

Abstract
Under diabetic conditions, sodium–glucose cotransporter 2 (SGLT2) for glucose uptake in proximal tubules (PTs) increases, whereas NAD+-dependent protein deacetylase silent mating type information regulation 2 homolog 1 (Sirtuin-1; SIRT1) for PT survival decreases. Therefore, we hypothesized that increased glucose influx by SGLT2 reduces SIRT1 expression. To test this hypothesis, db/db mice with diabetes and high-glucose (HG)-cultured porcine PT LLC-PK1 cells in a two-chamber system were treated with the SGLT2 inhibitor canagliflozin. We also examined SIRT1 and SGLT2 expression in human kidney biopsies. In db/db mice, SGLT2 expression increased with concomitant decreases in SIRT1, but was inhibited by canagliflozin. For determination of the polarity of SGLT2 and SIRT1 expression, LLC-PK1 cells were seeded into Transwell chambers (pore size, 0.4 µm; Becton Dickinson, Oxford, UK). HG medium was added to either or to both of the upper and lower chambers, which corresponded to the apical and basolateral sides of the cells, respectively. In this system, the lower chamber with HG showed increased SGLT2 and decreased SIRT1 expression. Canagliflozin reversed HG-induced SIRT1 downregulation. Gene silencing and inhibitors for glucose transporter 2 (GLUT2) blocked HG-induced SGLT2 expression upregulation. Gene silencing for the hepatic nuclear factor-1α (HNF-1α), whose nuclear translocation was enhanced by HG, blocked HG-induced SGLT2 expression upregulation. Similarly, gene silencing for importin-α1, a chaperone protein bound to GLUT2, blocked HG-induced HNF-1α nuclear translocation and SGLT2 expression upregulation. In human kidney, SIRT1 immunostaining was negatively correlated with SGLT2 immunostaining. Thus, under diabetic conditions, SIRT1 expression in PTs was downregulated by an increase in SGLT2 expression, which was stimulated by basolateral HG through activation of the GLUT2/importin-α1/HNF-1α pathway.
Introduction
Diabetic nephropathy (DN) is a complication of diabetes mellitus that causes end-stage renal disease1. Pathological manifestations of DN include glomerular and tubulointerstitial changes2. Tubules often represent the primary site for changes in DN3. Reduction in silent mating type information regulation 2 homolog 1 (Sirtuin-1; SIRT1) expression in proximal tubules (PTs) precedes that in podocytes, which constitutes an early event in patients with diabetes4. This downregulation of SIRT1 expression in PTs may be a prodrome of glomerular damages in DN. Nicotinamide mononucleotide, a SIRT1-related metabolite of nicotinic acid metabolism5, levels are reduced because of decreased SIRT1 levels in PTs, which damage podocytes. We describe this disease propagation process as “tubule-podocyte interplay.” We have previously reported that high-glucose (HG) medium reduced SIRT1 levels in cultured PTs4. The underlying mechanisms by which HG conditions regulate SIRT1 expression in PTs in diabetes remain unclear.
Sodium–glucose co-transporters (SGLT) mediate glucose reabsorption and cellular glucose entry. SGLTs are expressed on the apical site of PTs, with upregulated expression in db/db mice6. The sodium–glucose cotransporter (SGLT) comprises two isoforms: SGLT1 and SGLT2. The SGLT2 transporter mediates 90% renal glucose reabsorption and the remaining 10% occurs through SGLT1. Although SGLT2 plays a dominant role in glucose transport in PTs, SGLT1 also plays a significant role under the condition of SGLT2 inhibition or diabetic milieu. However, because SGLT1 is present in the intestinal and renal tissues, inhibition of this transporter has the potential to induce osmotic diarrhea7. Therefore, SGLT2 inhibitors were recently made available for clinical use as glucose-lowering reagents. Accumulating evidence suggests their protective effects on diabetic PT cells. In diabetes, SGLT2 inhibitors prevented increases in reactive oxidative species (ROS), causing apoptotic damage to PT cells8. Inhibition of SGLT2 restored decreased SIRT1 levels in diabetic PTs, preventing cellular senescence of PTs9. Therefore, we surmised that activation of SGLT2 reduces SIRT1 expression by inducing excessive glucose entry into PTs under diabetic conditions.
To elucidate a more detailed mechanism for the relationship between SGLT2 and SIRT1, we used obese-type db/db mice with diabetes treated with the SGLT2 inhibitor canagliflozin (Cana) and investigated SIRT1 and SGLT2 expressions. We also investigated the mechanism whereby HG regulates SGLT2 and SIRT1 expression in PT cells. Subsequently, we demonstrated the relationship between SGLT2 and SIRT1 expression in human kidney biopsy samples. SGLT2 inhibition may reverse reductions in SIRT1 in PT in DN, thereby providing a tissue-protective effect in DN.
Methods
Animal experiments
All mice were bred with a C57BL/6 genetic background. We purchased 7-week-old male db/db mice with diabetes (BKS.Cg-Leprdb/Leprdb) and db/m mice without diabetes (BKS.Cg-Leprdb/+) from CLEA Japan (Tokyo, Japan). All procedures were conducted in accordance with relevant guidelines and regulations; all protocols were approved by Keio University Animal Care and Use Committees. Throughout the study, mice were housed in individual cages and given water ad libitum. From arrival to use, the mice were fed a laboratory chow diet (CE-2 pellet; CLEA Japan, Inc; with the following composition: moisture, 9.3%; crude protein, 25.1%; crude fat, 4.8%; crude fiber, 4.2%; nitrogen-free extract, 50.0%; and crude ash, 6.7%). The animal room was maintained under controlled conditions (20 °C, 65% humidity, and a 12-h light/12-h dark photoperiod with lights on at 8:00 a.m.). Cana was provided by the Medicinal Chemistry Laboratory of Mitsubishi Tanabe Pharma Corporation (Osaka, Japan). Each group was given Cana mixed with their diet (powdered CE-2), which was provided ad libitum. The average daily dose of the drug (calculated from food intake and body weight) was as follows: Cana 0.02% w/w food admixture = 30.0 mg/kg and Cana 0.005% w/w food admixture = 7.5 mg/kg. Consistently, a recent study utilized Cana in in vivo settings at concentrations of 0.005%, 0.01%, and 0.03%10–13. Cana was administered to db/db mice (n = 8) for 8 weeks, beginning at 8 weeks or age. Powdered CE-2 was given to db/db mice (n = 8) and db/m mice (n = 8). At 16 weeks of age, the mice were anesthetized by pentobarbital injection (50 mg/kg) and exsanguinated through an incision in the cervical artery, under anesthesia. The kidneys were removed, weighed, and processed, as described previously14. The body weight of each mouse was measured every week. Plasma and urinary glucose levels were measured every 4 weeks.
Histopathological analysis
Immunohistochemistry, using specific antibodies, was performed as detailed in the Supplementary Materials.
Cell culture
We analyzed polarity of SGLT2 expression using LLC-PK1 porcine renal epithelial cells (ATCC, Manassas, VA, MD, USA; lot number: 59681631). The culture cells were cultivated at 37 °C, 5% CO2, in Dulbecco’s modified minimal essential medium supplemented with heat-inactivated 10% fetal bovine serum (Thermo Fisher Scientific, San Jose, CA, USA). LLC-PK1 cells at passage 8th–30th cells were used. We determined the effects of D-glucose on polarity of SGLT2 expression by seeding cells onto porous tissue culture inserts (pore size, 0.4 µm; Becton Dickinson, Oxford, UK). All experiments were performed using confluent monolayers of cells under serum-free conditions following growth arrest in serum-free medium. We used Cana at 100 and 500 nM, which effectively and selectively blocked SGLT2 expression without significantly inhibiting SGLT1 expression15. We determined these concentrations based on previous pharmacokinetics data16. In that study, when Cana was administered to patients with diabetes at a dosage of 100 mg/day, maximum serum concentrations reached 1126 ng/ml (2533 nM), equivalent to 43 nM, given that the protein-binding ratio of Cana was 98.3%. These cells were used for the immunofluorescence analysis of SGLT2 expression, incorporation of 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino]-2-deoxy- D-glucose (2-NBDG), immunoprecipitation analysis, and analysis of the effect of small interfering RNAs (siRNAs) on importin expression. The cells were cultured with 5.5 mmol/L glucose (normal glucose), 22.5 mmol/L glucose (high glucose), or 5.5 mmol/L glucose plus 17.0 mmol/L mannitol (mannitol); these concentrations were within the normal range that is relevant for humans with diabetes. Detailed methods are described in the Supplementary Materials.
Extraction of the nuclear fraction
Nuclear lysates were prepared using NE-PERTM Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific, San Jose, CA, USA), as described previously17. Briefly, cells were washed twice with ice-cold PBS buffer (1 mM KH2PO4, 155 mM NaCl, and 3 mM Na2HPO4-7H2O) and centrifuged at 500 × g for 3 min. The cell pellet was suspended in 200 μl of ice-cold cytoplasmic extraction reagent I by vortexing. The suspension was incubated on ice for 10 min, followed by addition of 11 μl of the second cytoplasmic extraction reagent II. After vortexing for 5 s, the suspension was incubated on ice for 1 min and centrifuged for 5 min at 16 000 × g. The supernatant fraction (cytoplasmic extract) was transferred to a pre-chilled tube. The insoluble pellet fraction, which contains crude nuclei, was resuspended in 100 μl of the nuclear extraction reagent by vortexing for 15 s and incubated on ice for 10 min, then centrifuged for 10 min at 16 000 × g. The resulting supernatant, constituting the nuclear extract, was used for subsequent experiments.
Immunoblotting and quantitative PCR of mouse kidney tissues and LLC-PK1 cells
Immunoblotting and real-time PCR were performed, as described previously4. Primer sequences and other details are described in the Supplementary Methods.
Human renal needle-biopsy specimens
We obtained renal needle-biopsy specimens from 11 patients with DN. Before study enrollment, written informed consent was obtained in all patients. Patient clinical data were acquired at the time of renal biopsy and are summarized in Table 1. The study was performed in accordance with the Declaration of Helsinki and the study protocol was approved by the human ethics review committee of the Department of Internal Medicine, School of Medicine, Keio University. Immunohistochemical analysis of SIRT1 and SGLT2 expression for each human sample was performed, as described previously18. Further details are provided in the Supplementary Methods.
Table 1.
Clinical parameters of patients with diabetic nephropathy (DN) and controls at the time of renal needle biopsy.
| Sample name | Sex | Age (years) | Serum creatine (mg/dL) | Proteinuria (g/day) | eGFR (ml/min/1.73 m2) | HbA1c (%) | Fasting blood glucose (mg/dl) |
|---|---|---|---|---|---|---|---|
| Diabetic Nephropathy | |||||||
| DN-1 | male | 83 | 1.81 | 1.6 | 28.5 | 6.6 | 191 |
| DN-2 | male | 42 | 1.13 | 4.9 | 58.1 | 6.3 | 139 |
| DN-3 | male | 62 | 1.73 | 1.5 | 32.6 | 6.1 | 97 |
| DN-4 | male | 47 | 1.82 | 5.4 | 33.4 | 6.1 | 98 |
| DN-5 | male | 80 | 1.23 | 10.2 | 44.0 | 6.0 | 97 |
| DN-6 | male | 56 | 2.93 | 12.0 | 18.9 | 6.4 | 144 |
| DN-7 | male | 75 | 1.57 | 1.4 | 33.6 | 6.1 | 205 |
| DN-8 | male | 59 | 1.88 | 0.5 | 71.2 | 5.8 | 136 |
| DN-9 | female | 64 | 1.89 | 2.9 | 36.3 | 6.1 | 167 |
| DN-10 | female | 51 | 0.54 | 4.6 | 91.0 | 5.9 | 135 |
| DN-11 | male | 61 | 0.69 | 7.7 | 89.0 | 7.2 | 155 |
| Controls | |||||||
| CO-1 | female | 39 | 0.61 | <0.1 | 86.2 | 5.2 | 84 |
| CO-2 | male | 27 | 0.87 | <0.1 | 88.1 | 4.9 | 99 |
| CO-3 | female | 51 | 0.64 | <0.1 | 76.7 | 5.3 | 101 |
| CO-4 | female | 35 | 0.66 | <0.1 | 84.9 | 5.0 | 75 |
| CO-5 | male | 42 | 0.83 | <0.1 | 81.4 | 5.5 | 91 |
Statistical analysis
Data are expressed as mean ± standard deviation. Differences were evaluated using one-way ANOVA, followed by Bonferroni’s multiple-comparison post hoc test (GraphPad Prism, version 4.0, GraphPad Software, La Jolla, CA, USA). The criterion for statistical significance was a P value < 0.05.
Results
Effects of canagliflozin treatment in db/db mice
To determine the effect of Cana in db/db mice, we measured physiological and metabolic parameters of four groups of mice (Fig. 1A). Mean body weights of the db/db groups were higher than those of the db/m group. Moreover, the mean body weight of the db/db group treated with 7.5 or 30.0 mg/kg/day Cana (db/db + 7.5 mg Cana group and db/db + 30.0 mg Cana group, respectively) was higher than that of the db/db group (Fig. 1B). Furthermore, daily food consumption of the Cana groups was greater than that of the db/m mice (Fig. 1C). Plasma fasting glucose levels and HbA1c increased in the db/db mice, although Cana significantly reduced these values compared with those of the db/db mice (Fig. 1D and E, respectively).
Figure 1.

Effect of canagliflozin on parameters of glucose metabolism. (A) Schedule for canagliflozin (Cana) treatment and experimental groups. Effects of Cana on body weight (B), food intake (C), fasting plasma glucose level (D) and glycated Hb levels (E) in db/db mice. HbA1c, hemoglobin A1c. Data represent the mean ± SEM (n = 8 mice/group). *P < 0.05 vs. db/m and §P < 0.05 vs. db/db.
Effects of Cana on renal SGLT2 expression
To determine effects of diabetes and Cana treatment on SGLT2 expression, we performed immunohistochemistry (Fig. 2A) and immunoblotting (Fig. 2B) analyses. Renal SGLT2 expression increased in db/db mice with diabetes compared with that in db/m mice, and this increase was attenuated by Cana treatment in db/db mice (Fig. 2A,B). Glucose transporter 2 (GLUT2) expression, which is expressed on the basolateral side of the PT cell, did not change in db/db mice or db/db mice treated with Cana (Supplementary Fig. 1). SIRT1 expression decreased in db/db mice with diabetes compared with that in db/m mice, which was rescued by Cana treatment in mice with diabetes (Fig. 2C,D). There was no significant difference in SGLT2 or SIRT1 expression between the db/db + 7.5 and db/db + 30.0 mg Cana groups.
Figure 2.

Effect of Cana on SGLT2 and SIRT1 expression in db/db mice. (A) Immunohistochemical analysis using a SGLT2-specific antibody. Representative kidney sections are shown for each group of mice. Quantitation of relative density is shown in the bar graph in the right panel. Scale bar, 100 µm. *P < 0.05 vs. db/m control and §P < 0.05 vs. db/db control (n = 8 mice/group) (B) The upper panel shows representative immunoblotting analysis of SGLT2 expression. The Bar graph in the lower panel indicates quantification of SGLT2 levels. Protein expression was normalized to that of α-tubulin. Relative protein levels are shown as the fold-change to the db/m (control) group. *P < 0.05 vs. db/m control and §P < 0.05 vs. db/db control. The results are representative of four independent experiments. (C) Immunohistochemical analysis using a SIRT1-specific antibody. Representative kidney sections are shown for each group of mice. Quantitation of relative density is shown in the bar graph. Scale bar, 50 µm. *P < 0.05 vs. db/m control and §P < 0.05 vs. db/db control (n = 8 mice/group). (D) The upper panel shows representative immunoblotting analysis of SIRT1 expression. The bar graph in the lower panel indicates quantification of SIRT1 levels. Protein expression was normalized to that of α-tubulin. Relative protein levels are shown as the fold-change to the db/m (control) group. *P < 0.05 db/m control and §P < 0.05 vs. db/db control. Results are representative of four independent experiments.
Basolateral HG stimulates SGLT2 expression
Given that PT cells retain membrane protein polarity, we examined how ambient glucose affects SGLT2 expression using a two-chamber culture system. Confluent LLC-PK1 cells that exhibit apical and basolateral polarity19 were serum-starved on porous tissue culture inserts and then exposed to normal (5.5 mM) or high (22.5 mM) D-glucose in the apical or basal chamber for 24 h (Fig. 3A). SGLT2 expression increased following the addition of 22.5 mM D-glucose to the basolateral, but not following that to the apical chamber (Fig. 3A). Next, we investigated intracellular signal transduction in PTs, directed from the basolateral to the apical side, that controls SGLT2 expression. Na-K ATPase20, GLUT221, angiotensin II type 1 receptor (AT1R)22, and AT2R23 are located within the basolateral membrane of PT cells and transduce signals elicited by HG. We added an inhibitor for each molecule, i.e., glibenclamide24, phloretin25, losartan26, and PD12331927, respectively, to the lower chamber. HG-induced increases in SGLT2 expression were inhibited by phloretin, a GLUT2 inhibitor (Fig. 3B). HG-induced SGLT2 expression upregulation was inhibited by a Glut2-specific siRNA (Fig. 3C).
Figure 3.

Effect of high glucose on SGLT2 expression in a two-chamber culture system. (A) Confluent growth-arrested cell monolayers were stimulated on the apical or basolateral side for up to 24 h, as described in the Methods section. Representative SGLT2 fluorescence of the four groups of culture conditions is shown. The bar graph represents the fluorescence intensity of each group (*P < 0.05 vs. the group with 5.5 mM D-glucose in the lower chamber and 5.5 mM D-glucose in the upper chamber, n = 4 independent experiments. Scale bar, 50 µm. (B) Effects of inhibitors for candidate signaling pathways on SGLT2 expression. LLC-PK1 cells were treated with a Na-K ATPase channel inhibitor (glibenclamide, 50 µM), GLUT2 inhibitor (phloretin, 100 µM), angiotensin II type 1 receptor (AT1R) inhibitor (losartan, 1 µM), and AT2R inhibitor (PD123319, 100 µM). Results are representative of four independent experiments. The bar graph represents the band intensity of each group (*P < 0.05 vs. NG group and §P < 0.05 vs. HG group, n = 4 independent experiments). (C) Effects of Glut2-siRNA (100 nmol/L) or non-targeting control siRNA (100 nmol/L) on SGLT2 expression. Results are representative of four independent experiments. The bar graph in the right panel indicates the quantification of SGLT2 levels. Protein expression was normalized to that of α-tubulin. Relative protein levels are shown as the fold-change to the NG group. *P < 0.05 vs. NG group, §P < 0.05 vs. HG group, and ¶P < 0.05 vs. HG with control siRNA, n = 4. (D) Subcellular fractionation and immunoblotting demonstrate HG-induced redistribution from the cytoplasm to the nucleus of HNF-1α. HG, high D-glucose (22.5 mM) condition; NG, normal D-glucose (5.5 mM) condition; N, nuclear fraction; C, cytosolic fraction. HNF-1α protein levels were determined by densitometry and are indicated for each fraction. The percent of HNF-1α in the nucleus relative to the total HNF-1α was calculated using values determined using a densitometry. *P < 0.05 vs. NG group. Results are representative of four independent experiments. (E) Effects of HNF-1α-siRNA (100 nmol/L) or non-targeting control siRNA (100 nmol/L) on SGLT2 expression. The bar graph in the right panel indicates quantification of SGLT2 levels. Relative protein levels are shown as the fold-change to the NG (control) group. *P < 0.05 vs. NG group, §P < 0.05 vs. HG group, and ¶P < 0.05 vs. HG with control siRNA, n = 4 independent experiments.
Next, we investigated the regulation of transcription factors that control SGLT2 expression. It is possible that hepatic nuclear factor-1α (HNF-1α) plays a key role because when translocated to the nucleus, it upregulates Sglt2 transcription28. HNF-1α was mainly detected in the cytoplasmic fraction of cells cultured under normal glucose concentrations (Fig. 3D). In contrast, HNF-1α translocated to the nucleus in the presence of HG (Fig. 3D), thereby suggesting that it can function as a transcription factor in HG conditions. Treatment with a specific siRNA for HNF-1α blocked HG-induced SGLT2 expression upregulation (Fig. 3E). Thus, basolateral HG upregulated SGLT2 expression by activating GLUT2 and transcription factor HNF-1α nuclear translocation.
Importin-α1 mediates basolateral HG stimulation for SGLT2 expression
We next explored the underlying mechanism by which HG initiates GLUT2-mediated signal transduction and HNF-1α nuclear translocation. GLUT2 acts as a metabolic sensor29. Importin-α, also known as karyopherin-α, is an intracellular transporter protein that binds to GLUT2, functioning as an intracellular signal transducer29,30. Therefore, we hypothesized that importin-α mediates GLUT2-induced signaling and HNF-1α activation in PTs31,32. First, we identified importin isoforms that predominated in PTs. The importin isoforms α1, α5, and α7 were abundant compared with isoforms α3, α4, and α6, with the importin-α1 mRNA level being the highest (Fig. 4A). Under HG conditions, the expression pattern of each isoform of importin-α did not change (Supplementary Fig. 2). We used specific siRNA for importins-α1, -α5, and -α7 (Fig. 4B) and examined their roles in HNF-1α nuclear translocation. Adding a specific siRNA in the lower chamber HG medium for importin-α1, but not for -α5 or -α7, inhibited nuclear accumulation of HNF-1α (Fig. 4C). In addition, a specific siRNA for importin-α1 also blocked HG-induced SGLT2 expression upregulation (Fig. 4D). Finally, HNF-1α was immunoprecipitated with importin-α1 from lysates prepared from cells cultured with HG, but not in those cultured with NG medium (Fig. 4E). We immunoprecipitated GLUT2 with importin-α1 from lysates prepared from cells cultured with NG, but not from those cultured with HG medium (Fig. 4F). Taken together, ambient basolateral HG activates GLUT2/importin-α1/HNF-1α signaling, which resulted in SGLT2 expression upregulation in PT cells (Fig. 4G).
Figure 4.

The GLUT2/importin-α1/HNF-1α pathway mediates high-glucose-induced SGLT2 expression. (A) Quantitative real-time PCR analysis of the relative abundance of mRNAs encoding importins in LLC-PK1 cells. Real-time PCR data were normalized to those of the GAPDH mRNA. Relative fold-differences were calculated using the mean value (n = 6) of importin-α6. (B) Immunoblotting confirmed the efficiency of siRNA knockdown of each of the representative importin-α isoform that was abundantly expressed in PTs. Results are representative of four independent experiments. (C) Subcellular fractionation and immunoblotting analysis of the localization of HNF-1α. LLC-PK1 cells were transfected with the indicated siRNA duplexes and 48 h later, cells were treated with media containing high glucose levels. Cytoplasmic and nuclear lysates were collected and analyzed using immunoblotting with an anti-HNF-1α antibody. Results are representative of four independent experiments. (D) LLC-PK1 cells were transiently transfected with siRNAs targeting importin-α1 or a non-targeting control siRNA. Whole cell lysates were prepared 48 h post-transfection and analyzed using immunoblotting. Results are representative of four independent experiments. The bar graph represents the band intensity of each group (*P < 0.05 vs. NG group and §P < 0.05 vs. HG group, n = 4). (E,F) Immunoprecipitation of nuclear extracts from LLC-PK1 cells that were treated with NG or HG. Importin-α1 formed complexes with HNF-1α (E), whereas it dissociated from GLUT2 (F) under HG conditions. Results are representative of four independent experiments. (G) Model for the molecular mechanism by which the GLUT2/importin-α1/HNF-1α pathway is involved in HG-induced SGLT2 expression upregulation.
Effects of Cana on SIRT1 expression in PT cells
Next, we studied effects of Cana on SGLT2 and SIRT1 expression using the two-chamber culture system. Because SGLT2 localizes to the apical side of PT cells, we added Cana to the upper chamber. Immunofluorescence and immunoblotting revealed that addition of HG to the basolateral chamber increased SGLT2 expression and 100 nM or 500 nM Cana did not inhibit this HG-induced increase in SGLT2 levels in LLC-PK1 cells (Fig. 5A,B). We monitored the entry of fluorescence-labeled glucose 2-NBDG in the upper chamber under the same experimental conditions33. Lower-chamber HG levels significantly increased glucose uptake from the upper chamber by LLC-PK1 cells, which was prevented by treatment with 100 or 500 nM Cana in the upper chamber (Fig. 5C). Lower-chamber HG levels significantly decreased SIRT1 expression. This process was inhibited by treatment with 100 or 500 nM Cana in the upper chamber (Fig. 5D).
Figure 5.

Effect of Cana on the expression of SIRT1 and its downstream signaling components. Confluent growth-arrested LLC-PK1 cell monolayers were stimulated with HG medium on the basolateral side for up to 24 h with or without pretreatment with Cana in the apical side. Immunofluorescence analysis (A) and immunoblotting (B) for SGLT2 expression in LLC-PK1 cells. The relative quantification of the SGLT2 immunofluorescence was measured and is indicated in the bar graphs. Scale bar, 50 µm. *P < 0.05 vs. NG group, n = 4 independent experiments. (C) The effect of SGLT2 inhibitors on cellular glucose entry in LLC-PK1 cells. LLC-PK1 cells were incubated in Dulbecco’s modified minimal essential medium containing 100 μM 2-NBDG for 15 min from the apical side of the cell. Cellular glucose entry was assessed as 2-NBDG entry into the cell, as described in the Methods section. Scale bar = 20 µm. *P < 0.05 vs. NG group and §P < 0.05 vs. HG without Cana, n = 4 independent experiments. NG, normal glucose (5.5 mM); HG, high glucose (22.5 mM); HG + Cana, HG with Cana treatment. (D) Immunoblotting analysis of SIRT1 expression in LLC-PK1 cell monolayers stimulated with HG medium on the basolateral side for up to 24 h with or without pretreatment with low or high doses of Cana. Quantification of immunoblotting images was normalized to α-tubulin. *P < 0.05 vs. NG group and §P < 0.05 vs. HG without Cana, n = 4 independent experiments.
Correlation between SGLT2 and SIRT1 expression in human kidney
The negative correlation between SGLT2 and SIRT1 expression under diabetic conditions was further examined using kidney tissue samples from patients with diabetes. We completed immunostaining for SGLT2 and SIRT1 of renal specimens obtained from 11 patients with DN (Fig. 6A). The intensity of staining of these proteins was negatively correlated (Fig. 6B), suggesting that glucose entry via SGLT2 also downregulates kidney SIRT1 expression.
Figure 6.
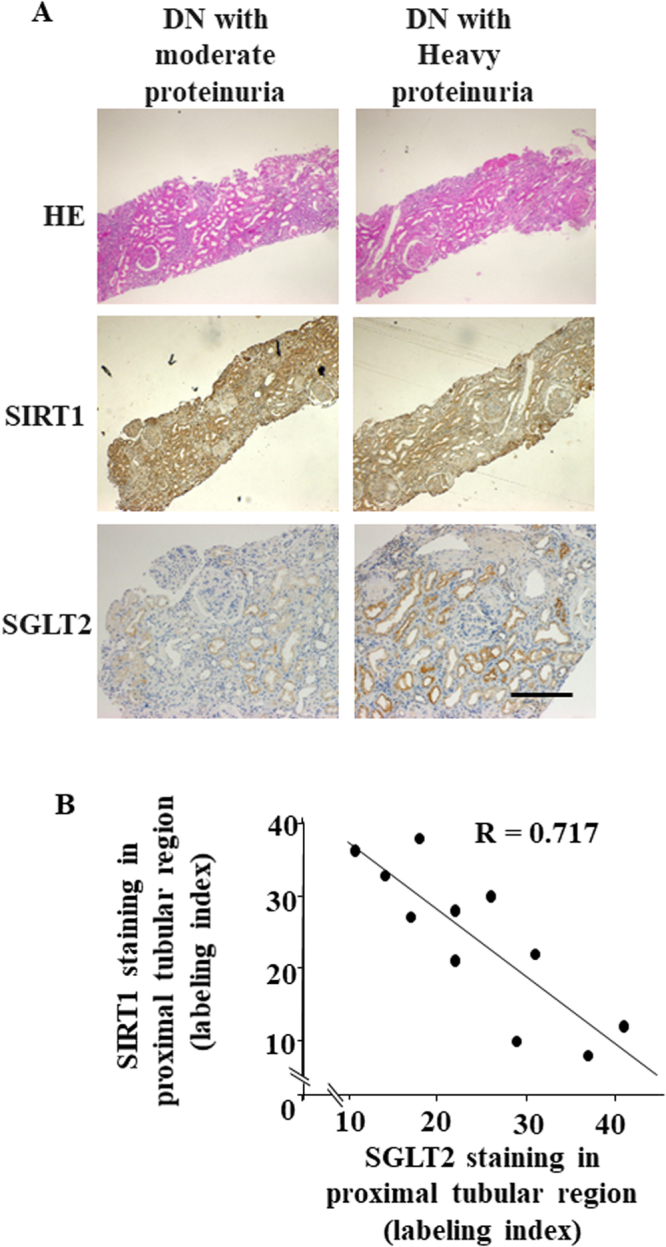
Immunohistochemical analysis of SIRT1 and SGLT2 expression in human renal biopsy specimens. (A) Representative photomicrographs of hematoxylin and eosin staining or immunostaining of SIRT1 and SGLT2 in renal needle-biopsy specimens of patients with diabetic nephropathy (DN) (DN-1 and -5, Table 1 for patient details). Scale bar, 50 nm. (B) The relationship between the intensity of immunostaining of SIRT1 and that of SGLT2 in the proximal tubular region in renal biopsy specimens from patients with DN, n = 11 subjects.
Discussion
In DN, gene expression in renal epithelial cells, particularly in PT cells, is altered even during early stages34. In this study, we demonstrated increased SGLT2 expression and decreased SIRT1 expression in db/db mice. Cana treatment inhibited SIRT1 expression downregulation, indicating a close association between these molecular changes. We also used a two-chamber tissue culture system to show that SGLT2 expression upregulation is caused by HG conditions in the lower chamber, corresponding to HG levels in the basolateral side of PT cells. HG conditions did not affect SGLT2 expression upregulation in the upper chamber, which corresponds to the apical side of PT cells. HG conditions dissociated the GLUT2/importin-α1 complex, enhancing formation of importin-α1/HNF-1α complexes. Importin-α1 led to HNF-1α nuclear translocation, promoting SGLT2 transcription. Basolateral HG also downregulated SIRT1 expression and this was ameliorated by apical side Cana treatment. Under diabetic conditions, PTs increased SGLT2 expression, resulting in increased glucose entry into PTs from glomerular filtrates and thus decreased SIRT1 expression. This relationship was also observed in renal biopsy samples from patients with diabetes. These results suggest that glucose entry from the apical side by SGLT2 helps downregulate SIRT1 expression in patients with DN.
Extracellular glucose elicits several intracellular signals, including redox signaling and angiotensin II receptor-mediated signaling22,23. Our pharmacological experiments suggest that GLUT2-mediated signaling is a relevant pathway20,21. On the basolateral side of PT cells, GLUT2, a low-affinity and high-capacity transporter, transports glucose absorbed from the urinary lumen by SGLT2 to the interstitial space and peritubular capillaries in the kidney. GLUT2 is also a membrane sugar receptor35. For instance, in hepatoma cells, there is close correlation between GLUT2 levels and glucose-sensitive genes36. In a pancreatic β-cell line, glucose-induced insulin secretion was mediated by glucose influx through GLUT237. This notion is supported by studies on GLUT2 null mice where the absence of GLUT2 impaired glucose-sensitive gene expression, including the pancreatic β-cell insulin gene38–40. GLUT2 appears to play a similar role in the glucose-sensitive SGLT2 gene expression in the kidney.
The molecular mechanism whereby GLUT2 directs intracellular signaling, resulting in the stimulation of glucose-sensitive gene transcription, was initially revealed in hepatocytes29. The large intracytoplasmic loop in the GLUT2 protein tethers the nuclear importer protein importin-α1, at the plasma membrane. Importin-α cooperates with importin-β and the cargo protein harboring the nuclear-localizing signal peptide as a nuclear import machinery, guiding target cargo molecules to shuttle through the nuclear pore41. We have identified several importin protein isoforms and this protein is a significant component of hepatocytes and pancreatic β-cells41. With extracellular glucose stimulation of mhAT3F hepatoma cells and pancreatic MIN-6 cells, massive amounts of importin-α1 accumulate in the cytoplasm42. These dynamic changes in the intracellular distribution were not detected in another liver cell line HepG2 or in a kidney cell line COS-742. In addition, the signal-transduction role of each importin-α isoform appears to be cell- and tissue-specific28. In LLC-PK1 cells, importin-α1, -α5 and -α7 are dominantly expressed and only the α1 isoform is associated with HNF-1α nuclear translocation. Other reports have described dysregulation of the importin system under diabetic conditions43. In diabetes, we observed increased renal expression of all importin isoforms, although the pathological relevance of this increase remains unclear. We found that by binding to HNF-1α, importin-a1 plays a critical role in activating HNF-1α. Ambient glucose stimuli enhanced this binding during dissociation from the GLUT2 molecule (Fig. 4G). Thus, in diabetes, importin-α1 upregulation may exist as an adaptation to increases in extracellular glucose levels, contributing to enhanced HNF-1α transport into the nucleus and resulting in SGLT2 expression upregulation.
The HNF-1α transcription factor acts as a chaperone protein for importin 1α. HNF-1α is required for glucose metabolism in the liver, pancreatic islets, kidneys, and intestines44. HNF-1α-deficient mice and patients suffering from maturity-onset type 3 diabetes develop type 2 diabetes and renal Fanconi syndrome characterized by increases in urinary glucose levels45. As demonstrated in this study, HNF-1α directly upregulates SGLT2 transcription, thereby contributing to renal glucose reabsorption. Although three regions within the HNF-1α gene encode NLS32 and HNF-1α shuttles between the nucleus and cytoplasm31, molecules that interact with NLS as well as the mechanisms for HNF-1α nuclear translocation remain unknown. This study is the first to demonstrate that importin-α1 transports HNF-1α to the nucleus, resulting in SGLT2 transcription upregulation (Fig. 4G).
Previous studies indicated that ambient HG decreases SIRT1 activity and expression in various cell types46–48, although detailed mechanisms are unknown. Increased oxidative stress, provoked by glucose entering the cell, plays a key role in this regulation. Increases in oxidative stress reduce cellular concentration of NAD+, leading to decreased SIRT1 activity. This, in turn, inactivates the forkhead box O1 (FoxO1) transcription factor, which then downregulates SIRT1 transcription49. Redox-sensitive transcription co-repressor CtBP may also inhibit SIRT1 transcription50. ROS degrade SIRT1 proteins through a proteasome-dependent mechanism51. In the present study, entry of glucose from the apical side was important for SIRT1 downregulation because upper chamber treatment with an SGLT2 inhibitor completely mitigated HG-induced downregulation of SIRT1. In other words, glucose entry because of HG in the lower chamber had little effect on SIRT1 expression, indicating that glucose entry from the basolateral side, through GLUT2, was marginal. Given that GLUT2 facilitates glucose transport, glucose entry occurs by simple diffusion and depends on the glucose concentration gradient inside and outside the cell. In contrast, SGLT2 is driven by electrochemical sodium ions gradients, and as long as sodium-potassium dependent ATPase pumps sodium out, glucose entry ensues, irrespective of the glucose gradient. Therefore, in renal PT cells, blocking SGLT2 prevents excessive glucose entry and glucose toxicity.
In diabetic conditions, clinical significance of restoring proximal tubular SIRT1 by SGLT2 inhibition merits comment. In addition to its plasma glucose-lowering effects, there are several beneficial renal effects of SGLT2 inhibition. SGLT2 inhibitors ameliorate DN by reducing oxidative stress in the renal tubules of db/db mice. We previously reported renal protective effects of SIRT1 through upregulation of the anti-oxidative stress molecule catalase in proximal tubular cells51. Several other mechanisms are involved in SIRT1-related anti-oxidative stress effects in the kidney52. SGLT2 inhibitors exhibit anti-inflammatory effects in mice with diabetes. By deacetylating the p65 subunit, SIRT1 also inhibits NFκB activity and kidney tissue inflammatory responses53. Moreover, SIRT1 protects against diabetic albuminuria by epigenetic podocyte claudin-1 downregulation4. SGLT2 inhibition also reduces albuminuria in mice with diabetes6. SIRT1 expression may explain several favorable renal effects demonstrated by SGLT2 inhibition.
As for the validity of a SGLT2 antibody used in this study, we have not perfiormed studies using SGLT2 knockout mice. However, we confirmed that there was no extra-renal SGLT2 expression (Supplementary Fig. 19A), supporting the specificity of the antibody. In addition, we assured antibody specificity using siRNA-mediated gene silencing (Supplementary Fig. 19B,C). There are several studies using this commercial antibody54–56.
Finally, SGLT2 inhibition reduces mortality in high-risk patients with diabetes57. SIRT1 is known as a “longevity gene,” and its introduction elongates the lifespan of yeast, hook worms, drosophila, rodents, and mammals58. SGLT2 inhibition effects on longevity may be related to SIRT1 activation, which favors tissue resistance to various stresses in renal and other SGLT2-expressing tissues.
In conclusion, SGLT2 expression was increased in the kidneys of db/db mice and humans with diabetes, whereas SIRT1 expression was decreased. SGLT2 expression upregulation results from basolateral glucose stimulation activating the GLUT2/importin-α1/HNF-1α signaling pathway. Cana-related inhibition of SGLT2 restored SIRT1 expression by preventing intracellular glucose entry from the apical side into the proximal tubular cells. Thus, Cana may be a beneficial treatment strategy against diabetic kidney disease.
Electronic supplementary material
Author Contributions
H.U. performed the experiments and drafted the manuscript. K.H., S.W., and H.I. designed the research, analyzed data and drafted the manuscript. H.M., H.M., T.K., T.K., and H.T. worked on the general coordination of the study.
Competing Interests
The authors declare no competing interests.
Footnotes
Hiroyuki Umino and Kazuhiro Hasegawa contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-25054-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Roxburgh SA, et al. Allelic depletion of grem1 attenuates diabetic kidney disease. Diabetes. 2009;58:1641–1650. doi: 10.2337/db08-1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tervaert TWC, et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 3.Bonventre JV. Can we target tubular damage to prevent renal function decline in diabetes? Semin. Nephrol. 2012;32:452–462. doi: 10.1016/j.semnephrol.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasegawa K, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013;19:1496–1504. doi: 10.1038/nm.3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wakino S, Hasegawa K, Itoh H. Sirtuin and metabolic kidney disease. Kidney Int. 2015;88:691–698. doi: 10.1038/ki.2015.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terami N, et al. Long-term treatment with the sodium glucose cotransporter 2 inhibitor, dapagliflozin, ameliorates glucose homeostasis and diabetic nephropathy in db/db mice. PLoS One. 2014;9:e100777. doi: 10.1371/journal.pone.0100777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, Griffen SC, Boulton DW, Leil TA. Use of systems pharmacology modeling to elucidate the operating characteristics of SGLT1 and SGLT2 in renal glucose reabsorption in humans. Front. Pharmacol. 2014;5:274. doi: 10.3389/fphar.2014.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatanaka T, et al. Inhibition of SGLT2 alleviates diabetic nephropathy by suppressing high glucose-induced oxidative stress in type 1 diabetic mice. Pharmacol Res Perspect. 2016;30:e00239. doi: 10.1002/prp2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kitada K, et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J. Diabetes Complications. 2014;28:604–611. doi: 10.1016/j.jdiacomp.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thrailkill KM, et al. SGLT2 inhibitor therapy improves blood glucose but does not prevent diabetic bone disease in diabetic DBA/2J male mice. Bone. 2016;82:101–107. doi: 10.1016/j.bone.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Watanabe Y, et al. Beneficial effect of canagliflozin in combination with pioglitazone on insulin sensitivity in rodent models of obese type 2 diabetes. PLoS One. 2015;10:e0116851. doi: 10.1371/journal.pone.0116851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naznin F, et al. Canagliflozin, a sodium glucose cotransporter 2 inhibitor, attenuates obesity-induced inflammation in the nodose ganglion, hypothalamus, and skeletal muscle of mice. Eur. J. Pharmacol. 2017;794:37–44. doi: 10.1016/j.ejphar.2016.11.028. [DOI] [PubMed] [Google Scholar]
- 13.Shiba K, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci. Rep. 2018;5:2362. doi: 10.1038/s41598-018-19658-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wen D, et al. Resveratrol attenuates diabetic nephropathy via modulating angiogenesis. PLoS One. 2013;8:e82336. doi: 10.1371/journal.pone.0082336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuriyama C, et al. Analysis of the effect of canagliflozin on renal glucose reabsorption and progression of hyperglycemia in zucker diabetic Fatty rats. J. Pharmacol. Exp. Ther. 2014;351:423–431. doi: 10.1124/jpet.114.217992. [DOI] [PubMed] [Google Scholar]
- 16.Iijima H, Kifuji T, Maruyama N, Inagaki N. Pharmacokinetics, pharmacodynamics, and safety of canagliflozin in Japanese patients with type 2 diabetes mellitus. Adv. Ther. 2015;32:768–782. doi: 10.1007/s12325-015-0234-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Penheiter SG, et al. Internalization-dependent and–independent requirements for transforming growth factor beta receptor signaling via the Smad pathway. Mol. Cell. Biol. 2002;22:4750–4759. doi: 10.1128/MCB.22.13.4750-4759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kamiyama M, et al. Detailed localization of augmented angiotensinogen mRNA and protein in proximal tubule segments of diabetic kidneys in rats and humans. Int. J. Biol. Sci. 2014;10:530–542. doi: 10.7150/ijbs.8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Slattery C, McMorrow T, Ryan MP. Overexpression of E2A proteins induces epithelial-mesenchymal transition in human renal proximal tubular epithelial cells suggesting a potential role in renal fibrosis. FEBS Lett. 2006;580:4021–4030. doi: 10.1016/j.febslet.2006.06.039. [DOI] [PubMed] [Google Scholar]
- 20.Ohtomo Y, Ono S, Zettergren E, Sahlgren B. Neuropeptide Y regulates rat renal tubular Na, K-ATPase through several signaling pathways. Acta Phsiol. Scand. 1996;158:97–105. doi: 10.1046/j.1365-201X.1996.508274000.x. [DOI] [PubMed] [Google Scholar]
- 21.Panchapakesan U, et al. Effects of SGLT2 inhibition in human kidney proximal tubular cells–renoprotection in diabetic nephropathy? PLoS One. 2013;8:e54442. doi: 10.1371/journal.pone.0054442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takao T, et al. Possible roles of tumor necrosis factor-α and angiotensin II type 1 receptor on high glucose-induced damage in renal proximal tubular cells. Ren. Fail. 2015;37:160–164. doi: 10.3109/0886022X.2014.959434. [DOI] [PubMed] [Google Scholar]
- 23.Brown RD, et al. Reduced sensitivity of the renal vasculature to angiotensin II in young rats: the role of the angiotensin type 2 receptor. Pediatr. Res. 2014;76:448–452. doi: 10.1038/pr.2014.121. [DOI] [PubMed] [Google Scholar]
- 24.Tiedge M, Lenzen S. Effects of glucose refeeding and glibenclamide treatment on glucokinase and GLUT2 gene expression in pancreatic B-cells and liver from rats. Biochem. J. 1995;308:139–144. doi: 10.1042/bj3080139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diogo LN, et al. Voluntary oral administration of losartan in rats. J. Am. Assoc. Lab. Anim. Sci. 2015;54:549–556. [PMC free article] [PubMed] [Google Scholar]
- 26.Liles C, et al. AT2R autoantibodies block angiotensin II and AT1R autoantibody-induced vasoconstriction. Hypertension. 2015;66:830–835. doi: 10.1161/HYPERTENSIONAHA.115.05428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zou TB, et al. The role of sodium-dependent glucose transporter 1 and glucose transporter 2 in the absorption of cyaniding-3-o-β-glucoside in Caco-2 cells. Nutrients. 2014;6:4165–4177. doi: 10.3390/nu6104165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pontoglio M, et al. HNF1alpha controls renal glucose reabsorption in mouse and man. EMBO Rep. 2000;1:359–365. doi: 10.1093/embo-reports/kvd071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guillemain G, et al. Karyopherinalpha2: a control step of glucose-sensitive gene expression in hepatic cells. Biochem. J. 2002;15:201–209. doi: 10.1042/bj3640201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pumroy RA, Cingolani G. Diversification of importin-α isoforms in cellular trafficking and disease states. Biochem. J. 2015;466:13–28. doi: 10.1042/BJ20141186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bjørkhaug L, Bratland A, Njølstad PR, Molven A. Functional dissection of the HNF-1alpha transcription factor: a study on nuclear localization and transcriptional activation. DNA Cell Biol. 2005;24:661–669. doi: 10.1089/dna.2005.24.661. [DOI] [PubMed] [Google Scholar]
- 32.Ye W, Lin W, Tartakoff AM, Tao T. Karyopherins in nuclear transport of homeodomain proteins during development. Biochim. Biophys. Acta. 2011;1813:1654–1662. doi: 10.1016/j.bbamcr.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blodgett AB, et al. A fluorescence method for measurement of glucose transport in kidney cells. Diabetes Technol. Ther. 2011;13:743–751. doi: 10.1089/dia.2011.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tucker BJ, Rasch R, Blantz RC. Glomerular filtration and tubular reabsorption of albumin in preproteinuric and proteinuric diabetic rats. J. Clin. Invest. 1993;92:686–694. doi: 10.1172/JCI116638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leturque A, Brot-Laroche E, Le Gall M. GLUT2 mutations, translocation, and receptor function in diet sugar managing. Am. J. Physiol. Endocrinol. Metab. 2009;296:E985–E992. doi: 10.1152/ajpendo.00004.2009. [DOI] [PubMed] [Google Scholar]
- 36.Antoine B, et al. Role of the GLUT 2 glucose transporter in the response of the L-type pyruvate kinase gene to glucose in liver-derived cells. J. Biol. Chem. 1997;272:17937–17943. doi: 10.1074/jbc.272.29.17937. [DOI] [PubMed] [Google Scholar]
- 37.Hughes SD, Johnson JH, Quaade C, Newgard CB. Engineering of glucose-stimulated insulin secretion and biosynthesis in non-islet cells. Proc. Natl. Acad. Sci. USA. 1992;89:688–692. doi: 10.1073/pnas.89.2.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gouyon F, et al. Simple-sugar meals target GLUT2 at enterocyte apical membranes to improve sugar absorption: a study in GLUT2-null mice. J. Physiol. 2003;552:823–832. doi: 10.1113/jphysiol.2003.049247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guillam MT, Burcelin R, Thorens B. Normal hepatic glucose production in the absence of GLUT2 reveals an alternative pathway for glucose release from hepatocytes. Proc. Natl. Acad. Sci. USA. 1998;95:12317–12321. doi: 10.1073/pnas.95.21.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guillam MT, et al. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997;17:327–330. doi: 10.1038/ng1197-327. [DOI] [PubMed] [Google Scholar]
- 41.Cautain B, Hill R, de Pedro N, Link W. Components and regulation of nuclear transport processes. FEBS J. 2015;282:445–462. doi: 10.1111/febs.13163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassany A, et al. A karyopherin alpha2 nuclear transport pathway is regulated by glucose in hepatic and pancreatic cells. Traffic. 2004;5:10–19. doi: 10.1046/j.1398-9219.2003.0143.x. [DOI] [PubMed] [Google Scholar]
- 43.Köhler M, et al. Increased importin alpha protein expression in diabetic nephropathy. Kidney Int. 2001;60:2263–2273. doi: 10.1046/j.1523-1755.2001.00069.x. [DOI] [PubMed] [Google Scholar]
- 44.Gu N, et al. Glucose regulation of dipeptidyl peptidase IV gene expression is mediated by hepatocyte nuclear factor-1 alpha in epithelial intestinal cells. Clin. Exp. Pharmacol. Physiol. 2008;35:1433–1449. doi: 10.1111/j.1440-1681.2008.05015.x. [DOI] [PubMed] [Google Scholar]
- 45.Yamagata K. Roles of HNF1α and HNF4α in pancreatic β-cells: lessons from a monogenic form of diabetes (MODY) Vitam. Horm. 2014;95:407–423. doi: 10.1016/B978-0-12-800174-5.00016-8. [DOI] [PubMed] [Google Scholar]
- 46.Rodgers JT, et al. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 47.Zheng Z, et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012;61:217–228. doi: 10.2337/db11-0416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Suchankova G, et al. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem. Biophys. Res. Commun. 2009;378:836–841. doi: 10.1016/j.bbrc.2008.11.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Motta MC, et al. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/S0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 50.Santos L, Escande C, Denicola A. Potential modulation of Sirtuins by oxidative stress. Oxid. Med. Cell Longev. 2016;2016:9831825. doi: 10.1155/2016/9831825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hasegawa K, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem. Biophys. Res. Commun. 2008;372:51–56. doi: 10.1016/j.bbrc.2008.04.176. [DOI] [PubMed] [Google Scholar]
- 52.Kume S, et al. Silent information regulator 2 (SIRT1) attenuates oxidative stress-induced mesangial cell apoptosis via p53 deacetylation. Free Radic. Biol. Med. 2006;40:2175–2182. doi: 10.1016/j.freeradbiomed.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 53.Jung YJ, et al. SIRT1 overexpression decreases cisplatin-induced acetylation of NF-kB p65 subunit and cytotoxicity in renal proximal tubule cells. Biochem. Biophys. Res. Commun. 2012;419:206–210. doi: 10.1016/j.bbrc.2012.01.148. [DOI] [PubMed] [Google Scholar]
- 54.Zhao Y, et al. Sodium intake regulates glucose homeostasis through the PPARδ/adiponectin-mediated SGLT2 pathway. Cell Metab. 2016;12(23(4)):699–711. doi: 10.1016/j.cmet.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 55.Ji W, et al. Effects of canagliflozin on weight loss in high-fat diet-induced obese mice. PLoS One. 2017;30(12(6)):e0179960. doi: 10.1371/journal.pone.0179960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shiba K, et al. Canagliflozin, an SGLT2 inhibitor, attenuates the development of hepatocellular carcinoma in a mouse model of human NASH. Sci Rep. 2018;5(8(1)):2362. doi: 10.1038/s41598-018-19658-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zinman B, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015;373:2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 58.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat. Rev. Mol. Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.