

Multiple invertebrates possess enzymes enabling de novo biosynthesis of essential omega-3 fatty acids.
Abstract
Marine ecosystems are responsible for virtually all production of omega-3 (ω3) long-chain polyunsaturated fatty acids (PUFA), which are essential nutrients for vertebrates. Current consensus is that marine microbes account for this production, given their possession of key enzymes including methyl-end (or “ωx”) desaturases. ωx desaturases have also been described in a small number of invertebrate animals, but their precise distribution has not been systematically explored. This study identifies 121 ωx desaturase sequences from 80 species within the Cnidaria, Rotifera, Mollusca, Annelida, and Arthropoda. Horizontal gene transfer has contributed to this hitherto unknown widespread distribution. Functional characterization of animal ωx desaturases provides evidence that multiple invertebrates have the ability to produce ω3 PUFA de novo and further biosynthesize ω3 long-chain PUFA. This finding represents a fundamental revision in our understanding of ω3 long-chain PUFA production in global food webs, by revealing that numerous widespread and abundant invertebrates have the endogenous capacity to make significant contributions beyond that coming from marine microbes.
INTRODUCTION
Marine ecosystems are responsible for virtually all natural production of omega-3 (ω3) long-chain (≥C20) polyunsaturated fatty acids (PUFA), which are essential dietary components for vertebrates that are required for neural development and health promotion while also having beneficial roles across a variety of human pathologies and disorders (1). With an expected increased demand for ω3 oil supplements prescribed for prevalent cardiovascular and inflammatory diseases in humans (1), there is intense interest in exploring novel sources of ω3 oils (2, 3). The current dogma is that photosynthetic marine microalgae, heterotrophic protists, and bacteria account for natural ω3 long-chain PUFA production (4–6), because they have all the enzymatic components necessary for de novo synthesis, unlike higher trophic organisms such as fish. Plants have the ability for de novo synthesis of PUFA of up to 18 carbons but limited elongation capacity to produce long-chain PUFA (7). Although some bacteria and lower eukaryotes can produce PUFA de novo via a polyketide synthase pathway (6), PUFA biosynthesis in most aquatic microbes requires fatty acid (FA) desaturases that catalyze the insertion of new double bonds (unsaturations) (8, 9). Δ9 desaturases mediate the insertion of the first unsaturation in stearic acid (18:0) to produce oleic acid (18:1ω9). Insertion of further unsaturations is achieved through sequential reactions driven by methyl-end (or “ωx”) desaturases that introduce a double bond between an existing unsaturation and the methyl end of the fatty acid (8). Thus, the action of an ω6 (or Δ12) desaturase enables the conversion of 18:1ω9 to 18:2ω6 [linoleic acid (LA)], the latter acting as an intermediate metabolite in de novo ω3 PUFA biosynthesis because it is subsequently converted to 18:3ω3 [α-linolenic acid (ALA)] by an ω3 (or Δ15) desaturase (4, 5). Although Δ9 desaturases are present in virtually all organisms (8), de novo ω3 PUFA production is only possible if both Δ12 and Δ15 desaturases coexist within an organism.
Vertebrates lack the ωx desaturases necessary for PUFA biosynthesis and thus require dietary LA and ALA to satisfy essential FA requirements (9, 10). The same condition has been largely accepted for all animals, with few exceptions reported. Two pulmonate molluscs (11) and some insects (12, 13) showed the ability to biosynthesize LA, but not ALA. Recently, the deep-sea tubeworm Riftia pachyptila (Annelida) has been shown to have an ω3 desaturase enabling the biosynthesis of ALA but not LA (14). Currently, only two terrestrial metazoans, the nematode Caenorhabditis elegans (15–17) and the arthropod Bemisia argentifolii (18), have been reported to have ωx desaturases with both Δ12 and Δ15 activities. If, contrary to current consensus, these few instances of metazoan ωx desaturases, in fact, reflect a prevalent occurrence of these enzymes across animals, then a major revision of our understanding of ω3 PUFA production in global food webs is required. The present study provides a comprehensive search of available sequence databases and analysis of the distribution of ωx desaturase genes across the Metazoa. We establish that, contrary to common perception, ωx desaturases are widely distributed across animal phyla. Furthermore, selected ωx desaturase genes from key lineages with widespread occurrence in aquatic ecosystems, namely, Cnidaria (Acropora millepora), Rotifera (Adineta vaga), Mollusca (Patella vulgata), Annelida (Platynereis dumerilii), and Arthropoda (Lepeophtheirus salmonis), were functionally characterized to clarify their role in PUFA biosynthesis. Our results demonstrate that multiple invertebrates can actively produce ω3 PUFA de novo. Moreover, we also establish additional desaturase capabilities in animal ωx desaturases that enable the biosynthesis of physiologically essential ω3 long-chain PUFA.
RESULTS AND DISCUSSION
Our search for ωx desaturase–like sequences showed the presence of these genes in both prokaryotes (Cyanobacteria) and eukaryotes including Archaeplastida (red and green algae, Viridiplantae), Excavata, SAR (stramenopiles, alveolates, and Rhizaria), Amoebozoa, Haptophyceae, Apusozoa, Heterolobosea, Cryptophyta, and Opisthokonta (divided into Fungi, Metazoa, and other Opisthokonta) (Fig. 1 and fig. S1). In Metazoa, we found hitherto unknown ωx desaturase diversity. Sequences with homology to ωx desaturases were retrieved from the phyla Cnidaria, Rotifera, Mollusca, Annelida, Arthropoda, and Nematoda. Overall, 121 ωx desaturase sequences from 80 animal species were identified (Fig. 1). A list of all genomes and transcriptomes searched, including those in which no ωx desaturase genes were found, can be accessed as described in the Data and materials availability section, below.
Fig. 1. Distribution of ωx desaturases.
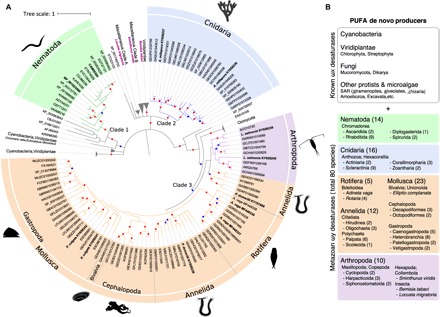
(A) A condensed phylogenetic tree depicting relationships among ωx desaturases from Cyanobacteria (8) and Eukaryota (279). Metazoan phyla (represented with 121 sequences) are named in the tree and assigned with colors for the following taxa: Nematoda (green), Cnidaria (blue), Arthropoda (purple), and Lophotrochozoa (orange). Nonmetazoan clades were collapsed and named according to their general taxonomic composition. “Miscellaneous Clade A” contains representatives of Amoeboza, Apusozoa, Haptophyceae, Heterolobosea, Nucleariidae, SAR, Chlorophyta, Rhodophyta, Cryptophyta, and the insect L. migratoria. Representatives of Excavata, Amoebozoa, Fungi, SAR, and the hexapod S. viridis compose “Miscellaneous Clade B.” The phylogenetic tree presented corresponds to the 80% rule consensus tree estimated in MrBayes. Colored dots on the nodes represent supporting values of 80% or above for MrBayes and PhyloBayes analyses (blue) or for all three (MrBayes, PhyloBayes, and RAxML) analyses (red). (B) Summary of taxonomic groups highlighting metazoans in which ωx desaturases have been identified in this study. Number of species is indicated in brackets. A list of all genomes and transcriptomes searched, including those in which no ωx desaturase genes were found, can be accessed as described in the Data and materials availability section.
Phylogenetic analysis using all recovered metazoan sequences, along with a representative subset of sequences from the remaining nonmetazoan groups, revealed at least three distinct ωx desaturase gene lineages (clades 1 to 3 in Fig. 1) in animals; most likely, these have independent evolutionary origins, because they do not form a single monophyletic animal clade. Clade 1 was composed exclusively of sequences from nematodes, including two C. elegans ωx desaturases (15–17), with an outgroup of nonmetazoan genes (for example, microalgae, a fungus, and an alveolate) (Fig. 1). Clade 2 comprised two distinct gene lineages in Cnidaria (Fig. 1), with sequences from stony corals, coral anemones, sea anemones, and zoanthids. The cnidarian sequences (clade 2) were nested within a broad and well-supported clade that included most of the eukaryotic nonmetazoan sequences of the data set, but the specific sister group to clade 2 was not well resolved (Fig. 1). Clade 3 included sequences from Lophotrochozoa (namely, Rotifera, Mollusca, and Annelida) and nonnematode Ecdysozoa (copepod arthropods). Oomycota sequences formed a well-resolved sister group to the clade 3 animal genes (Fig. 1). As in clade 2, there were frequently two genes in most clade 3 species. Beyond clades 1 to 3, three smaller sets of animal sequences from the arthropods Bemisia tabaci (Hemiptera), Locusta migratoria (Orthoptera), and Sminthurus viridis (Collembola) appeared scattered in the tree (Fig. 1). The division of most metazoan sequences into three main clades is also consistent with the amino acid compositions of the three histidine box motifs (H-box1–3) typical of desaturases (fig. S2) (19).
Horizontal gene transfer (HGT) can be inferred in some animal species and may explain the general phylogenetic pattern of metazoan ωx desaturase genes. For example, several ωx desaturase–like sequences from the silverleaf whitefly, B. tabaci, are most closely related to one of the Viridiplantae ωx desaturase clades in the phylogeny (Fig. 1 and fig. S1), which includes FAD2 plant desaturases functionally characterized as Δ12 desaturases (20). To exclude the possibility that these sequences might represent plant sequence contamination in B. tabaci, we examined the genomic neighborhoods of B. tabaci ωx desaturases. The B. tabaci sequences are genome-anchored and distributed across five different scaffolds (Fig. 2A). B. tabaci ωx desaturase sequences are flanked by genes that have clear orthologs in other metazoans (Fig. 2A). This genomic arrangement implies that the B. tabaci ωx desaturase sequences are bona fide components of the whitefly genome. Thus, HGT likely accounts for the occurrence of ωx desaturases in B. tabaci, with the genes possibly transferring from plants on which this whitefly feeds. Further cases of HGT appear to exist in a locust and a collembolan. Collembola have ωx desaturase sequences that are most closely related to sequences from Oomycota species that are plant pathogens, which are presumably prevalent in the soil habitat occupied by collembolans. Our data thus indicate that the described ability of some collembolans to synthesize LA is due to an endogenous capacity derived from the presence of ωx desaturases in this hexapod and not to any associated microbes (21). Furthermore, HGT has been suggested for the Fat-1 and Fat-2 ωx desaturases in C. elegans (15–17), with protists postulated as probable “donor taxa” (22). More generally, HGT between closely associated organisms has been established previously (23, 24), whereas other desaturase genes have been demonstrated to have arisen via HGT in arthropods, such as the two-spotted spider mite, Tetranychus urticae (25). Therefore, although additional evolutionary processes are also likely to be affecting metazoan ωx desaturase genes, it is reasonable to suppose that HGT could account for multiple ωx desaturases in metazoans. HGT is a particularly plausible scenario for aquatic invertebrates that are in direct contact with single-cell primary producers (6). Also consistent with this hypothesis is the fact that many of the ωx desaturase sequences from Cnidaria, Mollusca, Annelida, and Arthropoda are intronless genes (Fig. 2B), a feature suggesting a gene origin via RNA retrotransposition-mediated HGT (26).
Fig. 2. HGT accounts for the presence of ωx desaturase in some animal genomes.
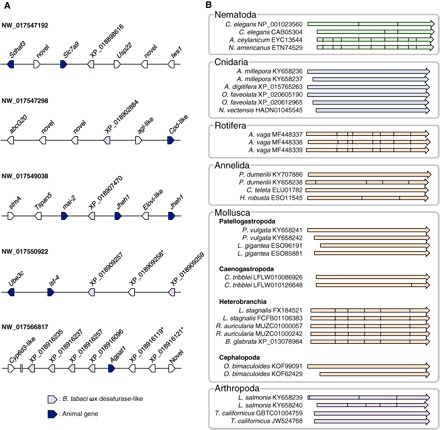
(A) Multiple copies of ωx desaturase–like sequences scattered among several loci of the B. tabaci genome. The ωx desaturase–like sequences are found in five different scaffolds and are flanked by animal genes. * indicates sequences that did not pass the filter (probability between 0.50 and 0.70). (B) Intron-exon structures in the ωx desaturase genes from selected metazoan species. The arrows indicate aligned coding sequences (CDS) extracted from the genomic assembly of each species. The positions of introns are indicated as solid lines within the arrows. The dotted line indicates shared intron-exon boundaries. All cnidarian genes and several genes from Annelida and Mollusca have an “intronless” organization. The CDS of the three ωx desaturase genes from A. vaga (Rotifera) share the same overall structure consisting of eight exons. In addition, all genes from three Heterobranchia species have the same structure with six exons in the CDS. Variable gene structures were found in Nematoda, Annelida, and Arthropoda. The arrow colors are consistent with those used to represent clades in Fig. 1.
Having established that ωx desaturase sequences are phylogenetically widespread across the Metazoa, it was a central aim to confirm that these genes are capable of de novo ω3 PUFA biosynthesis. Using a yeast heterologous expression system, we investigated the functions of two distinct ωx desaturases for each of five representative species of selected metazoan lineages including Cnidaria (A. millepora), Rotifera (A. vaga), Mollusca (P. vulgata), Annelida (P. dumerilii), and Arthropoda (L. salmonis) (Fig. 3 and Tables 1 and 2). With the exception of A. vaga, a fairly consistent pattern of ωx desaturase capabilities was found among representatives of Cnidaria, Mollusca, Annelida, and Arthropoda. Thus, one gene from the pair of ωx desaturases in each species exhibited Δ12 desaturase activity, enabling the biosynthesis of 18:2ω6 (Fig. 3B and Table 1). The second ωx desaturase of each pair exhibited Δ15 desaturation ability, being able to convert 18:2ω6 to 18:3ω3 (ALA) (Fig. 3 and Table 2) and thus completing all the desaturation steps required for de novo biosynthesis of ω3 PUFA. The capability for de novo biosynthesis of ω3 PUFA through the combined action of two distinct enzymes in representatives of Cnidaria, Mollusca, Annelida, and Arthropoda was also contained within each of the two ωx desaturases characterized from the rotifer A. vaga (Fig. 3 and Table 2). Our results contradict the currently accepted paradigm (6, 9, 27) because they show unequivocally that both Δ12 and Δ15 desaturase activities are present in multiple metazoans, enabling them to endogenously produce ω3 PUFA de novo. The yeast system used in this study thus confirms that the enzyme activities reported in in vivo experiments are due to endogenous metazoan genes and not microbes (28, 29). One of the ωx desaturases described in cnidarians, molluscs, annelids, and crustaceans was able to produce a variety of ω3 long-chain PUFA from their corresponding ω6 precursors (Table 2), in addition to 18:3ω3, confirming that these enzymes are true ω3 desaturases with coexisting Δ15, Δ17, and Δ19 activities. This represents long-chain PUFA–synthesizing capabilities previously unknown outside microbes (5, 14, 15).
Fig. 3. Functional characterization of metazoan ωx desaturases.
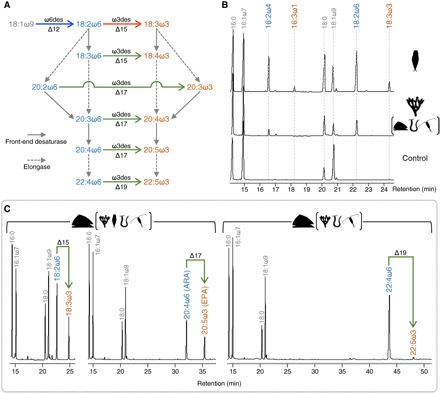
(A) The de novo production of ω3 PUFA requires both ω6 (Δ12) (blue arrow) and ω3 (Δ15) desaturases (red arrow). Both LA (18:2ω6) and ALA (18:3ω3) can be subsequently modified through the ω6 and ω3 long-chain PUFA biosynthesis pathways that proceed separately, at least in vertebrates (9), or can be interconnected by ω3 desaturases with Δ15, Δ17, or Δ19 activities (green arrows). (B) Chromatograms of FA methyl esters (FAME) from yeast expressing ωx desaturases from A. vaga (MF448339) (top) and A. millepora (KY658237) (middle) show Δ12/Δ15 and Δ12 activities, respectively. Further ωx desaturases with Δ12 activity were characterized from P. vulgata, P. dumerilii, and L. salmonis (Table 1). (C) Chromatograms of FAME from yeast expressing the ωx desaturase from P. vulgata (KY658241) and grown with supplemented ω6 substrates including 18:2ω6 (left), 20:4ω6 (middle), and 22:4ω6 (right) show the resulting ω3 products 18:3ω3, 20:5ω3, and 22:5ω3, respectively. Similar functions obtained for ωx desaturases from A. millepora, A. vaga, P. dumerilii, and L. salmonis are shown in Table 2. Molecular structures and diagnostic mass ions of 4,4-dimethyloxazoline derivatives from desaturation products are shown in figs. S3 and S4.
Table 1. Characterization of Δ12/Δ15 activities of metazoan ωx desaturases.
FA profiles of yeast transformed with empty pYES2 (control) were compared with those of yeast transformed with the corresponding ωx desaturase [A. millepora (KY658237), A. vaga (ωx1, MF448337; ωx2, MF448339), P. vulgata (KY658242), P. dumerilii (KY658238), and L. salmonis (KY658240)]. FA compositions (% of total FA) are given as means ± SEM of replicate samples (n = 6 for control and n = 4 for transgenic yeast). An asterisk indicates significant differences between control and yeast expressing the corresponding desaturase (Dunnett’s test; *P < 0.05). nd, not detected.
| FA | FA composition (% of total FA) | ||||||
| Control | A. millepora | A. vaga | P. vulgata | P. dumerilii | L. salmonis | ||
| ωx1 | ωx2 | ||||||
| 16:0 | 29.0 ± 1.0 | 31.7 ± 1.0 | 25.7 ± 0.6 | 24.4 ± 0.7* | 24.8 ± 0.4* | 28.5 ± 1.1 | 22.6 ± 1.3* |
| 16:1ω7† | 35.0 ± 1.2 | 23.3 ± 2.4* | 22.4 ± 0.3* | 19.5 ± 0.5* | 36.4 ± 0.6 | 34.5 ± 1.0 | 37.6 ± 0.8 |
| 16:2ω4 | nd | 2.8 ± 0.3* | 6.9 ± 0.3* | 10.4 ± 0.3* | 0.4 ± 0.1 | 0.2 ± 0.0 | 0.2 ± 0.0 |
| 16:3ω1 | nd | nd | 0.5 ± 0.0* | 1.5 ± 0.1* | nd | nd | nd |
| 18:0 | 10.1 ± 0.9 | 15.8 ± 0.6* | 15.8 ± 1.2* | 17.0 ± 1.0* | 10.6 ± 0.3 | 11.1 ± 0.4 | 11.7 ± 0.5 |
| 18:1ω9 | 25.6 ± 0.6 | 13.9 ± 2.2* | 13.0 ± 1.4* | 8.6 ± 0.4* | 24.1 ± 1.0 | 21.2 ± 0.4 | 24.6 ± 0.5 |
| 18:2ω6 | 0.3 ± 0.1 | 12.5 ± 0.7* | 13.8 ± 0.4* | 15.1 ± 0.5* | 3.8 ± 0.6* | 4.3 ± 0.2* | 3.3 ± 0.5* |
| 18:3ω3 | nd | nd | 1.8 ± 0.1* | 3.4 ± 0.2* | nd | nd | nd |
†Mostly 16:1ω7, although it also includes some 16:1ω9.
Table 2. Functional characterization of metazoan ω3 desaturases.
Transgenic yeast expressing the corresponding ω3 desaturases [A. millepora (KY658236), A. vaga (ωx1, MF448337; ωx2, MF448339), P. vulgata (KY658241), P. dumerilii (KY707886), and L. salmonis (KY658239)] were grown in the presence of exogenously added substrates 18:2ω6, 18:3ω6, 20:2ω6, 20:3ω6, 20:4ω6, and 22:4ω6. Conversions are expressed as a percentage of total FA substrate converted to desaturated product, with the corresponding activity (Δ) detected also shown. nd, not detected.
| Substrate | Product | Conversion (%) | Activity | |||||
| A. millepora | A. vaga | P. vulgata | P. dumerilii | L. salmonis | ||||
| ωx1 | ωx2 | |||||||
| 18:2ω6 | 18:3ω3 | 70.5 | 10.6 | 11.7 | 34.7 | 9.7 | 33.6 | Δ15 |
| 18:3ω6 | 18:4ω3 | 64.8 | 14.3 | 14.3 | 18.8 | 6.5 | 39.6 | Δ15 |
| 20:2ω6 | 20:3ω3 | 15.1 | 1.1 | 2.2 | 11.3 | 9.5 | 10.4 | Δ17 |
| 20:3ω6 | 20:4ω3 | 25.7 | 4.2 | 4.5 | 16.1 | 15.8 | 14.2 | Δ17 |
| 20:4ω6 | 20:5ω3 | 32.0 | 1.0 | 2.7 | 38.0 | 13.3 | 30.4 | Δ17 |
| 22:4ω6 | 22:5ω3 | 2.8 | nd | nd | 3.4 | 12.0 | 6.1 | Δ19 |
The present study shows that rather than merely converting dietary PUFA [like the “trophic upgrading” done by fish (30)], aquatic invertebrates can now be recognized as net producers of ω3 long-chain PUFA in agreement with studies (29), providing much more widespread biosynthesis of these compounds in the marine environment than previously appreciated. Consequently, models that estimate global production of ω3 long-chain PUFA, which are largely constructed on the assumption that single-cell microorganisms are the sole primary producers, need extensive revision (27, 31). ω3 long-chain PUFA production by metazoans will undoubtedly be significant due to the abundance that the animals that are now known to have ωx desaturases have in global ecosystems.
MATERIALS AND METHODS
Retrieval of ωx desaturase homologs
To examine the evolutionary diversity of ωx desaturases among metazoan phyla, we used a combination of strategies: extensive homologous sequence searches, rigorous ωx desaturase sequence selection, and phylogenetic reconstruction. Specifically, we conducted an extensive search of genomic and transcriptomic resources covering species from all domains of life, with a particular focus on Metazoa. Given the vast taxonomic and genetic variability of the “ωx desaturases” (8), homologous sequences were collected by applying similar but complementary methodologies, including HMMER, PSI-BLAST, and HHblits. Query sequences consisted of 113 representative prokaryotic and eukaryotic ωx desaturase protein sequences (19). First, the query sequences were aligned with hmmalign (HMMER software v3.1b2, hmmer.org) to the Pfam Fatty Acid Desaturase Family Profile (FA_desaturase PF00487.22). The resultant multiple sequence alignment (MSA) was then used to search for similar sequences through the HHMER3 web server (https://toolkit.tuebingen.mpg.de/#/tools/hmmer) (32) against the nonredundant GenBank protein database (nr, E-value < 1 × 10−10). Using this approach, around 14,000 distinct hits were collected. Second, the MSA was used to build a position-specific scoring matrix for a PSI-BLAST search (v.2.6.0+; E-value < 1 × 10−10, 11 iterations) against the nr database. In total, this search retrieved around 56,000 sequences. Third, the query sequences were used as input in a Homology detection by iterative HMM-HMM comparison (HHblits) (33) against the UniProt20 HMM database using the following nondefault options: maximum number of iterations = 8 and realign query-template alignments with Maximum Accuracy alignment algorithm (threshold ≥ 0.1). With HHblits, a total of 520 hits were collected. Finally, all hits were combined and duplicates were removed, resulting in an initial data set of 58,000 sequences.
Filtering out false-positive and incomplete ωx desaturase gene sequences
FA desaturase sequences are characterized by the presence of three conserved histidine box motifs (H-box1, H-box2, and H-box3) that typically contain eight histidines between them (19). The distance between histidine boxes (8) and their specific amino acid sequences are characteristic of each subfamily of membrane-bound desaturases including first, ωx, front-end, and sphingolipid desaturases (19). To identify putative ωx desaturase sequences, an automated genomic pipeline was built to filter out non–ωx desaturase sequences. According to the filter thresholds, sequences matching the following criteria were further considered: The FA_desaturase (PF00487.22) Pfam domain must be identified (P < 1 × 10−10) within the protein sequence, but the Pfam domain Lipid_DES (PF08557.8) should not be present because it is typically associated with sphingolipid desaturases; H-box1 had to be composed of five amino acid residues (HXXXH, which excluded Scd/Δ9 desaturases—HXXXXH); H-box2 had to be composed of five amino acid residues (HXXHH, which excluded front-end desaturases—HXXXHH); H-box3 had to consist also of five amino acid residues (HXXHH); the number of amino acid residues between the end of H-box1 and the start of H-box2 had to be between 30 and 32; H-box3 had to be located further apart close to the C-terminal region: the number of amino acids between the end of H-box2 and the start of H-box3 had to be between 120 and 250. To distinguish ωx from sphingolipid desaturases, the empirical frequency of amino acid occurrence within the histidine boxes of known eukaryotic sequences (19) was used to calculate the probability of a desaturase sequence being assigned to one of these two subfamilies. For example, the third and fourth amino acids in H-box1 of ωx and sphingolipid show very distinctive frequencies (19), with ωx desaturases typically having a cysteine followed by a glycine, whereas sphingolipid desaturases have an isoleucine and a serine. Only putative desaturase sequences with a probability ≥0.7 of being ωx (versus sphingolipid) were retained for further analyses. From the initial putative 58,000 homologous sequences, around 9000 passed all filtering steps (around 15.7%).
Metazoan ωx desaturase homolog search expansion
To expand the number of distinct metazoan ωx desaturase sequences, query metazoan sequences were searched against the following GenBank databases with BLAST (version 2.6.0+): whole genome shotgun (WGS), transcriptome shotgun assemblies (TSA) sets and tsa_nr. Specifically, the tblastn program was used against 515 WGS and 935 TSA projects from metazoan nonchordate species, and blastp was used to search metazoan ωx desaturase homologs against tsa_nr (E-value < 1 × 10−10). In addition, species-specific genome/transcriptome projects were also investigated (for example, P. vulgata). Genome WGS contigs containing significantly similar sequences to ωx desaturases were annotated with corresponding transcripts obtained from the TSA and Sequence Read Archive (SRA) except two Radix auricularia sequences (MUZC01000242 and MUZC01000057), which were annotated by tblastn using ωx desaturase homologs from other Heterobranchia species as queries. For each transcript, we selected the longest open reading frame (ORF) sequence with a minimum length of 159 amino acids (to guarantee that the three histidine boxes were present in the sequence). All selected TSA and tsa_nr sequences were then evaluated using the filters described above. In total, 41 additional metazoan sequences were found with this step.
Sequence number reduction: Clustering, isoforms, and identical sequence removal
To reduce the total number of sequences for the final data set, the software package SiLix v.1.2.10 (34) was applied to the nonmetazoan sequences (based on BLAST “all against all” pair of sequences alignments) for clustering sequences that shared a minimal pair-sequence identity and overlap of 70%. For nonmetazoan taxa, one representative of each taxonomic rank (for example, order or phylum) found in each cluster was selected for further analyses. The total number of sequences after clustering analysis was reduced to 910 and further reduced to 892 after removing identical sequences and splice isoforms (longest one for each species was kept). The sequences selected by IsoSVM (892) were then aligned with MAFFT v.7.309 (reference, parameters: --genafpair --maxiterate 1000), and 100 bootstrap maximum likelihood (ML)–based phylogenetic trees were inferred using ExaML tool v.3.0.18 (35). The ExaML trees were inferred given 100 precomputed starting trees using standard RAxML v.8.2.9 (36) following the ExaML manual instructions (https://github.com/stamatak/ExaML/blob/master/manual/ExaML.pdf). The majority-rule consensus ExaML tree was used to select sequences representing distinct well-supported clades of major taxonomic ranks (for example, keeping only major Dikaria fungal lineages) and to exclude potential contaminations. A last data set reduction (down to 287 sequences) was made by excluding the sequences with pairwise identity below 29% and overlap below 70%.
Phylogenetic analyses
The final sequences were aligned with MAFFT (parameters: --genafpair -- maxiterate 1000) and filtered with GUIDANCE v.2.0 (37) with the following parameters: 100 bootstrap replicates, sequence, column, and site masking cutoff threshold <0.5. The resultant MSA was then filtered to remove columns with gaps in 95% or more of the sequences. The best-fit model of protein evolution was selected using ProtTest v.3.3 (38) based on the Bayesian Information Criterion. The ML phylogenetic inference was performed using RAxML with 100 rapid bootstrap replicates and 20 ML searches. The Bayesian inference was applied using MrBayes v.3.2.1 (39) and PhyloBayes MPI v.1.7a (40). MrBayes was run with four Markov chains incrementally heated with the default values. Each chain started with a randomly generated tree and ran for 1x107 generations with sampling frequency of 1 tree for every 100 generations. The posterior distribution of trees, after discarding the first 25% as burn-in, was summarized in a 50% majority-rule consensus tree. Two independent replicates were conducted and inspected for consistency. The PhyloBayes analysis was executed applying distinct compositional profiles and one substitution rate matrix inferred from the data (CAT+GTR model). Similar to the MrBayes analysis, two independent chains were run in parallel, which terminated reaching moderate convergence after around 16,000 cycles with maximum discrepancy observed across all bipartitions of 0.28. The final protein sequence alignment and the various phylogenetic trees (text and image format) can be accessed as described in Data and materials availability.
Functional characterization of ωx desaturases from selected metazoan species
Full-length ORF sequences of putative ωx desaturase from A. millepora, P. vulgata, P. dumerilii, and L. salmonis were obtained from their transcriptome or genomic databases by tblastn searches using several amino acid sequences of ωx desaturase as queries (table S1). When necessary, extension of full-length ORF sequences was performed by rapid amplification of cDNA ends (RACE) to determine the full-length ORF sequence with conditions as described in table S2. All the primer sets used for polymerase chain reaction (PCR) are shown in table S3. The DNA sequences of PCR products (sequenced at GATC Biotech) were aligned to produce full-length ORF sequences. The synthetic nucleotide ORF sequences from the P. dumerilii (KY707886) and A. vaga (MF448337 and MF448339) ωx desaturases were purchased from Integrated DNA Technologies Inc. and Eurofins Genomics K.K., respectively.
The function of ωx desaturases was characterized in the yeast Saccharomyces cerevisiae (41). Briefly, the ORF of the two ωx desaturase–like genes retrieved in each selected species were amplified by PCR using primers containing restriction enzyme sites for further cloning into the yeast expression vector pYES2 (table S3). The pYES2 constructs containing the ωx desaturase ORF sequences were sequenced (GATC Biotech) prior to being used to transform InvSc1 (Invitrogen) yeast as described previously (41). Transgenic yeast expressing each ωx desaturase were grown in the absence of exogenously added FA substrates or in the presence of 18:2ω6, 18:3ω6, 20:2ω6, 20:3ω6, 20:4ω6, and 22:4ω6 supplemented at 0.5 (C18), 0.75 (C20), and 1 mM (C22) to compensate for uptake efficiency varying with chain length (41). Control treatments consisted of yeast transformed with the empty pYES2 and run under the exact same conditions as above. After 48 hours, yeast cells were collected and FAME derivatives were prepared from total lipids (41). Functions of the ωx desaturases were established by comparing the FA profiles of ωx desaturase transformed yeast with those of the controls. For exogenously added FA substrates, desaturase conversions were calculated as [product areas/(product areas + substrate area)] × 100 (41). Identity of the desaturation products was determined by comparing their retention times with FA contained in commercial standards (Marine Oil FAME Mix, Restek Corporation; Supelco 37 Component FAME Mix, Sigma-Aldrich). Positions of double bonds were determined by preparing 4,4-dimethyloxazoline (DMOX) derivatives of FAME samples followed by gas chromatography–mass spectrometry analysis (41).
Reagents
For functional characterization assays, all FA substrates were purchased from Nu-Chek Prep Inc. except 20:4ω6, which was from Cayman Chemical. Chemicals used to prepare the S. cerevisiae minimal medium uracil were from Sigma-Aldrich, except for the bacteriological agar obtained from Oxoid Ltd. Thin-layer chromatography (20 × 20 cm × 0.25 mm) plates precoated with silica gel 60 (without fluorescent indicator) were purchased from Merck. All solvents were HPLC (high-performance liquid chromatography)–grade and were from Fisher Scientific. DMOX derivatization reagents (2-amino-2-methyl-1-propanol) were purchased from Sigma-Aldrich.
Statistics
Comparisons of means of FA composition between control yeast (n = 6) and each transgenic yeast (n = 4) were carried out using Dunnett’s test with P < 0.05 indicating statistical significance. All statistical analyses were performed in R 3.4.1. (www.r-project.org).
Supplementary Material
Acknowledgments
We thank A. Magurran and J. Napier for comments on the manuscript and R. Ruivo for drawings in Figs. 1 and 3. Funding: This work received funding from the MASTS pooling initiative (The Marine Alliance for Science and Technology for Scotland) funded by the Scottish Funding Council (grant reference HR09011), and their support is gratefully acknowledged. Access to the Institute of Aquaculture laboratories was funded by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 262336 (AQUAEXCEL), Transnational Access Project Number 0095/06/03/13. Author contributions: M.M.F., D.E.K.F., L.K.B., and L.F.C.C. designed and performed the overall sequence screening strategy. N.K., D.S.F., and D.E.K.F. retrieved the sequences of functionally characterized genes. M.M.F. performed the phylogenetic analyses. Ó.M., N.K., M.M.F., D.E.K.F., L.F.C.C., and D.R.T. designed the experiments, interpreted the results, and wrote the manuscript. N.K. and Ó.M. performed the cDNA cloning and functional characterization assays. N.K. and J.C.N. analyzed the yeast FAs. All authors contributed to the manuscript with intellectual input and approved the final version. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The list of animal WGS (515) and TSA (935) projects searched with BLAST, the list of final data set sequences used, the protein alignment, and all phylogenetic trees are accessible through figshare.com (https://figshare.com/s/7e518eab3e63496f9281) and the CIIMAR website (https://box.ciimar.up.pt/owncloud/index.php/s/0dWVl52b7jmsXFH). The sequences functionally characterized in this work have been deposited in GenBank with the accession codes listed in table S1. All other data are available from the corresponding authors upon reasonable request.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaar6849/DC1
fig. S1. Complete view of the phylogenetic tree presented in Fig. 1A, with major metazoan taxonomic clades colored as in Fig. 1.
fig. S2. Summary tree (cladogram) showing relationships between major well-supported clades together with sequence logo representation of conserved histidine box motifs in the same clades.
fig. S3. Mass spectra of DMOX derivatives obtained from desaturation products as shown in Table 1.
fig. S4. Mass spectra of DMOX derivatives obtained from desaturation products as shown in Table 2.
table S1. List of databases and query sequences for retrieval of functionally characterized genes.
table S2. PCR conditions for the RACE and full-length ORF amplifications.
table S3. List of primers used for the cloning process and amplification of full-length ORF of ωx desaturases.
REFERENCES AND NOTES
- 1.Swanson D., Block R., Mousa S. A., Omega-3 fatty acids EPA and DHA: Health benefits throughout life. Adv. Nutr. 3, 1–7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruiz-Lopez N., Haslam R. P., Napier J. A., Sayanova O., Successful high-level accumulation of fish oil omega-3 long-chain polyunsaturated fatty acids in a transgenic oilseed crop. Plant J. 77, 198–208 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue Z., Sharpe P. L., Hong S.-P., Yadav N. S., Xie D., Short D. R., Damude H. G., Rupert R. A., Seip J. E., Wang J., Pollak D. W., Bostick M. W., Bosak M. D., Macool D. J., Hollerbach D. H., Zhang H., Arcilla D. M., Bledsoe S. A., Croker K., McCord E. F., Tyreus B. D., Jackson E. N., Zhu Q., Production of omega-3 eicosapentaenoic acid by metabolic engineering of Yarrowia lipolytica. Nat. Biotechnol. 31, 734–740 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Nichols D. S., Prokaryotes and the input of polyunsaturated fatty acids to the marine food web. FEMS Microbiol. Lett. 219, 1–7 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Khozin-Goldberg I., Iskandarov U., Cohen Z., LC-PUFA from photosynthetic microalgae: Occurrence, biosynthesis, and prospects in biotechnology. Appl. Microbiol. Biotechnol. 91, 905–915 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Pereira S. L., Leonard A. E., Mukerji P., Recent advances in the study of fatty acid desaturases from animals and lower eukaryotes. Prostaglandins Leukot. Essent. Fatty Acids 68, 97–106 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Leonard A. E., Pereira S. L., Sprecher H., Huang Y.-S., Elongation of long-chain fatty acids. Prog. Lipid Res. 43, 36–54 (2004). [DOI] [PubMed] [Google Scholar]
- 8.Sperling P., Ternes P., Zank T. K., Heinz E., The evolution of desaturases. Prostaglandins Leukot. Essent. Fatty Acids 68, 73–95 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Castro L. F. C., Tocher D. R., Monroig Ó., Long-chain polyunsaturated fatty acid biosynthesis in chordates: Insights into the evolution of Fads and Elovl gene repertoire. Prog. Lipid Res. 62, 25–40 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Guillou H., Zadravec D., Martin P. G. P., Jacobsson A., The key roles of elongases and desaturases in mammalian fatty acid metabolism: Insights from transgenic mice. Prog. Lipid Res. 49, 186–199 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Weinert J., Blomquist G. J., Borgeson C. E., De novo biosynthesis of linoleic acid in two non-insect invertebrates: The land slug and the garden snail. Experientia 49, 919–921 (1993). [Google Scholar]
- 12.Zhou X.-R., Horne I., Damcevski K., Haritos V., Green A., Singh S., Isolation and functional characterization of two independently-evolved fatty acid Δ12-desaturase genes from insects. Insect Mol. Biol. 17, 667–676 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Malcicka M., Visser B., Ellers J., An evolutionary perspective on linoleic acid synthesis in animals. Evol. Biol. 45, 15–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu H., Wang H., Cai S., Zhang H., A novel ω3-desaturase in the deep sea giant tubeworm Riftia pachyptila. Mar. Biotechnol. 19, 345–350 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Spychalla J. P., Kinney A. J., Browse J., Identification of an animal ω-3 fatty acid desaturase by heterologous expression in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 94, 1142–1147 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peyou-Ndi M. M., Watts J. L., Browse J., Identification and characterization of an animal Δ12 fatty acid desaturase gene by heterologous expression in Saccharomyces cerevisiae. Arch. Biochem. Biophys. 376, 399–408 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Zhou X.-R., Green A. G., Singh S. P., Caenorhabditis elegans Δ12-desaturase FAT-2 is a bifunctional desaturase able to desaturate a diverse range of fatty acid substrates at the Δ12 and Δ15 positions. J. Biol. Chem. 286, 43644–43650 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buckner J. S., Hagen M. M., Triacylglycerol and phospholipid fatty acids of the silverleaf whitefly: Composition and biosynthesis. Arch. Insect Biochem. Physiol. 53, 66–79 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Hashimoto K., Yoshizawa A. C., Okuda S., Kuma K., Goto S., Kanehisa M., The repertoire of desaturases and elongases reveals fatty acid variations in 56 eukaryotic genomes. J. Lipid Res. 49, 183–191 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Covello P. S., Reed D. W., Functional expression of the extraplastidial Arabidopsis thaliana oleate desaturase gene (FAD2) in Saccharomyces cerevisiae. Plant Physiol. 111, 223–226 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malcicka M., Ruther J., Ellers J., De novo synthesis of linoleic acid in multiple Collembola species. J. Chem. Ecol. 43, 911–919 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Crisp A., Boschetti C., Perry M., Tunnacliffe A., Micklem G., Expression of multiple horizontally acquired genes is a hallmark of both vertebrate and invertebrate genomes. Genome Biol. 16, 50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boto L., Horizontal transfer in the acquisition of novel traits by metazoans. Proc. Biol. Sci. 281, 20132450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen W., Hasegawa D. K., Kaur N., Kliot A., Pinheiro P. V., Luan J., Stensmyr M. C., Zheng Y., Liu W., Sun H., Xu Y., Luo Y., Kruse A., Yang X., Kontsedalov S., Lebedev G., Fisher T. W., Nelson D. R., Hunter W. B., Brown J. K., Jander G., Cilia M., Douglas A. E., Ghanim M., Simmons A. M., Wintermantel W. M., Ling K.-S., Fei Z., The draft genome of whitefly Bemisia tabaci MEAM1, a global crop pest, provides novel insights into virus transmission, host adaptation, and insecticide resistance. BMC Biol. 14, 110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bryon A., Kurlovs A. H., Dermauw W., Greenhalgh R., Riga M., Grbić M., Tirry L., Osakabe M., Vontas J., Clark R. M., Van Leeuwen T., Disruption of a horizontally transferred phytoene desaturase abolishes carotenoid accumulation and diapause in Tetranychus urticae. Proc. Natl. Acad. Sci. U.S.A. 114, E5871–E5880 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Syvanen M., Horizontal gene transfer: Evidence and possible consequences. Annu. Rev. Genet. 28, 237–261 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Hixson S. M., Arts M. T., Climate warming is predicted to reduce omega-3, long-chain, polyunsaturated fatty acid production in phytoplankton. Glob. Chang. Biol. 22, 2744–2755 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Farkas T., Kariko K., Csengeri I., Incorporation of [1-14C] acetate into fatty acids of the crustaceans Daphnia magna and Cyclops strenus in relation to temperature. Lipids 16, 418–422 (1981). [Google Scholar]
- 29.S. Pairohakul, “Evidence for polyunsaturated fatty acid biosynthesis in the ragworm (Nereis virens) and the lugworm (Arenicola marina)”, thesis, Newcastle University (2013). [Google Scholar]
- 30.Monroig Ó., Tocher D. R., Navarro J. C., Biosynthesis of polyunsaturated fatty acids in marine invertebrates: Recent advances in molecular mechanisms. Mar. Drugs 11, 3998–4018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Budge S. M., Devred E., Forget M-H., Stuart V., Trzcinski M. K., Sathyendranath S., Platt T., Estimating concentrations of essential omega-3 fatty acids in the ocean: Supply and demand. ICES J. Mar. Sci. 71, 1885–1893 (2014). [Google Scholar]
- 32.Alva V., Nam S. Z., Söding J., Lupas A. N., The MPI bioinformatics Toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 44, W410–W415 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remmert M., Biegert A., Hauser A., Söding J., HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 9, 173–175 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Miele V., Penel S., Duret L., Ultra-fast sequence clustering from similarity networks with SiLiX. BMC Bioinformatics 12, 116 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kozlov A. M., Aberer A. J., Stamatakis A., ExaML Version 3: A tool for phylogenomic analyses on supercomputers. Bioinformatics 31, 2577–2579 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stamatakis A., RAxML Version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sela I., Ashkenazy H., Katoh K., Pupko T., GUIDANCE2: Accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 43, W7–W14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Darriba D., Taboada G. L., Doallo R., Posada D., ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Hohna S., Larget B., Liu L., Suchard M. A., Huelsenbeck J. P., MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lartillot N., Rodrigue N., Stubbs D., Richer J., PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 62, 611–615 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Kabeya N., Sanz-Jorquera A., Davie A., Monroig Ó., Biosynthesis of polyunsaturated fatty acids in sea urchins: Molecular and functional characterisation of three fatty acyl desaturases from Paracentrotus lividus (Lamark 1816). PLOS ONE 12, e0169374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/5/eaar6849/DC1
fig. S1. Complete view of the phylogenetic tree presented in Fig. 1A, with major metazoan taxonomic clades colored as in Fig. 1.
fig. S2. Summary tree (cladogram) showing relationships between major well-supported clades together with sequence logo representation of conserved histidine box motifs in the same clades.
fig. S3. Mass spectra of DMOX derivatives obtained from desaturation products as shown in Table 1.
fig. S4. Mass spectra of DMOX derivatives obtained from desaturation products as shown in Table 2.
table S1. List of databases and query sequences for retrieval of functionally characterized genes.
table S2. PCR conditions for the RACE and full-length ORF amplifications.
table S3. List of primers used for the cloning process and amplification of full-length ORF of ωx desaturases.