

Abstract
We show that a chiral copper hydride (CuH) complex catalyzes C-C bond-forming dearomatization of pyridines and pyridazines at room temperature. The catalytic reaction operates directly on free heterocycles and generates the nucleophiles in situ, eliminating the need for stoichiometric preactivation of either reaction partner; further, it is one of very few methods available for the enantioselective 1,4-dearomatization of heteroarenes. Combining the dearomatization with facile derivatization steps enables one-pot syntheses of enantioenriched pyridines and piperidines.
Graphical abstract
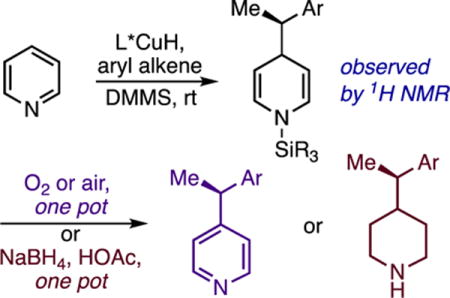
Dearomatization of electron-deficient heteroarenes with carbon nucleophiles is an essential transformation for the synthesis of pyridines and piperidines, which are the two most common azaheterocycles in FDA-approved small-molecule drugs.1,2a–2e However, direct dearomative addition to pyridine generally requires harsh conditions2c,3b and has limited compatibility with complex, functionalized molecules. Most C–C bond-forming pyridine dearomatizations employ activated substrates generated through stoichiometric functionalization of the heterocyclic nitrogen with strong electrophiles.2–7 While useful, this approach has a number of limitations. For instance, many methods require pre-synthesis of the activated heterocycle2c or prior formation of the nucleophile, and separate deprotection steps are commonly required to cleave the activating group from the dihydropyridine (DHP) product. Further, whereas numerous methods exist for asymmetric 1,2-dearomatization,4a–4e achieving stereocontrol in 1,4-selective variants has proven much more challenging. The latter transformation is seldom possible without auxiliaries or pre-formed chiral nucleophiles (Figure 1, A and B);5,6a–6c asymmetric catalysis of pyridine 1,4-dearomatization7a–7d was unknown until very recently, 2c and highly enantioselective catalytic reactions (Figure 1, C)7b–7d are currently only possible with a narrow set of multiply activated cationic substrates. A number of catalyzed additions of hydride or silyl nucleophiles have been reported that operate directly on pyridine rather than on stoichiometrically activated derivatives,8a–8j but there are no reactions of this type that achieve either C-C bond-formation or asymmetric induction. In this communication, we show that a chiral copper complex catalyzes C–C bond-forming dearomatization under mild conditions without requiring activation of the heterocycle, pre-formation of the nucleophile, or protecting group manipulations (Figure 1, D). Moreover, the reaction is a very rare example of highly enantioselective catalytic 1,4-dearomatization, it succeeds with a broad selection of substituted pyridines and pyridazines, and it generates DHPs that can be converted to enantioenriched pyridines or piperidines in the same pot.
 |
(eq 1) |
Figure 1.
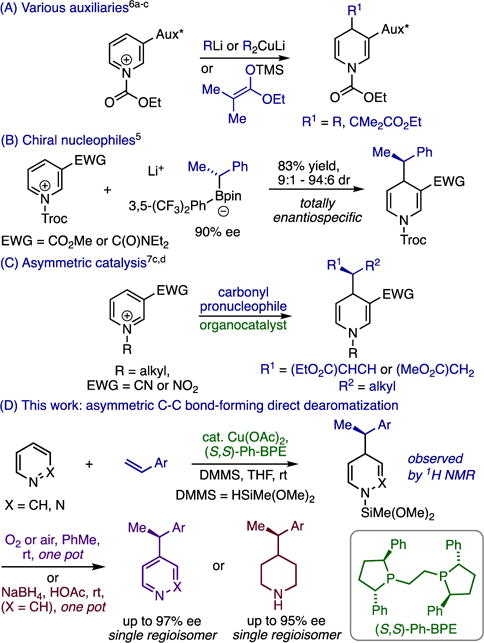
Methods for stereocontrolled 1,4-dearomatization; (A) with chiral auxiliaries; (B) with chiral nucleophiles; (C) using asymmetric catalysis; (D) this work: asymmetric direct catalytic dearomatization.
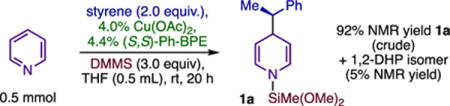
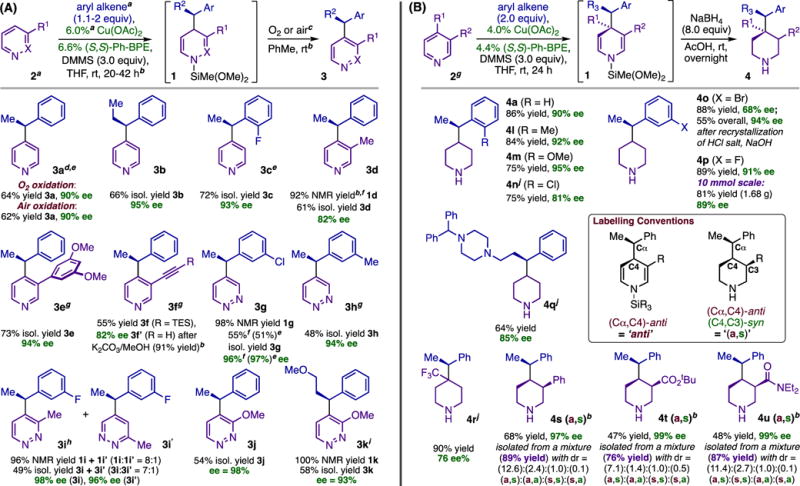
We hypothesized that pyridine could be activated toward nucleophilic dearomatization in an asymmetric hydrofunctionalization reaction,9,10 thereby permitting direct dearomatization of the heterocycle while replacing pre-formed nucleophiles with abundant olefin precursors. Subjecting a mixture of pyridine and styrene to (Ph-BPE)CuH, prepared as before (eq 1),11,12 gave 97% conversion (1H NMR) to a mixture of dihydropyridines strongly biased in favor of the 1,4-adduct 1a (22:1 average 1,4:1,2). Treating crude DHPs with O23c provided efficient rearomatization and enabled a one-pot synthesis of functionalized pyridines (generically, 3; Table 1, A). Chiral analysis of pyridine 3a (90% ee) indicated that the dearomatization had occurred with high enantioselectivity and that the aerobic rearomatization was stereospecific.13 Applying the dearomatization to the synthesis of enantioenriched piperidines also proved to be straightforward: adding NaHB(OAc)3 to crude DHPs provided good yields of piperidines (generically, 4; Table 1, B) in a one-pot operation14 that could be conducted on gram-scale without appreciable loss of yield or selectivity (4p; Table 1, B).
Table 1.
Asymmetric Dearomative Syntheses of Functionalized Pyridines (A) and Piperidines (B).
|
Reactions used 1 mmol 2, 2 equiv olefin except where noted;
see SI for details;
O2 was used for 3b-k.;
yields and ee’s are averages for two runs except where noted;
reaction used 4.0% Cu(OAc)2, 4.4% Ph-BPE;
one measurement;
used 0.5 mmol 2;
used 1.5 equiv olefin;
used 1.1 equiv olefin;
ee determination used the N-BOC derivative.
The dearomatization/reoxidation protocol succeeded with pyridines, pyridazines, and a variety of C3-substituted derivatives thereof. The enantioselectivity obtained with pyridazines (3g-k; Table 1, A) was consistently excellent and insensitive to the presence of electron-donor groups; in contrast, the ee’s obtained with pyridines were moderately depressed by electron-releasing groups (e.g., 3d; Table 1, A) and enhanced by aryl and π-acceptor substituents (3e, 4s-u; Table 1, A and B). Substituted pyridines were also viable substrates for dearomatization/reduction, and stereochemical analysis of products 4s-u (Table 1, B) revealed that both transformations exert control over the endocyclic stereocenters they respectively generate, leading to mixtures of diastereomeric piperidines having a major bias for the 4 (a,s) diastereomers [see inset in Table 1, B for explanation of nomenclature]. The major diastereomers were isolable in stereochemically pure form, and thus the dearomatization/reduction protocol enabled selective preparation of piperidines containing three contiguous stereocenters starting from prochiral substrates. Our work with this series provided key insights into the stereochemical properties of the asymmetric dearomatization. Single-crystal X-ray diffraction analysis of 4s (a,s)•HCl revealed its absolute configuration,15,16 making it clear that dearomative addition is retentive with respect to the benzylic stereocenter set during hydrocupration and selective for (Cα,C4)-anti DHPs, whereas the reduction is selective for (C3,C4)-syn piperidines. The basis for anti-selective dearomative addition is unclear at present, but it appears to be general.15,17 Notably, retention of the phenethylcopper stereocenter contrasts with the clean inversion Aggarwal observed in the addition of chiral phenethylboronates to acylpyridiniums (Figure 1, B);5 our result is mechanistically interesting given that the organocopper nucleophiles involved here do undergo invertive addition in other transformations.12c
Unlike C3 substituents, groups at C4 are only accommodated in special cases (as in 4r; Table 1, B), and substitution at C2 is not tolerated even for substituents that increase the intrinsic electrophilicity of the free heterocycle (e.g., CF3, CO2Me). Further, examples 3i-k (Table 1, A) show that organocopper nucleophiles preferentially add para to the less hindered nitrogen even when this entails attack on the more encumbered and more electron-rich of the two activated sites. These observations concerning C2 substitution can be readily rationalized if one invokes coordination of the heterocycle to a sterically demanding Lewis acid (e.g., Cu) in the dearomative addition step.
The dearomatization is compatible with various aryl alkene substituents, but certain trends involving this reactant were very surprising. Dearomatization exhibits useful levels of selectivity for a variety of olefin β substituents (3b, 3k, 4q; Table 1, A and B), and ortho substitution is broadly tolerated (3c, 4l-n; Table 1, A and B). But surprisingly, when groups such as F, Me and OMe are present at the para position of the styrene, they completely suppress dearomatization. This observation runs counter to all our previous experience with styrene hydrofunctionalization12a–12e and leads us to propose that para substituents incur a destabilizing interaction unique to the dearomative addition transition state. Consistent with this, we observed some sensitivity to the steric demand of the meta substituent; thus, 3-methylstyrene is problematic for pyridine but not pyridazine (3g, 3h; Table 1), while meta-halides are tolerated with both (3g, 3i, 4o-p; Table 1, A and B).
Figure 2 illustrates one plausible mechanism for the Cu-catalyzed direct dearomatization. Activation of the heterocycle occurs through formation of dative complex I (step i), which undergoes dearomatization with an organocopper nucleophile (II) (step iii).12a,18 The resulting N-cuprated DHP intermediate (III) could then furnish product IV and the regenerated catalyst via σ-bond metathesis with the silane (step iv), similarly to transmetalation processes implicated in other catalytic hydrofunctionalizations.12,18–19 We are currently undertaking a detailed mechanistic investigation directed at elucidating how activation and addition occur in this reaction.
Figure 2.
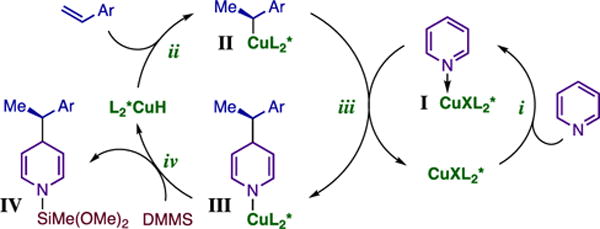
Plausible Mechanism for the Direct Dearomatization.
In summary, we have demonstrated that pyridine and pyridazine undergo direct asymmetric dearomatization in the presence of a chiral CuH catalyst. This unique reaction eliminates the need for extraneous activation and nucleophile-formation steps, and it permits one-pot syntheses of highly enantioenriched C4-functionalized heterocycles. We expect that our ongoing mechanistic investigations will shed light on the unusual reactivity trends we observe and aid in the discovery of more general dearomative transformations.
Supplementary Material
Acknowledgments
The National Institutes of Health under award numbers GM46059 and R35-GM122483 supported research reported in this publication. We thank Richard Liu and Andy Thomas (MIT) for advice on the preparation of this manuscript, Charlene Tsay (MIT) for X-ray crystallographic analysis, Bruce Adams (MIT) for assistance with NMR structure-determination, and the National Institutes of Health for a supplemental grant for the purchase of supercritical fluid chromatography (SFC) equipment (GM058160-17S1).
Footnotes
Supporting Information. The supporting Information is available free of charge on the ACS Publications website. Experimental procedures and characterization data for all compounds (PDF, CIF)
CIF
Notes
No competing financial interests have been declared
References
- 1.Vitaku E, Smith DT, Njardson JT. J Med Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- 2.For reviews on pyridine dearomatization and dihydropyridine chemistry, see:; (a) Lavilla R. J Chem Soc, Perkin Trans. 2002;1:1141–1156. [Google Scholar]; (b) Ahamed M, Todd MH. Eur J Org Chem. 2010;2010:5935–5942. [Google Scholar]; (c) Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d. [DOI] [PubMed] [Google Scholar]; (d) Zhuo CX, Zhang W, You SL. Angew Chem, Int Ed. 2012;51:12662–12686. doi: 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]; (e) Ding Q, Zhou X, Fan R. Org Biomol Chem. 2014;12:4807–4815. doi: 10.1039/c4ob00371c. [DOI] [PubMed] [Google Scholar]
- 3.For examples of 1,4-dearomatizations using copper, see:; (a) Piers E, Soucy M. Can J Chem. 1974;52:3563–3564. [Google Scholar]; (b) Comins DL, Abdullah AH. J Org Chem. 1982;47:4315–4319. [Google Scholar]; (c) Akiba KY, Iseki Y, Wada M. Bull Chem Soc Jpn. 1984;57:1994–1999. [Google Scholar]
- 4.For examples of asymmetric catalysis using activated substrates, see:; (a) Sun Z, Yu S, Ding Z, Ma D. J Am Chem Soc. 2007;129:9300–9301. doi: 10.1021/ja0734849. [DOI] [PubMed] [Google Scholar]; (b) Black DA, Beveridge RE, Arndtsen BA. J Org Chem. 2008;73:1906–1910. doi: 10.1021/jo702293h. [DOI] [PubMed] [Google Scholar]; (c) Fernández-Ibáñez MA, Maciá B, Pizzuti MG, Minnaard AJ, Feringa BL. Angew Chem, Int Ed. 2009;48:9339–9341. doi: 10.1002/anie.200904981. [DOI] [PubMed] [Google Scholar]; (d) Lutz JP, Chau ST, Doyle AG. Chem Sci. 2016;7:4105–4109. doi: 10.1039/c6sc00702c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mohiti M, Rampalakos C, Feeney K, Leonori D, Aggarwal VK. Chem Sci. 2014;5:602–607. [Google Scholar]
- 6.For representative examples, see:; (a) Meyers AI, Natale NR, Wettlaufer DG, Rafii S, Clardy J. Tetrahedron Lett. 1981;22:5123–5126. [Google Scholar]; (b) Mangeney P, Gosmini R, Raussou S, Commerçon M, Alexakis A. J Org Chem. 1994;59:1877–1888. [Google Scholar]; (c) Yamada S, Morita C. J Am Chem Soc. 2002;124:8184–8185. doi: 10.1021/ja0203317. [DOI] [PubMed] [Google Scholar]
- 7.(a) Mancheño OG, Asmus S, Zurro M, Fischer T. Angew Chem, Int Ed. 2015;54:8823–8827. doi: 10.1002/anie.201502708. [DOI] [PubMed] [Google Scholar]; (b) Bertuzzi G, Sinisi A, Caruana L, Mazzanti A, Fochi M, Bernardi L. ACS Catal. 2016;6:6473–6477. [Google Scholar]; (c) Bertuzzi G, Sinisi A, Pecorari D, Caruana L, Mazzanti A, Bernardi L, Fochi M. Org Lett. 2017;19:834–837. doi: 10.1021/acs.orglett.6b03824. [DOI] [PubMed] [Google Scholar]; (d) Flanigan DM, Rovis T. Chem Sci. 2017;8:6566–6569. doi: 10.1039/c7sc02648j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Oshima K, Ohmura T, Suginome M. J Am Chem Soc. 2011;133:7324–7327. doi: 10.1021/ja2020229. [DOI] [PubMed] [Google Scholar]; (b) Gutsulyak DV, van der Est A, Nikonov GI. Angew Chem, Int Ed. 2011;50:1384–1387. doi: 10.1002/anie.201006135. [DOI] [PubMed] [Google Scholar]; (c) Hill MS, Kociok-Köhn G, MacDougall DJ, Mahon MF, Weetman C. Dalton Trans. 2011;40:12500–12509. doi: 10.1039/c1dt11235j. [DOI] [PubMed] [Google Scholar]; (d) Königs CDF, Klare HFT, Oestreich M. Angew Chem, Int Ed. 2013;52:10076–10079. doi: 10.1002/anie.201305028. [DOI] [PubMed] [Google Scholar]; (e) Dudnik AS, Weidner VL, Motta A, Delferro M, Marks TJ. Nat Chem. 2014;6:1100–1107. doi: 10.1038/nchem.2087. [DOI] [PubMed] [Google Scholar]; (f) Gandhamsetty N, Park S, Chang S. J Am Chem Soc. 2015;137:15176–15184. doi: 10.1021/jacs.5b09209. [DOI] [PubMed] [Google Scholar]; (g) Intemann J, Bauer H, Pahl J, Maron L, Harder S. Chem – Eur J. 2015;21:11452–11461. doi: 10.1002/chem.201501072. [DOI] [PubMed] [Google Scholar]; (h) Fan X, Zheng J, Li ZH, Wang H. J Am Chem Soc. 2015;137:4916–4919. doi: 10.1021/jacs.5b03147. [DOI] [PubMed] [Google Scholar]; (i) Kaithal A, Chatterjee B, Gunanathan C. Org Lett. 2016;18:3402–3405. doi: 10.1021/acs.orglett.6b01564. [DOI] [PubMed] [Google Scholar]; (j) Zhang F, Song H, Zhuang X, Tung CH, Wang W. J Am Chem Soc. 2017;139:17775–17778. doi: 10.1021/jacs.7b11416. [DOI] [PubMed] [Google Scholar]
- 9.Kanai has suggested that hydrometallation and dearomative addition steps occur in the mechanism of Co-catalyzed C-H functionalization of pyridine, although they do not describe observing the DHP; rather, they suggest that (presumably fast) rearomatization is intrinsic to the catalytic cycle.; Andou T, Saga Y, Komai H, Matsunaga S, Kanai M. Angew Chem, Int Ed. 2013;52:3213–3216. doi: 10.1002/anie.201208666. [DOI] [PubMed] [Google Scholar]
- 10.After our work was completed, a paper appeared by Yu, et al. showing that phenethylcopper species generated in asymmetric hydrocupration effect reductive ortho C-H-functionalization of quinoline N-oxides, probably via an initially dearomative mechanism.; Yu S, Sang HL, Ge S. Angew Chem, Int Ed. 2017;56:15896–15900. doi: 10.1002/anie.201709411. [DOI] [PubMed] [Google Scholar]
- 11.For detailed information about the safe handling of dimethoxy(methyl)silane (DMMS), see the Supporting Information.
- 12.See, e.g.,; (a) Bandar JS, Pirnot MT, Buchwald SL. J Am Chem Soc. 2015;137:14812–14818. doi: 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang YM, Buchwald SL. J Am Chem Soc. 2016;138:5024–5027. doi: 10.1021/jacs.6b02527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Yang Y, Perry IB, Buchwald SL. J Am Chem Soc. 2016;138:9787–9790. doi: 10.1021/jacs.6b06299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gribble MW, Jr, Pirnot MT, Bandar JS, Liu RY, Buchwald SL. J Am Chem Soc. 2017;139:2192–2195. doi: 10.1021/jacs.6b13029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhou Y, Bandar JS, Buchwald SL. J Am Chem Soc. 2017;139:8126–8129. doi: 10.1021/jacs.7b04937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The rearomatization causes some degradation of the DHPs through a pathway that regenerates some of the starting heterocycle – probably fragmentation of benzyl radical from a dihydropyridine radical cation. See the mechanism described in; Ludvík J, Volke J, Klíma J. Electrochim Acta. 1987;32:1063–1071. [Google Scholar]
- 14.The reduction step is similar to that in; Suryavanshi PA, Sridharan V, Maiti S, Menéndez JC. Chem – Eur J. 2014;20:8791–8799. doi: 10.1002/chem.201402607. [DOI] [PubMed] [Google Scholar]
- 15.See supporting information for details.
- 16.The (a,s) Relative stereochemistry of products 4t and 4u was confirmed through NMR-based structure-determination. See SI.
- 17.For examples s-u, 1H NMR assignments of anti-1 and syn-1 were possible using the known dr’s for 4s-u. Assignments made by analogy to these indicated that anti-1 was also major for every other DHP we analyzed by 1H NMR. (NMR estimates for the dr are 25:1 for 1d, 13:1 for 1k, 11:1 for 1s, 7:1 for 1t, 4.6:1 for 1u; Table 1 A and B.) See the supporting information for details.
- 18.Xi Y, Hartwig JF. J Am Chem Soc. 2017;139:12758–12772. doi: 10.1021/jacs.7b07124. [DOI] [PubMed] [Google Scholar]
- 19.See entry 19 on page S4 of the supporting information for reference 12c.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.