

Abstract
This study aims to investigate the role of miR-203-HOTAIR interaction in the suppression of renal cell carcinoma (RCC). We employed series of in vitro assays such as proliferation, invasion, migration and colony formation along with in vivo tumor xenograft model. Profiling of miR-203 and HOTAIR expression revealed that miR-203 was significantly under-expressed whereas HOTAIR was overexpressed in RCC cell-lines and clinical specimens compared to normal cell line and tissue. Both miR-203 and HOTAIR expression significantly distinguished malignant from normal tissues and significantly correlated with clinicopathological characteristics of patients. Overexpression of miR-203 significantly inhibited proliferation, migration and invasion with an induction of apoptosis and cell cycle arrest. Whereas, HOTAIR suppression resulted in the similar functional effects in the same RCC cell lines. In silico RNA-22 algorithm showed a binding site for miR-203 in HOTAIR. We observed a direct interaction between miR-203 and HOTAIR by RNA-immunoprecipitation (RIP) and luciferase reporter assays. We show that miR-203-HOTAIR interaction resulted in the inhibition of epithelial to mesenchymal transition (EMT) and metastatic genes as indicated by induction of key metastasis-suppressing proteins E-cadherin, claudin (epithelial markers) and PTEN along with induction of tumor suppressor genes p21 and p27. A significant decrease in vimentin (mesenchymal-marker), KLF4 and nanog (stemness-markers) was also observed. This is the first report demonstrating miR-203 mediated regulation of HOTAIR induces tumor suppressor effects in RCC by regulating EMT and metastatic pathway genes. Thus, the study suggests that therapeutic regulation of HOTAIR by miR-203 overexpression may provide an opportunity to regulate RCC growth and metastasis.
Introduction
Renal cell carcinoma (RCC) is the most common kidney malignancy and a leading cause of cancer death worldwide (1,2). The prevalence of RCC has increased in the United States accounting for 3% to 4% of all adult malignant diseases with approximately 64,000 new cases and 14,400 deaths annually (2). Majority of Clear cell Renal cell carcinoma (ccRCC), the most common form of renal malignancy, are diagnosed in the advanced metastatic stage resulting in dramatic decrease of five year relative survival rate (3). Compared to other malignancies, RCC is found to be resistant to both chemotherapy and hormone therapy (4). The advanced aggressive stage of this disease has inadequate therapeutic options and poor prognosis. Aggressiveness of cancer is highly associated with epithelial-to-mesenchymal transition (EMT) which promotes tumorigenic progression of epithelial cells with increased cell migration and invasion, ‘stemness’, and inhibition of apoptosis and senescence (5–7). The most critical step of EMT is loss of cell to cell adhesion of epithelial cells with a gain of mesenchymal components leading to the initiation of migratory and invasion phenotype. Emerging evidence shows that acquisition of EMT and induction of cancer stem cell (CSC) like phenotype are mechanistically linked and confer drug resistance and tumor recurrence (8–10). Understanding signaling mechanism that controls RCC progression, metastasis and stemness is a key to develop better therapeutic and diagnostic interventions for the disease.
Long non coding RNA (lncRNAs) and miRNAs play important roles in development and progression of diseases (11–16), but their interaction in the regulation of biological function in normal and cancer cells remain unknown. HOTAIR, a lncRNA, is highly expressed in multiple tumors, and has been established as a predictor of metastasis and poor outcome (9) and a potential biomarker for lymph node metastasis in hepatocellular carcinoma. The oncogenic role of HOTAIR and its function as a negative prognostic factor as well in pancreatic cancer has been reported (8). Recent studies also demonstrate that lncRNA HOTAIR is a target of treatment in prostate and renal cancer (17–19). Similarly, miRNA-203, located at chromosome 14q32 in human (20) and identified in skin keratinocytes (21,22), has been described as tumor suppressor miRNA in rhabdomyosarcoma cells, thereby promoting myogenic differentiation by inhibiting the Notch and the JAK1/STAT1/STAT3 pathways (23), in laryngeal squamous cell carcinoma (24), lung cancer cells (1), and in esophageal cancer (25). A recent study by Mingxi et al has focused on FGF2 as the target of miR-203 in renal cancer (26).
The role of miR-203 in the regulation of HOTAIR has never been investigated. In the present study, we performed functional and mechanistic investigation of miR-203-HOTAIR interaction in RCC. Here we report that, (i) miR-203 is significantly under expressed in RCC cell lines and clinical tissues compared to non-malignant cell line and normal samples. An inverse phenomenon is observed in case of HOTAIR with overexpression in cancer cell lines compared to normal cell line; (ii) miR-203 and HOTAIR have potential to independently distinguish malignant from normal tissues, both of them are significantly correlated to clinicopathological characteristics; (iii) miR-203 directly binds to HOTAIR in a sequence specific manner and regulates its expression; (iv) functionally, overexpression of miR-203 impaired cell proliferation, migration and invasion of RCC cells with induction of apoptosis and cell cycle arrest. Reciprocally, attenuation of HOTAIR resulted in the similar functional effects as that of miR-203 overexpression; (v) miR-203 mediated regulation of HOTAIR led to the inhibition of EMT as we observed induction of E-cadherin and Claudin (epithelial markers) with a concomitant decrease in Vimentin (mesenchymal marker). We also observed an increase in the expression of PTEN, p21 and p27; (v) finally, we observed inhibition of tumor growth by local injection of miR-203 mimic in nude mice xenografts in vivo.
Materials and methods
Cell Culture
Normal renal epithelial cells HK-2 (ATCC number: CRL-2190) and renal cancer cell lines ACHN (ATCC number: CRL-1611) and Caki-1 (ATCC number HTB-46) were purchased from the American Type Culture Collection (ATCC) (Manassas, VA) in the year 2016 and grown according to ATCC protocol. These human-derived cell lines were authenticated by DNA short-tandem repeat analysis by ATCC. Cell line experiments were performed within 5-6 months of their procurement/resuscitation. ACHN and Caki-1were cultured in MEM and MaCoy’s 5A medium, respectively, supplemented with 10% fetal bovine serum and 1X antibiotics (penicillin and streptomycin). HK-2 cells were cultured in keratinocyte serum free media (GIBCO/Invitrogen, Carlsbad, CA, USA). Cells were cultured at 37°C in a humidified atmosphere with 5% CO2.
Patient samples
Clinical Formaldehyde-fixed-paraffin-embedded (FFPE) samples of 24 patients with pathologically confirmed clear cell RCC (cc-RCC) were obtained from the department of Pathology of Veterans Affair Medical Center, San Francisco (VAMCSF), CA, USA. A board-certified pathologist has reviewed all slides and identified malignant and adjacent normal tissue. Written informed consent was obtained from all patients and the study was in accordance with recognized ethical guidelines (IRB approval no: 16-18555).
Transient transfection
In order to overexpress miR-203 or knockdown HOTAIR, cells were transfected with mirVana miR-203 Mimics (10 nM) and 25 nM of siHOTAIR (Thermo Fisher Scientific), respectively, using Lipofectamine RNAi Max (Thermo Fisher Scientific). mirVana miRNA Mimic Negative Control #1 and siRNA control (Thermo Fisher Scientific) were used at the same concentration in each transfection experiment to verify efficiency.
Cell viability and colony formation assay
Cell viability was determined at 24, 48 and 72 hours using a CellTiter 96 Aqueous Solution Cell Proliferation Assay kit (Promega, Madison, WI) according to the manufacturer’s instructions. After transfection for 72 hours, cells were seeded at a low density (1000 cells/plate) for colony formation assay and were allowed to grow until visible colonies were formed. Plates were then stained with giemsa and colonies were counted.
Quantitative real-time PCR
Total RNA was extracted from tissue samples, cell lines and tumor samples using a miRNeasy FFPE kit and miRNeasy mini kit (Qiagen), respectively. RNA and miRNA were reverse-transcribed into cDNA with the High capacity cDNA reverse transcription kit (Thermo Fisher). Quantitative real-time RT-PCR was performed in duplicate with QuantStudio 7 Flex-Real Time PCR System (Applied Biosystem) using TaqMan universal PCR master mix according to the manufacture’s protocol (Applied Biosystems Inc., Foster City, CA, USA), TaqMan probes and primers were from Applied Biosystems. Human GAPDH and RNU48 were used as endogenous controls for gene expression and miRNA respectively. Relative expression of RNA and miRNA were calculated using comparative Ct.
Migration and invasion assays
Culture inserts of 8-μm pore size (Transwell; Costar) were placed into the wells of 24-well culture plates and used for migration and invasion assay. For invasion assay, inserts were coated with Matrigel (BD Biosciences) (100μg/well). In the lower chamber, 500 μl of media containing 10% FBS was added, and 1×105 cells (without FBS) were seeded to the upper chamber. After 48-72 hours of incubation at 37 °C with 5% CO2, cells migrated or invaded through the pores were fixed with 4% formalin and stained with 0.05% crystal violet. Crystal violet was solubilized with methanol, and absorbance at 540 nm was measured by a kinetic microplate reader (Spectra MAX 190; Molecular Devices). Data are the mean ± S.E. of three independent experiments.
Cell cycle analysis and apoptosis assay
Cells transfected with miR-203 mimic and negative control were harvested using accutase (Corning) and washed with cold PBS. For cell cycle, PBS washed cells were fixed in cold 70% ethanol overnight at −20°C. Fixed cells were further washed with PBS, stained with PI/RNase Staining Buffer (BD Pharmingen) and incubated for 30 minutes at room temperature in the dark. DNA content was analyzed using BD FACS Verse (BD Pharmingen). For apoptosis, cold PBS washed cells were resuspended in 1X binding buffer and stained with Annexin V-FITC and 7AAD viability dye (Annexin V-FITC/7AAD kit, Beckman Coulter). Cells were then incubated for 30 minutes at room temperature in the dark and analyzed using BD FACSVerse (BD Pharmingen).
Luciferase assays
The wild type (WT) region of HOTAIR containing target site sequences complementary to the seed sequence of miR-203 were cloned downstream of the luciferase gene in the pMIR-REPORT luciferase vector (Ambion, Cambridge MA), and named HOTAIR-WT. An off-target sequence of HOTAIR was cloned in the same vector and named HOTAIR-Mut. For reporter assays, ACHN and Caki-1 cells were transiently co-transfected with wild-type or mutant plasmid and miR-203 or control-miR. Firefly luciferase activities were measured using the Dual Luciferase Assay (Promega, Madison, WI) 24 hours after transfection and the results were normalized with Renilla luciferase. Each reporter plasmid was transfected at least two times and each sample was assayed in triplicate.
RNA immunoprecipitation (RIP) Assay
RNA immunoprecipitation (RIP) was performed to investigate the binding of miR-203 to lncRNA HOTAIR. An imprint RIP kit was used as per manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO, USA). Ago2 and IgG (control) antibodies were used for immunoprecipitation. The RIP RNA fraction was reverse transcribed to cDNA using High capacity cDNA reverse transcription kit (Thermo Fisher). Final analysis was performed using RT-qPCR and shown as fold enrichment of HOTAIR to Ago2 with respect to IgG.
Western blot analysis
Cells were lysed with NP-40 (Thermo Scientific) plus Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Scientific). Protein concentration was determined using BCA Protein Assay (Thermo Fisher Scientific). Total proteins (15-20 μg) were separated by NuPAGE 4-12% Bis-Tris Protein Gels (Invitrogen) and subsequently transferred onto nitrocellulose membrane using the iblot2 Dry Blotting System (Invitrogen). Prior to incubation with 1:1000 fold diluted primary antibodies overnight at 4°C, blots were blocked in Odyssey blocking buffer (Li-Cor) for an hour. The following primary antibodies were used: PTEN (Cell Signaling, 9552), E-cadherin (Thermo Fisher Scientific, MA5-11496), claudin (Thermo Fisher Scientific, 37-4900), vimentin (Cell Signaling, 3390), p21 (Cell Signaling, 2946), p27 (Cell Signaling, 2552), KLF4 (cell signaling 4038), nanog (cell signaling 4903) and ß-actin (Cell Signaling, 3700). After washing the membranes, either goat anti-rabbit IgG (H+L) 800 W or goat anti-mouse IgG (H+L) 680RD was applied for 45 minutes at room temperature (1∶20,000, LI-COR). Membranes were again washed with PBS containing Tween 20. Blots were scanned using an Odyssey Infrared Imaging System Scan and quantification was carried out with the LI-COR Odyssey® scanner and software (LI-COR Biosciences).
Immunofluorescence
Treated cells were fixed in 4% paraformaldehyde for 15 minutes. Prior to overnight incubation with 1:100 fold diluted primary antibody, cells were blocked with blocking buffer (1X PBS/5% normal goat serum/0.3% Triton X-100) for 1 hour. After washing with PBS, cells were treated with 1:100 fold diluted secondary antibody for 2 hours and counter stained with 0.5μg/ml of 4′,6-diamidino-2-phenylindole (DAPI) for 5 minutes. Cells were then mounted using prolong gold antifade reagent and images were captured using Zeiss microscope (model: Axio Imager.D2).
In vivo intratumoral delivery of miR-203 mimic and control
Local administration of miRVANA miRNA mimic and control were done in established palpable nude mouse tumors to study the anti-tumorigenic effect of overexpression of miR-203 as compared to the mouse group injected with control miRNA. Each mouse was injected subcutaneously with 1 × 107 ACHN renal cancer cells and after eleven days palpable tumors developed. 6.25 μg of synthetic miR-203 mimic or miR-mimic-negative control (control) was complexed with 1.6 μl siPORT Amine transfection reagent (Ambion, Austin, TX) in 50 μl PBS and was delivered intratumorally at an interval of 3 days for a total of eight times. In total, 5 mice received miR-203 mimic and 5 mice received control miR. Tumor growth was followed for 28 days from the first injection of miR-mimic and control. The mice were sacrificed on day 39. Tumors were excised, snap frozen and stored at −80°C for further biochemical assays. All animal care was in accordance with recognized ethical guidelines (IACUC approval no: 16-004).
Statistical Analysis
All data were derived from at least two or three independent experiments. Statistical analysis was performed and values were presented as mean ± standard error (S.E). Significant differences between the groups were determined using the Student’s t-test. A p value of < 0.05 was considered as an indication of statistical significance. ROC curve analysis was performed using MedCalc software showing area under curve (AUC) and 95% confidence interval. Chi-square test was made to determine the correlation between targets and clinicopathological characteristics.
Results
Expression of miR-203 and HOTAIR in renal cancer
Expression levels of miR-203 were noticeably lower in RCC cell lines (ACHN and Caki-1) and in cancer tissue samples (n=24) compared to their respective normal counterpart as seen by real time RT-PCR (Figure 1A–1B). However, quantitative RT-PCR results revealed that lncRNA HOTAIR expression was markedly higher in renal cancer cell lines ACHN and Caki-1 compared to normal renal epithelial cell line HK-2 (Figure 1C). Similarly, higher levels of HOTAIR expression was also noticed in cancer tissue samples (n=24) as compared to their matched normal controls (Figure 1D).
Figure 1. Expression levels and clinical significance of miR-203 and HOTAIR in renal cancer.

(A) and (C) Expression levels of miR-203 and HOTAIR, respectively in ACHN, Caki-1 and HK2. (B) and (D) Expression levels of miR-203 and HOTAIR in tissue samples (n=24). (E and F) Receiver operating curve (ROC) analysis showing ability of miR-203 and HOTAIR to distinguish between malignant and non-malignant tissue samples. (G and H) Chi-square test showing correlation of clinicopathological variables with miR-203 and HOTAIR expression.
Clinical significance of miR-203 and HOTAIR in renal cancer
Receiver operating curve (ROC) analyses were performed to evaluate the ability of miR-203 and HOTAIR expression to discriminate between normal and tumor tissues based on 24 patient sample data. An area under the ROC curve (AUC) of 0.944 (P <0.0001; 95% CI = 0.838 to 0.990) (Figure 1E) was obtained suggesting that miR-203 expression can discriminate between malignant and non-malignant tissues. Similarly, HOTAIR expression can also discriminate between malignant and non-malignant tissues having an AUC of 0.923 (P value = <0.0001; 95% CI = 0.781 - 0.986) (Figure 1F). Hence both miR-203 and HOTAIR have the potential to be used as a diagnostic marker, though it needs to be validated in a larger independent cohort. We also determined the correlation of miR-203 and HOTAIR expression with clinicopathological variables such as Fuhrman grade, pathological stage (pT). Correlation of miR-203 and HOTAIR with clinicopathological characteristics in our clinical cohort as shown in Figure 1G and Figure 1H revealed that cases with low miR-203 and high HOTAIR expression increase from low grade, low pathological stage to high grade and high pathological stage.
miR-203 overexpression impairs cell proliferation, migration and invasion with induction of apoptosis and cell cycle arrest in RCC cell lines
To determine the functional significance of miR-203 in renal cancer, we transiently transfected ACHN and Caki-1 cells with miR-203 mimic and control for 72 hours which resulted in overexpression of miR-203 (Figure 2A). Transient transfection of miR-203 mimic in ACHN and Caki-1 cells caused reduced cell proliferation (Figure 2B–2C) and marked decrease in colony formation compared to controls (Figure 2D). Significant reduction in cell migration and invasion were also observed in miR-203 overexpressing renal cancer cells (Figure 2E). Overexpression of miR-203 induced apoptosis in renal cancer cell lines as compared to control (Figure 2F–2G). Cell cycle analysis after 72 hours’ transfection revealed a significant increase in G0/G1 cell population in cells transfected with miR-203 mimic compared to control with a decrease in the S and G2/M phase population (Figure 2H).
Figure 2. Effects of miR-203 overexpression in RCC cell-lines.

(A) Overexpression of miR-203 in ACHN and Caki-1 cells when transfected with 10nM of miR-203 mimic and its corresponding control for 72 hours. (B-C) Cell viability of ACHN and Caki-1 cells after transfecting with miR-203 mimic and control (D) Graphical representation of colony formation shows overexpression of miR-203 significantly decreased colony formation in ACHN and Caki-1 cells. (E) Graphical representation showing decrease in migration and invasion in both ACHN and Caki-1 cells after miR-203 overexpression compared to control treatment. (F-G) Both ACHN and Caki-1 cell lines show significant induction of apoptosis with respect to control after miR-203 overexpression. (H) Cell cycle analysis shows G0/G1 arrest in both ACHN and Caki-1 cells with miR-203 overexpression. Values in the boxes are the average of three experiments ± standard error (SE).
Direct binding of miR-203 to a target specific HOTAIR site
Overexpression of miR-203 caused significant knockdown (60%-65%) of HOTAIR expression (Figure 3A). Thus, we used an in silico computational algorithm RNA22 (https://cm.jefferson.edu/rna22/) and identified a single predicted binding site for miR-203 in the HOTAIR sequence (Figure 3B). Luciferase reporter assays with miR-203-overexpressing ACHN and Caki-1 cells revealed that miR-203 repressed luciferase activity, whereas no effect was observed with control-miR. An off-target site had decreased response to miR-203 (Figure 3B), indicating that miR-203 binds to HOTAIR in a sequence-specific manner. Binding of miR-203 to HOTAIR was further confirmed by RIP assay using Ago2 antibody and IgG antibody as control. Results showed 10-15% fold enrichment of HOTAIR to Ago2 vs IgG in miR-203 overexpressed ACHN and Caki-1 cells compared to controls (Figure 3C).
Figure 3. Binding of miR-203 to HOTAIR and effects of HOTAIR in RCC cell-lines.

(A) Knockdown of HOTAIR after transfecting ACHN and Caki1 cells with miR-203 mimic and control. (B) Complimentary binding site for miR-203 in HOTAIR gene. Luciferase assays showing decreased reporter activity after co-transfection of either wild type HOTAIR or off target part of HOTAIR with miR-203 mimic and control in renal cancer cells. (C) RIP assay using Ago2 antibody and IgG antibody (control) showing fold enrichment of HOTAIR to Ago2 w.r.t IgG in miR-203 overexpressing ACHN and Caki-1 cells compared to their controls. (D) Reduced expression of HOTAIR in ACHN and Caki-1 cells after transfecting with 25nM HOTAIR siRNA. (E) Graphical representation of colony formation shows knockdown of HOTAIR significantly decreased colonies in ACHN and Caki-1 cells. (F) Knockdown of HOTAIR shows decrease in cell proliferation of ACHN and Caki-1 cells. (G) Reduced migration and invasion in HOTAIR siRNA transfected cells compared to control treatment. (H-I) Both ACHN and Caki-1 cell lines show significant induction of apoptosis compared to control after knockdown of HOTAIR.
Knockdown of HOTAIR mimics the functional effects of miR-203 overexpression
Since miR-203 directly binds to HOTAIR and overexpression of miR-203 caused decreased HOTAIR levels, we checked the effect of HOTAIR knockdown in renal cancer cells. We transiently transfected ACHN and Caki-1 cells with HOTAIR siRNA and control for 72 hours which showed significant knockdown of HOTAIR expression in both cell-lines (Figure 3D). It also inhibited colony formation as compared to controls (Figure 3E) and caused a significant decrease in cell proliferation (Figure 3F). Knockdown of HOTAIR decreased cell migration, invasion (Figure 3G) and induced apoptosis (Figure 3H–3I).
Overexpression of miR-203 mediated suppression of HOTAIR inhibits epithelial-to-mesenchymal transition and stemness in RCC
Ectopic expression of miR-203 in RCC cells also resulted in an increase in epithelial marker E-cadherin and claudin with a concomitant decrease in the mesenchymal marker vimentin in both mRNA and protein levels (Figure 4A–4E). Immunofluorescence studies in miR-203 overexpressing cells show induced expression of E-cadherin (red) and its increased localization in plasma membrane, typical of the pattern observed in epithelial cells, compared to their controls (Figure 4F, upper panel). Further, immunofluorescent staining for mesenchymal marker vimentin revealed that its expression (green) is reduced in miR-203 overexpressing ACHN and Caki-1 cell-lines with respect to control (Figure 4F, lower pannel). Similarly, attenuation of HOTAIR also resulted in upregulation of E-cadherin, claudin and simultaneous decrease in vimentin expression (supplementary Figure S1). Moreover, CSC, like EMT, is also found to play critical role in tumor metastasis and cancer invasion. Thus, we were curious to check the expression levels of KLF4 and nanog (stemness markers). Ectopic expression of miR-203 in both ACHN and Caki-1 cell lines resulted in decreased levels of KLF4 and nanog proteins compared to control suggesting decreased stemness with overexpression of miR-203 (Figure 4G).
Figure 4. Overexpression of miR-203 inhibits epithelial-to-mesenchymal transition and stemness in RCC.

(A–D) qRT-PCR analysis showing significant increase in the mRNA levels of E-cadherin, claudin and decrease in mRNA levels of vimentin. (E) Western blot analysis showing protein levels of E-cadherin, claudin, vimentin and beta actin (control) in ACHN and Caki-1 cells after overexpression of miR-203. (F) E-cadherin (red) and vimentin (green) immunostaining counterstained with DAPI (blue) in ACHN and Caki1 cells after transfections with miR-Control or miR-203 mimic, scale bar: 500 μm (right bottom). (G) Western blot analysis showing expression of stemness marker protein levels of KLF4, nanog and beta actin (control) in ACHN and Caki-1 cells after overexpression of miR-203.
Overexpression of miR-203 or attenuation of HOTAIR induces the tumor suppressor PTEN gene and downstream effectors involved in proliferation and survival pathways
We next sought to determine the effect of miR-203-mediated suppression of HOTAIR on downstream pathway genes. Both real-time PCR data and immunoblot studies indicated that suppression of HOTAIR by overexpression of miR-203 led to upregulation of PTEN, a master regulator of PI3K/Akt pathway (Figure 5A–5B) and upregulation of downstream molecules p21, and p27 at both the mRNA and protein levels. On the other hand, knockdown of HOTAIR also resulted in the induction of these genes (supplementary Figure S2). These results suggest that miR-203 mediated regulation of HOTAIR induces the expression of downstream tumor suppressor genes important for the functional regulation of RCC.
Figure 5. Overexpression of miR-203 induces tumor suppressor PTEN gene and its down-stream effectors involved in proliferation and survival pathways.

(A) qRT-PCR analysis shows overexpression of miR-203 induces PTEN, p27 and p21 expression in mRNA levels in ACHN (upper panel) and Caki-1 (lower panel). (B) Immunoblot analysis showing upregulation of PTEN, p21, p27 protein levels which are involved in renal cancer cell proliferation and survival.
In vivo tumor growth inhibition in a mouse xenograft model
A significant decrease in tumor growth was observed by local administration of miR-203 mimic and control in established tumors over the course of the experiment. Average tumor volume in controls was 153 mm3 compared to 53.9 mm3 in mice that received miR-203 mimic (Figure 6A–6B). Total RNA was subsequently extracted from excised tumors and was analyzed for miR-203 and HOTAIR expressions. As expected, miR-203 levels were significantly higher (Figure 6C) with reduced expression of HOTAIR (Figure 6D) in the tumor samples excised from mice that received miR-203 mimic compared to that of the control group.
Figure 6. Decrease in tumorigenicity in vivo by intra-tumoral delivery of miR-203 mimic.
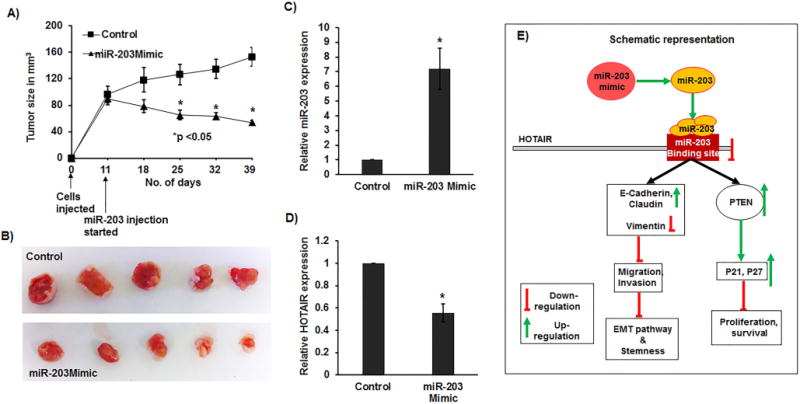
(A) Tumor volume after intratumoral injection of control or miR-203 mimic into established tumors (starting from day 11). Data represents the mean of each group and error bars are S.E.M. (B) Pictures of tumors at the day of termination of experiment (day 39). qRT-PCR analysis showing (C) significant increase in the levels of miR-203 and (D) significant decrease of HOTAIR expression in excised mouse tumor samples. (E) Schematic representation showing knockdown of HOTAIR due to overexpression of miR-203 resulting in inhibition of proliferation/survival/invasion and migration pathways.
Discussion
Long noncoding RNAs play important roles in the regulation of gene expression. A lncRNA HOTAIR, known to interact with polycomb repressive complex, has been widely reported to promote malignancy of different cancers (12,27). Recent studies also emphasize the critical role of miRNAs in various processes including cell development, differentiation, proliferation and apoptosis (28,29). Dysregulation in the expression of miRNA also plays key roles in a wide spectrum of physiologic and pathologic processes (30,31). Accruing data shows that miRNAs can function as oncogene or tumor suppressor genes (32), and play pivotal roles in various cancers (33). miR-203 has been shown to be a tumor suppressor in different cancers (23,24) by acting on multiple signaling pathways and targeting various proteins (1,25,26,34). Both HOTAIR and miR-203 have independent roles in the progression and metastasis of cancer, but the exact mechanism of how they interact with each other remains to be elucidated.
In our present study, we focused on the role of miR-203 in the regulation of lncRNA HOTAIR and elucidated the effect of their interaction on downstream EMT and metastatic pathway in renal carcinoma cells. Expression levels of miRNA-203 in RCC cell lines and patient tumor samples was low compared to that of HK-2 cells or normal tissue samples respectively. In contrast, HOTAIR had higher expression levels in renal cancer cell lines and kidney cancer clinical samples compared to their matched normal tissues or normal cell line. ROC curve analysis confirms that expression of both miR-203 and HOTAIR can distinguish between malignant and normal tissues, indicating their clinical significance in RCC.
Mechanistic studies revealed that, overexpression of miR-203 impaired cell viability, colony formation, migration, invasion and initiated apoptosis along with induction of G0/G1 arrest in RCC cell lines. Similarly, HOTAIR knockdown manifested same functional changes mimicking the effects of miR-203 overexpression in renal cancer cells. These results confirm that overexpression of miR-203 and depletion of HOTAIR causes suppression of RCC tumor growth and progression.
MicroRNAs exhibit their functional effects by regulating gene expression either by binding to specific binding sites in their target genes or by inhibiting gene translation (35). We used the computational algorithm (RNA22) to find potential miR-203 binding sites in HOTAIR. Interestingly, expression of HOTAIR was found to be suppressed in ACHN and Caki-1 cells overexpressing miR-203 suggesting a direct involvement of miRNA-203 in HOTAIR regulation. Indeed, luciferase reporter assay in ACHN and Caki-1 cells showed that miR-203 binds to HOTAIR in a sequence specific manner. This fact was further confirmed by RIP assay analysis wherein immunoprecipitated Ago2 showed increased fold enrichment of HOTAIR with respect to IgG in miR-203 overexpressing ACHN and Caki-1 cells compared to controls. These results confirm a direct interaction between miR-203 and HOTAIR in RCC.
Emerging lines of evidence suggest that the major obstacle in the treatment of cancer are tumor recurrence and metastasis. EMT, a conserved embryologic genetic program, is a critical early event in invasion and metastasis of cancer cells. During EMT, the expression of E-cadherin, referred to as the ‘caretaker’ of the epithelial phenotype, decreases, resulting in loss of cell—cell adhesion and increased migration (36). In our study, the phenotypic effects with ectopic expression of miR-203 were consistent with the inhibition of EMT. Thus, we found that miR-203 overexpression in RCC cells resulted in an increase in the epithelial markers E-cadherin and claudin with a concomitant decrease in the mesenchymal marker vimentin at both the mRNA and protein levels (Figure 6E). In addition, PTEN (phosphatase and tensin homolog) is one of the most frequently altered tumor suppressor genes in cancer (37,38). Inactivation of PTEN results in upregulation of PI3K/AKT pathway that drives cancer progression to metastatic stage (39–45). We found induction of PTEN after ectopic expression of miR-203 in both ACHN and Caki1 cells resulting in cell cycle arrest, increased apoptosis and decreased migration and invasion. Moreover, miR-203 mimic mediated induction of PTEN resulted in the induction of p21 and p27 in ACHN and Caki-1 cells that are involved in cell survival, proliferation, apoptosis, migration and invasion (39,40).
To examine the biological significance of decreased miR-203 expression, we also performed in vivo experiments using renal cancer xenografts in nude mice model. Our results show that ectopic miR-203 expression significantly attenuates tumor growth and decreases expression of HOTAIR in nude mice confirming the in vitro tumor suppressor effects of miR-203 in vivo.
In summary, our findings have revealed a novel role of lncRNA HOTAIR-miR-203 interaction in regulating EMT and metastasis in RCC. Expression of both have clinical significance in RCC since each of them can independently distinguish malignant from normal tissues. Our study also identified miR-203 as a regulatory microRNA that binds specifically to a target site and attenuates HOTAIR expression in renal cancer cells. In addition, miR-203 mediated regulation of HOTAIR inhibited tumorigenic attributes of RCC cell lines. Thus, this report envisages that miR-203 and HOTAIR may be useful in RCC therapeutics.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Dr. Roger Erickson for his support and assistance with the preparation of the manuscript. This study was supported by the Department of Veterans Affairs through VA Merit Review number I01BX001123 (awarded to Dr. Rajvir Dahiya) and the National Institutes of Health/National Cancer Institute through grant number RO1CA199694 (awarded to Dr. Rajvir Dahiya), RO1CA196848 (awarded to Dr. Soichiro Yamamura).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interests.
References
- 1.Wang N, Liang H, Zhou Y, Wang C, Zhang S, Pan Y, et al. MiR-203 Suppresses the Proliferation and Migration and Promotes the Apoptosis of Lung Cancer Cells by Targeting SRC. In: Cheng JQ, editor. PLoS One Public Library of Science. Vol. 9. 2014. p. e105570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.van Vlodrop IJH, Joosten SC, De Meyer T, Smits KM, Van Neste L, Melotte V, et al. A Four-Gene Promoter Methylation Marker Panel Consisting of GREM1, NEURL, LAD1, and NEFH Predicts Survival of Clear Cell Renal Cell Cancer Patients. Clin Cancer Res. 2017;23 doi: 10.1158/1078-0432.CCR-16-1236. [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Mazumdar M, Bacik J, Russo P, Berg WJ, Metz EM. Effect of cytokine therapy on survival for patients with advanced renal cell carcinoma. J Clin Oncol American Society of Clinical Oncology. 2000;18:1928–35. doi: 10.1200/JCO.2000.18.9.1928. [DOI] [PubMed] [Google Scholar]
- 5.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009;139:871–90. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 6.Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cell Mol Life Sci. 2012;69:3429–56. doi: 10.1007/s00018-012-1122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Larsen JE, Nathan V, Osborne JK, Farrow RK, Deb D, Sullivan JP, et al. ZEB1 drives epithelial-to-mesenchymal transition in lung cancer. J Clin Invest. 2016;126:3219–35. doi: 10.1172/JCI76725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mani SA, Guo W, Liao M-J, Eaton EN, Ayyanan A, Zhou AY, et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Jiao M, Wu K, Li L, Zhu G, Wang X, et al. TNF-α induced epithelial mesenchymal transition increases stemness properties in renal cell carcinoma cells. Int J Clin Exp Med e-Century Publishing Corporation. 2014;7:4951–8. [PMC free article] [PubMed] [Google Scholar]
- 10.Jung H-Y, Yang J. Unraveling the TWIST between EMT and cancer stemness. Cell Stem Cell. 2015;16:1–2. doi: 10.1016/j.stem.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene. 2013;32:1616–25. doi: 10.1038/onc.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–6. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, et al. Long Noncoding RNA HOTAIR Regulates Polycomb-Dependent Chromatin Modification and Is Associated with Poor Prognosis in Colorectal Cancers. Cancer Res. 2011;71:6320–6. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Liu M, Feng Y, Xu Y-F, Huang Y-F, Che J-P, et al. Downregulated miR-646 in clear cell renal carcinoma correlated with tumour metastasis by targeting the nin one binding protein (NOB1) Br J Cancer Nature Publishing Group. 2014;111:1188–200. doi: 10.1038/bjc.2014.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Faragalla H, Youssef YM, Scorilas A, Khalil B, White NMA, Mejia-Guerrero S, et al. The Clinical Utility of miR-21 as a Diagnostic and Prognostic Marker for Renal Cell Carcinoma. J Mol Diagnostics. 2012;14:385–92. doi: 10.1016/j.jmoldx.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 16.Mikhaylova O, Stratton Y, Hall D, Kellner E, Ehmer B, Drew AF, et al. VHL-Regulated MiR-204 Suppresses Tumor Growth through Inhibition of LC3B-Mediated Autophagy in Renal Clear Cell Carcinoma. Cancer Cell. 2012;21:532–46. doi: 10.1016/j.ccr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chiyomaru T, Fukuhara S, Saini S, Majid S, Deng G, Shahryari V, et al. Long Non-coding RNA HOTAIR Is Targeted and Regulated by miR-141 in Human Cancer Cells. J Biol Chem. 2014;289:12550–65. doi: 10.1074/jbc.M113.488593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chiyomaru T, Yamamura S, Fukuhara S, Yoshino H, Kinoshita T, Majid S, et al. Genistein inhibits prostate cancer cell growth by targeting miR-34a and oncogenic HOTAIR. In: Rishi A, editor. PLoS One. Vol. 8. 2013. p. e70372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu Y, Liu J, Zheng Y, You L, Kuang D, Liu T. Suppressed expression of long non-coding RNA HOTAIR inhibits proliferation and tumourigenicity of renal carcinoma cells. Tumour Biol. 2014;35:11887–94. doi: 10.1007/s13277-014-2453-4. [DOI] [PubMed] [Google Scholar]
- 20.Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, Santos J, Calin GA, Cigudosa JC, et al. Genetic and Epigenetic Silencing of MicroRNA-203 Enhances ABL1 and BCR-ABL1 Oncogene Expression. Cancer Cell. 2008;13:496–506. doi: 10.1016/j.ccr.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 21.Yi R, Poy MN, Stoffel M, Fuchs E. A skin microRNA promotes differentiation by repressing “stemness”. Nature. 2008;452:225–9. doi: 10.1038/nature06642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonkoly E, Wei T, Pavez Loriè E, Suzuki H, Kato M, Törmä H, et al. Protein Kinase C-Dependent Upregulation of miR-203 Induces the Differentiation of Human Keratinocytes. J Invest Dermatol. 2010;130:124–34. doi: 10.1038/jid.2009.294. [DOI] [PubMed] [Google Scholar]
- 23.Diao Y, Guo X, Jiang L, Wang G, Zhang C, Wan J, et al. MiR-203, a Tumor Suppressor Frequently Down-regulated by Promoter Hypermethylation in Rhabdomyosarcoma. J Biol Chem. 2014;289:529–39. doi: 10.1074/jbc.M113.494716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tian L, Li M, Ge J, Guo Y, Sun Y, Liu M, et al. MiR-203 is downregulated in laryngeal squamous cell carcinoma and can suppress proliferation and induce apoptosis of tumours. Tumor Biol Springer Netherlands. 2014;35:5953–63. doi: 10.1007/s13277-014-1790-7. [DOI] [PubMed] [Google Scholar]
- 25.Zhang F, Yang Z, Cao M, Xu Y, Li J, Chen X, et al. MiR-203 suppresses tumor growth and invasion and down-regulates MiR-21 expression through repressing Ran in esophageal cancer. Cancer Lett. 2014;342:121–9. doi: 10.1016/j.canlet.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 26.Xu M, Gu M, Zhang K, Zhou J, Wang Z, Da J. MiR-203 inhibition of renal cancer cell proliferation, migration and invasion by targeting of FGF2. Diagn Pathol. 2015;10:24. doi: 10.1186/s13000-015-0255-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsai M-C, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science (80-) 2010;329:689–93. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartel DP. MicroRNAs: Target Recognition and Regulatory Functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nat Struct Mol Biol. 2012;19:586–93. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 30.Sayed D, Abdellatif M. MicroRNAs in Development and Disease. Physiol Rev. 2011;91:827–87. doi: 10.1152/physrev.00006.2010. [DOI] [PubMed] [Google Scholar]
- 31.Hatziapostolou M, Polytarchou C, Iliopoulos D. MiRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol Metab. 2013;24:361–73. doi: 10.1016/j.tem.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Ventura A, Jacks T. MicroRNAs and Cancer: Short RNAs Go a Long Way. Cell. 2009;136:586–91. doi: 10.1016/j.cell.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 34.Saini S, Majid S, Yamamura S, Tabatabai L, Suh SO, Shahryari V, et al. Regulatory Role of mir-203 in Prostate Cancer Progression and Metastasis. Clin Cancer Res. 2011;17:5287–98. doi: 10.1158/1078-0432.CCR-10-2619. [DOI] [PubMed] [Google Scholar]
- 35.Majid S, Dar AA, Saini S, Yamamura S, Hirata H, Tanaka Y, et al. MicroRNA-205-directed transcriptional activation of tumor suppressor genes in prostate cancer. Cancer. 2010;116:5637–49. doi: 10.1002/cncr.25488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene NIH Public Access. 2008;27:6920–9. doi: 10.1038/onc.2008.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 38.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–7. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 39.Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14. doi: 10.1016/j.cell.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 40.Li DM, Sun H. PTEN/MMAC1/TEP1 suppresses the tumorigenicity and induces G1 cell cycle arrest in human glioblastoma cells. Proc Natl Acad Sci U S A. 1998;95:15406–11. doi: 10.1073/pnas.95.26.15406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, et al. PTEN controls tumor-induced angiogenesis. Proc Natl Acad Sci. 2001;98:4622–7. doi: 10.1073/pnas.081063798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by PTEN. Biochem Soc Trans. 2005;33:1507. doi: 10.1042/BST0331507. [DOI] [PubMed] [Google Scholar]
- 44.Yamada KM, Araki M. Tumor suppressor PTEN: modulator of cell signaling, growth, migration and apoptosis. J Cell Sci. 2001;114:2375–82. doi: 10.1242/jcs.114.13.2375. [DOI] [PubMed] [Google Scholar]
- 45.Trotman LC, Pandolfi PP. PTEN and p53: who will get the upper hand? Cancer Cell. 2003;3:97–9. doi: 10.1016/s1535-6108(03)00022-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.