

A bacterial histone-like protein regulates the formation of privileged, drug-tolerant subpopulations of mycobacteria.
Abstract
There is increasing evidence that phenotypically drug-resistant bacteria may be important determinants of antibiotic treatment failure. Using high-throughput imaging, we defined distinct subpopulations of mycobacterial cells that exhibit heritable but semi-stable drug resistance. These subpopulations have distinct transcriptional signatures and growth characteristics at both bulk and single-cell levels, which are also heritable and semi-stable. We find that the mycobacterial histone-like protein HupB is required for the formation of these subpopulations. Using proteomic approaches, we further demonstrate that HupB is posttranslationally modified by lysine acetylation and lysine methylation. Mutation of a single posttranslational modification site specifically abolishes the formation of one of the drug-resistant subpopulations of cells, providing the first evidence in prokaryotes that posttranslational modification of a bacterial nucleoid-associated protein may epigenetically regulate cell state.
INTRODUCTION
A major clinical challenge in the tuberculosis field is that long courses of treatment are required to clear infection. One compelling explanation for the long treatment duration is that within a population of genetically identical mycobacterial cells, there are subpopulations of cells that are significantly less susceptible to antibiotics and that prolonged drug exposure is required to clear these bacteria. The most commonly invoked subpopulation of phenotypically drug-tolerant cells are so-called persister cells. Persister cells were first identified as rare, nongrowing cells that are able to withstand drug concentrations many times higher than the minimum inhibitory concentration (MIC) (1). When persister cells are regrown in media without drug, the resulting progeny are killed with the same kinetics as the starting population (2). They are described as “phenotypically” drug-tolerant, in contradistinction to genetically encoded alterations in drug susceptibility. “Drug tolerance” indicates that the bacterium survives but not does grow in the presence of drug, where growth in the presence of drug is considered “drug resistance” (3).
More recent work in both model organisms and mycobacteria suggests that persister cells should be viewed as the end of a continuum of physiologic states and drug susceptibilities that exist within genetically homogeneous bacterial populations. Recent studies of mycobacteria have identified subpopulations of phenotypically drug-resistant cells that arise at a relatively high frequency (4–10). Here, phenotypic drug resistance tracked between sister cells, suggesting that drug resistance was epigenetically regulated in these actively growing subpopulations (8). However, the molecular mechanisms underlying the emergence of phenotypically drug-resistant populations of mycobacterial cells remain unclear.
To address this question, we used an adaptation of a previously established high-throughput dynamic imaging technique to identify and characterize bacterial subpopulations that grow in the presence of drug (11). This approach allowed us to define subpopulations of mycobacterial cells that grow in the presence of the first-line antitubercular antibiotic isoniazid (INH). Using comprehensive transcriptional profiling and live-cell imaging, we define two subpopulations, large and small colony variants (LCVs and SCVs), with distinct transcriptional signatures and growth properties. The drug resistance and growth characteristics of these subpopulations are heritable and semi-stable, suggesting that they are epigenetically regulated. We find that the nucleoid-associated protein HupB is required for the formation of these phenotypically drug-resistant subpopulations. HupB is posttranslationally methylated and acetylated at lysine residues, and previous work demonstrates that HupB lysine acetylation alters chromatin structure (12). We demonstrate that mutation of a single HupB posttranslational modification site results in the specific loss of the SCV subpopulation. These findings support a model whereby posttranslational modification of HupB heritably alters gene expression and phenotypic state in a subpopulation of mycobacterial cells, akin to epigenetic regulation through the posttranslational modification of eukaryotic histones.
RESULTS
Growth in the presence of drug defines mycobacterial subpopulations
To identify subpopulations of mycobacteria that grow in the presence of drug, we used an adaptation of ScanLag, a technique that combines plating with high-throughput dynamic imaging to identify rare bacterial subpopulations with distinct growth properties (11). We used Mycobacterium smegmatis (mc2155), a model mycobacterium with a relatively rapid doubling time that is used as a tractable model for Mycobacterium tuberculosis. M. smegmatis was grown in standard media (7H9 with OADC, Tween 80, and glycerol) without drug and then plated on medium containing the bactericidal antibiotic INH at a range of concentrations around the MIC established in broth culture (10) and on plates (Fig. 1A). In broth culture, as the drug concentration approaches MIC90, there is a slowing but not complete inhibition of the bacterial population’s growth. We reasoned that at MIC90 concentrations of drug, the entire population might be growing slowly but homogeneously, and in this case, all the cells plated would form colonies slowly at the same time. Alternatively, if slow bulk growth reflects the average of growth inhibition for the majority of the bacterial population mixed with a rare, faster-growing subpopulation, only the subset of growing cells would form colonies. We observe the latter; as drug concentration increases, a subset of the plated cells forms colonies (Fig. 1A). When M. smegmatis is plated on medium at the concentration of INH, at which ~10% of cells form colonies (4 μg/ml), these colonies emerge at two distinct time points (Fig. 1, B, D, and E), ultimately generating a bimodal distribution of colony sizes (Fig. 1E). This suggests the presence of two distinct cell lineages that grow in the presence of drug. These findings are qualitatively similar to the colony variants observed in M. tuberculosis, even in the absence of drug exposure (Fig. 1C and fig. S1), although there was a broader range of colony sizes observed in M. tuberculosis than in M. smegmatis. In the setting of drug exposure, as INH concentrations increase toward MIC90, slowed bulk population growth also appears to reflect the selection of a subpopulation of cells able to form colonies on drug (Fig. 1C and fig. S1).
Fig. 1. Identification of mycobacterial subpopulations that grow in the presence of drug.
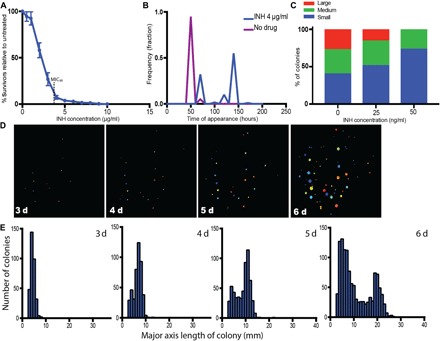
(A, B, D, and E) M. smegmatis. (C) M. tuberculosis. (A) Colonies formed on INH plates with indicated concentrations as a percentage of colonies formed on plates without drug. (B) Time of initial detection of colonies, as determined by CellProfiler, in the presence and absence of INH (4 μg/ml). (C) Distribution of M. tuberculosis colony sizes after 22 days of growth on INH. (D) Example series of one plate tracked over time with CellProfiler (INH, 4 μg/ml). Plates were run through custom program, and colored circles indicate the actual size of the detected colonies. (E) Histogram of major axis length of colonies for all colonies plated over time (INH, 4 μg/ml).
Phenotypic drug resistance is heritable and semi-stable
The cells that grow on concentrations of INH near the MIC are able to form colonies, indicating that their drug resistance is at least partially heritable. To determine the stability of the drug-resistant phenotype, SCVs and LCVs of M. smegmatis were picked into liquid medium without drug and then replated on INH after various periods of outgrowth. Upon immediate reexposure to antibiotics, the progeny of both the SCVs and LCVs exhibited significantly increased INH resistance as compared to the unselected starting population of M. smegmatis cells (Fig. 2A). With increasing time in culture, this increased resistance was gradually lost such that after 96 hours of culture, the cultured LCV and SCV cells recapitulated the drug susceptibility of the starting population (Fig. 2A).
Fig. 2. Phenotypic drug resistance is heritable and semi-stable.
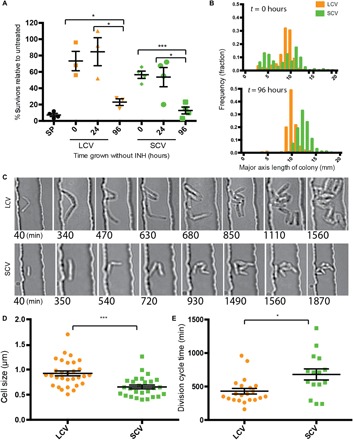
(A) Small and large colonies were picked, resuspended in liquid media, and plated immediately (t = 0) and then passaged through liquid media without drug and plated at indicated time points. Ratio of colonies on INH plates/plates without INH was measured. SP, starting population. Significance was determined by unpaired t test. (B) LCVs and SCVs were picked from INH plates, resuspended in 7H9 media, and replated onto plates without drug. The histograms of the resulting colony sizes from each colony type after immediate replating are shown. (C) Large and small colonies were picked from INH plates, and cells were loaded into a microfluidic device and imaged for ~48 hours in media with INH (4 μg/ml). (D) Quantification of cell size at birth. Significance was determined by unpaired t test. (E) Quantification of time from birth to division for individual cells. Significance was determined by unpaired Student’s t test. For all panels, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Variants have distinct growth properties
In addition to demonstrating semi-stable drug resistance, the colony morphotypes are also heritable and semi-stable. Thus, when cells from small colonies are replated, they produce more small colonies than cells from large colonies (Fig. 2B). However, after 96 hours of culture, the distribution of colony sizes from LCV and SCV cells is indistinguishable. These data suggest that the different colony morphotypes arise from cells with different growth parameters. To test this hypothesis, the growth characteristics of LCVs and SCVs were assessed at a single-cell level in the presence of INH using live-cell imaging (Fig. 2C). Both cells from LCVs and SCVs grow and divide in the presence of INH at 4 μg/ml (Fig. 2C), unlike cells from the starting population and consistent with the plating data. Cells from LCVs are significantly longer (Fig. 2D) and have shorter division times (Fig. 2E) compared to cells from small colonies.
Variants have distinct transcriptional signatures
We hypothesized that differences in transcriptional state underlie the distinct growth properties of the cells forming LCVs and SCVs. To test this hypothesis, we created pools of LCVs and SCVs and performed RNA sequencing (RNA-seq). DESeq analysis identified extensive transcriptional differences between the LCVs and SCVs (Fig. 3, A and B), suggestive of broad transcriptional repression in the small colony cells (13–15). Five hundred thirty-three genes were found to be significantly down-regulated (fold change > 2, P < 0.01) in the SCVs, including ~20 genes that were down-regulated more than 10-fold (Fig. 3C and table S2). The differentially expressed genes included genes implicated in metabolic functions [such as MSMEG_5568 (clavaldehyde dehydrogenase), MSMEG_0894 (dihydrodipicolinate reductase), and MSMEG_3362 (enoyl–coenzyme A hydratase)] and transcriptional regulation (such as arsR, marR, and tetR), consistent with broad differences in cellular physiology (accession numbers SRR5507179, SRR5507182, SRR5507184, and SRR5507185). Neither katG nor inhA was differentially expressed in SCVs, suggesting that the mechanism of phenotypic drug resistance is not differential transcription of the activator or target of INH.
Fig. 3. Variants have distinct transcriptional signatures.
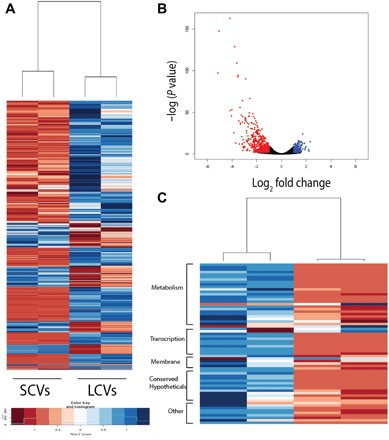
Pools of LCVs and SCVs were assayed in duplicate. (A) Hierarchical clustering analysis of genes differentially expressed more than threefold. (B) Volcano plot analysis of RNA-seq data comparing gene expression profiles of SCVs and LCVs. Dots indicated in red and blue are genes that are down-regulated and up-regulated, respectively, in the SCVs more than twofold with a P value of <0.01. (C) Gene categories of genes differentially expressed >10-fold in LCVs versus SCVs.
HupB is required for the formation of drug-resistant variants
The frequency at which these INH-resistant cells arise and revert was too high to be due to known mutational mechanisms in M. smegmatis (16). To assess the possibility that they arise through an alternative mutation mechanism that generates high-frequency genetic variation, whole-genome sequencing (WGS) of LCVs and SCVs was performed. There were no individual mutations or mutated genes in recognizable pathways associated exclusively with either variant type (accession numbers SRR5507164, SRR507165, SRR5507174, SRR507175, SRR5507176, and SRR5507177). Thus, we hypothesized that a novel epigenetic mechanism governs the formation of these drug-resistant subpopulations.
HupB is a nucleoid-associated protein that has previously been implicated in growth regulation and survival after INH exposure (17–19). Deletion of hupB does not alter the growth of M. smegmatis in 7H9 medium (fig. S2), although it is essential for bacterial survival in M. tuberculosis (20). Previous work using bulk assays including alamar blue and an agar diffusion test has shown that a hupB deletion mutant is more susceptible to INH treatment (17, 18). Here, we quantified the number of wild-type (WT) and hupB mutant colonies on increasing concentrations of INH. Consistent with the previously published work, we found that the hupB deletion mutant is significantly less able to survive when plated on concentrations of INH near the MIC (Fig. 4A). The slope of the growth inhibition curve at these INH concentrations was steeper in the hupB deletion mutant as compared to the WT strain. This finding suggests that deletion of hupB results in the loss of a subpopulation of phenotypically drug-resistant cells, rather than affecting the growth of the entire population. These defects can be complemented with an integrated copy of WT hupB expressed from its native promoter (Fig. 4A). The hupB deletion mutant has defects in the formation of both LCVs and SCVs (Fig. 4B).
Fig. 4. Loss of HupB results in significant reduction of colony formation in the presence of INH and alters gene expression.
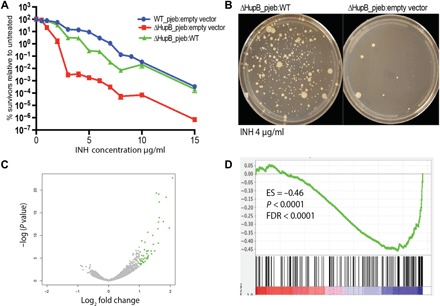
(A) Percent survivors were calculated from the number of colonies counted on the plates with drug as compared to the number of colonies on the plates without drug. (B) Representative images of ΔHupB_pjeb:WT and ΔHupB_pjeb:empty vector plated on INH (4 μg/ml). (C) RNA-seq analysis comparing gene expression patterns in ΔHupB_pjeb:WT and ΔHupB_pjeb:empty. Dots indicated in green are up-regulated in the ΔHupB_pjeb:empty strain. Strains were collected at late stationary phase. (D) GSEA of genes differentially expressed in ΔHupB_pjeb:empty strain versus ΔHupB_pjeb:WT as compared to the genes identified as down-regulated more than fourfold with a P value of <0.01 using DESeq analysis in comparison to SCV versus LCV. ES, enrichment score.
The impact of hupB on gene expression has not previously been determined in mycobacteria, although work on homologues in other organisms suggests that it is responsible for the maintenance and plasticity of the bacterial chromosome (21–24). Consistent with this model, RNA-seq analysis of the hupB deletion mutant as compared to WT cells demonstrated that loss of hupB results in derepression of 77 genes (Fig. 4C and table S3; BioSample accession numbers SAMN06770114 and SAMN06770117). Gene set enrichment analysis (GSEA) demonstrated that the genes that are up-regulated in the hupB deletion mutant significantly overlap with those that are down-regulated in the SCVs (Fig. 4D), including genes involved in metabolism (MSMEG_2659 and MSMEG_3198) and transcriptional regulation (MSMEG_3960 and MSMEG_6451), suggesting a model whereby HupB-mediated repression of gene expression through its action on chromatin state might bifurcate the population into SCVs and LCVs (25).
Mutation of a HupB modification site alters gene expression and results in specific loss of small variant subpopulation
Published studies have demonstrated that mycobacterial HupB has an unusual structure, composed of an N-terminal domain that is homologous to other prokaryotic HU proteins and a C-terminal domain that contains a tetrapeptide repeat characteristics of eukaryotic histone H1 proteins (26, 27). Studies have identified lysine acetylation and serine/threonine phosphorylation sites in the N-terminal domain of M. tuberculosis HupB and have further shown that in vitro acetylation of HupB alters its chromatin binding capacity (12, 28). We performed a whole-cell proteomic screen using WT M. smegmatis grown in 7H9 and found evidence that these modification sites are conserved in M. smegmatis (fig. S3) (28, 29). To confirm these modification sites, we purified a His-tagged version of HupB from M. smegmatis and performed liquid chromatography–tandem mass spectrometry (MS) on the purified protein, confirming six methylated and acetylated sites (Fig. 5, A and C, and fig. S4). Strikingly, three of six of the modified sites were identified as putative DNA binding sites in the published crystal structure of the N-terminal domain of HupB (Fig. 5B) (30), suggesting that modification of these sites might modulate the DNA binding capacity of HupB and thereby alter gene expression.
Fig. 5. Posttranslational modification sites on HupB.
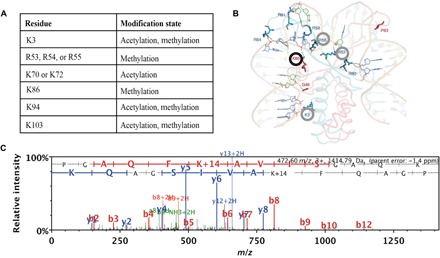
(A) Modified amino acids and posttranslational modification sites on HupB identified by whole-cell proteomics. (B) Predicted structure of M. tuberculosis HupB reprinted with permission from Bhowmick et al. (30). Modified amino acid residues identified in this study are circled. The K86 residue, the spectrum of which is depicted in (C), is circled in black. (C) MS/MS of tryptic peptide identified after a Mascot database search, where methylation on Lys86 can be inferred from the peptide mass/charge ratio (m/z) and the ion fragmentation pattern.
We used a genetic approach to test the importance of the HupB modification sites for the HupB-dependent formation of INH-resistant colonies because many of these sites appear to be the target of several modifications, which made quantitative comparison of modification levels challenging. We complemented the hupB deletion mutant with hupB alleles in which we systematically mutated the lysine modification sites to arginine or alanine. We first assessed the impact of these mutations on protein abundance as determined by Western blot analysis. Many of the mutations appear to grossly destabilize HupB, but we identified two highly conservative mutations, K86R and R55K, that do not alter protein abundance (fig. S5). We focused on HupB K86R for further characterization because mycobacteria carrying this mutation display WT growth characteristics when grown in 7H9 medium (fig. S2). RNA-seq analysis of the ΔHupB_pjeb:WT and ΔHupB_pjeb:K86R strains demonstrated that the K86R mutation does not alter gene expression at a bulk level (accession numbers SAMN06770114 and SAMN06770115).
We sought to assess the effect of disrupting the K86 modification site on the ability of M. smegmatis to survive when exposed to concentrations of INH near the MIC. We compared the ability of the M. smegmatis hupB deletion mutant complemented with WT hupB or the K86R variant to form colonies on INH. The K86R mutant forms significantly fewer colonies when plated on INH (Fig. 6A). Strikingly, although the K86R mutant strain forms LCVs at the same rate as the WT, this mutation results in the specific loss of SCV formation (Fig. 6, C to E).
Fig. 6. K86R mutation results in the preferential loss of small colonies.
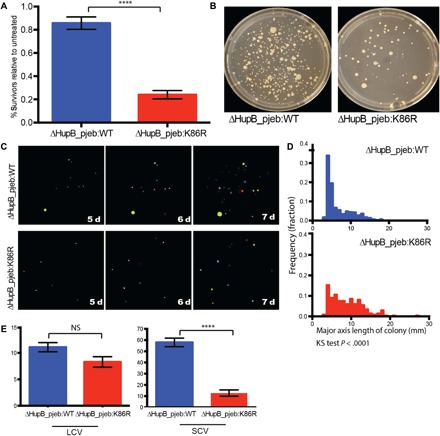
(A) Total colony formation in the presence of INH (4 μg/ml) as a percentage of colonies formed on plates without drug. Colonies were counted after 7 days. (B) Representative plates of indicated strains plated on INH (4 μg/ml) at day 7. (C) Representative plates tracked over time using CellProfiler. (D) Histograms of colony sizes on INH plates (4 μg/ml) at day 7. Frequency distribution between ΔHupB_pjeb:WT and ΔHupB_pjeb:K86R is significantly different as determined by the Kolmogorov-Smirnov (KS) test. (E) Number of SCVs and LCVs at day 7 on INH plates (4 μg/ml). In (A) and (E), significance is determined by the Mann-Whitney test and is indicative of over 10 independent biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. NS, not significant.
DISCUSSION
In M. tuberculosis infection, there is a biphasic response to antibiotic treatment, where roughly 90% of bacteria are killed within the first few days after treatment, whereas the last 10% of the population exhibit a significantly slower rate of clearance (31). The recalcitrance of this subpopulation to antibiotics is thought to be partially responsible for the extended treatment periods required to fully clear M. tuberculosis infection (31). The makeup of this slowly eliminated drug-tolerant bacterial subpopulation is largely unknown, although transcriptional adaptation has been hypothesized to play a role (31).
These observations have generated interest in understanding the mechanisms and drug susceptibilities of phenotypically distinct subpopulations of mycobacterial cells. Until recently, bacterial subpopulations have largely been understood in terms of the persister cell paradigm. However, recent studies have demonstrated that in mycobacterial populations, there is an array of phenotypically distinct cells, recognizable because of differences in drug susceptibilities. They arise and revert at different frequencies and are characterized by different growth properties and the magnitude of their drug resistances. The relative importance of any given subpopulation for treatment failure has been impossible to assess, however, because there is no mechanistic understanding of their formation or genetic path to modulate their emergence.
We find that treating M. smegmatis with concentrations of INH near the MIC kills most, but not all, of the bacteria in the population. The surviving cells are composed of two distinct, epigenetically regulated lineages of bacteria, distinguishable as SCVs and LCVs, which continue to grow in the presence of drug. The cells in these variant populations have distinct transcriptional profiles, growth properties, and altered drug susceptibility that are semi-heritable and cannot be explained by underlying genetic mutations.
Variable colony phenotypes have previously been observed in response to stressors in other organisms. SCVs in Staphylococcus aureus were first discovered in the early 1900s. Interest in them increased in the last couple of decades due to growing evidence that SCVs are associated with chronic, persistent infections that are notoriously difficult to treat (32, 33). SCVs have since been identified in other bacteria, including Pseudomonas aeruginosa, and occur in response to a variety of in vitro and in vivo stress conditions (34–36).
Still, the term SCV is descriptive and does not imply a common molecular mechanism. The SCVs that have been identified under this array of conditions have fallen into two broad classes: (i) stable SCVs that do not revert to the normal colony phenotype upon subculture and are caused by an underlying genetic mutation, and (ii) unstable SCVs that rapidly revert to the normal colony phenotype upon subculturing. Stresses such as antibiotics and starvation have been shown to increase lag times of individual cells (37). Therefore, it has remained unclear whether unstable SCVs are formed by a founding cell population with increased lag times, or if the SCVs are composed of a lineage of cells that heritably grow differently in the presence of the applied stress. These two alternative models imply different underlying mechanisms and biological consequences, and thus, discriminating between them is important to improve our understanding of bacterial survival strategies. Here, we show that in mycobacteria, two distinct colony variant types grow in the presence of INH. These SCV and LCV subpopulations are composed of cells with distinct, heritable, growth properties and gene expression profiles, consistent with the latter model. These subpopulations together likely contribute to the overall INH tolerance of M. smegmatis that is observed with population-level assays.
We also demonstrate that the mycobacterial histone-like protein HupB is required for the formation of these drug-resistant subpopulations. Our experiments do not formally determine whether HupB-regulated subpopulations are preexisting in the population or whether they arise de novo in the face of drug treatment. The high rate at which we detect these resistant populations in the presence of drug (up to 10% of the population) suggests that antibiotic stress may increase the capacity of the bacteria to form these variants; however, future experiments must be done to discriminate between these possibilities.
We and others have also found that HupB is highly posttranslationally modified at lysine, serine, and threonine residues (12, 28). We find both modified and unmodified peptides in our analysis, suggesting that these modifications are present on only a subset of the HupB proteins in a bacterial cell culture. We demonstrate that mutation of a single lysine, which appears to have the potential to be both acetylated and methylated, results in the specific loss of the SCV subpopulation. Our studies do not resolve which modification of K86 is causally related to SCV formation. Our efforts to use quantitative proteomics to compare levels of modified K86 between SCV and LCV failed to demonstrate significant differences, suggesting more complex models whereby combinations of HupB modifications or modification at earlier stages of cellular differentiation drives SCV formation.
Previously established mechanisms of epigenetic inheritance in bacteria, such as DNA methylation and positive feedback loops, typically involve regulation of only one or a few genes (38–40). Our findings suggest the existence of a fundamentally distinct form of epigenetic regulation in prokaryotes, whereby modification of a histone-like protein has widespread effects on gene expression that are inherited across generations and bifurcates the populations into two distinct cell types. In eukaryotes, it is well established that histones can package chromosomal DNA into domains of varying compaction and transcriptional activity. A similar role for bacterial histone-like proteins in the structural organization of the bacterial nucleoid has more recently begun to be appreciated (24, 41, 42). Accordingly, a recent study showed that acetylation alters the ability of mycobacterial HupB to bind and compact DNA, suggesting a plausible mechanism by which these heritable changes in cell state and gene expression could be implemented (12). This form of heritable epigenetic regulation mirrors the role of histone modifications in eukaryotes and is the first indication that an analogous mechanism may exist in bacteria. Histone modifications were recently shown to drive the induction of a reversibly drug-tolerant state in subpopulations of cancer cells, suggesting unexpected parallels of drug survival strategies across these distant cell types (43).
Although here we used sublethal antibiotics to isolate these phenotypic variants, colony size variation has been observed in mycobacteria in a variety of conditions. At a single-cell level, semi-heritable phenotypic variation has been observed in M. tuberculosis in response to antibiotic and immune stress (44). These findings, together with our work, suggest that perhaps SCVs and LCVs may be privileged, epigenetically regulated subpopulations that are selected for under a variety of conditions and contribute to the recalcitrance of M. tuberculosis to immune control and antibiotic treatment.
MATERIALS AND METHODS
Bacterial plating assay
M. smegmatis cultures were started from individual colonies in triplicate and grown to stationary phase (approximately 2 days) in supplemented 7H9 broth (7H9 with OADC, 0.05% Tween 80, and 0.2% glycerol). Cultures were then normalized, back-diluted, and grown to an optical density (OD) of ~1.0. Cultures were then diluted from 10 × 10−3 to 10 × 10−5, and 100-μl aliquots were plated on 7H10 plates [7H10 supplemented with OADC (Middlebrook) and 0.05% Tween 80]. INH was diluted in water at 5 mg/ml and added to plates at indicated concentrations. Plates were incubated at 37°C for the indicated periods of time. Plates were imaged at defined time points following plating, and images were analyzed using a custom CellProfiler pipeline to count and measure the size of colonies.
M. tuberculosis drug responses
M. tuberculosis H37Rv was grown to mid-log phase in supplemented 7H9. The culture was gravity-filtered through a 5-μm syringe filter (Millipore) and further diluted in the same media before plating on 7H10 containing varying concentrations of INH. Plates were incubated for 22 days at 37°C before measurement of colony size.
Live-cell imaging
Small and large colonies were picked from 7H10 plates with INH (4 μg/ml) and resuspended in 1 ml of supplemented 7H9 containing INH (4 μg/ml) and sonicated. Cells were loaded onto a custom microfluidic device and imaged on a DeltaVision microscope, as previously described (10). Images were acquired every 10 min for 48 hours.
Reversion assays
Small and large colonies were picked from 7H10 plates with INH (4 μg/ml), resuspended in 1 ml of supplemented 7H9, sonicated, diluted as indicated, and plated either directly or after the indicated period of growth in supplemented 7H9 medium without drug. Cells were plated on 7H10 plates with and without INH.
RNA-seq analysis
Cells were harvested and resuspended in 1 ml of TRIzol and disrupted via bead beating. RNA was extracted using Direct-zol columns (Zymo Research), and ribosomal RNA was removed using a magnetic Ribo-Zero Bacteria kit (Illumina). Libraries were prepared with the KAPA mRNA-Seq Kit and sequenced on the MiSeq (Illumina). Reads were aligned with Burrows-Wheeler Aligner (BWA) using the BWA-MEM algorithm (45). Alignments were processed with SAMtools (46). Read counts were obtained with HTseq-count FIX using the M. smegmatis mc2155 reference sequence (NC_008596) (15). Differential gene expression analysis was done with the R package DESeq2 (47). GSEA was performed as previously described (25). Differentially expressed genes in between the indicated strains were preranked on the basis of log-fold change and then compared via GSEA to genes down-regulated fourfold or greater in the small colonies.
Whole-genome sequencing
Small and large colonies were isolated from three independent cultures and grown in 5 ml of supplemented 7H9 overnight. Cells were pelleted and DNA was extracted as described previously (48). Libraries were prepared with the Nextera XT Kit and sequenced on the MiSeq at a depth of ~2 million reads per sample. To identify polymorphisms, reads were aligned with BWA using the BWA-MEM algorithm (45). Alignments were processed with Pilon (49).
Unbiased proteomic analysis
Data from our previously published whole-cell proteomic analysis of M. tuberculosis H3Rv were reanalyzed to identify posttranslationally modified peptides (50). Spectra were searched against a database that contained the predicted open reading frames (ORFs) in the genome of H37Rv supplemented with common contaminates using SEQUEST (Thermo Fisher Scientific). Carbamidomethylation on cysteine was specified as fixed modification, and oxidation on methionine, acetylation on lysine, and acetylation on protein N terminus were specified as variable modifications. Peptides were filtered at a 1% false discovery rate with PeptideProphet and grouped into proteins with ProteinProphet with a cutoff of 0.95.
Construction of hupB deletion and mutant strains
A hupB deletion strain was constructed using previously published protocols to make unmarked deletion mutants by allelic exchange (51). To generate complementation constructs in which hupB is expressed under its native promoter, the hupB ORF and 200 base pairs upstream of the ORF were cloned into the integrating mycobacterial expression vector pJEB402 using Xba I and Hind III site-directed mutagenesis performed using the QuikChange protocol (Agilent). To generate His-tagged hupB, a 6× His tag was cloned at the C terminus of hupB.
HupB purification
His-tagged hupB was expressed in M. smegmatis under its native promoter, as described above. To purify the protein, cells expressing His-tagged hupB or negative control cells containing an empty vector were cultured in supplemented 7H9 with kanamycin (25 μg/ml) to an OD of ~1.0. Cells from 1 liter of culture were pelleted, and pellets were resuspended in lysis buffer [200 mM NaCl, 50 mM tris-HCl (pH 7.9), 0.1 mM EDTA, 0.1 mM dithiothreitol, and 5% glycerol]. Cells were lysed using a French press, and debris was removed by centrifuging for 10 min at 10,000 rpm. Supernatant was added to cobalt beads (TALON) and incubated overnight at 4°. Beads were spun down at low speed and liquid was removed, and then beads were washed three times for 10 min in wash buffer [50 mM tris-HCl (pH 7.9), 0.1 mM EDTA, 500 mM NaCl, and 10 mM imidazole]. HupB was eluted with 1 ml of elution buffer [50 mM tris-HCl (pH 7.9), 0.1 mM EDTA, 400 mM NaCl, and 500 mM imidazole]. Eluate was mixed with 2× tricine SDS–polyacrylamide gel electrophoresis buffer (Life Technologies) and run on a 10 to 20% tricine gel (Life Technologies). Expressed HupB was identified by Coomassie staining at the expected molecular weight and not present in the negative control. The HupB band was cut out, and in-gel digestion with trypsin was performed (52).
A 4.0-μl aliquot was directly injected onto a custom-packed 2 cm × 100 μm C18 Magic 5-μm particle trap column. Peptides were then eluted and sprayed from a custom-packed emitter (75 μm × 25 cm C18 Magic 3-μm particle) with a linear gradient from 95% solvent A (0.1% formic acid in water) to 35% solvent B (0.1% formic acid in acetonitrile) in 60 min at a flow rate of 300 nl/min on a Waters nanoACQUITY UPLC system. Data-dependent acquisitions were performed on a Q Exactive mass spectrometer (Thermo Fisher Scientific) according to an experiment, where full MS scans from 300 to 1750 m/z were acquired at a resolution of 70,000 followed by 10 MS/MS scans acquired under high-energy collisional dissociation fragmentation at a resolution of 17,500 with an isolation width of 1.6 Da. Raw data files were processed with Proteome Discoverer (version 1.4) before searching with Mascot Server (version 2.5) against the M. smegmatis database. Search parameters used were tryptic with two missed cleavages, parent mass tolerances of 10 parts per million, and fragment mass tolerances of 0.05 Da. A fixed modification of carbamidomethyl cysteine, variable modifications of acetyl (protein N-term and lysine) and pyroglutamic for N-term glutamine, oxidation of methionine, and mono-, di-, and trimethylation of lysine and arginine were considered. Search results were loaded into the Scaffold Viewer (Proteome Software Inc.) for assessment of protein identification probabilities and label-free quantitation.
Supplementary Material
Acknowledgments
Funding: This work was supported by Burroughs Wellcome Foundation and National Institute of Allergy and Infectious Diseases grants U19 AI107774, U19 AI109755, T32 AI 49928-8 (appointment, 1 September 2010 to 31 August 2011), T32 AI 49928-9 (appointment, 1 September 2011 to 31 August 2012), and T32 AI 49928-10 (appointment, 1 September 2012 to 31 August 2013). Author contributions: A.S., S.M.F., G.H.B., and T.R. designed the experiments. A.S., G.H.B., and A.D. executed experiments. M.R.C. analyzed RNA-seq and WGS data. J.L. conducted and analyzed MS data. B.B. assisted with analysis of MS data. A.S., G.H.B., and S.M.F. wrote the manuscript. Contributions to figures/tables are as follows: Fig. 1, A.D. and G.H.B.; Fig. 2, A.S.; Fig. 3, A.S. and M.R.C.; Fig. 4, A.S. and M.R.C.; Fig. 5, A.S. and J.L.; Fig. 6, A.S.; table S1, A.S.; table S2, A.S. and M.R.C.; table S3, A.S. and M.R.C.; fig. S1, G.H.B.; fig. S2, A.S.; fig. S3, A.S. and J.L.; fig. S4, J.L.; and fig. S5, A.S. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao1478/DC1
table S1. Bacterial strains used in this study.
table S2. Genes down-regulated >10-fold in small versus large colonies.
table S3. Genes up-regulated more than fivefold in HupBΔ versus WT.
fig. S1. M. tuberculosis colony sizes and numbers plated on increasing concentrations of INH.
fig. S2. M. smegmatis strains expressing WT and mutant HupB alleles grown at WT rates.
fig. S3. Modifications identified in previous study and current work.
fig. S4. His-tagged HupB was purified from M. smegmatis, and modifications were identified by MS.
fig. S5. Effects of disrupting HupB modification sites on HupB protein abundance.
REFERENCES AND NOTES
- 1.Bigger J. W., Treatment of staphylococcal infections with penicillin by intermittent sterilisation. Lancet 244, 497–500 (1944). [Google Scholar]
- 2.Lewis K., Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Aldridge B. B., Keren I., Fortune S. M., The spectrum of drug susceptibility in mycobacteria. Microbiol. Spectr. 2, 711–725 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Rego E. H., Audette R. E., Rubin E. J., Deletion of a mycobacterial divisome factor collapses single-cell phenotypic heterogeneity. Nature 546, 153–157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Su H.-W., Zhu J.-H., Li H., Cai R.-J., Ealand C., Wang X., Chen Y.-X., Kayani M. u. R., Zhu T. F., Moradigaravand D., Huang H., Kana B. D., Javid B., The essential mycobacterial amidotransferase GatCAB is a modulator of specific translational fidelity. Nat. Microbiol. 1, 16147 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Certain L. K., Way J. C., Pezone M. J., Collins J. J., Using engineered bacteria to characterize infection dynamics and antibiotic effects in vivo. Cell Host Microbe 22, 263–268.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Adams K. N., Takaki K., Connolly L. E., Wiedenhoft H., Winglee K., Humbert O., Edelstein P. H., Cosma C. L., Ramakrishnan L., Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145, 39–53 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wakamoto Y., Dhar N., Chait R., Schneider K., Signorino-Gelo F., Leibler S., McKinney J. D., Dynamic persistence of antibiotic-stressed mycobacteria. Science 339, 91–95 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Claudi B., Spröte P., Chirkova A., Personnic N., Zankl J., Schürmann N., Schmidt A., Bumann D., Phenotypic variation of Salmonella in host tissues delays eradication by antimicrobial chemotherapy. Cell 158, 722–733 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Aldridge B. B., Fernandez-Suarez M., Heller D., Ambravaneswaran V., Irimia D., Toner M., Fortune S. M., Asymmetry and aging of mycobacterial cells lead to variable growth and antibiotic susceptibility. Science 335, 100–104 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levin-Reisman I., Gefen O., Fridman O., Ronin I., Shwa D., Sheftel H., Balaban N. Q., Automated imaging with ScanLag reveals previously undetectable bacterial growth phenotypes. Nat. Methods 7, 737–739 (2010). [DOI] [PubMed] [Google Scholar]
- 12.Ghosh S., Padmanabhan B., Anand C., Nagaraja V., Lysine acetylation of the Mycobacterium tuberculosis HU protein modulates its DNA binding and genome organization. Mol. Microbiol. 100, 577–588 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Lee J.-H., Ammerman N. C., Nolan S., Geiman D. E., Lun S., Guo H., Bishai W. R., Isoniazid resistance without a loss of fitness in Mycobacterium tuberculosis. Nat. Commun. 3, 753 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kucukyildirim S., Long H., Sung W., Miller S. F., Doak T. G., Lynch M., The rate and spectrum of spontaneous mutations in Mycobacterium smegmatis, a bacterium naturally devoid of the postreplicative mismatch repair pathway. G3 6, 2157–2163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiteford D. C., Klingelhoets J. J., Bambenek M. H., Dahl J. L., Deletion of the histone-like protein (Hlp) from Mycobacterium smegmatis results in increased sensitivity to UV exposure, freezing and isoniazid. Microbiology 157, 327–335 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Niki M., Niki M., Tateishi Y., Ozeki Y., Kirikae T., Lewin A., Inoue Y., Matsumoto M., Dahl J. L., Ogura H., Kobayashi K., Matsumoto S., A novel mechanism of growth phase-dependent tolerance to isoniazid in mycobacteria. J. Biol. Chem. 287, 27743–27752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewin A., Baus D., Kamal E., Bon F., Kunisch R., Maurischat S., Adonopoulou M., Eich K., The mycobacterial DNA-binding protein 1 (MDP1) from Mycobacterium bovis BCG influences various growth characteristics. BMC Microbiol. 8, 91 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeJesus M. A., Gerrick E. R., Xu W., Park S. W., Long J. E., Boutte C. C., Rubin E. J., Schnappinger D., Ehrt S., Fortune S. M., Sassetti C. M., Ioerger T. R., Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio 8, e02133–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kar S., Edgar R., Adhya S., Nucleoid remodeling by an altered HU protein: Reorganization of the transcription program. Proc. Natl. Acad. Sci. U.S.A. 102, 16397–16402 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luijsterburg M. S., Noom M. C., Wuite G. J. L., Dame R. T., The architectural role of nucleoid-associated proteins in the organization of bacterial chromatin: A molecular perspective. J. Struct. Biol. 156, 262–272 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Swinger K. K., Rice P. A., IHF and HU: Flexible architects of bent DNA. Curr. Opin. Struct. Biol. 14, 28–35 (2004). [DOI] [PubMed] [Google Scholar]
- 24.Wang W., Li G.-W., Chen C., Xie X. S., Zhuang X., Chromosome organization by a nucleoid-associated protein in live bacteria. Science 333, 1445–1449 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee A., Bhattacharyya G., Grove A., The C-terminal domain of HU-related histone-like protein Hlp from Mycobacterium smegmatis mediates DNA end-joining. Biochemistry 47, 8744–8753 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Kushwaha A. K., Grove A., C-terminal low-complexity sequence repeats of Mycobacterium smegmatis Ku modulate DNA binding. Biosci. Rep. 33, 175–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta M., Sajid A., Sharma K., Ghosh S., Arora G., Singh R., Nagaraja V., Tandon V., Singh Y., HupB, a nucleoid-associated protein of Mycobacterium tuberculosis, is modified by serine/threonine protein kinases in vivo. J. Bacteriol. 196, 2646–2657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pethe K., Bifani P., Drobecq H., Sergheraert C., Debrie A.-S., Locht C., Menozzi F. D., Mycobacterial heparin-binding hemagglutinin and laminin-binding protein share antigenic methyllysines that confer resistance to proteolysis. Proc. Natl. Acad. Sci. U.S.A. 99, 10759–10764 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhowmick T., Ghosh S., Dixit K., Ganesan V., Ramagopal U. A., Dey D., Sarma S. P., Ramakumar S., Nagaraja V., Targeting Mycobacterium tuberculosis nucleoid-associated protein HU with structure-based inhibitors. Nat. Commun. 5, 4124 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Jindani A., Doré C. J., Mitchison D. A., Bactericidal and sterilizing activities of antituberculosis drugs during the first 14 days. Am. J. Respir. Crit. Care Med. 167, 1348–1354 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Proctor R. A., von Eiff C., Kahl B. C., Becker K., McNamara P., Herrmann M., Peters G., Small colony variants: A pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Kahl B. C., Small colony variants (SCVs) of Staphylococcus aureus—A bacterial survival strategy. Infect. Genet. Evol. 21, 515–522 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Leimer N., Rachmühl C., Palheiros Marques M., Bahlmann A. S., Furrer A., Eichenseher F., Seidl K., Matt U., Loessner M. J., Schuepbach R. A., Zinkernagel A. S., Nonstable Staphylococcus aureus small-colony variants are induced by low pH and sensitized to antimicrobial therapy by phagolysosomal alkalinization. J. Infect. Dis. 213, 305–313 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Mirani Z. A., Aziz M., Khan S. I., Small colony variants have a major role in stability and persistence of Staphylococcus aureus biofilms. J. Antibiot. 68, 98–105 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Tuchscherr L., Medina E., Hussain M., Völker W., Heitmann V., Niemann S., Holzinger D., Roth J., Proctor R. A., Becker K., Peters G., Löffler B., Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 3, 129–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fridman O., Goldberg A., Ronin I., Shoresh N., Balaban N. Q., Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 513, 418–421 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Veening J.-W., Smits W. K., Kuipers O. P., Bistability, epigenetics, and bet-hedging in bacteria. Annu. Rev. Microbiol. 62, 193–210 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Shell S. S., Prestwich E. G., Baek S.-H., Shah R. R., Sassetti C. M., Dedon P. C., Fortune S. M., DNA methylation impacts gene expression and ensures hypoxic survival of Mycobacterium tuberculosis. PLOS Pathog. 9, e1003419 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sánchez-Romero M. A., Cota I., Casadesús J., DNA methylation in bacteria: From the methyl group to the methylome. Curr. Opin. Microbiol. 25, 9–16 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Dillon S. C., Dorman C. J., Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Dame R. T., The role of nucleoid-associated proteins in the organization and compaction of bacterial chromatin. Mol. Microbiol. 56, 858–870 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Sharma S. V., Lee D. Y., Li B., Quinlan M. P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M. A., Wong K.-K., Brandstetter K., Wittner B., Ramaswamy S., Classon M., Settleman J., A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 141, 69–80 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Manina G., Dhar N., McKinney J. D., Stress and host immunity amplify Mycobacterium tuberculosis phenotypic heterogeneity and induce nongrowing metabolically active forms. Cell Host Microbe 17, 32–46 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Li H., Durbin R., Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hung J.-H., Weng Z., Mapping short sequence reads to a reference genome. Cold Spring Harb. Protoc. 2017, pdb.prot093161 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Anders S., Huber W., Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ford C. B., Lin P. L., Chase M. R., Shah R. R., Iartchouk O., Galagan J., Mohaideen N., Ioerger T. R., Sacchettini J. C., Lipsitch M., Flynn J. L., Fortune S. M., Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 43, 482–486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C. A., Zeng Q., Wortman J., Young S. K., Earl A. M., Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Millington K. A., Fortune S. M., Low J., Garces A., Hingley-Wilson S. M., Wickremasinghe M., Kon O. M., Lalvani A., Rv3615c is a highly immunodominant RD1 (Region of Difference 1)-dependent secreted antigen specific for Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. U.S.A. 108, 5730–5735 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pelicic V., Jackson M., Reyrat J.-M., Jacobs W. R., Gicquel B., Guilhot C., Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 94, 10955–10960 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shevchenko A., Tomas H., Havli J., Olsen J. V., Mann M., In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaao1478/DC1
table S1. Bacterial strains used in this study.
table S2. Genes down-regulated >10-fold in small versus large colonies.
table S3. Genes up-regulated more than fivefold in HupBΔ versus WT.
fig. S1. M. tuberculosis colony sizes and numbers plated on increasing concentrations of INH.
fig. S2. M. smegmatis strains expressing WT and mutant HupB alleles grown at WT rates.
fig. S3. Modifications identified in previous study and current work.
fig. S4. His-tagged HupB was purified from M. smegmatis, and modifications were identified by MS.
fig. S5. Effects of disrupting HupB modification sites on HupB protein abundance.