; Enderlin G.; Andersen N. K.; Len C.; Hervé G.; Sartori G.; Nielsen P. High-Affinity RNA Targeting by Oligonucleotides Displaying Aromatic Stacking and Amino Groups in the Major Groove. Comparison of Triazoles and Phenyl Substituents. J. Org. Chem. 2014, 79, 2854–2863. 10.1021/jo4025896. [DOI] [PubMed] [Google Scholar]
- Zayas J.; Annoual M. D.; Das J. K.; Felty Q.; Gonzalez W. G.; Miksovska J.; Sharifai N.; Chiba A.; Wnuk S. F. Strain Promoted Click Chemistry of 2- or 8-Azidopurine and 5-Azidopyrimidine Nucleosides and 8-Azidoadenosine Triphosphate with Cyclooctynes. Application to Living Cell Fluorescent Imaging. Bioconjugate Chem. 2015, 26, 1519–1532. 10.1021/acs.bioconjchem.5b00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raindlová V.; Pohl R.; Šanda M.; Hocek M. Direct Polymerase Synthesis of Reactive Aldehyde-Functionalized DNA and Its Conjugation and Staining with Hydrazines. Angew. Chem., Int. Ed. 2010, 49, 1064–1066. 10.1002/anie.200905556. [DOI] [PubMed] [Google Scholar]
- Raindlová V.; Pohl R.; Hocek M. Synthesis of Aldehyde-Linked Nucleotides and DNA and Their Bioconjugations with Lysine and Peptides through Reductive Amination. Chem. - Eur. J. 2012, 18, 4080–4087. 10.1002/chem.201103270. [DOI] [PubMed] [Google Scholar]
- Borsenberger V.; Howorka S. Diene-modified nucleotides for the Diels–Alder-mediated functional tagging of DNA. Nucleic Acids Res. 2009, 37, 1477–1485. 10.1093/nar/gkn1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch J.; Wiessler M.; Jäschke A. Post-Synthetic Modification of DNA by Inverse-Electron-Demand Diels-Alder Reaction. J. Am. Chem. Soc. 2010, 132, 8846–8847. 10.1021/ja102871p. [DOI] [PubMed] [Google Scholar]
- Rieder U.; Luedtke N. W. Alkene-Tetrazine Ligation for Imaging Cellular DNA. Angew. Chem., Int. Ed. 2014, 53, 9168–9172. 10.1002/anie.201403580. [DOI] [PubMed] [Google Scholar]
- Naik A.; Alzeer J.; Triemer T.; Bujalska A.; Luedtke N. W. Chemoselective Modification of Vinyl DNA by Triazolinediones. Angew. Chem., Int. Ed. 2017, 56, 10850–10853. 10.1002/anie.201702554. [DOI] [PubMed] [Google Scholar]
- Dadová J.; Orság P.; Pohl R.; Brázdová M.; Fojta M.; Hocek M. Vinylsulfonamide and Acrylamide Modification of DNA for Cross-linking with Proteins. Angew. Chem., Int. Ed. 2013, 52, 10515–10518. 10.1002/anie.201303577. [DOI] [PubMed] [Google Scholar]
- Dadová J.; Vrabel M.; Adamik M.; Brazdova M.; Pohl R.; Fojta M.; Hocek M. Azidopropylvinylsulfonamide as a New Bifunctional Click Reagent for Bioorthogonal Conjugations: Application for DNA-Protein Cross-Linking. Chem. - Eur. J. 2015, 21, 16091–16102. 10.1002/chem.201502209. [DOI] [PubMed] [Google Scholar]
- Olszewska A.; Pohl R.; Brazdova M.; Fojta M.; Hocek M. Chloroacetamide-Linked Nucleotides and DNA for Cross-Linking with Peptides and Proteins. Bioconjugate Chem. 2016, 27, 2089–2094. 10.1021/acs.bioconjchem.6b00342. [DOI] [PubMed] [Google Scholar]
- Hocek M. Synthesis of Base-Modified 2′-Deoxyribonucleoside Triphosphates and Their Use in Enzymatic Synthesis of Modified DNA for Applications in Bioanalysis and Chemical Biology. J. Org. Chem. 2014, 79, 9914–9921. 10.1021/jo5020799. [DOI] [PubMed] [Google Scholar]
- Xu W.; Chan K. M.; Kool E. T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. 10.1038/nchem.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivatsan S. G.; Tor Y. Fluorescent Pyrimidine Ribonucleotide: Synthesis, Enzymatic Incorporation, and Utilization. J. Am. Chem. Soc. 2007, 129, 2044–2053. 10.1021/ja066455r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramlich P. M. E.; Warncke S.; Gierlich J.; Carell T. Click-Click-Click: Single to Triple Modification of DNA. Angew. Chem., Int. Ed. 2008, 47, 3442–3444. 10.1002/anie.200705664. [DOI] [PubMed] [Google Scholar]
- Noé M. S.; Sinkeldam R. W.; Tor Y. Oligodeoxynucleotides Containing Multiple Thiophene-Modified Isomorphic Fluorescent Nucleosides. J. Org. Chem. 2013, 78, 8123–8128. 10.1021/jo4008964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows D. C.; Gervay-Hague J. Vinyl sulfones: Synthetic preparations and medicinal chemistry applications. Med. Res. Rev. 2006, 26, 793–814. 10.1002/med.20074. [DOI] [PubMed] [Google Scholar]
- Pathak T. Vinyl sulfone-modified carbohydrates: an inconspicuous group of chiral building blocks. Tetrahedron 2008, 64, 3605–3628. 10.1016/j.tet.2008.01.069. [DOI] [Google Scholar]
- Fang Y.; Luo Z.; Xu X. Recent advances in the synthesis of vinyl sulfones. RSC Adv. 2016, 6, 59661–59676. 10.1039/C6RA10731A. [DOI] [Google Scholar]
- For the synthesis and reactions of β-chloro (or bromo)vinyl sulfones see:; a Shainyan B. A.; Mirskova A. N. Nucleophilic reactions at a vinyl center. XXII. Synthesis and nucleophilic vinyl substitution in (E)-aryl β-fluorovinyl sulfones. Zh. Org. Khim. 1989, 25, 68–74. [Google Scholar]; b Višņevska J.; A̅bele E. Synthesis and Reactions of 2-Chlorovinyl Sulfones. Latv. J. Chem. 2011, 50, 22–31. 10.2478/v10161-011-0051-y. [DOI] [Google Scholar]; c Višņevska J.; Belyakov S.; Shestakova I.; Gulbe A.; Jaschenko E.; Abele E. Evaluation of (E)-2-chlorovinylsulfones as novel class of cytotoxic agents and highly (E)-stereoselective addition of N-, S- and Se-nucleophiles to (E)-2-chlorovinylsulfones under phase transfer catalysis conditions. Heterocycl. Lett. 2012, 2, 245–252. [Google Scholar]; d Aziz J.; Messaoudi S.; Alami M.; Hamze A. Sulfinate derivatives: dual and versatile partners in organic synthesis. Org. Biomol. Chem. 2014, 12, 9743–9759. 10.1039/C4OB01727G. [DOI] [PubMed] [Google Scholar]; e Iwasaki M.; Fujii T.; Yamamoto A.; Nakajima K.; Nishihara Y. Palladium-catalyzed regio- and stereoselective chlorothiolation of terminal alkynes with sulfenyl chlorides. Chem. - Asian J. 2014, 9, 58–62. 10.1002/asia.201301295. [DOI] [PubMed] [Google Scholar]; f Xie M.; Wang J.; Fang K.; Wang S.; Yan L. Regio- and stereoselective synthesis of (E)-β-halo alkenyl ketones/sulfones via haloallylation of alkynyl ketones/sulfones. Tetrahedron Lett. 2015, 56, 4388–4391. 10.1016/j.tetlet.2015.05.098. [DOI] [Google Scholar]
- Kumar R.; Dwivedi V.; Sridhar Reddy M. Metal-Free Iodosulfonylation of Internal Alkynes: Stereodefined Access to Tetrasubstituted Olefins. Adv. Synth. Catal. 2017, 359, 2847–2856. 10.1002/adsc.201700576. [DOI] [Google Scholar]
- Bi W.; Ren C.; Jia L.; Xia X.; Chen X.; Chen X.; Zhao Y. Synthesis of (E)-β-iodovinyl sulfones via DTBP/I2 promoted difunctionalization of alkynes with sodium benzenesulfinates. Phosphorus, Sulfur Silicon Relat. Elem. 2017, 192, 391–396. 10.1080/10426507.2016.1259622. [DOI] [Google Scholar]
- Xiang Y.; Kuang Y.; Wu J. Generation of beta-Halo Vinylsulfones through a Multicomponent Reaction with Insertion of Sulfur Dioxide. Chem. - Eur. J. 2017, 23, 6996–6999. 10.1002/chem.201701465. [DOI] [PubMed] [Google Scholar]
- McCarthy J. R.; Matthews D. P.; Stemerick D. M.; Huber E. W.; Bey P.; Lippert B. J.; Snyder R. D.; Sunkara P. S. Stereospecific method to (E) and (Z) terminal fluoroolefins and its application to the synthesis of 2′-deoxy-2′-fluoromethylenenucleosides as potential inhibitors of ribonucleoside diphosphate reductase. J. Am. Chem. Soc. 1991, 113, 7439–7440. 10.1021/ja00019a061. [DOI] [Google Scholar]
- Wnuk S. F.; Garcia P. I. Jr.; Wang Z. Radical-Mediated Silyl- and Germyldesulfonylation of Vinyl and (a-Fluoro)vinyl Sulfones: Application of Tris(trimethylsilyl)silanes and Tris(trimethylsilyl)germanes in Pd-Catalyzed Couplings. Org. Lett. 2004, 6, 2047–2049. 10.1021/ol049312n. [DOI] [PubMed] [Google Scholar]
- Andrei D.; Wnuk S. F. Synthesis of the Multisubstituted Halogenated Olefins via Cross-Coupling of Dihaloalkenes with Alkylzinc Bromides. J. Org. Chem. 2006, 71, 405–408. 10.1021/jo051980e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markitanov Y. M.; Timoshenko V. M.; Shermolovich Y. G. β-Keto sulfones: preparation and application in organic synthesis. J. Sulfur Chem. 2014, 35, 188–236. 10.1080/17415993.2013.815749. [DOI] [Google Scholar]
- Saraiva M. T.; Costa G. P.; Seus N.; Schumacher R. F.; Perin G.; Paixão M. W.; Luque R.; Alves D. Room-Temperature Organocatalytic Cycloaddition of Azides with β-Keto Sulfones: Toward Sulfonyl-1,2,3-triazoles. Org. Lett. 2015, 17, 6206–6209. 10.1021/acs.orglett.5b03196. [DOI] [PubMed] [Google Scholar]
- Lu Q.; Zhang J.; Zhao G.; Qi Y.; Wang H.; Lei A. Dioxygen-Triggered Oxidative Radical Reaction: Direct Aerobic Difunctionalization of Terminal Alkynes toward β-Keto Sulfones. J. Am. Chem. Soc. 2013, 135, 11481–11484. 10.1021/ja4052685. [DOI] [PubMed] [Google Scholar]
- Lai C.; Xi C.; Jiang Y.; Hua R. One-pot approach for the regioselective synthesis of β-keto sulfones based on acid-catalyzed reaction of sulfonyl chlorides with arylacetylenes and water. Tetrahedron Lett. 2005, 46, 513–515. 10.1016/j.tetlet.2004.10.176. [DOI] [Google Scholar]
- Mendieta L.; Pico A.; Tarrago T.; Teixido M.; Castillo M.; Rafecas L.; Moyano A.; Giralt E. Novel peptidyl aryl vinyl sulfones as highly potent and selective inhibitors of cathepsins L and B. ChemMedChem 2010, 5, 1556–1567. 10.1002/cmdc.201000109. [DOI] [PubMed] [Google Scholar]
- Xiang J.; Ipek M.; Suri V.; Tam M.; Xing Y.; Huang N.; Zhang Y.; Tobin J.; Mansour T. S.; McKew J. β-Keto sulfones as inhibitors of 11β-hydroxysteroid dehydrogenase type I and the mechanism of action. Bioorg. Med. Chem. 2007, 15, 4396–4405. 10.1016/j.bmc.2007.04.035. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Suzol S. H.; Wen Z.; Artiles A. G.; Mathivathanan L.; Raptis R. G.; Wnuk S. F. Uracil Nucleosides with Reactive Group at C5 Position: 5-(1-Halo-2-sulfonylvinyl)uridine Analogues. Org. Lett. 2016, 18, 1418–1421. 10.1021/acs.orglett.6b00346. [DOI] [PubMed] [Google Scholar]
- Wen Z.; Suzol S. H.; Peng J.; Liang Y.; Snoeck R.; Andrei G.; Liekens S.; Wnuk S. F. Antiviral and Cytostatic Evaluation of 5-(1-Halo-2-sulfonylvinyl)- and 5-(2-Furyl)uracil Nucleosides: 5-(1-Halo-2-sulfonylvinyl)- and 5-(2-Furyl)uracil Nucleosides. Arch. Pharm. 2017, 350, e1700023 10.1002/ardp.201700023. [DOI] [PubMed] [Google Scholar]
- For stereoselective synthesis of (Z)-sulfonylvinylamines from (arylsulfonyl)acetonitrile or oxime acetates and their conversion to β-keto sulfones see:; a Tsui G. C.; Glenadel Q.; Lau C.; Lautens M. Rhodium(I)-Catalyzed Addition of Arylboronic Acids to (Benzyl-/Arylsulfonyl)acetonitriles: Efficient Synthesis of (Z)-β-Sulfonylvinylamines and β-Keto Sulfones. Org. Lett. 2011, 13, 208–211. 10.1021/ol102598p. [DOI] [PubMed] [Google Scholar]; b Tang X.; Huang L.; Xu Y.; Yang J.; Wu W.; Jiang H. Copper-catalyzed coupling of oxime acetates with sodium sulfinates: an efficient synthesis of sulfone derivatives. Angew. Chem., Int. Ed. 2014, 53, 4205–4208. 10.1002/anie.201311217. [DOI] [PubMed] [Google Scholar]
- Kada R.; Knoppová V.; Jurášek A.; Kováč J. Furan derivatives-LXXVII. Tetrahedron 1976, 32, 1411–1414. 10.1016/0040-4020(76)85021-1. [DOI] [Google Scholar]
- Metal-free alpha-sulfanylation of ketones catalyzed by proline has been recently developed:Vaquer A. F.; Frongia A.; Secci F.; Tuveri E. RSC Adv. 2015, 5, 96695–96704. 10.1039/C5RA17913K. [DOI] [Google Scholar]
- Suryakiran N.; Srikanth Reddy T.; Venkateswarlu Y. Iodination of β-keto-sulfones using molecular iodine and hydrogen peroxide in aqueous medium: facile synthesis of α-iodomethyl sulfones. J. Sulfur Chem. 2007, 28, 471–476. 10.1080/17415990701595865. [DOI] [Google Scholar]
- Kovács T.; Ötvös L. Simple synthesis of 5-vinyl- and 5-ethynyl-2′-deoxyuridine-5′-triphosphates. Tetrahedron Lett. 1988, 29, 4525–4528. 10.1016/S0040-4039(00)80537-7. [DOI] [Google Scholar]
- Jacobsen H.; Klenow H.; Overoaard-Hansen K. The N-Terminal Amino-Acid Sequences of DNA Polymerase I from Escherichia coli and of the Large and the Small Fragments Obtained by a Limited Proteolysis. Eur. J. Biochem. 1974, 45, 623–627. 10.1111/j.1432-1033.1974.tb03588.x. [DOI] [PubMed] [Google Scholar]
- Beard W. A.; Wilson S. H. Structure and mechanism of DNA polymerase beta. Biochemistry 2014, 53, 2768–2780. 10.1021/bi500139h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobek M.; Kavai I.; Sharma R. A.; Grill S.; Dutschman G.; Cheng Y. C. Acetylenic nucleosides. 4. 1-(.beta.-D-Arabinofuranosyl)-5-ethynylcytosine. Improved synthesis and evaluation of biochemical and antiviral properties. J. Med. Chem. 1987, 30, 2154–2157. 10.1021/jm00394a039. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Prasad R.; Beard W. A.; Hou E. W.; Horton J. K.; McMurray C. T.; Wilson S. H. Coordination between polymerase beta and FEN1 can modulate CAG repeat expansion. J. Biol. Chem. 2009, 284, 28352–28366. 10.1074/jbc.M109.050286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M.; Lai Y.; Torner J.; Zhang Y.; Zhang Z.; Liu Y. Base excision repair of oxidative DNA damage coupled with removal of a CAG repeat hairpin attenuates trinucleotide repeat expansion. Nucleic Acids Res. 2014, 42, 3675–3691. 10.1093/nar/gkt1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.