

SUMMARY
Aging is a major risk factor for many human diseases, and in vitro generation of human neurons is an attractive approach for modeling aging-related brain disorders. However, modeling aging in differentiated human neurons has proved challenging. We generated neurons from human donors across a broad range of ages, either by iPSC-based reprogramming and differentiation or by direct conversion into induced neurons (iNs). While iPSCs and derived neurons did not retain aging-associated gene signatures, iNs displayed age-specific transcriptional profiles and revealed age-associated decreases in the nuclear transport receptor RanBP17. We detected an age-dependent loss of nucleocytoplasmic compartmentalization (NCC) in donor fibroblasts and corresponding iNs and found that reduced RanBP17 impaired NCC in young cells, while iPSC rejuvenation restored NCC in aged cells. These results show that iNs retain important aging-related signatures, thus allowing modeling of the aging process in vitro, and they identify impaired NCC as an important factor in human aging.
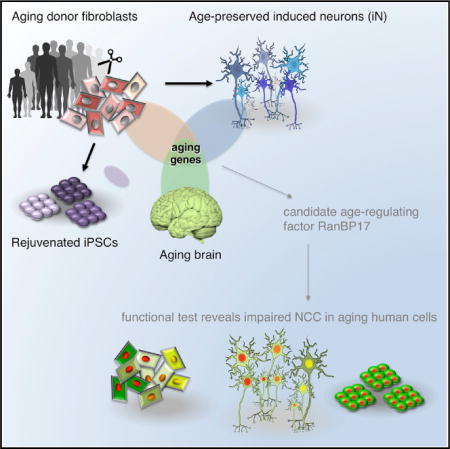
Graphical abstract
In Brief Mertens and colleagues compare transcriptomes of human fibroblasts, induced neurons (iNs), iPSCs, iPSC-derived neurons, and brain samples from a broad range of aged donors, finding that iNs retain donor aging signatures, while iPSCs are rejuvenated. RanBP17 was consistently decreased during aging, leading to compromised nucleocytoplasmic compartmentalization in aged human cells.
INTRODUCTION
The inevitable process of aging affects all tissues of the body and determines the quality and length of life. Human aging is by far the most critical risk factor for the development of several diseases that appear to exclusively affect the elderly, due to mostly unknown reasons (Cummings, 2008; Gladyshev, 2013). While aggressive familial early onset versions of fatal neurodegenerative diseases like Alzheimer’s or Parkinson’s disease can emerge in mid-life, the overwhelming majority of cases develop sporadically in old age, without known genetic causes. Neurons are a prime target for cellular aging. Unlike most other cell types, neurons are mainly born during embryogenesis and then face a demand for life-long performance. Progressive aging also leads to declines in neuronal plasticity and cognitive performances in the majority of healthy people, suggesting that neurons in the brain might decay over their lifetime (Burke and Barnes, 2006; Yankner et al., 2008). Interestingly, transcriptional profiling of different tissues has revealed similar age-related changes across different tissues, including genes involved in stress response, inflammation, and Ca2+ homeostasis, whereas tissue-specific changes of the aging human cortex were detected in genes controlling synaptic functions (Adler et al., 2007; Fraser et al., 2005; Lu et al., 2004; Murphy, 2006).
To experimentally assess how progressive human age can cause persistent cellular alterations that eventually emerge as decreased functionality, vital human cells, particularly neurons from donors of a broad range of ages, would be highly desirable. However, due to the inaccessibility of live human brain tissue, most studies have been limited to animal models that, while yielding important insights, have also revealed limitations regarding transferability to human physiology and lifespan. Human patient-specific induced pluripotent stem cell (iPSC)-based disease models have provided fascinating insights into disease-relevant mechanisms and pre-clinical drug evaluation directly in functional human neurons (Israel et al., 2012; Mertens et al., 2013b). However, preservation of human age as a major pathogenic risk factor would seem unlikely, given that cells must transit the embryo-like iPSC state, which likely rejuvenates old somatic cells back into an embryonic state (Lapasset et al., 2011; Maherali et al., 2007; Meissner et al., 2008). Furthermore, the numerous cell divisions required for the reprogramming process and differentiation may dilute any accumulated macromolecular damage. The direct transcription factor-based conversion of fibroblasts into induced neurons (iNs) represents an alternative avenue for generating human neurons in vitro (Pang et al., 2011; Vierbuchen et al., 2010). Induction of only two transcription factors in combination with a cocktail of small molecular enhancers was shown to directly yield functional iNs from human fibroblasts with high efficiencies (Ladewig et al., 2012; Liu et al., 2013). As iNs circumvent the pluripotent state as well as any cell division, we hypothesize that direct conversion preserves the cellular signatures of aging and results in neurons that show age equivalence with their human donor. In this study, we set out to analyze primary cells from young and old human donors to identify the key factors relevant to human aging and to subsequently program these cells into iNs to generate an age-equivalent in vitro model for neuronal cell aging.
RESULTS
iPSC Reprogramming Erases Transcriptomic Signatures of Aging Present in Primary Fibroblasts
Increasing evidence suggests that iPSC reprogramming of somatic cells into the embryo-like state largely resets their epigenetic state and either permanently deletes or temporarily conceals any memory of their age of origin. To test this paradigm in the context of human aging, we obtained skin fibroblasts from 19 individuals ranging in age from 0 to 89 years (Table S1) and reprogrammed 16 lines into iPSCs by overexpression of the four Yamanaka factors (Figure 1A) (Takahashi et al., 2007). Clonally derived iPSCs showed typical morphologies of human pluripotent cells and nuclear expression of Nanog (Figure 1B); they were analyzed by G-banding to be karyotypically normal (Figure 1C). All iPSC lines possessed an expression profile typical of pluripotent stem cells, with Lin28A/B, MycN, Nanog, Tert, Oct4, Nodal, Dppa2/4, and Otx2 all being more than 1,0003× upregulated as compared to fibroblasts (Figure 1D; Table S2). To investigate the extent to which iPSCs can retain the aging signatures of their donors, we performed high-throughput whole transcriptome RNA sequencing (RNA-seq) and compared the gene expression patterns of old and young fibroblasts and of derived iPSCs. The expression profiles of fibroblasts and iPSC samples were clearly distinguishable and clustered by overall expression similarity (Figure 1E). The independent clonal iPSC lines from the same donor typically, but not always, clustered together (Figure 1E). To identify putative aging genes, we performed differential expression analysis between cells derived from donors <40 years and cells from donors >40 years of age and found that, in fibroblasts, 78 genes were significantly differentially expressed, with a false discovery rate (FDR)-adjusted p value (padj) of <0.05 (Figure 1F; Table S3) (Benjamini and Hochberg, 1995). In contrast, iPSCs showed fewer overall expression variations and only one gene was determined to be significantly differentially expressed (Figure 1G). When only retrovirus-based iPSCs were compared, no genes were found to be significantly differentially expressed (data not shown). These data thus confirm that the age-dependent gene expression signatures present in primary fibroblasts become largely erased during reprogramming into iPSCs.
Figure 1. Age-Dependent Transcriptome Signatures in Fibroblasts and Derived iPSCs.
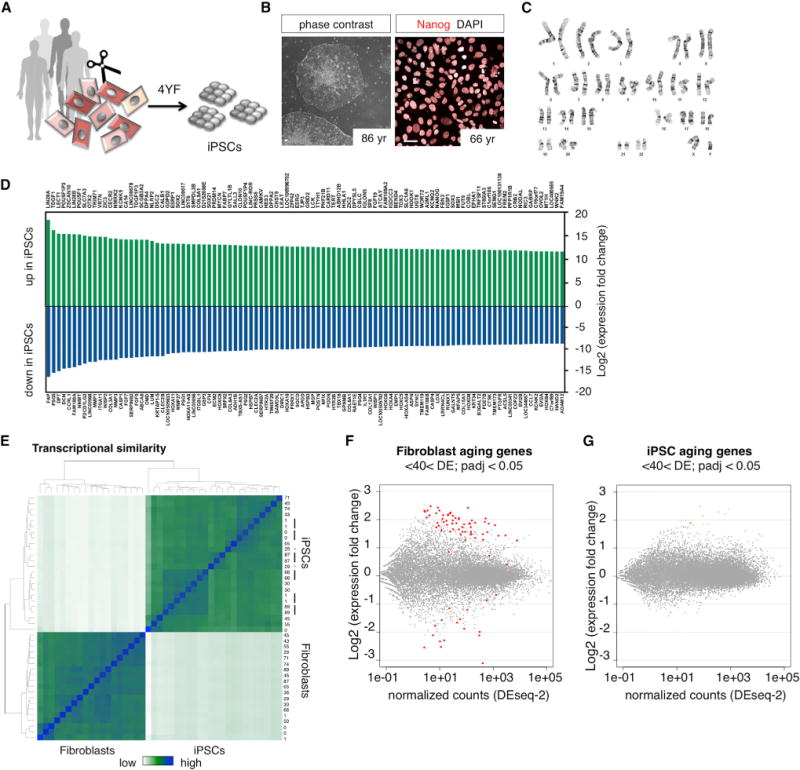
(A) iPSC reprogramming of young and old donor fibroblasts using the four Yamanaka-factors (4YF).
(B) Phase contrast images and immunocytochemical characterization of human iPSCs stained with Nanog. The scale bars represent 20 μm.
(C) Representative image for karyotype analysis of iPSC clones with G-banding (all included lines tested normal).
(D) Differentially expressed genes between fibroblasts and iPSCs. The top 100 up and downregulated genes are shown (extended data in Table S2).
(E) Heatmap showing hierarchical clustering of transcriptome similarity of fibroblasts and derived iPSCs. The black bars indicate multiple iPSC clones from the same donor that cluster together, and the * indicates lines from the same donor that do not cluster together.
(F and G) MA plot shows differential expression between young (<40 year) and old (>40 year) fibroblasts and iPSCs (one clone per donor used for analysis). The colored dots indicate significant aging genes (padj < 0.05) (extended data in Table S3).
Direct iN Conversion Yields Functional iNs and Is Equally Efficient for Young- and Old-Derived Fibroblasts
To generate neurons directly from fibroblasts, we optimized an Ascl1/Ngn2 (AN)-based and small molecule-enhanced iN protocol (Figure 2A) (Ladewig et al., 2012; Liu et al., 2013). Fibroblast identity was verified and the cells were lentivirally transduced to express rtTA and a 2A-peptide-linked transcript coding for Ngn2 and Ascl1, resulting in transgenic, but silent and expandable, AN fibroblasts (Figures 2B and S1). For iN conversion, cells were transferred to conversion media containing small molecular inhibitors for TGFβ/SMAD and GSK3β signaling as well as enhancers of intracellular cyclic AMP (cAMP) (Figure 2B). Under these conditions, fibroblasts underwent marked morphological changes, with typical neuronal morphologies appearing around week 2 of conversion and becoming more pronounced after 3 to 6 weeks. Neuronal cells stained positive for βIII-tubulin, phosphorylated human Tau (hTau), MAP2ab, and the mature neuronal marker NeuN, and most cells showed punctate staining for the glutamateric synaptic marker vGlut1 (Figure 2C). Non-neuronal cells showed typical fibroblast morphologies and Vimentin expression; cell division was not involved in the iN process (Figure S1). Following gentle relocation onto a layer of astrocytes during week 4 of conversion, iNs displayed pronounced human synapsin-I promoter-driven red fluorescent protein (RFP) fluorescence (LV-hSyn∷RFP), mature neuronal morphologies, and punctate staining of synapsin-I at the intersections of neurites (Figure 2D). They also showed both evoked and spontaneous action potential firing (Figure 2E). We next applied this iN paradigm to our fibroblast cohort and found that cells from all ages efficiently converted into iNs that showed solid neuronal marker expression within 3 weeks (Figure 2F). Quantification revealed that, independent of donor age, typically over 50% of all DAPI-positive cells were positive for βIII-tubulin and hTau, around 40% expressed NeuN, and over 30% were consistently positive for MAP2ab (Figures 2G and S1). Equally, iNs generated from all tested donors (n = 13) displayed Na+/K+ channel-mediated inward/outward currents as well as multiple evoked action potentials with no apparent differences between iNs derived from young and old donors’ fibroblasts (Figure 2H). Quantification of neuronal subtype markers revealed that a consistent majority of the neurons adopted a glutamatergic fate and usually 15%–20% of the cells were gamma aminobutyric acid (GABA)-positive, whereas dopaminergic or serotoninergic markers were only sporadically observed (Figures 2I and S1). Using this iN protocol, functional iNs can be efficiently generated from fibroblasts of all ages with no apparent differences in their basic cellular and functional properties.
Figure 2. Direct Conversion of Young and Old Human Fibroblasts into Functional iNs Is Efficient Regardless of Donor Age.
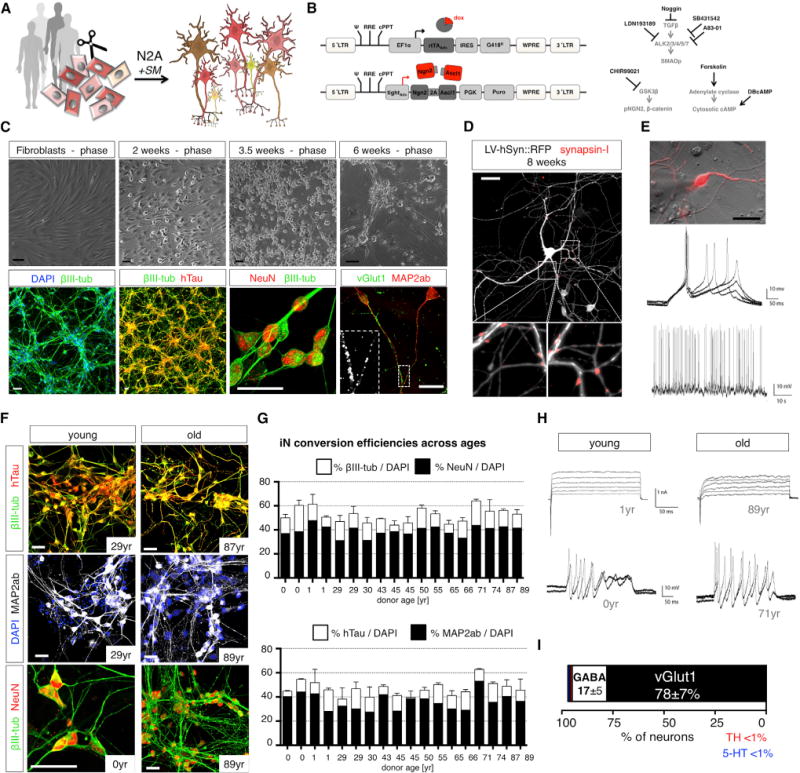
(A) Direct iN conversion of young and old donor fibroblasts using Ngn2-2A-Ascl1 (N2A) and small molecular enhancers (SM).
(B) Schematic of lentiviral system for inducible overexpression of N2A and SM for iN conversion.
(C) Phase contrast images of progressively converting fibroblasts, and immunofluorescence images of 6-week converted fibroblasts stained with βIII-tubulin, hTau, NeuN, Map2ab, and vGlut1.
(D) iNs co-cultured on astrocytes labeled with LV-hSyn∷RFP and stained for synapsin-I following 8 weeks of conversion.
(E) Electrophysiological characterization of iNs shows multiple evoked and spontaneous action potentials.
(F) Immunocytochemical characterization of iN from young and old donors after 3 weeks of conversion. All of the scale bars represent 20 μM.
(G) Quantification of neuronal yields per DAPI from 18 donors following 3 weeks of conversion. The bar graphs show mean + SEM.
(H) Electrophysiological characterization of young- and old-derived iNs shows Na+/K+ channel-mediated inward/outward currents in response to depolarizing voltage steps (upper) and multiple evoked action potentials (lower) without apparent differences between young and old (n = 13 donors).
(I) Quantification of neuronal subtype markers based on immunocytochemical analysis (extended data in Figure S2).
Also see related Figure S1.
Fluorescence-Activated Cell Sorting-Based Purification of PSA-NCAM-Positive iNs for Whole Transcriptome RNA-Seq
To analyze the transcriptome of iNs via RNA-seq, neuronal RNA needs to be pure, with minimal fibroblast contamination that might confound analysis regarding cell aging. As live iNs express PSA-NCAM on the cell surface, we established a fluorescence-activated cell sorting (FACS)-based protocol for purification of neurons (Figures 3A and 3B). Following sorting and plating on astrocytes, over 90% of human cells expressed βIII-tubulin, with over 95% of them also being hTau-positive (Figure 3C). Following prolonged culture, almost all human cells showed mature neuronal morphologies and marker expression (Figure 3D). We next performed RNA-seq analyses over the time course of conversion and observed dramatic gradual changes in gene expression (Figure 3E). Neuronal genes such as Dcx, NeuroD2, and Mapt, and voltage-gated Na+ and K+ channels, synaptic proteins, and neurotransmitter receptors were strongly upregulated, whereas fibroblast genes such as dermokine (Dmkn), collagen, keratin, and cell-cycle genes such as Ccna2, Ccnb2, Nuf2, and Anln were dramatically downregulated (Figure S2; Table S4). Neighboring time points clustered together, indicating a gradual process of cell type conversion. Gene ontology (GO) term analysis of the 200 most strongly increased genes revealed highly significant enrichment of interacting factors that control neuron generation and function (Figures 3F and S2; Table S5). As expected, FACS-puri fied neurons showed marked enrichment of neuronal genes such as neuronal Ca2+ and K+ channels, glutamate receptors, synaptic proteins, and neuronal cytoskeletal markers as compared to their unsorted counterparts (Figures 3E and 3G), indicating efficient purification of iNs from remaining fibroblasts for RNA-seq.
Figure 3. FACS-Based Purification and Transcriptome Analysis of iNs.
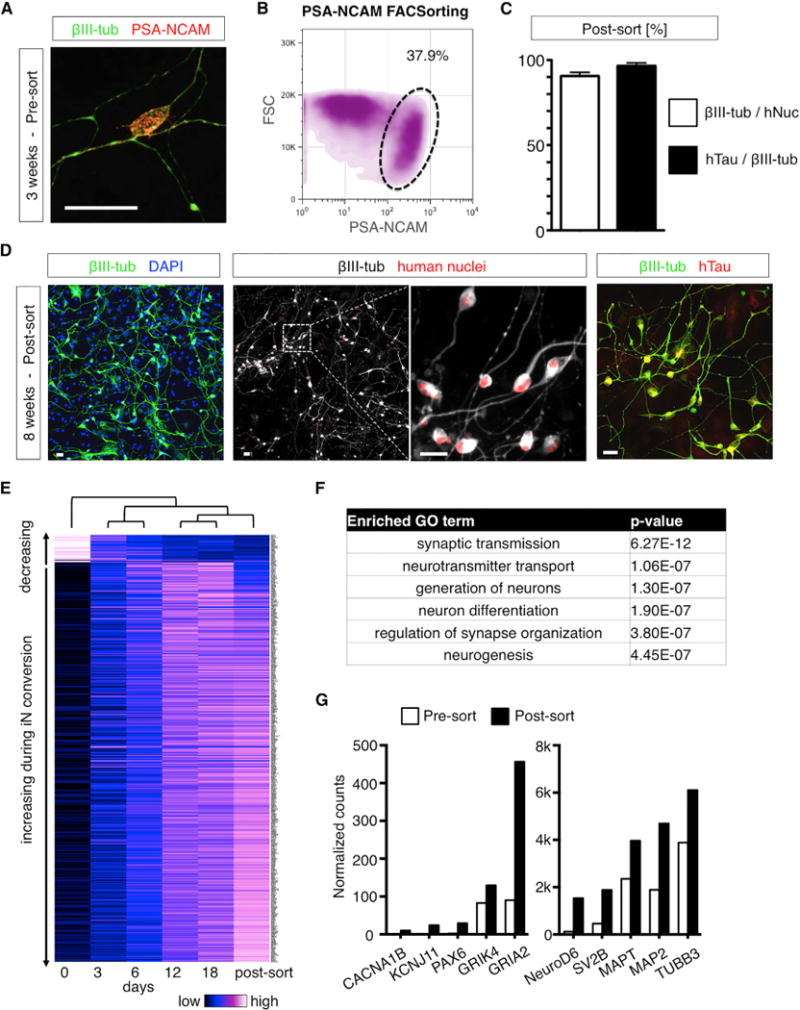
(A) Immunocytochemical staining of iNs with PSA-NCAM shows punctate staining on the surface of neurons.
(B) Density plot of PSA-NCAM-stained iN cultures during FACSorting.
(C) Quantification of βIII-tubulin and hTau-positive neurons over human nuclei (hNuc) 1 week after FACS purification and replating. The bar graphs showmean + SEM.
(D) Immunocytochemical analysis of 3-week-old sorted neurons following a total of 8 weeks of conversion stained with βIII-tubulin, hTau, and human nuclei (hNuc). The scale bars represent 20 μm.
(E) Heatmap of time course RNA-seq expression analysis of progressively converting iN cells and FACS-purified iNs (extended data in Table S4).
(F) GO term analysis of the 200 most strongly (fold change) upregulated genes following 18 days of iN conversion (extended data in Table S5).
(G)Gene expressionanalysis before andafterPSA-NCAMFACSpurification. The bars show normalized counts of day 18bulk (white) and day 18sorted (black) samples.
Also see related Figure S2.
iNs from Young and Old Donors Show Specific Transcriptional Aging Signatures
To explore our hypothesis that fibroblast-derived iNs retain information about their donor’s age, we performed RNA-seq analysis of iNs FACS-purified at 3 weeks of conversion (n = 22 samples from 18 donors). Unsupervised hierarchical clustering based on overall gene expression showed high similarity between all iN samples, and independently repeated iN conversions from the same fibroblasts reliably clustered together (Figure 4A). Strikingly, differential expression analysis between young (<40 years) and old (>40 years) fibroblasts and iNs revealed profound age-dependent gene expression in the iNs, with 202 genes significantly differentially expressed (FDR < 0.05; Figures 4B and 4C). In addition, linear correlation of normalized gene expression values with donor age confirmed that both fibroblasts and iNs showed a subset of genes progressively increasing or decreasing with age (Figure S3). Surprisingly, only seven of the differentially expressed genes overlapped between fibroblasts and iNs, as both cell types appeared to show cell-type-specific, age-dependent expression profiles (Figure 4D; Table S3). GO term and KEGG pathway analysis revealed that, in fibroblasts, genes involved in wound healing, stress response, and Ca2+ signaling—which are categories that have been previously been implicated in skin aging—were significantly altered (Figure 4E; Table S6) (Adler et al., 2007; Fraser et al., 2005; Lu et al., 2004; Murphy, 2006; Peterson et al., 1985; Rando and Chang, 2012). Enriched gene categories in aging iNs related to functional neuronal categories such as Ca2+ homeostasis, neuron projection, and regulation of synaptic plasticity (Figure 4F). Most interestingly, several genes from these categories, namely calcitonin (Calca), the cation channel Trpc6, the endothelin receptor Ednrb, and the catenin Ctnna1 also relate to the GO term “aging” (GO:0007568), which was also enriched in our aging iN system (Table S6).
Figure 4. Age-Dependent Gene Expression in Human Fibroblasts and iNs.
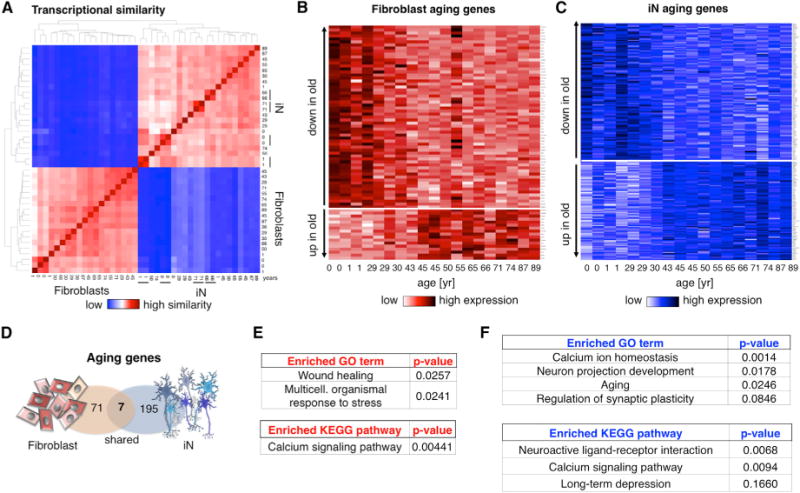
(A) Heatmap showing hierarchical clustering of transcriptome similarity of fibroblasts and derived iNs. The black bars indicate multiple independent conversions from the same donor.
(B and C) Heatmaps of significantly differentially expressed genes between young (<40 year) and old (>40 year) FACS-purified fibroblasts (red; 78 genes) and iNs (blue; 202 genes) with an FDR-adjusted p value < 0.05 (extended data in Table S3).
(D) Schematic Venn diagram showing the overlap of fibroblast and iN aging genes.
(E and F) GO term and KEGG pathway analysis of the 78 fibroblast and 202 iN aging genes (extended data in Table S6).
Also see related Figure S3.
Expression of the Nuclear Pore-Associated Transport Protein RanBP17 during Aging
To further explore aging-related changes in gene expression, we performed RNA-seq analysis of 14 human prefrontal cortex samples from 0 up to 89 years of age and analyzed gene expression. First, we noticed that cortical samples consisted of RNA from multiple cell types as they contain large numbers of glial, neuronal, endothelial, and hematopoietic cell-derived transcripts, with neuron-derived RNA being only a minor fraction (Figure S4). We analyzed for differentially expressed aging genes and detected 49 highly significant (padj < 0.05) genes that overlapped between aging iNs and cortex, an overlap 7-fold higher than between iNs and fibroblasts. Further, cortical aging genes were enriched for several similar functional GO/KEGG categories like aging iNs, but also showed enrichment for genes involved in “learning and memory”, “Alzheimer’s disease”, “intracellular transport”, and other categories (Figure S4). Interestingly, we detected only three significant aging genes, LAMA3, PCDH10, and RANBP17, to be shared between aging fibroblasts, iNs, and the brain (Figure 5A). We reasoned that potential master regulators of aging might reveal themselves in this category. We became particularly interested in the nuclear pore-associated transport receptor RanBP17, which we found significantly downregulated in the elderly (Figure 5B). For validation of the RNA-seq data for RanBP17, we performed quantitative (q) PCR and western blotting, which both confirmed progressive reduction of RanBP17 with age (Figures 5B and S4). RanBP17 belongs to the importin-β family, which are involved in the transport of nuclear localization signal (NLS)-containing cargo proteins through the nuclear pore complex by binding FG-repeat-containing nucleoporins (Koch et al., 2000; Lee et al., 2010). In line with this, RanBP17 localized as a rim around the nucleus in both fibroblasts and neurons (Figure 5C). To further explore protein levels of RanBP17 in the aging human brain, we paraffin sectioned and immunostained ten of the cortex samples. All of them showed clear staining for βIII-tubulin in cortical neurons in the different layers (Figures 5D and 5E). Immunostaining for RanBP17 showed strong expression in layer II and III neurons of the young adult cortex, whereas it was often barely detectable above background in cortical neurons of old brains (Figures 5E and 5F). Quantification of neuronal RanBP17 immunofluorescence over background revealed significantly lower levels of RanBP17 in old brains compared to young (Figure 5G), and RanBP17 levels as determined by western blot analysis inversely correlated with age (Figures 5H and S4), thus confirming the decreasing expression RanBP17 protein with age in vivo. In addition, several independent “:omics” studies showed RanBP17 consistently among the very top age-dependent genes in 519 samples of glioblastoma multiforme (Broad Institute TCGA Genome Data Analysis Center, 2014), 480 samples of kidney renal clear cell carcinoma (Broad Institute TCGA Genome Data Analysis Center, 2013a), and 226 samples of thyroid adenocarcinoma (Broad Institute TCGA Genome Data Analysis Center, 2013b; summarized in Figure S5).
Figure 5. Expression of RanBP17 in the Aging Human Prefrontal Cortex.
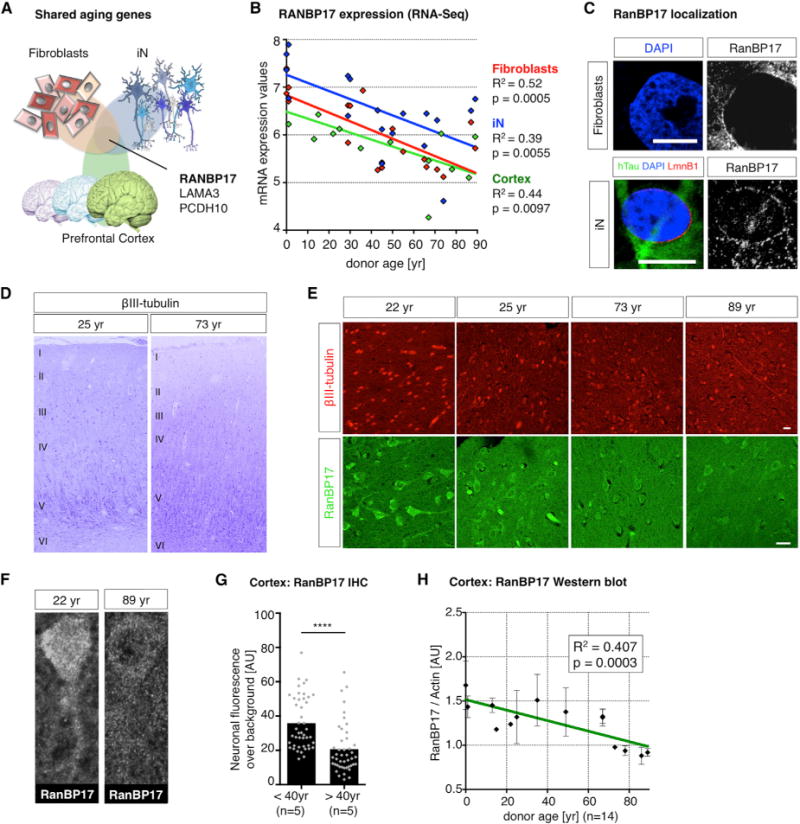
(A) Schematic Venn diagram showing the overlap of postmortem cortex, fibroblast, and iN aging genes.
(B) Correlation of RanBP17 expression with age in fibroblasts (red), purified iNs (blue), and RNA extracted from the human prefrontal cortex (green, n = 14 donors; Table S1). The data points indicate single donors, and the lines depict linear regression function (expression values: VST normalized counts).
(C) Immunocytochemical analysis of subcellular RanBP17 localization in fibroblasts (upper) and iNs (lower) co-stained with hTau and Lamin B1. The scale bars represent 10 μm.
(D) Formalin-fixed human cortices were embedded in paraffin and sectioned for immunohistochemistry to identify cortical neurons and layers. The βIII-tubulin-positive cortical neurons were brightly stained in cortices from young adult and old brains (look-up-table, LUT, colored; numbers indicate cortical layer structure).
(E) Immunohistochemical analysis of young adult and old brains for βIII-tubulin and age-dependent decrease in RanBP17 in neurons. The scale bar represents 20 μm.
(F) Representative images of RanBP17-stained cortical neurons show loss of RanBP17 from the neuronal somata and nuclei in old brains.
(G) Quantification of neuronal fluorescence intensity comparing young and old brains (n = 10 donors). The bar graph shows means and dots individual cells (significance values: ****p < 0.001).
(H) Quantification of western blot analysis for RanB17 protein over actin. The graph shows a correlation of cortical RanBP17 over actin with age. The dots indicate values of single donors ± SEM, and the line depicts linear regression.
Also see related Figures S4 and S5.
Impaired Nucleocytoplasmic Compartmentalization in Old Fibroblasts and Age-Equivalent Mature iNs
Based on these data, we reasoned that the decrease in RanBP17 might indicate a possible functional impairment of the nuclear pore in the aged. As the most prominent function of the nuclear pore is to maintain proper nucleocytoplasmic compartmentalization (NCC), we established a reporter assay to measure NCC using lentiviral delivery of a dual reporter system (2Gi2R). This reporter consists of a fused double-GFP containing an NES sequence (2G:NES) and an internal ribosome entry site (IRES)-linked double-RFP containing an NLS sequence (2R:NLS) (Figure 6A). Increased false localization of green fluorescence in the nucleus along with a simultaneous loss of nuclear red fluorescence (GFPnuc/RFPnuc), thus represents a measure for loss of NCC (Figure 6A). To test whether there is a detectable difference in NCC between cell populations, we measured GFPnuc/RFPnuc in nuclei of fibroblasts using automated region of interest (ROI) selection based on binarized DAPI images from confocal sections (Figure 6B). To first test the applicability of our NCC assay, we overexpressed the HIV-1 protein Vpr in young fibroblasts since Vpr is suspected of altering the integrity of the nuclear envelope (Blömer et al., 1997; Guenzel et al., 2014). Vpr overexpression resulted in a marked increase in GFPnuc/RFPnuc ratios (Figure S6). Treatment of cells with the nuclear export inhibitor Leptomycin B resulted in a progressive increase in green fluorescence in the nucleus over time, paralleled by increasing GFPnuc/RFPnuc ratios, which were solely attributed to increasing mislocalization of 2G:NES over time (Figure S6). Most interestingly, fibroblasts derived from old donors showed relatively more GFP in the nucleus and more RFP in the cytoplasm (Figure 6C). Quantification revealed a significant impairment of NCC in old compared to young and middle-aged donor-derived cells (Figure 6D). Loss of NCC significantly correlated with donor age as well as with RanBP17 expression levels (Figures 6E and S6). Thus, using 2Gi2R as a sensitive NCC reporter, we were able to identify an age-dependent impairment of NCC in old donor-derived human fibroblasts. Further, as 3-week-old iNs showed aging transcriptome signatures, including decreased RanBP17 levels in the old, we asked whether mature and functional iNs from old donors possessed altered NCC. We transduced iNs with the 2Gi2R reporter and let them mature on astrocytes until week 6 (Figure 6F). Mature neurons showed complex morphologies and solid expression of the NCC reporter proteins (Figure 6G). Strikingly, iNs from the oldest donors showed significantly increased GFPnuc/RFPnuc ratios compared to both middle-aged (29 year–50 year) and young donor-derived iNs (Figure 6H). Strongly affected neurons showed obvious compartmentalization defects; the extent of the detected loss of neuronal NCC significantly correlated with donor age and RanBP17 expression (Figure S6). These data show that, in addition to transcriptomic changes after 3 weeks of conversion, mature human iNs show functional NCC impairment, thus implicating NCC as an important functional signature of old human neurons. To confirm age-related NCC impairment, we next applied fluorescence loss in photobleaching (FLIP) as a second assay to measure compartmentalization to our aging fibroblasts. For this, we performed live-cell analysis of a shuttling 2xGFP:NLS:NES probe in young and old fibroblasts. FLIP revealed significantly longer half-life times in old donor-derived cells, indicating less efficient shuttling of the 2xGFP:NLS:NES probe (Figure S6). This observation represents an additional experimental measure supporting observed NCC defects as detected using the 2Gi2R assay.
Figure 6. NCC in Young and Old Fibroblasts and Age-Equivalent iNs.
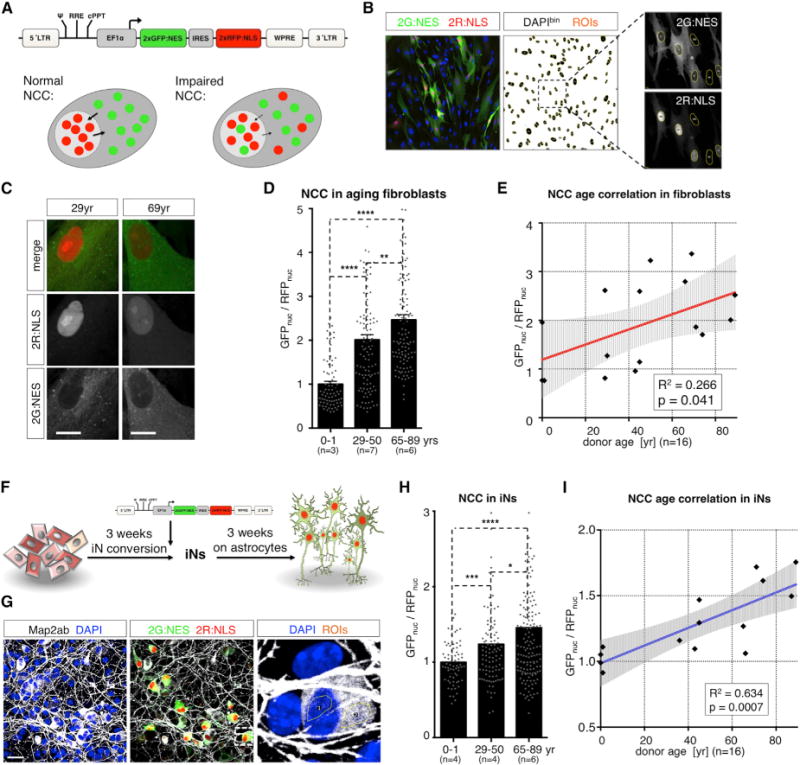
(A) 2Gi2R: lentiviral vector for expression of the IRES-linked NCC reporters 2xGFP:NES and 2xRFP:NLS. The experimental rationale for NCC measurement is that increasing GFPnuc/RFPnuc ratios indicate NCC defects and RFPnuc/RFPcyt and GFPnuc/GFPcyt ratios divide into import- or export-related phenotypes, respectively.
(B) ROI selection of confocal sections for the measurement of nuclear GFP and RFP in cultured fibroblasts.
(C) Representative florescence images of 2Gi2R reporter fibroblasts from a young adult (29 year) and old (69 year) donor. The scale bars represent 20 μm.
(D)GFPnuc/RFPnuc, ratios in aging fibroblasts (n = 16 donors; normalized to young group). The bar graphs show mean + SEM and the dots indicate single cells (significance values: *p < 0.05; **p < 0.01; ***p < 0.005; and ****p < 0.001).
(E) Correlation of NCC values of individual donors with age (black dots). The red line depicts linear regression fit, and the shaded area is the 95% confidence interval.
(F) Experimental design: 3-week-old iNs were transduced with NCC reporter virus and relocated on a layer of astrocytes for maturation.
(G) NCC was assessed by measuring neuronal GFPnuc/RFPnuc ratios in confocal sections.
(H) GFPnuc/RFPnuc ratios in aging iNs (n = 14 donors). The data is normalized to the young group. The bar graphs show mean, and the dots indicate single cells (significance values: *p < 0.05; **p < 0.01; ***p < 0.005; and ****p < 0.001).
(I) Correlation of NCC values of individual donors with age (black dots). The blue line depicts linear regression fit, and the shaded area is the 95% confidence interval.
Also see related Figure S6.
RanBP17 Decrease Causes Loss of NCC in Young Cells, and iPSC Rejuvenation Restores NCC in Old Cells
To examine whether the age-dependent loss of RanBP17 plays an active role in the age-dependent loss of NCC, we generated lentiviruses carrying short hairpin (sh)RNAs against RanBP17 (iR#1 and iR#2; Figure 7A). Efficient knock down of RanBP17 was achieved with both constructs on protein and mRNA levels (Figures 7B and 7C), but cells transduced with iR#2 showed a stronger mRNA decrease and signs of toxicity after 5 days (data not shown). Moderate knock down using iR#1 resulted in a marked increase in GFPnuc/RFPnuc in different cell lines, suggesting a causative role for RanBP17 in the loss of NCC in old cells (Figure 7D). Interestingly, knock down of RanBP17 in young fibroblasts (1 year) changed the expression of 68% of the 78 identified fibroblast aging genes in the same direction, as was observed in progressive aging, further strengthening the idea that RanBP17 plays an upstream role for the cellular aging phenotypes (Figure 7E). Next, since iPSCs from old donors appeared to rejuvenate their transcriptome and restored RanBP17 expression (Figure 7F), we wondered whether iPSC reprogramming might reconstitute proper NCC in old donor-derived cells. To assess NCC in iPSCs that normally grow in dense colonies, we transferred 11 reporter iPSC lines to the PluriPro monolayer culture system and measured NCC (Figure 7G). As anticipated, we found that GFPnuc/RFPnuc ratios from young, middle-aged, and old donors were indeed very similar with regard to compartmentalization and no detectable age-dependent impairment was detectable (Figures 7H and 7I). These data indi cate that loss of RanBP17 is sufficient to impair NCC, and that iPSC reprogramming functionally restores NCC in old-derived cells.
Figure 7. RanBP17 Decrease Causes Loss of NCC and iPSC Reprogramming Restores NCC in Old Donor-Derived Cells.
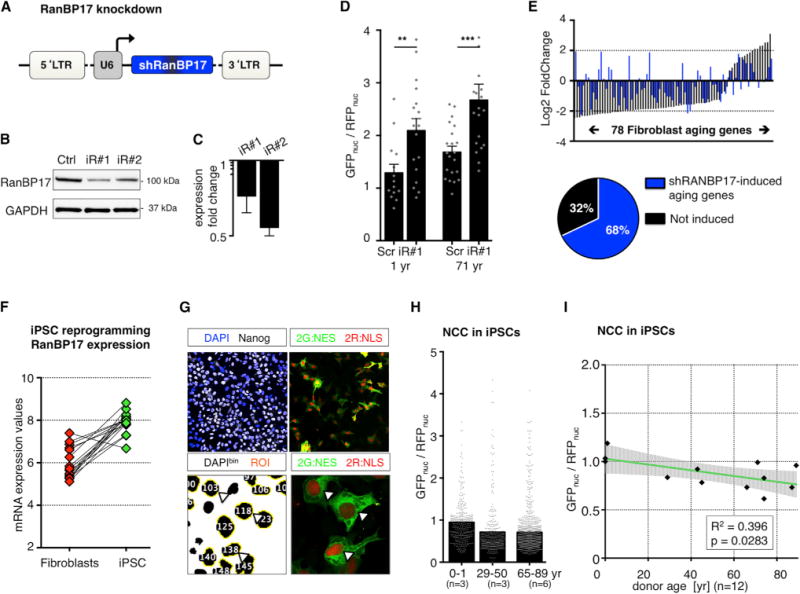
(A) Lentiviral vector for RanBP17 knockdown (shRNAs: iR#1 and iR#2).
(B) Western immunoblotting for RanBP17 in cells expressing iR#1, iR#2, or scrambled control shRNA.
(C) qPCR analysis for iR#1 and iR#2 compared to scrambled control.
(D) GFPnuc/RFPnuc ratios in young (1 year, left) and old (71 year, right) RanBP17 knockdown cells. The bar graphs show mean, and the dots indicate single cells (significance values: *p < 0.05; **p < 0.01; ***p < 0.005, and ****p < 0.001).
(E) Fibroblast aging genes in response to RanBP17 knockdown. The graph shows expression changes (old versus young) of the 78 fibroblast aging genes in response to aging (black bars) and in response to RanBP17 knockdown in young fibroblasts (1 year). The RanBP17 knockdown caused 68% of the aging genes to change in the same direction as in aging.
(F) RanBP17 mRNA levels in fibroblasts (red) and corresponding iPSCs (green; VST normalized counts).
(G) Monolayer PluriPro culture of iPSCs for the measurement of NCC.
(H) GFPnuc/RFPnuc ratios in iPSCs reprogrammed from young and old donors (n = 12 donors). The bars represent means, and the dots represent individual cells.
(I) Correlation of NCC with donor age in iPSCs. The black dots represent individual donor iPSCs. The green line represents the linear regression fit, and the shaded area is the 95% confidence interval.
Also see related Figure S7.
iPSC-Derived iNs Remain Transcriptionally Rejuvenated
To test if potentially concealed transcriptomic aging signatures reappeared following neuronal differentiation of iPSCs, we directly converted iPSCs from a subset of donors into neurons using the same N2A-based approach, PSA-NCAM-based FACS purification, and RNA-seq (Figure S7). As expected, iPSC-iNs showed very little significant transcriptomic aging signs. Differential expression analysis showed a 19-fold lower number of age-dependent genes in iPSC-iNs than in the corresponding fibroblast-derived iNs from the exact same subset of donors (12 versus 227 genes, respectively; Figure S7). None of these genes overlapped with any of the fibroblasts or iN aging genes and only one gene overlapped with the cortex aging genes. These data suggest that, following differentiation, iPSC derivatives, at least the iNs, remain in a largely rejuvenated state.
DISCUSSION
Modeling Aging with Human iNs In Vitro
The advent of iPSC technology has allowed for the investigation of human diseases directly in the affected human cell type (Marchetto et al., 2011; Mertens et al., 2013a). While this approach appears highly useful for the study of neurodegenerative disorders in patient-specific iPSC derivatives, age-related physiological changes might be one of the most important factors in the development of disease-associated pathologies. Fibroblasts from centenarians can be reprogrammed into iPSCs with telomere size, oxidative damage, and mitochondrial metabolism indistinguishable from embryonic stem cells (Lapasset et al., 2011; Suhr et al., 2010; Takahashi et al., 2007). Further, mesenchymal stem cells reprogrammed into iPSCs and subsequently redifferentiated toward mesenchymal stem cells showed that age-related DNA methylation patterns were erased during reprogramming. In line with our transcriptomic observations in iPSC-derived iNs, iPSC-derived mesenchymal stem cell (MSCs) remained rejuvenated on the methylome level, suggesting that aging signatures might be constantly erased rather than temporarily concealed in iPSCs (Frobel et al., 2014). However, the question of whether the cell-rejuvenating aspect and the dedifferentiation aspect of iPSC reprogramming can be uncoupled remains relevant. Interestingly, some rejuvenating aspects of the four Yamanaka factors appear to impact early during reprogramming, and aspects of rejuvenation might be possible without full dedifferentiation, as senescence-associated nucleocytoplasmic mobility of the epigenetic modifier HP1β could be restored in old cells within days (Manukyan and Singh, 2014). Taking all this into account, it can be expected that neurons differentiated from, for example, Alzheimer’s or Parkinson’s patient-specific iPSCs are phenotypically young. Consistently, attempts to model age-related neurodegenerative states in iPSC-derived neurons have made use of additional stressors such as excitotoxicity (Koch et al., 2011), reactive oxygen species (Mertens et al., 2013a; Nguyen et al., 2011; Reinhardt et al., 2013), or overexpression of disease- or aging-related mutant proteins to accelerate pathological processes in these otherwise rejuvenated neuronal cells (Koch et al., 2012; Miller et al., 2013). An issue with such approaches is that selected stressors, unlike intrinsic cell aging, usually act exogenously on the cells and mimic only limited, if any, aspects of cellular aging. In contrast, iN conversion represents a technology that circumvents the early embryonic and pre-germline state of iPSCs, and we demonstrated that direct conversion yields authentic human neurons that reflect important aspects of cellular age. In addition, the fact that iN goes directly from one fibroblast to one neuron and does not involve or require cell divisions that might dilute or induce repair of macromolecular damage might contribute to the fact that aging phenotypes present in cultured primary fibroblasts become translated to a neuronal context during iN (Hennekam, 2006; Liu et al., 2013; Toyama and Hetzer, 2013). Consistent with earlier transcriptome studies on the aging brain, we found gene categories differentially expressed in aging iNs that point to age-dependent differential regulation of synaptic function, projection development, and Ca2+ homeostasis, which in turn impacts functions such as synaptic plasticity and projection (Burke and Barnes, 2006; Fraser et al., 2005; Lu et al., 2004). Interestingly, while only less than 4% of the iN aging genes overlapped with the fibroblast aging genes, 7-fold more overlapped with the postmortem cortex, suggesting the presence of an aging transcriptome signature relevant for the aging brain. Our findings also raise the question of how cellular age is encoded during the fibroblasts-to-neurons transition. A possible explanation, in addition to preserved macromolecular damage, is that only a small number of genes function as master regulators of aging; if perturbed, they impair important downstream processes that eventually unfold as phenotypical aging. RanBP17 emerged as being one of these genes identified in this study.
NCC and Aging
Progeria syndromes such as Hutchinson-Gilford progeria syndrome (HGPS) have provided unique opportunities to study cellular processes in the context of accelerated aging (Gordon et al., 2014). The mutations in the nuclear envelope protein LaminA/C (progerin) result in nuclear stiffness and severely altered nuclear architecture, leading to a body-wide phenotype of premature aging (Hennekam, 2006; Scaffidi and Misteli, 2006). While HGPS pathology involves DNA damage and stress-response pathways, the cellular mechanisms that are primarily impaired by progerin, and how they might mimic old cellular age in various tissues remain unknown. Apart from genetically encoded aging, growing evidence has drawn attention to the nuclear pore complex as a primary target for aging in organisms with long lifespans. Nucleoporins have been identified as extremely long-lived low-turnover proteins with very limited capacity for renewal and repair, especially in postmitotic cells (Savas et al., 2012; Toyama et al., 2013). Thus, one may speculate that heavily damaged nuclear pore complexes would likely impair the function of their interaction partners. By studying normal progressive human aging in fibroblasts, iNs, and the cortex, we have shown that RanBP17 is significantly downregulated with age in several human systems. Strikingly, our results have been further confirmed by several independent GDAC analyses based on a combined 1,200+ samples that also found RanBP17 to be one of the most significantly age-regulated genes in human glioblastomas, kidney, and thyroid carcinomas (summarized in Figure S6). Furthermore, an analysis detected RanBP17 among 14 genes that showed both significant age-correlation and age-dependent differential expression in three independent gene expression platforms (Bozdag et al., 2013). However, the questions of how progressive aging might affect RanBP17 expression, as well as the exact mechanisms of how RanBP17 loss disturbs NCC remain to be explored. While not directly assessed here, in view of the accelerated aging phenotype observed in HGPS, it appears conceivable that a progerin-impaired nuclear envelope structure might allow nuclear leakiness, which could lead to phenotypical cell aging, including DNA damage and deficient response pathways (Musich and Zou, 2009). Most likely, nuclear malformations would also affect the envelope-embedded nuclear pore complex and its interaction partners, which are the main gatekeepers of compartmentalization (Capelson and Hetzer, 2009). Our results would thus favor a model where NCC becomes impaired in old cells as a consequence of altered gene expression and altered nuclear pore function (D’Angelo et al., 2009).
The identification of NCC impairment in aging holds serious implications for nuclear integrity related to cell aging. For example, genetic polymorphisms within the FOXO3 gene have been associated with human longevity (Flachsbart et al., 2009; Willcox et al., 2008). Interestingly, FOXO3a protein mediates oxidative defense and cell survival, and its activity is regulated by nucleocytoplasmic shuttling through its NLS and NES sequences; thus it might represent a potential downstream effector of impaired NCC in old cells (Lehtinen et al., 2006). Similarly, the anti-neurogenic repressor element 1-silencing transcription factor (REST) is an important neuroprotective factor in the old brain that, when lost from the nucleus, might permit neurodegeneration (Lu et al., 2014). Furthermore, injury-induced and Ca2+-mediated HDAC5 export from the nucleus is important for the regeneration of peripheral nerves (Cho et al., 2013), and impor-tin-β itself has been shown to play a role in transducing NLS-containing damage signals back from the injured axon to the nucleus (Hanz et al., 2003). Together with our data, all these findings point to NCC as potentially a key process in the age-related loss of neuronal performance and stress resistance in the old brain. Given that RanBP17 decrease appears in different tissues, NCC defects may also be present in other aging cell types, contributing to a range of age-related phenotypes in different organs. However, the functional relationship, if any, of age-related NCC impairment with potential downstream effectors, including the ones mentioned above, needs to be determined.
We here provide a set of data showing that, unlike in rejuvenated iPSCs and their derivatives, human aging can be modeled through direct cell type conversion into iNs that preserve transcriptomic features of their donors age. Based on transcriptome profiling of aging fibroblasts, derived iNs, and the aging human cortex, we identified the nuclear pore-associated transport protein RanBP17 as a significant factor in cell aging that goes along with an impairment of protein localization between the nucleus and cytoplasm in different human somatic cell types, including neurons.
EXPERIMENTAL PROCEDURES
Direct Conversion of Adult Human Fibroblasts into iNs
Primary human dermal fibroblasts were obtained from the Coriell Institute Cell Repository, the University Hospital in Erlangen, DNA and Cell Bank of ICM in Paris, and ATCC (Table S1). Protocols were previously approved by the Salk Institute Institutional Review Board and informed consent was obtained from all subjects. Cells were cultured in DMEM containing 15% tetracycline-free fetal bovine serum and 0.1% NEAA (Life Technologies), transduced with lentiviral particles for EtO (Ladewig et al., 2012; Mertens et al., 2013a) and XTP-Ngn2:2A:Ascl1 (N2A), and expanded in the presence of G418 (200 μg/ml; Life Technologies) and puromycin (1 μg/ml; Sigma Aldrich). For iN conversion, fibroblasts were pooled into high densities and after 24 hr the medium was changed to neuron conversion (NC) medium based on DMEM:F12/Neurobasal (1:1) for 3 weeks. NC contains the following supplements: N2 supplement, B27 supplement (both 13; GIBCO), doxycycline (2 μg/ml; Sigma Aldrich), Laminin (1 μg/ml; Life Technologies), dibutyryl cAMP (500 μg/ml; Sigma Aldrich), human recombinant Noggin (150 ng/ml; Preprotech), LDN-193189 (0.5 μM; Cayman Chemicals) and A83-1 (0.5 μM; Stemgent), CHIR99021 (3 μM; LC Laboratories), Forskolin (5 μM; LC Laboratories), and SB-431542 (10 μM; Cayman Chemicals). Medium was changed every third day. For further maturation, iNs were switched to DMEM:F12/Neurobasal-based neural maturation media (NM) containing N2, B27, GDNF, BDNF (both 20 ng/ml; R&D), dibutyryl cAMP (500 μg/ml; Sigma Aldrich), doxycycline (2 μg/ml; Sigma-Aldrich), and laminin (1 μg/ml; Life Technologies). For maturation on astrocyes, iNs were dislodged during week 4 using TrypLE and replated on a feeder layer of mouse astrocytes and cultured in NM media containing 1% knockout serum replacement (KOSR) (Life Technologies).
FACS Purification of iNs and RNA Isolation
Following 3 weeks of iN conversion, iNs were detached using TrypLE and stained for PSA-NCAM (1:100) for 45 min at 4°C in sorting buffer (250 mM myo-inositol and 5 μg/ml polyvinyl alcohol, PVA, in PBS) containing 1% KOSR. Cells were washed and stained with Alexa-747 anti mouse IgM for 30 min at 4°C, resuspended in sorting buffer containing EDTA and DNase, and filtered using a 40 μm cell strainer. The Alexa647-positive population was sorted directly into Trizol-LS, and RNA was isolated according to the manufacturer’s instructions and digested with TURBO DNase (Life Technologies). RNA integrity (RIN) numbers were assessed using the Agilent TapeStation before library preparation.
NCC Assay
Cells were transduced with the LV-XEP-2Gi2R carrying the IRES-linked sequences for 2xGFP:NES and 2xRFP:NLS. We used the HIV-Rev NES (LQLPPLERLTL) and the SV40 Large-T NLS (PKKKRKV). For imaging, cells were transferred to tissue culture-treated ibidi μ-slides coated with astrocytes for iNs, PluriPro matrix (Cell Guidance Systems) for iPSCs, and uncoated for fibroblasts. Cell were fixed with 4% PFA for 20 min at room temperature and mounted in PVA-DABCO (Sigma Aldrich). Confocal images were taken on Zeiss LSM710 or LSM780 series confocal microscope. All data for one NCC experiment were acquired from cells cultured, transduced, and processed in parallel and images were taken on the same microscope with exactly the same settings reused. For analysis, 2 μm confocal sections through the nuclear layer were acquired from three confocal z stacks. Image J software was used to identify nuclear ROIs based on binarized DAPI channels and green and red fluorescence means were measured in the ROIs. Because of the morphological complexity of the astrocyte iN co-cultures, neuronal nuclear ROIs were selected manually. To quantify the integrity of cellular NCC, GFPnuc/RFPnuc were calculated in transgenic cells using Microsoft Excel and further processes in GraphPad Prism 6.
Supplementary Material
Highlights.
Human iPSCs erase aging signatures and hiPSC-derived neurons remain rejuvenated
Directly converted iNs preserve donor age-dependent transcriptomic signatures
Nuclear transport receptor RanBP17 is decreased in aged human cells and iNs
Aged and RanBP17-depleted cells show nucleocytoplasmic compartmentalization defects
Acknowledgments
We thank Ken Diffenderfer and the Salk STEM core facility for technical support with iPSC generation; Caz O’Connor and Conor Fitzpatrick for FACSorting; and Lynne Moore, Neal Nathan, Eric Konkel, T.J. Eames, and Kim McIntyre for technical support. We thank the DNA and Cell Bank of ICM in Paris for fibroblast banking; Leah Boyer, Carol M. Marchetto, Andres Paucar, Alexandra Durr, Olga Corti, and Alexis Brice for their contribution to fibroblast collection; Peter Davies for antibodies; and Mary Lynn Gage for editorial comments. Human prefrontal cortex samples were provided by the NICHD Brain Bank. The study was supported by the G. Harold & Leila Y. Mathers Charitable Foundation, the JPB Foundation, the Leona M. and Harry B. Helmsley Charitable Trust grant #2012-PG-MED002, Annette Merle-Smith, CIRM (TR2-01778), the German Federal Ministry of Education and Research (BMBF, FKZ: 1315874, #01GN0979), the Bavarian State of Ministry of Education, Science and the Arts in the framework ForIPS, and the Glenn Foundation Center For Aging Research.
Footnotes
ACCESSION NUMBERS
RNA-seq data have been deposited at EMBL-EBI ArrayExpress: E-MTAB-3037.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.stem.2015.09.001.
AUTHOR CONTRIBUTIONS
Conception, design, and writing of the manuscript: J.M. and F.H.G. Cell type conversions and cellular and molecular assays: J.M. Bioinformatics analysis: J.M. and A.C.M.P. Electrophysiology: J.Y. Library preparation and sequencing: M.K. Data interpretation, contribution to experimental design, and editing of the manuscript: J.M., A.C.M.P., J.Y., M.K., L.B., S.L., S.M., E.H., H.L., J.R.H., Y.K., J.T.G., T.T., M.H., and F.H.G. Provision of fibroblasts and iPSCs: B.C. and J.W.
References
- Adler AS, Sinha S, Kawahara TLA, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007;21:3244–3257. doi: 10.1101/gad.1588507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B. 1995;57:289–300. [Google Scholar]
- Blömer U, Naldini L, Kafri T, Trono D, Verma IM, Gage FH. Highly efficient and sustained gene transfer in adult neurons with a lentivirus vector. J Virol. 1997;71:6641–6649. doi: 10.1128/jvi.71.9.6641-6649.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdag S, Li A, Riddick G, Kotliarov Y, Baysan M, Iwamoto FM, Cam MC, Kotliarova S, Fine HA. Age-specific signatures of glioblastoma at the genomic, genetic, and epigenetic levels. PLoS ONE. 2013;8:e62982. doi: 10.1371/journal.pone.0062982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center. Correlation between mRNAseq expression and clinical features. Broad Institute of MIT and Harvard; 2013a. http://dx.doi.org/10.7908/C1J67F8Q. [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center. Correlation between gene methylation status and clinical features. Broad Institute of MIT and Harvard; 2013b. http://dx.doi.org/10.7908/C12B8W55. [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center. Correlation between mRNA expression and clinical features. Broad Institute of MIT and Harvard; 2014. http://dx.doi.org/10.7908/C1445K69. [Google Scholar]
- Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nat Rev Neurosci. 2006;7:30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- Capelson M, Hetzer MW. The role of nuclear pores in gene regulation, development and disease. EMBO Rep. 2009;10:697–705. doi: 10.1038/embor.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V. Injury-induced HDAC5 nuclear export is essential for axon regeneration. Cell. 2013;155:894–908. doi: 10.1016/j.cell.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J. The black book of Alzheimer’s disease, part 1. Prim Psychiatry. 2008;15:66–76. [Google Scholar]
- D’Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284–295. doi: 10.1016/j.cell.2008.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachsbart F, Caliebe A, Kleindorp R, Blanché H, von Eller-Eberstein H, Nikolaus S, Schreiber S, Nebel A. Association of FOXO3A variation with human longevity confirmed in German centenarians. Proc Natl Acad Sci USA. 2009;106:2700–2705. doi: 10.1073/pnas.0809594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Khaitovich P, Plotkin JB, Pääbo S, Eisen MB. Aging and gene expression in the primate brain. PLoS Biol. 2005;3:e274. doi: 10.1371/journal.pbio.0030274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frobel J, Hemeda H, Lenz M, Abagnale G, Joussen S, Denecke B, Ŝarić T, Zenke M, Wagner W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Reports. 2014;3:414–422. doi: 10.1016/j.stemcr.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladyshev VN. The origin of aging: imperfectness-driven non-random damage defines the aging process and control of lifespan. Trends Genet. 2013;29:506–512. doi: 10.1016/j.tig.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Rothman FG, López-Otín C, Misteli T. Progeria: a paradigm for translational medicine. Cell. 2014;156:400–407. doi: 10.1016/j.cell.2013.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenzel CA, Hérate C, Benichou S. HIV-1 Vpr-a still “enigmatic multitasker”. Front Microbiol. 2014;5:127. doi: 10.3389/fmicb.2014.00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- Hennekam RCM. Hutchinson-Gilford progeria syndrome: review of the phenotype. Am J Med Genet A. 2006;140:2603–2624. doi: 10.1002/ajmg.a.31346. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van Gorp S, Nazor KL, Boscolo FS, et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482:216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, Bohlmann I, Schäfer M, Hansen-Hagge TE, Kiyoi H, Wilda M, Hameister H, Bartram CR, Janssen JWG. Identification of a novel putative Ran-binding protein and its close homologue. Biochem Biophys Res Commun. 2000;278:241–249. doi: 10.1006/bbrc.2000.3788. [DOI] [PubMed] [Google Scholar]
- Koch P, Breuer P, Peitz M, Jungverdorben J, Kesavan J, Poppe D, Doerr J, Ladewig J, Mertens J, Tüting T, et al. Excitation-induced ataxin-3 aggregation in neurons from patients with Machado-Joseph disease. Nature. 2011;480:543–546. doi: 10.1038/nature10671. [DOI] [PubMed] [Google Scholar]
- Koch P, Tamboli IY, Mertens J, Wunderlich P, Ladewig J, Stüber K, Esselmann H, Wiltfang J, Brüstle O, Walter J. Presenilin-1 L166P mutant human pluripotent stem cell-derived neurons exhibit partial loss of γ-secretase activity in endogenous amyloid-β generation. Am J Pathol. 2012;180:2404–2416. doi: 10.1016/j.ajpath.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Ladewig J, Mertens J, Kesavan J, Doerr J, Poppe D, Glaue F, Herms S, Wernet P, Kögler G, Müller FJ, et al. Small molecules enable highly efficient neuronal conversion of human fibroblasts. Nat Methods. 2012;9:575–578. doi: 10.1038/nmeth.1972. [DOI] [PubMed] [Google Scholar]
- Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez JM, De Vos J, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25:2248–2253. doi: 10.1101/gad.173922.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Zhou S, Smas CM. Identification of RANBP16 and RANBP17 as novel interaction partners for the bHLH transcription factor E12. J Cell Biochem. 2010;111:195–206. doi: 10.1002/jcb.22689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Yuan Z, Boag PR, Yang Y, Villén J, Becker EBE, DiBacco S, de la Iglesia N, Gygi S, Blackwell TK, Bonni A. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- Liu ML, Zang T, Zou Y, Chang JC, Gibson JR, Huber KM, Zhang CL. Small molecules enable neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat Commun. 2013;4:2183. doi: 10.1038/ncomms3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature. 2014;507:448–454. doi: 10.1038/nature13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, Sridharan R, Xie W, Utikal J, Eminli S, Arnold K, Stadtfeld M, Yachechko R, Tchieu J, Jaenisch R, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Manukyan M, Singh PB. Epigenome rejuvenation: HP1β mobility as a measure of pluripotent and senescent chromatin ground states. Sci Rep. 2014;4:4789. doi: 10.1038/srep04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetto MC, Brennand KJ, Boyer LF, Gage FH. Induced pluripotent stem cells (iPSCs) and neurological disease modeling: progress and promises. Hum Mol Genet. 2011;20(R2):R109–R115. doi: 10.1093/hmg/ddr336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–770. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Stüber K, Poppe D, Doerr J, Ladewig J, Brüstle O, Koch P. Embryonic stem cell-based modeling of tau pathology in human neurons. Am J Pathol. 2013a;182:1769–1779. doi: 10.1016/j.ajpath.2013.01.043. [DOI] [PubMed] [Google Scholar]
- Mertens J, Stüber K, Wunderlich P, Ladewig J, Kesavan JC, Vandenberghe R, Vandenbulcke M, van Damme P, Walter J, Brüstle O, Koch P. APP processing in human pluripotent stem cell-derived neurons is resistant to NSAID-based γ-secretase modulation. Stem Cell Reports. 2013b;1:491–498. doi: 10.1016/j.stemcr.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT. Using whole-genome transcriptional analyses to identify molecular mechanisms of aging. Drug Discov Today Dis Mech. 2006;3:41–46. [Google Scholar]
- Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany, NY) 2009;1:28–37. doi: 10.18632/aging.100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schüle B, Dolmetsch RE, Langston W, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, Citri A, Sebastiano V, Marro S, Südhof TC, Wernig M. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson C, Gibson GE, Blass JP. Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N Engl J Med. 1985;312:1063–1065. doi: 10.1056/NEJM198504183121618. [DOI] [PubMed] [Google Scholar]
- Rando TA, Chang HY. Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell. 2012;148:46–57. doi: 10.1016/j.cell.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P, Schmid B, Burbulla LF, Schöndorf DC, Wagner L, Glatza M, Höing S, Hargus G, Heck SA, Dhingra A, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12:354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Savas JN, Toyama BH, Xu T, Yates JR, 3rd, Hetzer MW. Extremely long-lived nuclear pore proteins in the rat brain. Science. 2012;335:942–942. doi: 10.1126/science.1217421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhr ST, Chang EA, Tjong J, Alcasid N, Perkins GA, Goissis MD, Ellisman MH, Perez GI, Cibelli JB. Mitochondrial rejuvenation after induced pluripotency. PLoS ONE. 2010;5:e14095. doi: 10.1371/journal.pone.0014095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Toyama BH, Hetzer MW. Protein homeostasis: live long, won’t prosper. Nat Rev Mol Cell Biol. 2013;14:55–61. doi: 10.1038/nrm3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama BH, Savas JN, Park SK, Harris MS, Ingolia NT, Yates JR, 3rd, Hetzer MW. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell. 2013;154:971–982. doi: 10.1016/j.cell.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willcox BJ, Donlon TA, He Q, Chen R, Grove JS, Yano K, Masaki KH, Willcox DC, Rodriguez B, Curb JD. FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci USA. 2008;105:13987–13992. doi: 10.1073/pnas.0801030105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.