

SUMMARY
Cardiac development requires coordinated and large-scale rearrangements of the epigenome. The roles and precise mechanisms through which specific epigenetic modifying enzymes control cardiac lineage specification, however, remain unclear. Here we show the H3K4 methyltransferase SETD7 controls cardiac differentiation by reading H3K36 marks independently of its enzymatic activity. Through ChIP-seq, we found SETD7 targets distinct sets of genes to drive their stage-specific expression during cardiomyocyte differentiation. SETD7 associates with different co-factors at these stages, including SWI/SNF chromatin remodeling factors during mesodermal formation and the transcription factor NKX2.5 in cardiac progenitors to drive their differentiation. Further analyses revealed that SETD7 binds methylated H3K36 in the bodies of its target genes to facilitate RNA PolII-dependent transcription. Moreover, abnormal SETD7 expression impairs functional attributes of terminally-differentiated cardiomyocytes. Together, these results reveal how SETD7 acts at sequential steps in cardiac lineage commitment and provide insights into crosstalk between dynamic epigenetic marks and chromatin-modifying enzymes.
eTOC BLURB
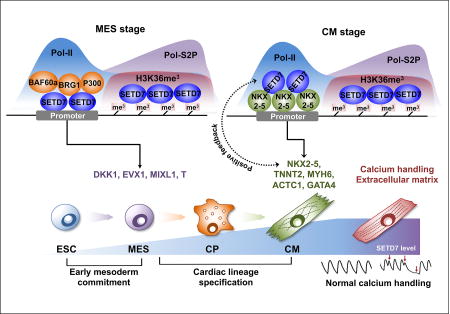
Wu and colleagues define SETD7 as a key regulator of cardiac lineage commitment. SETD7 regulates the expression of lineage-specific target genes and interacts with various co-factors during cardiomyocyte differentiation. SETD7 associates with H3K36me3 histone modification which required for the transcriptional activation.
INTRODUCTION
Generation of cardiomyocytes (CMs) from human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) has emerged as the state-of-the-art approach to study human cardiac development and pathobiology of adult and congenital heart disease. Cardiac differentiation is regulated by temporal expression of genes critical for mesodermal and cardiac lineage specification, which we modulate in vitro to efficiently differentiate human ESCs and iPSCs to functional CMs (Burridge et al., 2012, 2015). In addition, recent studies have demonstrated that coordinated transition of epigenetic modifications is required for stage-specific gene expression during cardiac differentiation (Paige et al., 2012; Wamstad et al., 2012). Genome-wide profiling of active histone markers (H3K4me3 and H3K36me3) and repressive histone marker (H3K27me3) revealed the expression of key regulators for CM differentiation is associated with the transition between H3K4me3 and H3K27me3, whereas the cardiac structural genes showed changes only in H3K4me3. By contrast, H3K36me3 marker was enriched in both regulatory and cardiac structural genes during the entire duration of CM differentiation.
Epigenetic modifying enzymes play a critical role in epigenetic regulation, with important clinical implications. Recent studies showed impaired function of epigenetic modifying enzymes contributes to congenital heart disease (Chang and Bruneau, 2012; Vallaster et al., 2012), such as in morphological heart defects caused by loss of H3K36 methyltransferase WHSC1 (Nimura et al., 2009). Mutation in histone methyltransferase MLL2 leads to congenital heart defects in Kabuki syndrome (Ng et al., 2010) and the deficiency of SMYD1 expression during heart development induces malformation of right ventricle (Li et al., 2013). Chromatin remodeling factors such as SWI/SNF complex are also critical for early heart development by controlling transcription of cardiac-specific genes (Hang et al., 2010). While these studies provide robust evidence that epigenetic modifying enzymes are essential for normal heart development, the mechanisms by which the epigenetic modifying enzymes regulate the complex gene expression networks of cardiac differentiation through a coordinated transition of epigenetic modifications remain poorly understood.
SETD7 was originally identified as a methyltransferase for histone 3 (H3) lysine 4 residue by mediating mono-methylation (H3K4me1) (Wang et al., 2001). SETD7 diminished its activity for histone assembled in nucleosome but showed high activity with free histone substrates (Castaño et al., 2016). Thus previous studies have largely focused on the role of SETD7 on non-histone proteins such as p53 (Chuikov et al., 2004), TAF10 (Kouskouti et al., 2004), DNMT1 (Estève et al., 2009), ERα (Subramanian et al., 2008), E2F1 (Kontaki and Talianidis, 2010), SOX2 (Fang et al., 2014), YAP (Oudhoff et al., 2013), LIN28A (Kim et al., 2014), and HIF1-alpha (Kim et al., 2016). Only a handful of studies showed SETD7 can bind on the promoter regions and regulate individual gene expression (Chen et al., 2012; Kofent et al., 2016). Although these data suggest SETD7 is directly involved in gene transcription, the question of whether SETD7-mediated H3K4me1 is essential for the transcriptional regulation of its target gene remains controversial.
Dynamic changes in SETD7 expression during developmental processes have been described, in particular during cardiac differentiation (Xie et al., 2013; Gifford et al., 2013). Recent studies have demonstrated that SETD7 is required for normal morphogenesis of zebrafish heart (Kim et al., 2015) and interacts with cardiac transcription factor Tbx1 in early cardiac development (Chen et al., 2012). While these findings suggest SETD7 plays a role in cardiac development, specific mechanisms by which SETD7 regulates CM differentiation have not been thoroughly investigated.
Here we show that SETD7 is a critical factor of cardiac differentiation. We found SETD7 expression was significantly up-regulated during cardiac differentiation and modulated transcription of key regulatory genes for lineage commitment. ChIP-seq of SETD7 in each stage of cardiac differentiation identified previously unknown genome-wide binding patterns of SETD7 and revealed distinct SETD7 target genes at each stage of differentiation. In addition, our results identified stage-specific co-factors of SETD7, which are required for transcriptional regulation of the target genes. Cross-analysis with epigenetic dynamics elucidated unidentified interactions between SETD7 and H3K36me in cardiac differentiation. Finally, loss of SETD7 resulted in irregular contractile activity of terminally differentiated CMs.
RESULTS
SETD7 is induced during human cardiac differentiation
To study the role of epigenetic modifying enzymes in cardiac differentiation, we employed direct differentiation of hESCs to CMs as a model system of human cardiac development. Monolayers of CMs were successfully generated using a chemically-defined protocol as previously described (Wu et al., 2015). Beating CMs exhibited normal Ca2+ handling properties and organized sarcomeric structures (Figure S1A–S1C). The expression of cardiac-specific genes appeared on day 5 of differentiation and reached maximal expression level on day 9 (Figure S1D). Based on the observed expression patterns and on previous studies (Paige et al., 2012), we selected four stages of differentiation that correspond to particular cell types: undifferentiated human embryonic stem cells (hESCs, differentiation day 0), mesodermal cells (MESs, differentiation day 2), cardiac progenitor cells (CPs, differentiation day 5), and beating cardiomyocytes (CMs, differentiation day 9).
To identify the expression pattern of epigenetic modifying enzymes during cardiac differentiation, we performed RNA sequencing (RNA-seq) at each stage of CM differentiation. Cross-analysis with RNA-seq data of murine CM differentiation showed that SETD7 was significantly up-regulated during CM differentiation in both human and mouse (Figure 1A). Analysis of the Expression Atlas Database revealed SETD7 was highly expressed in adult human and olive baboon (Papio anubis) heart tissue (Figure 1A). To confirm the expression of SETD7 in CMs, we generated CMs from hESCs and measured the expression of SETD7 during CM differentiation. Quantitative PCR (qPCR) of hESC-CMs showed a significant expression of SETD7 and TNNT2 (Figure 1B–D). Immunoblotting (IB) analysis demonstrated an increased expression of SETD7 during cardiac differentiation (Figure 1E). Immunocytochemistry (ICC) with SETD7 and TNNT2 antibodies revealed that the majority of TNNT2+ cells exhibited nuclear localization of SETD7 (Figure 1F).
Figure 1. Dynamic target genes of SETD7 during hESC-CM differentiation.

(A) RNA-seq expression level of histone methyltransferases at each stage of CM differentiation in hESCs (human), mESCs (mouse), and heart tissues of human and olive baboon (Papio anubis). Heart tissue RNA-seq data were acquired from the Expression Atlas database. (B) Flow cytometry analysis of TNNT2+ cells in H7 hESCs and hESC-CMs (mean ± SEM; n=5). (C–D) RT-qPCR of TNNT2 and SETD7 in hESCs and hESC-CMs. *P < 0.05; **P < 0.01 (one-way ANOVA; mean ± SEM; n=4). (E) Immunoblot analysis of cell lysates at indicated differentiation stages. (F) Immuno-staining of TNNT2, SETD7 in CMs (left). Cell counting of SETD7+ and/or TNNT2+ cells from immune-stained images (right). Scale bar, 10 µm. (G) K-means clustering of SETD7 enrichment during CM differentiation. Y-axis indicates log transformed fold-changes based on day 0 enrichment (Upper) or log transformed fold-changes based on RNA expression at day 0 (Lower). (H–I) Genome browser screenshots of ChIP-seq and RNA-seq for ACTC1 and EOMES. (J) GO analysis of Cluster 4 and Cluster 5. Color code indicates negative log transformed multiple testing adjusted P-value. (K–L) Gene-concept network displaying the gene names associated with the top signaling pathways identified in Cluster 4 and Cluster 5. See also Figure S1 and Table S1.
Dynamic occupancy of SETD7 on stage-specific target genes of cardiac differentiation
To investigate the role of SETD7 in cardiac development, we performed ChIP-seq for SETD7 during the four stages of CM differentiation. Unsupervised K-means clustering identified five groups with distinct SETD7 enrichment patterns (Figure 1G and Table S1). Cluster 1 and Cluster 3 sequentially lost SETD7 enrichment during CM differentiation, while Cluster 2 retained SETD7 enrichment in hESC and mesoderm stages with reduced enrichment thereafter (Figure 1G). Cluster 4 transiently gained SETD7 enrichment, but was attenuated after the mesoderm stage. Cluster 5 exhibited a sequential increase in SETD7 expression (Figure 1G). Average profiles of SETD7 enrichment on these target genes showed unique binding patterns of SETD7 on promoter and genebody regions (Figure 1H, 1I and S1E–S1H). RNA expression of each cluster correlated with SETD7 enrichment, suggesting SETD7 deposition is associated with active gene transcription with unique functions at each stage of the CM differentiation (Figures 1G–1I). Gene Ontology (GO) enrichment analyses of SETD7 target genes showed important cardiac development pathways were up-regulated at each stage of differentiation (Figure 1J). For example, genes in Cluster 4 were enriched for embryo development pathways, including pattern specification process, somite and mesoderm development, NOTCH, and WNT signaling pathways (Figures 1J, S1I and S1J). In contrast, genes in Cluster 5 were enriched for mature development pathways such as heart morphogenesis, cardiac development, and cardiac muscle cell differentiation. A similar contrast between Cluster 4 and Cluster 5 genes was elucidated by gene-concept network analysis in which a number of genes related to cardiac development predominated in Cluster 5 while mesoderm-specific genes predominated in Cluster 4 (Figures 1K and 1L). Interestingly, GO enrichment analyses of Cluster 1 and Cluster 3 also showed genes enriched for regulation of stem cells, such as transcriptional regulation of pluripotent stem cells and expression of proliferation-enhancing genes POU5F1 (OCT4), SOX2, and NANOG (Figure S1J). Though SETD7 expression continuously increased during differentiation, SETD7 occupancy and expression of these target genes were significantly decreased. These data suggest SETD7 in hESC stage is a transcriptional activator of these genes but may negatively affect the early stages of CM differentiation. Together, our ChIP-seq and RNA-seq data suggest there exist distinguishable functions for SETD7 at each stage of cardiac differentiation via stage-specific binding on its target genes.
Depletion of SETD7 expression results in impaired cardiac differentiation
To understand the specific mechanisms by which SETD7 regulates CM differentiation, we used transcription activator-like effector nuclease (TALEN)-mediated gene knock-out system (Karakikes et al., 2017) to disrupt SETD7 expression in a HES3NKX2-5eGFP/w hESC line that encodes enhanced GFP (eGFP) under a NKX2-5 exon (Elliott et al., 2011). After screening clones transfected with a TALEN pair that targets start codon of SETD7, we successfully isolated heterozygous (SETD7+/−) and homozygous (SETD7−/−) clones. The SETD7−/− clone showed a complete depletion of SETD7 expression in undifferentiated hESCs and day 14 of CM differentiation (Figure 2A). In contrast to the SETD7+/+ clone, the SETD7−/− clone showed a significant decrease in NKX2-5-eGFP+ population, as well as a reduction of cardiac gene expression such as MYH6 and TBX20 during CM differentiation (Figures 2B and 2C). We confirmed these results in H7 hESCs stably expressing shRNA for SETD7, suggesting that SETD7 is necessary for CM differentiation (Figure 2D). We then generated an inducible shRNA system to study the effect of SETD7 specifically on CM differentiation. We introduced doxycycline (DOX) inducible shRNA in H7 and HES3NKX2-5eGFP/w lines and successfully generated stable clones carrying SETD7 and scramble shRNAs. In the presence of DOX, expression of cardiac genes was significantly down-regulated during CM differentiation, which correlated with suppression of NKX2-5-eGFP (Figure 2E–G and S2A). No significant differences were detected in the scramble shRNA line. Interestingly, we did not detect any positive effects of SETD7 overexpression on the efficiency of CM differentiation, suggesting SETD7 alone was insufficient to facilitate CM differentiation (Figure S2B and S2C). Together, these results demonstrate SETD7 expression is necessary for CM differentiation.
Figure 2. SETD7 is necessary for CM differentiation.

(A) Immunoblot analysis of cell lysates from HES3NKX2-5eGFP/w hESC clone transfected with TALEN targeting start codon of SETD7 gene (upper). Immunoblot analysis of cell lysate from SETD7+/+ and SETD7−/− HES3NKX2-5eGFP/w-derived CMs (lower) (n=3). (B) NKX2-5-GFP expression in SETD7+/+ and SETD7−/− HES3NKX2-5eGFP/w-derived CMs (left). Flow cytometry of GFP+ cells (right). Scale bar, 100 µm. (C) RT-qPCR of TBX20 and MYH6 in SETD7+/+ and SETD7−/− HES3NKX2-5eGFP/w-derived CMs. (D) mRNA levels of SETD7, TNNT2, and MYH6 in control and SETD7 shRNA hESC-CMs. hESCs were infected with lentivirus carrying scramble or SETD7 shRNA. (E–F) Expression level of cardiac-related genes in hESCs and hESC-CMs expressing doxycycline-inducible scramble or SETD7 shRNA. (G) Representative fluorescence microscopy images of hESC HES3NKX2-5eGFP/w-derived CMs. Scale bar, 100 µm. Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.005 (mean ± SEM; n=4). See also Figure S2.
SETD7 is required for mesoderm lineage commitment through transcriptional regulation of key regulatory genes
ChIP-seq of SETD7 during CM differentiation revealed its dynamic target genes in each stage of differentiation. To examine how SETD7 regulates transcription of its target genes during the differentiation process, we first measured the effects of SETD7 knock-out on the expression of mesoderm-specific genes. IB analysis confirmed up-regulation of SETD7 expression during mesoderm specification in SETD7+/+ line, whereas the SETD7−/− line showed a depletion of SETD7 expression (Figure 3A). Expression of key regulatory genes for early mesoderm lineage commitment (T, EVX1, DKK1, MIXL1, EOMES, and NODAL) and cardiac mesoderm lineage genes (GATA3 and MESP1) was significantly diminished in the SETD7−/− line (Figure 3B). GO analysis of RNA-seq at the mesoderm stage also showed down-regulation of genes in the SETD7−/− line was associated with early cardiac differentiation pathways, such as ventricular cardiac muscle tissue development and ventricular cardiac muscle tissue morphogenesis (Figure S2D). In addition, rescue of SETD7 expression in the SETD7−/− line using a lentivirus-based over-expression system resulted in increased expression of mesoderm-specific genes (Figure 3C and 3D). To confirm the indispensable role of SETD7 in mesodermal lineage commitment, we next generated embryoid bodies (EBs) that randomly differentiated into three germ layers (Figure S2E). EBs from the SETD7−/− line showed down-regulation of endomesoderm-specific genes and up-regulation of ectoderm-specific genes compared to the SETD7+/+ line (Figures 3E and 3F). Down-regulation of endomesoderm genes was also observed in Dox-treated EBs carrying Tet-inducible shRNA system for SETD7 (Figure S2F and S2G). Moreover, ChIP-seq analysis demonstrated SETD7 enriched on key regulatory genes for mesoderm specification in mesoderm stage of CM differentiation (Cluster 4). These data show SETD7 plays a key role in early cardiac differentiation through transcriptional regulation of mesoderm-specific genes.
Figure 3. SETD7 is required for transcriptional regulation of lineage specific genes during CM differentiation.

(A) Immunoblot analysis of cell lysates from days 0, 1 and 2 of CM differentiation of SETD7+/+, SETD7+/− and SETD7−/− hESC lines. (B) RT-qPCR of mesoderm specific genes in mesoderm lineage cells derived from SETD7+/+, SETD7+/− and SETD7−/− hESC lines. (C) Immunoblot analysis of cell lysates from hESCs and mesoderm lineage cells of SETD7−/− lines carrying control or SETD7 overexpression plasmid. Stable cell lines were generated using lentiviral overexpression system. (D) RT-qPCR of mesoderm specific genes in hESCs and mesoderm lineage cells of SETD7−/− lines carrying control or SETD7 overexpression plasmid. (E) Immunoblot analysis of embryoid body cell lysate from SETD7+/+, SETD7+/−, and SETD7−/− hESC lines. α-tubulin was used as a loading control. (F) RT-qPCR of endomesoderm and ectoderm related genes in embryoid bodies from SETD7+/− and SETD7−/− hESC line compared with SETD7+/+. Total mRNA was extracted from day 14 of embryoid body formation. (G) Representative fluorescence microscopy images of eGFP signal of hESC HES3NKX2-5eGFP/w-derived CMs (day 14) Doxycycline was added to cell media on days 0, 3, and 5 of hESC-CM differentiation. Scale bar, 100 µm. (H) Immunoblot analysis of SETD7 protein levels in hESC HES3NKX2-5eGFP/w-derived CMs (day 14) doxycycline-inducible shRNA for SETD7 or scramble (left). Doxycycline was added to cell media on days 0, 3, and 5 of hESC-CM differentiation. Relative signal intensity of SETD7 (right). (I) RT-qPCR of cardiac specific genes, and SETD7 expression levels in hESC HES3NKX2-5eGFP/w-derived CMs (day 14) doxycycline-inducible shRNA for SETD7 or scramble. Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.005 (mean ± SEM; n=3). See also Figure 2, Figure 3, and Table S2.
SETD7 is necessary for cardiac lineage specification
Next, we investigated the effects of SETD7 on cardiac lineage specification after mesoderm stage. We suppressed SETD7 expression at different time points using Tet-inducible shRNA system (Figure S2H). Consistent with our previous observations, differentiation efficiency was severely impaired when SETD7 suppression was initiated at day 0 of CM differentiation (Figures 2E, 3G and 3H). Knock-down (KD) of SETD7 at day 3 and day 5 (early cardiac specification) also impaired differentiation efficiency in comparison to scramble shRNA line or control treatment group (Figure 3G). In addition, down-regulation of cardiac specific genes was observed in condition of SETD7 suppression at day 3 and day 5 (Figure 3I). In particular, major cardiac transcription factors (TFs) such as NKX2-5, TBX5, and GATA4, identified as target genes of SETD7 (Cluster 5), were significantly down-regulated in SETD7 KD (Figure 3I). The expression of cardiac specific genes such as MYH6 and TNNT2 was similarly attenuated in with SETD7 KD (Figure 3I). These data suggest SETD7 expression in cardiac commitment is required for transcriptional activation of cardiac specific genes.
Effects of SETD7 on lineage-specific gene expression of non-cardiomyocyte cell types
As the IB analysis of SETD7 in human fetal tissues showed high expression level of SETD7 in various tissue types, we tested whether SETD7 affects lineage differentiation processes of cell types other than CMs (Figure S2I and S2J). First, we differentiated SETD7+/+ and SETD7−/− hESC lines to neuron precursor cells (NPCs, ectoderm) and hepatocyte-like cells (endoderm). Using currently available protocols (Song et al., 2009), we generated NPCs and hepatocyte-like cells expressing stage specific markers from both SETD7−/− and SETD7+/+ lines (Figure S3A–S3G). Interestingly, we observed an abnormal expression pattern of cell type-specific genes such as PAX6 (early NPC marker), NEUROG1 (late NPC marker), SOX17 (early endoderm lineage), NHE-4 (hepatocyte precursor), AFP (hepatocyte), and ALB (hepatocyte) (Figure S3C and S3F). Moreover, we observed SETD7 is highly up-regulated during both NPC and hepatocyte-like cell differentiation process (Figure S3A and S3E). These data suggest SETD7 possesses unidentified roles in transcriptional regulation of certain lineage-specific genes during endodermal and ectodermal commitment.
We also tested whether depletion of SETD7 affects differentiation of endothelial cells (ECs), a cell type that shares early lineage path with CMs at the mesodermal and cardiac progenitor cell stages (Figure S3H–S3P). Using the monolayer-based, embryoid body-free differentiation protocol previously described (Gu et al., 2017), we differentiated SETD7+/+ and SETD7−/− hESC lines to ECs, which displayed cobblestone-like morphology representative of bona fide ECs (Figure. S3H). The number of ECs obtained from the differentiation was also not altered, suggesting SETD7 deficiency does not adversely affect generation of bona fide ECs from hESCs (Figure S3I). However, mRNA expression levels of several endothelial functional genes were attenuated in absence of SETD7. Endothelial markers CDH5, PECAM1, and VWF were significantly down-regulated in SETD7−/− hESC-ECs (Figure S3J–S3L). NOS3 expression was similarly down-regulated, indicating SETD7−/− hESC-ECs could not generate endothelial nitric oxide, a hallmark phenotype of endothelial dysfunction (Figure S3M). Expression level of Endocan (ESM1) gene was also significantly reduced, indicating SETD7−/− hESC-ECs were less angiogenic than WT (Figure S3N). Expression levels of cell surface adhesion molecules E-Selectin (SELE) and VCAM1 were not altered in the absence of inflammatory cytokines (Figure S3O and S3P). These data suggest SETD7 is required in hESC-EC differentiation for normal expression of genes critical for endothelial cell function and homeostasis.
SETD7 associates with stage-specific co-factors for transcriptional regulation of their target genes during cardiac differentiation
Next, we investigated the underlying mechanisms of SETD7 on transcriptional regulation of its target genes during CM differentiation. Previous studies have shown SETD7 binds to BAF60a (mouse heart tissue) and BRG1 (endothelial cells), factors known to be essential components of the SWI/SNF chromatin remodeling complex (Chen et al., 2012; Okabe et al., 2012). Furthermore, SETD7 is reported to interact with p300/CBP-associated factor (PCAF), another known co-factor of the SWI/SNF chromatin remodeling complex (Huang, 2003; Ogiwara et al., 2011). Based on these findings, we tested whether SETD7 is also associated with these co-factors during cardiac mesoderm commitment. Immunoprecipitation (IP) assay showed the interaction of SETD7 with p300, BRG1, and BAF60a in MES stage cells (Figure 4A). We also detected the co-occupancy of p300, BRG1, and SETD7 on the promoter regions of DKK1, MIXL1, and T genes in the cells of MES stage in the SETD7+/+ line (Figure 4B). Interestingly, binding of p300 and BRG1 on target genes was significantly reduced in the SETD7−/− line, suggesting SETD7 recruited these co-transcription factors to the promoter regions of target genes (Figure 4B). The interaction between SETD7 and chromatin remodeling factors was not detected at the CM stage, consistent with a recent study showing that BRG1 expression was significantly decreased after cardiac mesoderm specification (Figure S4A) (Alexander et al., 2015). These data suggest SETD7 possesses different binding partners in CM stages.
Figure 4. SETD7 associates with stage specific co-factors for active gene transcription of its target genes.

(A) Immunoblot analysis of SETD7 immunoprecipitates (IP line) and cell lysates (Input line) from mesoderm lineage cells derived from hESCs. (B) Genome browser screenshots of SETD7 ChIP-seq and RNA-seq profiles in DKK1, MIXL1 and T genes at the mesodermal stage of CM differentiation (upper). ChIP-qPCR assay of SETD7, BRG1, and p300 at promoter regions of DKK1, MIXL1, and T genes during CM differentiation (lower). The sequence and genomic location of each primer were described in Table S2. (C) Relative enrichment levels of SETD7 (in human) and Nkx2-5 (in mouse) on TSS of ortholog coding genes in CMs. Heatmaps were ranked by SETD7 enrichment. ChIP-seq data adapted from He et al., 2011. (D) Immunoblot analysis of SETD7 immunoprecipitates (IP line) and cell lysates (Input line) from hESC-CMs. (E) Genome browser screenshots of SETD7 ChIP-seq profiles in MYH6 and NKX2-5 regions in four stages of CM differentiation (upper). ChIP-qPCR of SETD7 and NKX2-5 enrichment at promoter regions of MYH6 and NKX2-5 genes during CM differentiation (lower). Orange bar indicates region targeted by qPCR. (F) Genome browser screenshot of Setd7 gene with a conserved NKX2-5 motif in the promoter region. Enrichment of Nkx2-5, Tbx5, and Gata4 on the promotor region of Setd7 promoter in mouse CMs. ChIP-seq data were adapted from Luna-Zurita et al., 2016. (G) ChIP-qPCR of NKX2-5 enrichment at promoter regions of NANOG, SETD7, NKX2-5, TNNT2, and MYH6 in hESC-CMs. (H) RT-qPCR of mRNA expression of SETD7, NKX2-5, TBX5, and MYH6 genes in CMs transfected with two different sets of siRNA for SETD7 and NKX2-5. (I) ChIP-qPCR of SETD7 and IgG at TSS regions of TNNT2 genes in NKX2-5 knock-down CMs. Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.005 (mean ± SEM; n=3). See also Figure S4.
ChIP-seq analysis showed SETD7 is targeting well-known cardiac TFs of NKX2-5, TBX5, and GATA4 in CP and CM stages (Cluster 5). Because these cardiac TFs bind together forming a complex that regulates transcription of cardiac-specific genes during CM differentiation (Luna-Zurita et al., 2016), we speculate that SETD7 interacts with these cardiac TFs to regulate cardiac lineage specification. To test this hypothesis, we cross-analyzed SETD7 enrichment data with ChIP-seq of Nkx2-5, Mef2a, Gata4, Tbx5, and Srf in mouse tissue (He et al., 2011). We found the majority of SETD7 target genes were shared with cardiac TFs, indicating a close association of SETD7 with the cardiac TF network (Figure 4C and S4B). IP analysis of SETD7 in CMs showed the interaction of SETD7 and NKX2-5 (Figure 4D). ChIP-qPCR identified co-occupancy of SETD7 and NKX2-5 on MYH6 and NKX2-5 promoters in a stage-dependent manner, revealing a coordinated network between SETD7 and NKX2-5 for transcriptional regulation of the target genes (Figure 4E). We also identified a conserved binding motif of NKX2-5 in the promoter region of SETD7 (Figure 4F). ChIP-seq of Nkx2-5, Tbx5, and Gata4 showed that Nkx2-5 binds to the promoter region of Setd7 in mouse CMs (Figure 4F). ChIP-qPCR with primers targeting NKX2-5-binding region confirmed the occupancy of NKX2-5 in the promoter of human SETD7 (Figure 4G). KD of NKX2-5 in CMs resulted in down-regulation of SETD7 expression (Figure 4H). Consistent with these observations, loss of SETD7 suppressed NKX2-5 expression in CMs, suggesting a positive feedback loop between NKX2-5 and SETD7 (Figure 4H). TBX5 and MYH6 expression was attenuated with KD of NKX2-5 or SETD7. In addition, binding of SETD7 on TNNT2 promoter region was significantly decreased by the KD of NKX2-5, confirming that SETD7 and NKX2-5 are required for their shared target gene expression (Figure 4I). Taken together, these results demonstrate SETD7 interacts with stage-specific co-factors during human and mouse cardiac differentiation for transcriptional regulation of the target genes.
Correlation of SETD7 enrichment with active chromatin modifications during CM differentiation
We next tested whether enzymatic activity of SETD7 is necessary for CM differentiation, given the role of SETD7 as H3K4me1 methyltransferase. As a potent inhibitor of SETD7, PFI-2 suppressed the enzymatic activity of SETD7 on mono-methylation of H3K4 (Barsyte-Lovejoy et al., 2014) (Figure S4A). Treatment of PFI-2 during the entire CM differentiation process showed no significant differences in TNNT2+ cell population at day 14 of differentiation, whereas the depletion of SETD7 itself severely reduced the TNNT2+ cell population (Figure 5A and 5B). The expression of mesoderm-specific genes was not decreased by PFI-2 treatment during mesoderm specification (Figures 5C and 5D). Interestingly, the global level of H3K4me1 also was not altered by either PFI-2 treatment or depletion of SETD7 (Figures 5A and 5C). These data suggest enzymatic activity of SETD7 is not necessary for CM differentiation, whereas an unidentified role of SETD7 in epigenetic regulation exists.
Figure 5. SETD7 is linked with H3K36 methylation on its target genes.

(A) Immunoblot analysis of cell lysates from SETD7+/+ and SETD7−/− lines at day 14 of CM differentiation. PFI-2 and DMSO were treated during entire CM differentiation. (B) Flow cytometry of TNNT2+ cells of SETD7+/+ and SETD7+/- at day 14 of CM differentiation. PFI-2 and DMSO were treated during entire differentiation process (mean ± SEM; n=8 for SETD7+/+ with DMSO and SETD7−/−; n=6 for SETD7+/+ with PFI-2). (C) Immunoblot analysis of mesodermal cells during CM differentiation. DMSO or PFI was treated for 2 days. (D) RT-qPCR of DKK1, NODAL, T, and KDR expression levels in mesodermal cells during CM differentiation. DMSO or PFI was treated for two days. (E) Enrichment levels of histone markers and SETD7 at target genes in each of five clusters and of RNA expression. (F) Spot intensities of two peptide chip arrays. Each peptide chip contained 384 different histone modifications incubated with SETD7 protein. Blue dots represent mean signal intensities from two different peptide arrays. (G) Peptide pull-down assay of histone peptides with SETD7 protein. Biotinylated histone peptides were incubated with GST-SETD7 protein and pulled down with streptavidin coated beads. (H) Immunoblot analysis of SETD7 immunoprecipitates (IP line) and cell lysates (Input line) from hESC-CMs. (I) Genome browser screenshots of H3K36me3 and SETD7 ChIP-seq profiles in T at four stages of CM differentiation (upper). ChIP-qPCR assay of H3K36me3 and SETD7 enrichment at the two gene body regions (dashed orange lines) of the T gene in four stages of CM differentiation (lower). (J) Genome-wide heatmaps of SETD7 and H3K36me3 enrichment in target gene bodies. RNA expression of target genes shown in the right-most heatmap. K-means clustering of SETD7 enrichment used to sort heatmaps. (K) ChIP-qPCR for relative enrichment of SETD7 and H3K36me3 on gene body regions of MYH6, TNNT2 and NANOG genes in left ventricular tissue of healthy individuals (n=5). Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.005 (mean ± SEM; n=3 (panel B); n=5 (panel K)). See also Figure S5 and Figure S6.
To investigate the relationship between SETD7 and the epigenetic dynamics of CM differentiation, we next performed ChIP-seq at each stage of CM differentiation using antibodies against H3K27ac, followed by cross-analyses with public ChIP-seq data sets for H3K4me3, H3K27me3, and H3K36me3 (Paige et al., 2012). Spearman correlation analysis showed SETD7 genomic binding sites are enriched for active histone markers (H3K27ac, H3K4me3, and H3K36me3) but not for repressive markers (H3K27me3) (Figure S5B and S5C). We further tested the dynamic transitions of histone markers in individual SETD7 target genes. The enrichment of active histone markers showed similar profiles with SETD7 enrichment, whereas the repressive histone marker had an opposite pattern of enrichment compared to SETD7 (Figure 5E). As expected, RNA expression profiles of target genes were highly similar to SETD7 and active histone marker enrichment (Figure 5E). ChIP-qPCR confirmed co-occupancy of both SETD7 and active histone marker H3K4me3 in the cardiac genes during CM differentiation (Figure S5D–S5F). However, SETD7 enrichment was not detected on non-cardiac genes such as SLC3A2 and NANOG, which also showed higher H3K4me3 modifications (Figure S5D–S5F). These data indicate SETD7 enrichment is highly correlated with transition of active histone markers of its target genes during CM differentiation.
SETD7 recognizes and associates H3K36 methylation on gene body region of its target genes during cardiac differentiation
We next determined which histone modifications are directly associated with SETD7. We performed a histone peptide array with active SETD7 protein purified from baculovirus infected sf9 cells. SETD7 showed a strong affinity with H3K36me3, H3K36me2, H3K36me1, H3K36ac, and unmodified-H3K36 (Figure 5F). Peptide array using active SETD7 tagged with GST and FLAG confirmed binding of SETD7 on H3K36 peptides (Figure S5A). SETD7 was pulled down with H3K36-methylated peptide and non-methylated peptide, whereas no binding was detected with control peptide H3K79 (Figure 5G). Moreover, the interaction between SETD7 and H3K36me3 was confirmed by IP assay in CMs, indicating strong interactions between SETD7 and H3K36me3 in CMs (Figure 5H).
We further explored the correlation between H3K36 methylation and SETD7 enrichment on its target genes during CM differentiation. Consistent with the genome-wide enrichment pattern of H3K36me3, which was located on the genebody regions of actively transcribing genes (Bannister et al., 2005), ChIP-seq analysis of SETD7 showed broad an occupancy of SETD7 on the genebody regions of target genes during CM differentiation (Figure S1E–H). Genome-wide correlation analysis also confirmed SETD7 enrichment was linked to H3K36me3 modification (Figure S6B). ChIP-qPCR with specific primers targeting gene body regions of T, MIXL1, NKX2-5, MYH6, and TNNT2 further demonstrated the co-occupancy of SETD7 and H3K36me3 in a stage-specific pattern (Figure 5I and Figure S6C–S6F). Whole genome analysis revealed increased transcription of genes with enrichment of both SETD7 and H3K36me3, suggesting co-occupancy of SETD7 and H3K36me3 can enhance transcription of their target genes (Figure 5J).
To test whether SETD7 is associated with H3K36me3 under physiological conditions, we next collected human left ventricular (LV) heart tissues from five healthy individuals and performed ChIP of SETD7 and H3K36me3. ChIP-qPCR showed co-occupancy of SETD7 and H3K36me3 in genebody regions of MYH6 and TNNT2, which was observed in human ESC-CMs (Figure 5K). We confirmed these data in two different fetal heart tissues (Figure S6G–S6I). Taken together, our findings demonstrate SETD7 is associated with H3K36me3 modifications on its target genes during CM differentiation.
SETD7 is not necessary for methylation of H3K36, but required for Pol II-mediated gene transcription
We next tested whether SETD7 exerts its enzymatic activity on H3K36me3. In vitro methylation assay using active SETD7 protein showed SETD7 was not able to mediate H3K36me3 on H3K36 unmodified, H3K36me1, H3K36me2 peptide, and histone 3 protein (Figure 6A, 6B, and S7A). As expected, SETD7 mediated methylation of H3K4me1 on free histone 3 and octamer, but not on poly-nucleosome (Figure 6B). In addition, ChIP-seq and ChIP-qPCR analyses of H3K36me3 in the mesoderm stage of SETD7+/+ and SETD7−/− lines revealed no significant decrease of H3K36me3 enrichment on its target genes (Figure 6C–6E and S7B). However, H3K36me3 level was suppressed by KD of SETD2, a major H3K36me3 methylase (Venkatesh et al., 2012) (Figure 6F and 6G). We found SETD7 enrichment on target genes was correlated with decreased H3K36me3 level by SETD2 KD (Figure 6G). These data indicate SETD7 is not necessary for H3K36me3, but binding of SETD7 on its target genes is highly depended on H3K36me3 status.
Figure 6. SETD7 is not necessary for H3K36me3 but required for Pol II-mediated gene transcription.

(A) Immunoblot analysis of SETD7 and H3K36me3. In vitro methylation assay performed with histone peptides and active SETD7-FLAG protein. H3K36me3 peptide used as positive control. (B) Immunoblot analysis of SETD7, H3K4me1, and H3K36me3. In vitro methylation assay was performed with histone H3, histone octamer, polynucleosome and active SETD7-FLAG protein. (C) Genome browser screenshots of H3K36me3 ChIP-seq profiles in T and EOMES genes at mesodermal stages of SETD7+/+ and SETD7−/− lines. (D) Genome-wide heatmaps of H3K36me3 enrichment on genebody region in SETD7+/+ and SETD7−/−(E) ChIP-qPCR of H3K36 enrichment on genebody regions of T and DKK1 genes at mesoderm stage. (F) RT-qPCR of SETD2, SETD7, TNNT2, and MYH6 expression levels in SETD2 knock-down CMs. (G) ChIP-qPCR of SETD7 and H3K36 enrichment on genebody regions of TNNT2 and MYH6 genes in SETD2 knock-down CMs. (H) Average profiles of PolII-S2P enrichment in genebody regions of Cluster 4 genes (left) and genome-wide level (right). (I) Genome browser screenshots of SETD7, H3K36me3, Pol II, and PolII-S2P ChIP-seq profiles on Brachyury (T) gene at mesoderm lineage of SETD7+/+ and SETD−/− lines. (J–K) ChIP-qPCR of PolII and PolII-S2P enrichment at TES region of T and DKK1 genes in SETD7+/+ and SETD7−/− lines at mesoderm stage. Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01 (mean ± SEM; n=3). See also Figure S7.
To further investigate the underlying mechanisms by which SETD7 controls the active function of H3K36me3 in transcriptional regulation of its target gene, we performed ChIP-seq with Pol II-S2P, Pol II in SETD7+/+, and SETD7−/− lines. The occupancy of Pol II-S2P on the genebody region of SETD7 target genes (Cluster 4), especially near the TES site, is markedly impaired by depletion of SETD7 (Figure 6H, 6I, and S7C). ChIP-qPCR with specific primers detecting the TES sites of T and MIXL1 confirmed our findings (Figure 6J and 6K). In genome-wide level, SETD7 also influenced the occupancy of Pol II-S2P and Pol II on the TES region (Figure 6H and Figure S7C). We further analyzed the occupancy of Pol II-S2P of each cluster of SETD7 enrichment. Upon SETD7 depletion, we found decreased Pol II-S2P occupancy in the TES site of genes, mostly with SETD7-H3K36me3 highly enriched clusters (Cluster B and Cluster D) (Figure S7E). Together, these data suggest SETD7 is necessary to recruit and facilitate Pol II-derived transcription of its target genes.
SETD7 is critical for calcium handling properties of mature cardiomyocytes
Given the high level of expression of SETD7 in hESC-CMs and in fetal and adult myocardial tissue (Figure 7A–7D), we next investigated the function of SETD7 in mature CMs (differentiation day 30~40). We generated highly pure CMs using a glucose starvation method (Sharma et al., 2015) and cultured them to day 30. We next introduced shRNA that targets SETD7 with a lentivirus system and infected CMs were purified with puromycin to generate stable KD CMs. No cell death was observed in SETD7 KD CMs (Figure 7E). However, motion vector detection analysis (Hayakawa et al., 2014; Kopljar et al., 2017) showed increased standard deviation of peak interval in SETD7 KD CMs compared to scramble CMs (Figure 7F). In addition, abnormal beating was observed in SETD7 KD CMs (Figure 7G and 7H).
Figure 7. SETD7 is critical for calcium handling properties of mature cardiomyocytes.

(A) SETD7 expression at various days of hESC-CM differentiation (n=4). (B–C) RT-qPCR of SETD7 and TNNT2 expression levels in hESC, hESC-CMs, fetal heart, and adult heart tissues. (D) Immunoblot analysis of SETD7 in control and SETD7 knock-down lines. (E) Cell viability assay of control and SETD7 knock-down lines. (F) Standard deviation of peak intervals of control and SETD7 knock-down lines. Motion vector from movie images was recorded and analyzed with SONY SI8000 cell motion imaging system (n=18). (G) Total number of wells with arrhythmic-beating hESC-CMs from control and SETD7 knock-down lines. (H) Representative traces of the motion velocity of hESC-CMs infected with scramble or SETD7 shRNA. (I) Immuno-staining of Cxn43, α-Actinin and TNNT2 in control and SETD7 knock-down CMs. (J) Representative ratio-metric calcium imaging traces of control and SETD7 knock-down hESC-CMs. (K–Q) Calcium handling parameters of scramble and SETD7 shRNA lines (n>30). (R–T) Relative RNA expression of SETD7 and calcium handling genes in scramble and shRNA SETD7 groups. (U) ChIP-qPCR of SETD7 and IgG enrichment on TNNT2, MYH6, and CASQ2 genes in hESC-CMs (n=3). Statistical significance obtained by one-way ANOVA. *P < 0.05; **P < 0.01; ***P < 0.005 (mean ± SEM).
In SETD7 KD CMs, no abnormal alignment of sarcomeric protein or mis-localization of connexin 43 was detected, a phenotype frequently observed in hypertrophic iPSC-CMs or in damaged CMs (Figure 7I) (Burridge et al., 2016; Kostin et al., 2003). SETD7 KD did, however, result in irregular patterns of calcium transients, a major physiologic indicator of myocardial function (Figure 7J). While the diastolic calcium ratios were similar in scramble and SETD7 shRNA lines, the peak calcium ratios were significantly attenuated in SETD7 shRNA group, indicating a decreased calcium amplitude (Figures 7K and 7L). The temporal parameters such as decay tau, time to peak, and transient durations were significantly elongated in SETD7 KD lines compared to the scramble line (Figures 7M–7O). Similarly, reduced calcium maximum rise rate (Figure 7P) and irregular calcium transients were observed in the SETD7 KD line (Figure 7Q). These data indicate SETD7 affects calcium handling of terminally differentiated CMs.
To explore the role of SETD7 in modulation of calcium handling properties, we measured the expression level of major calcium handling genes in hESC-CMs. RT-qPCR showed the sarcoplasmic reticulum (SR) calcium binding protein CASQ2 was significantly reduced in SETD7 KD CMs compared to control CMs (Figures 7R and 7S). CASQ2, by binding to the free calcium ion in SR, regulates calcium load in the sarcoplasmic reticulum and contribute to regular calcium handling in CMs. The expression of ryanodine receptor 2 (RYR2), a major regulator of calcium release from SR, was increased in SETD7 KD hESC-CMs (Figure 7T). Lower CASQ2 level in the SR down-regulated the SR calcium storage capacity, while higher RYR2 expression level contributed to spontaneous calcium release events from SR. Both the changes enhanced the probability of arrhythmia-like calcium handling events as we observed in SETD7-KD iPSC-CMs. To confirm the direct transcriptional regulation of SETD7 on CASQ2 gene, we further measured the direct binding of SETD7 to the CASQ2 genomic region using ChIP-qPCR. Our results showed a greater binding capacity of SETD7 on the genebody region of CASQ2 compared to that of IgG control, but lesser than the binding of SETD7 on MYH6 and TNNT2 (Figure 7U). These data suggest that SETD7 can partially interfere CASQ2 expression in CMs. Taken together, our data show that SETD7 modulates calcium handling properties of terminally differentiated CMs by regulating SR-related genes such as CASQ2 and RYR2.
DISCUSSION
Our study identifies SETD7 as a key regulator of CM differentiation. ChIP-seq analysis of SETD7 at each stage of CM differentiation identified its stage-specific target genes and co-factors. We found SETD7 is essential for the transcriptional regulation of key regulatory genes for cardiac lineage commitment, demonstrating the indispensable role of SETD7 in CM differentiation.
As co-factors in the SWI/SNF chromatin remodeling complex, BAF60 and BRG1 are known to be crucial activators of cardiac regulatory genes during early cardiac development (Chen et al., 2012; Takeuchi et al., 2011). However, a recent study showed BRG1 possesses a dual role for cardiac mesoderm specification by repressing non-mesoderm genes through recruitment of the polycomb complex to target promoters (Alexander et al., 2015). These studies indicate the function of BRG1 in early cardiac development may differ by its co-factors. Our study showed SETD7 specifically interacts with BRG1 in the mesoderm stage and recruits it to the promoters of key regulatory genes for mesoderm lineage commitment.
Moreover, we show SETD7 partially shares its target genes with other cardiac TFs in the CM stages. We found a strong interaction between SETD7 and NKX2-5 and co-occupancy on the promoter region of target genes in CMs. A positive feedback loop between SETD7 and NKX2-5 was observed, indicating SETD7 serves as a critical component of major cardiac TF complexes and contributes to the transcriptional regulation of cardiac specific genes during CM differentiation.
H3K36 methylation is known to play a critical role in gene transcription activation (Bannister et al., 2005) and is considered a marker of gene transcription. H3K36 methylation is present in major cellular processes such as gene silencing, DNA damage repair, and DNA replication and its diverse function is dependent on its binding protein (Brien et al., 2012; Musselman et al., 2012; Suzuki et al., 2016). Here we show SETD7 recognizes H3K36 methylation during the hESC-CM differentiation. Peptide array and IP assay demonstrate SETD7 has a high affinity with H3K36me3. The co-occupancy of SETD7 and H3K36me3 on the genebody region is highly correlated with gene activation during cardiac differentiation, suggesting SETD7 recognizes H3K36 methylation and regulates the transcription of target genes. We did not detect methylation activity of SETD7 on H3K36 residues, KD or knock-out of SETD7 showed no effect on H3K36me3 level of its target genes. These data suggest SETD7 recognizes H3K36 residue as a binding target but not as a substrate for methylation.
At the transcriptional level, H3K36me3 is mainly mediated by SETD2 that conjugates with active Pol II during transcription elongation. This enrichment of H3K36me3 resulted in the suppression of cryptic transcription of target genes. However, KD of SETD2 remarkably reduced H3K36me3 levels of most of genes, whereas the effects of reduced H3K36me3 on gene transcription varied. In addition, H3K36me3 enrichment on the endoderm-specific gene is necessary to transcription of its target gene during endoderm specification (Zhang et al., 2014). H3K36me3 may thus be involved not only in the suppression of cryptic transcription, but also in the regulation of active gene transcription. In this study, we found the depletion of SETD7 exerted no significant effects on H3K36me3 level but instead reduced binding of Pol II-S2P on genebody and TES regions of SETD7 target genes. Taken together, SETD7 acts as a key regulator factor for active function of H3K36me3 in transcriptional regulation.
Given the critical role of SETD7 in CM differentiation, we next examined whether it modulates physiological function of terminally differentiated CMs in vitro and in adult human heart in vivo. Here, we found the loss of SETD7 resulted in abnormal calcium handling of CMs, driven by abnormal expression of CASQ2 and RYR2 expression. In the heart tissue of healthy donors, we confirmed SETD7 co-localizes with the H3K36me3 marker in the cardiac genes as we observed in hESC-CMs, suggesting SETD7 expression may be required for the maintenance of cardiac specific genes in the adult heart.
In addition to the critical function of SETD7 in hESC-CM differentiation, our data indicate a role of SETD7 as a regulator of general transcriptional activation. We found SETD7 is highly expressed in other tissues and cell types such as hepatocyte-like cells and NPCs. Interestingly, SETD7 is continuously up-regulated during the differentiation process of hepatocytes and NPCs and modulates expression of lineage-specific genes in both cell types. Similarly, ablation of SETD7 disrupted expression of endothelial genes in ESC-ECs, suggesting a role of SETD7 in endothelial function. Moreover, our ChIP-seq of SETD7 in hESC stage showed genomic binding of SETD7 on broad genebody regions despite its low expression in hESC stage.
In conclusion, our study elucidates the multifaceted role of SETD7 in cardiac development and identify SETD7 as a co-factor for H3K36me3 in transcriptional regulation of cardiac genes. We describe the role of epigenetic modifying enzyme in complex transcriptional regulatory mechanisms of cardiac gene expression, which can be utilized to develop therapeutic solutions for congenital heart disease and to improve methodologies for generating chamber-specific human pluripotent stem cell-derived CMs.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph Wu (joewu@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary human samples
The protocols used in this study were approved by the Institutional Review Board (IRB) at Stanford University. The written consent was obtained from all the patients involved in this
| Sample | Gender | Age | Condition | Tissue quired |
|---|---|---|---|---|
| * Fetal tissue samples | ||||
|
| ||||
| Fetus #1 (F1) | Male | 16.4 week | abortion | Brain, Pancreas, Liver, Stomach, Kidney, Heart, Skeletal muscle, |
| Fetus #2 (F2) | Female | 17.0 week | abortion | Brain, Pancreas, Liver, Stomach, Kidney, Heart, Skeletal muscle, |
|
| ||||
| * Adult tissue samples | ||||
|
| ||||
| Individual #1 (A1) | Male | 45 | CVAa | Left Ventricle |
| Individual #2 (A2) | Female | 58 | CVA | Left Ventricle |
| Individual #3 (A3) | Male | 22 | CVA | Left Ventricle |
| Individual #4 (A4) | Female | 74 | CVA | Left Ventricle |
| Individual #5 (A5) | Female | 45 | CVA | Left Ventricle |
| Individual #6 (A6) | Male | 67 | CVA | Left Ventricle |
CVA: Cerebrovascular accident
study.
hESC lines and culture condition
H7 hESC line was obtained from WiCell Research Institute (WA07). Dr. Edouard G Stanley kindly provided HES3NKX2-5eGFP/w human ESC line. Human ESC lines (H7 and HES3) were maintained on Matrigel-coated plates (BD Biosciences) in Essential 8 Medium (GIBCO, Life Technology). The medium was changed every day.
Transcription activator-like effector nuclease (TALEN)-mediated gene knock-out ESC lines
TALEN pair vectors were designed to target start codon of SETD7 and constructed using rapid TALEN assembly system as previously described (Ding et al., 2013). Both TALEN pair vectors were delivered into HES3NKX2-5eGFP/w hESC line by nucleofection using P3 Primary Cell 4D-Nucleofector X Kit (Lonza). After 48 hrs following nucleofection, transfected cells were enriched using fluorescence-activated cell sorting for double positive cells (GFP+/RFP+). Subsequently, sorted cells were expanded, manually picked and genotyped by direct sequencing of the PCR products and TOPO-cloned PCR products.
Lentivirus production and generation of stable cell line
The lentiviral shRNA plasmid sets–pLKO control, pLKO-shSETD7 (Clone ID: 030648.2-985s21c1), pTRIPZ shControl, pTRIPZ SETD7 sh1 (Clone ID: V2THS_230143) and pTRIPZ SETD7 sh2 (Clone ID: V2THS_99174)–were purchased from Dharmacon and Sigma-Aldrich. The HEK293T cells were plated in 10 cm dish and transfected with target plasmid and packaging plasmids (pMD2G and pPAX2) using Lipofectamine 2000 according to the manufacturer’s protocol (Life technologies; 11668019). The transfected HEK293T cells incubated for 3 days and medium was collected every day. Collected medium was centrifuged at 3000 g for 15 min to remove cells and cell debris. Supernatant containing virus was concentrated by PEG-it virus concentrator solution according to the manufacturer’s protocol (System Bioscience; LV810A). H7 or HES3NKX2-5eGFP/w hESCs were seeded onto 6-well plates and infected with lentivirus containing the shRNA for SETD7. Cells expressing control or SETD7 shRNA were selected by puromycin (2 µg/ml) treatment for 2–3 days.
METHOD DETAILS
Differentiation of human PSCs to cardiomyocytes
For cardiac differentiation, a chemically defined monolayer differentiation protocol was used as previously described (Ebert et al., 2014; Wu et al., 2015). Briefly, iPSCs at ~90% confluence were incubated with differentiation basal medium comprising RPMI 1640 medium (Invitrogen) and B27 supplement minus insulin (Invitrogen). CHIR99021, a selective glycogen synthase kinase 3β inhibitor, was added to the differentiation basal medium. On day 2, medium was removed and replaced with differentiation basal medium minus CHIR99021. On day 3, the Wnt antagonist, IWR-1, was added to the medium. After 48 hrs, medium was removed and replaced with differentiation basal medium without any inhibitors. On day 7, the cells were incubated with complete cardiomyocyte medium consisting of RPMI 1640 medium and B27 supplement plus insulin (Invitrogen). The medium was changed every 2 days. Monolayers of cardiomyocytes derived from iPSCs (iPSC-CMs) were cultured for ~30 days and subsequently dissociated for experimental use using TrypLE Express (Life Technologies). Specific day time point of selected four different stages are day 0 (hESC), day 2 (MES), day 5 (CP), day 9 (CM), and day 30~40 (purified CM).
Immunoblotting and immunoprecipitation
For immunoblotting, each sample was subjected to electrophoresis on 4–12% NuPAGE Bis–Tris gradient gels (Life Technology; NP0335) and proteins were transferred to polyvinylidene difluoride membranes using wet-based transfer system (Bio-Rad). Membranes were incubated overnight with the indicated primary antibodies, followed by incubation for 1 hr with horseradish peroxidase-conjugated secondary antibody (Abcam; ab131366). Signals were detected by chemiluminescence. For immunoprecipitation, cells were harvested and lysed in IP lysis buffer (Thermo Scientific; 87788). Lysates were centrifuged for 10 min at 10,000 g at 4 °C. Specific antibodies conjugated with magnetic protein G beads (Life Technology; 10004D) (or agarose beads (Active Motif; 53039)) were added to supernatants for overnight incubation at 4 °C. Subsequently, beads were pelleted for 1 min at 7,000 r.p.m. and washed twice with IP lysis buffer. Proteins were eluted in 2× Laemmli buffer (Bio-Rad) or 4× LDS sample buffer (Life Technology; NP0007) by boiling for 10 min. Immunoprecipitated proteins were subjected to immunoblotting using horseradish peroxidase-conjugated primary antibodies or VeriBlot of IP Detection Reagent (Abcam; ab131366).
RNA extraction and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using RNeasy Plus Mini Kit or miRNeasy Mini Kit (Qiagen; 74134, 217004). Reverse transcription was performed using iScript™ cDNA Synthesis Kit (Bio-Rad; 1708890) and qRT-PCR was performed using iQ® SYBR™ Green Supermix (Bio-Rad; 170-8880) or TaqMan® Universal PCR Master Mix (Applied Biosystems; 4304437) on CFX96 real-time PCR detector (Bio-Rad). Relative mRNA levels were normalized to those of 18S or GAPDH mRNA in each reaction. Three or Four biological replicates per group were used for qRT-PCR.
Chromatin immunoprecipitation and ChIP-seq
Antibodies were used as described above. Each antibody was incubated with Dynabeads (Life Technology; 10003D) for 12 hr at 4 °C. A small portion of the crosslinked, sheared chromatin was saved as Input and the remainder was employed for the immunoprecipitation using antibody conjugated Dynabeads. After overnight incubation at 4 °C, incubated beads were rinsed with sonication buffer (50 mM Hepes pH 7.9, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 0.5 mM PMSF), high salt buffer (50 mM Hepes pH 7.9 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 0.5 mM PMSF) and LiCl buffer (20 mM Tris, pH 8.0, 1 mM EDTA, 250 mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate, 0.5 mM PMSF). Washed beads were incubated with elution buffer (50 mM Tris, pH 8.0, 1 mM EDTA, 1% SDS, 50 mM NaHCO3) for 1 hr at 65 °C and de-crosslinked with 5 M NaCl for overnight at 65 °C. Immunoprecipitated DNA was treated with Rnase A and Proteinase K and purified by ChIP DNA clean and concentrator (Zymo Research; D5205).
Intracellular calcium imaging
Single iPSC-CMs were plated onto Matrigel-coated coverglass (CS-24/50, Warner Instruments, Inc.) at a density of ~10,000 cells per square centimeter. Cells were allowed to recover for 3–4 days and loaded with 5 µM Fura-2 AM (Thermo Fisher Scientific) in Tyrode’s solution (140 mM NaCl, 5.4 mM KCl, 1 mM MgCl2, 10 mM glucose, 1.8 mM CaCl2, and 10 mM HEPES, pH adjusted to 7.4 with NaOH at room temperature) for 30 min at room temperature. Cells were imaged on a customized Ti-S/L 100 Inverted Microscope based imaging platform with 40x oil immersion objective (CFI SUPER FLUOR, NA 1.30 WD .22). Bipolar pulse was used to pace cells at 0.5 and 1 HZ. Cells were kept at 37 °C while recording. Fura-2 signals were captured in high frame rate video recording mode (512 × 512 pixels) at a speed of 50 frames per second. Videos were analyzed with NIS Elements: Advanced Research Software (Nikon) and raw ratio-pair data were further processed with custom-made script based on Interactive Digital Language.
Immunofluorescence staining and confocal microscopy
hESC-CMs were fixed in 4% paraformaldehyde in phosphate buffered saline. Following permeabilization with 0.3% Triton X-100, CMs were stained with primary antibodies against cardiac troponin T type 2 (TNNT2; Thermo Scientific and Abcam) and SETD7 (Abcam). After reaction with the primary antibodies, cells were incubated with the appropriate Alexa Fluor-conjugated secondary antibodies (Santa Cruz Biotechnology or Life Technologies). Images of the stained cells were obtained under a brightfield microscope (Leica). Confocal images were taken by using a 63× Plan-Apochromat oil immersion objective (Carl Zeiss) and a LSM 510 Meta confocal microscope (Carl Zeiss). Images were analyzed by using ZEN software (Carl Zeiss) and ImageJ software (National Institutes of Health).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses for each experiment are described in the figure legends or in the appropriate text. Multiple group comparisons were calculated using one-way ANOVA. Pairwise comparisons were carried out using the two-tailed unpaired Student’s t-test. *P < 0.05; **P < 0.01; ***P < 0.005. All error bars are defined as S.E.M. unless indicated.
Chromatin immunoprecipitation and ChIP-seq analysis
ChIP-seq data were aligned to the human genome (hg19) or mouse genome (mm10) by Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) with only uniquely aligned reads kept. A;l duplicate reads were removed by SAMtools (http://www.htslib.org/). FastQC was applied for quality control of sequencing (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). PhantomPeak was applied for quality control of ChIP-seq (https://code.google.com/archive/p/phantompeakqualtools/). For all samples generated, normalized strand coefficient (NSC) was above 1.05 and relative strand correlation (RSC) was above 0.8. The aforementioned pre-processing scripts are available online (https://github.com/ny-shao/chip-seq_preprocess). The genebody annotations of protein coding genes were extracted from ENSEMBL V75 and bedtools were applied to extract the aligned reads in genebody regions for all ChIP-seq samples (http://bedtools.readthedocs.org/en/latest/). The counts tables of SETD7 and the histone marks of coding genes were fed to DESeq for estimation of fold changes of reads enrichment in time series and genes with significant enrichment changes (adjusted P-value < 0.1, fold-change > 2) in pairwise comparisons were selected for downstream analyses (http://bioconductor.org/packages/release/bioc/html/DESeq.html). Fold changes were clustered by K-means (k=5) clustering in R. Functional enrichment analyses were implemented by GeneAnswer package of Bioconductor (https://www.bioconductor.org/packages/release/bioc/html/GeneAnswers.html). Gene regulatory network was generated by iRegulon package (http://iregulon.aertslab.org/) in Cytoscape (http://www.cytoscape.org/). Average profile and heatmaps of the genomic features of ChIP-seq were generated by ngs.plot (https://github.com/shenlab-sinai/ngsplot) (Shen et al., 2014). Orthologs of human and mouse were downloaded from Mouse Genome Informatics (MGI, http://www.informatics.jax.org/homology.shtml) and genes were sorted based on the enrichment density of SETD7 calculated by ngs.plot normalized to reads per millions. The tracks screenshots of sequencing data were generated by IGV browser (https://www.broadinstitute.org/igv/). Corrgram package in R software was used to calculate and visualize correlation among different chromatin remodelers and histone marks.
RNA-seq analysis
RNA-seq data generated by Illumina sequencer were assembled by STAR (https://github.com/alexdobin/STAR) to the human genome (hg19). The Ampliseq data were processed by Ion Torrent server. The transcriptome annotation was downloaded from GENCODE project (V19) (http://www.gencodegenes.org/). Fold changes of expression levels were estimated by DESeq as aforementioned in ChIP-seq analyses. Functional enrichment analyses were implemented by GeneAnswers package.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq and ChIP-seq data generated in this paper is GSE107785.
Supplementary Material
Table S1 (Related to Figure 1). Clustered gene list with relative SETD7 enrichment values.
Table S2 (Related to Figure 4). Primer sequence and genomic location.
Document S1. Figures S1–S7
Highlights.
SETD7 targets distinct gene sets at discrete stages of cardiomyocyte specification
SETD7 interacts with diverse co-factors during cardiomyocyte differentiation
SETD7 reads H3K36me3 on gene bodies
SETD7 is required for calcium handling in terminally-differentiated cardiomyocytes
Acknowledgments
We would like to thank Edouard G Stanley for NKX2-5eGFP/w hESCs lines. We also thank Ates Akgun for generating hESC-CMs, Angelos Oikonomopoulos and Chun Liu for protocol of hepatocyte differentiation, Soah Lee for Cxn43 staining, and Sang-Ging Ong for helpful comments. This study is supported by the American Heart Association 17MERIT33610009, National Institutes of Health R01 HL113006, NIH R01 HL126527, NIH R01 HL128170, NIH R01 HL130020 (JCW); NIH T32 EB009035 (DTP); California Institute of Regenerative Medicine GC1R-06673-A (MPS) and RT3-07798 (JCW); and National Research Foundation of Korea 2012R1A6A3A03039821 (JL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONs
Conceptualization, J.L. and J.C.W; Methodology, V.T.; Formal Analysis, N.S., J.C. and Y.Z.; Investigation, J.L., M.Z., H.G., Y.K., T.L, H.W. and D.T.P.; Data Curation, N.S., J.C. and Y.Z.; Writing – Original Draft, J.L.; Writing – Review & Editing, J.L., D.T.P. and K.D.W.; Resources, K.D.W.; Supervision, M.S. and J.C.W.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Alexander JM, Hota SK, He D, Thomas S, Ho L, Pennacchio LA, Bruneau BG. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development. 2015;142:1418–1430. doi: 10.1242/dev.109496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Spatial Distribution of Di- and Tri-methyl Lysine 36 of Histone H3 at Active Genes. J. Biol. Chem. 2005;280:17732–17736. doi: 10.1074/jbc.M500796200. [DOI] [PubMed] [Google Scholar]
- Barsyte-Lovejoy D, Li F, Oudhoff MJ, Tatlock JH, Dong A, Zeng H, Wu H, Freeman SA, Schapira M, Senisterra GA, et al. (R)-PFI-2 is a potent and selective inhibitor of SETD7 methyltransferase activity in cells. Proc. Natl. Acad. Sci. 2014;111:12853–12858. doi: 10.1073/pnas.1407358111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brien GL, Gambero G, O’Connell DJ, Jerman E, Turner SA, Egan CM, Dunne EJ, Jurgens MC, Wynne K, Piao L, et al. Polycomb PHF19 binds H3K36me3 and recruits PRC2 and demethylase NO66 to embryonic stem cell genes during differentiation. Nat. Struct. Mol. Biol. 2012;19:1273–1281. doi: 10.1038/nsmb.2449. [DOI] [PubMed] [Google Scholar]
- Burridge PW, Keller G, Gold JD, Wu JC. Production of De Novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell Stem Cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Sharma A, Wu JC. Genetic and epigenetic regulation of human cardiac reprogramming and differentiation in regenerative medicine. Annu. Rev. Genet. 2015;49:461–484. doi: 10.1146/annurev-genet-112414-054911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burridge PW, Li YF, Matsa E, Wu H, Ong S-G, Sharma A, Holmström A, Chang AC, Coronado MJ, Ebert AD, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016;22:547–556. doi: 10.1038/nm.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño J, Morera C, Sesé B, Boue S, Bonet-Costa C, Martí M, Roque A, Jordan A, Barrero MJ. SETD7 regulates the differentiation of human embryonic stem cells. PLoS ONE. 2016;11:e0149502. doi: 10.1371/journal.pone.0149502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-P, Bruneau BG. Epigenetics and cardiovascular development. Annu. Rev. Physiol. 2012;74:41–68. doi: 10.1146/annurev-physiol-020911-153242. [DOI] [PubMed] [Google Scholar]
- Chen L, Fulcoli FG, Ferrentino R, Martucciello S, Illingworth EA, Baldini A. Transcriptional control in cardiac progenitors: Tbx1 interacts with the BAF chromatin remodeling complex and regulates Wnt5a. PLoS Genet. 2012;8:e1002571. doi: 10.1371/journal.pgen.1002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- Ding Q, Lee Y-K, Schaefer EAK, Peters DT, Veres A, Kim K, Kuperwasser N, Motola DL, Meissner TB, Hendriks WT, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12:238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Kodo K, Liang P, Wu H, Huber BC, Riegler J, Churko J, Lee J, Almeida Pde, Lan F, et al. Characterization of the molecular mechanisms underlying increased ischemic damage in the aldehyde dehydrogenase 2 genetic polymorphism using a human induced pluripotent stem cell model system. Sci. Transl. Med. 2014;6:255ra130. doi: 10.1126/scitranslmed.3009027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DA, Braam SR, Koutsis K, Ng ES, Jenny R, Lagerqvist EL, Biben C, Hatzistavrou T, Hirst CE, Yu QC, et al. NKX2-5eGFP/w hESCs for isolation of human cardiac progenitors and cardiomyocytes. Nat. Methods. 2011;8:1037–1040. doi: 10.1038/nmeth.1740. [DOI] [PubMed] [Google Scholar]
- Estève P-O, Chin HG, Benner J, Feehery GR, Samaranayake M, Horwitz GA, Jacobsen SE, Pradhan S. Regulation of DNMT1 stability through SET7-mediated lysine methylation in mammalian cells. Proc. Natl. Acad. Sci. 2009;106:5076–5081. doi: 10.1073/pnas.0810362106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Zhang L, Wei W, Jin X, Wang P, Tong Y, Li J, Du JX, Wong J. A methylation-phosphorylation switch determines Sox2 stability and function in ESC maintenance or differentiation. Mol. Cell. 2014;55:537–551. doi: 10.1016/j.molcel.2014.06.018. [DOI] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Shao N-Y, Sa S, Li D, Termglinchan V, Ameen M, Karakikes I, Sosa G, Grubert F, Lee J, et al. Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell. 2017;20:490–504.e5. doi: 10.1016/j.stem.2016.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, Zhou B, Chang C-P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Kunihiro T, Ando T, Kobayashi S, Matsui E, Yada H, Kanda Y, Kurokawa J, Furukawa T. Image-based evaluation of contraction-relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: Correlation and complementarity with extracellular electrophysiology. J. Mol. Cell. Cardiol. 2014;77:178–191. doi: 10.1016/j.yjmcc.2014.09.010. [DOI] [PubMed] [Google Scholar]
- He A, Kong SW, Ma Q, Pu WT. Co-occupancy by multiple cardiac transcription factors identifies transcriptional enhancers active in heart. Proc. Natl. Acad. Sci. U. S. A. 2011;108:5632–5637. doi: 10.1073/pnas.1016959108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z-Q. A role for cofactor-cofactor and cofactor-histone interactions in targeting p300, SWI/SNF and Mediator for transcription. EMBO J. 2003;22:2146–2155. doi: 10.1093/emboj/cdg219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakikes I, Termglinchan V, Cepeda DA, Lee J, Diecke S, Hendel A, Itzhaki I, Ameen M, Shrestha R, Wu H, et al. A comprehensive TALEN-based knockout library for generating human induced pluripotent stem cell-based models for cardiovascular diseases. Circ. Res. 2017;120:1561–1571. doi: 10.1161/CIRCRESAHA.116.309948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-D, Kim E, Koun S, Ham H-J, Rhee M, Kim M-J, Huh T-L. Proper activity of histone H3 lysine 4 (H3K4) methyltransferase is required for morphogenesis during zebrafish cardiogenesis. Mol. Cells. 2015;38:580–586. doi: 10.14348/molcells.2015.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-K, Lee H, Han K, Kim SC, Choi Y, Park S-W, Bak G, Lee Y, Choi JK, Kim T-K, et al. SET7/9 methylation of the pluripotency factor LIN28A is a nucleolar localization mechanism that blocks let-7 biogenesis in human ESCs. Cell Stem Cell. 2014;15:735–749. doi: 10.1016/j.stem.2014.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Nam HJ, Lee J, Park DY, Kim C, Yu YS, Kim D, Park SW, Bhin J, Hwang D, et al. Methylation-dependent regulation of HIF-1α stability restricts retinal and tumour angiogenesis. Nat. Commun. 2016;7:10347. doi: 10.1038/ncomms10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofent J, Zhang J, Spagnoli FM. The histone methyltransferase Setd7 promotes pancreatic progenitor identity. Development. 2016;143:3573–3581. doi: 10.1242/dev.136226. [DOI] [PubMed] [Google Scholar]
- Kontaki H, Talianidis I. Lysine methylation regulates E2F1-induced cell death. Mol. Cell. 2010;39:152–160. doi: 10.1016/j.molcel.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Kopljar I, De Bondt A, Vinken P, Teisman A, Damiano B, Goeminne N, Van den Wyngaert I, Gallacher DJ, Lu HR. Chronic drug-induced effects on contractile motion properties and cardiac biomarkers in human induced pluripotent stem cell-derived cardiomyocytes. Br. J. Pharmacol. 2017;174:3766–3779. doi: 10.1111/bph.13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klövekorn W-P, Bauer EP, Schaper J. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol. Cell. Biochem. 2003;242:135–144. [PubMed] [Google Scholar]
- Kouskouti A, Scheer E, Staub A, Tora L, Talianidis I. Gene-specific modulation of TAF10 function by SET9-mediated methylation. Mol. Cell. 2004;14:175–182. doi: 10.1016/s1097-2765(04)00182-0. [DOI] [PubMed] [Google Scholar]
- Li H, Zhong Y, Wang Z, Gao J, Xu J, Chu W, Zhang J, Fang S, Du SJ. Smyd1b is required for skeletal and cardiac muscle function in zebrafish. Mol. Biol. Cell. 2013;24:3511–3521. doi: 10.1091/mbc.E13-06-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna-Zurita L, Stirnimann CU, Glatt S, Kaynak BL, Thomas S, Baudin F, Samee MAH, He D, Small EM, Mileikovsky M, et al. Complex interdependence regulates heterotypic transcription factor distribution and coordinates cardiogenesis. Cell. 2016;164:999–1014. doi: 10.1016/j.cell.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Avvakumov N, Watanabe R, Abraham CG, Lalonde M-E, Hong Z, Allen C, Roy S, Nuñez JK, Nickoloff J, et al. Molecular basis for H3K36me3 recognition by the Tudor domain of PHF1. Nat. Struct. Mol. Biol. 2012;19:1266–1272. doi: 10.1038/nsmb.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin M, Gildersleeve H, Beck AE, Tabor HK, Cooper GM, Mefford HC, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat. Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimura K, Ura K, Shiratori H, Ikawa M, Okabe M, Schwartz RJ, Kaneda Y. A histone H3 lysine 36 trimethyltransferase links Nkx2-5 to Wolf-Hirschhorn syndrome. Nature. 2009;460:287–291. doi: 10.1038/nature08086. [DOI] [PubMed] [Google Scholar]
- Ogiwara H, Ui A, Otsuka A, Satoh H, Yokomi I, Nakajima S, Yasui A, Yokota J, Kohno T. Histone acetylation by CBP and p300 at double-strand break sites facilitates SWI/SNF chromatin remodeling and the recruitment of non-homologous end joining factors. Oncogene. 2011;30:2135–2146. doi: 10.1038/onc.2010.592. [DOI] [PubMed] [Google Scholar]
- Okabe J, Orlowski C, Balcerczyk A, Tikellis C, Thomas MC, Cooper ME, El-Osta A. Distinguishing hyperglycemic changes by Set7 in vascular endothelial cells. Circ. Res. 2012;110:1067–1076. doi: 10.1161/CIRCRESAHA.112.266171. [DOI] [PubMed] [Google Scholar]
- Oudhoff MJ, Freeman SA, Couzens AL, Antignano F, Kuznetsova E, Min PH, Northrop JP, Lehnertz B, Barsyte-Lovejoy D, Vedadi M, et al. Control of the Hippo pathway by Set7-dependent methylation of Yap. Dev. Cell. 2013;26:188–194. doi: 10.1016/j.devcel.2013.05.025. [DOI] [PubMed] [Google Scholar]
- Paige SL, Thomas S, Stoick-Cooper CL, Wang H, Maves L, Sandstrom R, Pabon L, Reinecke H, Pratt G, Keller G, et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell. 2012;151:221–232. doi: 10.1016/j.cell.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Li G, Rajarajan K, Hamaguchi R, Burridge PW, Wu SM. Derivation of highly purified cardiomyocytes from human induced pluripotent stem cells using small molecule-modulated differentiation and subsequent glucose starvation. J. Vis. Exp. 2015:e52628. doi: 10.3791/52628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, Nestler E. ngs. plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014;15:284. doi: 10.1186/1471-2164-15-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Cai J, Liu Y, Zhao D, Yong J, Duo S, Song X, Guo Y, Zhao Y, Qin H, et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:cr2009107. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- Subramanian K, Jia D, Kapoor-Vazirani P, Powell DR, Collins RE, Sharma D, Peng J, Cheng X, Vertino PM. Regulation of estrogen receptor a by the SET7 lysine methyltransferase. Mol. Cell. 2008;30:336–347. doi: 10.1016/j.molcel.2008.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Kato H, Suzuki Y, Chikashige Y, Hiraoka Y, Kimura H, Nagao K, Obuse C, Takahata S, Murakami Y. Histone H3K36 trimethylation is essential for multiple silencing mechanisms in fission yeast. Nucleic Acids Res. 2016;44:4147–4162. doi: 10.1093/nar/gkw008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, Delgado-Olguín P, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nat. Commun. 2011;2:187. doi: 10.1038/ncomms1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallaster M, Vallaster CD, Wu SM. Epigenetic mechanisms in cardiac development and disease. Acta Biochim. Biophys. Sin. 2012;44:92–102. doi: 10.1093/abbs/gmr090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh S, Smolle M, Li H, Gogol MM, Saint M, Kumar S, Natarajan K, Workman JL. Set2 methylation of histone H3 lysine 36 suppresses histone exchange on transcribed genes. Nature. 2012;489:452–455. doi: 10.1038/nature11326. [DOI] [PubMed] [Google Scholar]
- Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, Ding H, Wylie JN, Pico AR, Capra JA, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151:206–220. doi: 10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Cao R, Xia L, Erdjument-Bromage H, Borchers C, Tempst P, Zhang Y. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell. 2001;8:1207–1217. doi: 10.1016/s1097-2765(01)00405-1. [DOI] [PubMed] [Google Scholar]
- Wu H, Lee J, Vincent LG, Wang Q, Gu M, Lan F, Churko JM, Sallam KI, Matsa E, Sharma A, et al. Epigenetic regulation of phosphodiesterases 2A and 3A underlies compromised β-adrenergic signaling in an iPSC model of dilated cardiomyopathy. Cell Stem Cell. 2015;17:89–100. doi: 10.1016/j.stem.2015.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–1148. doi: 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xie S, Zhou Y, Xie Y, Liu P, Sun M, Xiao H, Jin Y, Sun X, Chen Z, et al. H3K36 histone methyltransferase Setd2 is required for murine embryonic stem cell differentiation toward endoderm. Cell Rep. 2014;8:1989–2002. doi: 10.1016/j.celrep.2014.08.031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 (Related to Figure 1). Clustered gene list with relative SETD7 enrichment values.
Table S2 (Related to Figure 4). Primer sequence and genomic location.
Document S1. Figures S1–S7