

Abstract
Noneosinophilic airway inflammation occurs in approximately 50% of patients with asthma. It is subdivided into neutrophilic or paucigranulocytic inflammation, although the proportion of each subtype is uncertain because of variable cut-off points used to define neutrophilia. This article reviews the evidence for noneosinophilic inflammation being a target for therapy in asthma and assesses clinical trials of licensed drugs, novel small molecules and biologics agents in noneosinophilic inflammation. Current symptoms, rate of exacerbations and decline in lung function are generally less in noneosinophilic asthma than eosinophilic asthma. Noneosinophilic inflammation is associated with corticosteroid insensitivity. Neutrophil activation in the airways and systemic inflammation is reported in neutrophilic asthma. Neutrophilia in asthma may be due to corticosteroids, associated chronic pulmonary infection, altered airway microbiome or delayed neutrophil apoptosis. The cause of poorly controlled noneosinophilic asthma may differ between patients and involve several mechanism including neutrophilic inflammation, T helper 2 (Th2)-low or other subtypes of airway inflammation or corticosteroid insensitivity as well as noninflammatory pathways such as airway hyperreactivity and remodelling. Smoking cessation in asthmatic smokers and removal from exposure to some occupational agents reduces neutrophilic inflammation. Preliminary studies of ‘off-label’ use of licensed drugs suggest that macrolides show efficacy in nonsmokers with noneosinophilic severe asthma and statins, low-dose theophylline and peroxisome proliferator-activated receptor gamma (PPARγ) agonists may benefit asthmatic smokers with noneosinophilic inflammation. Novel small molecules targeting neutrophilic inflammation, such as chemokine (CXC) receptor 2 (CXCR2) antagonists reduce neutrophils, but do not improve clinical outcomes in studies to date. Inhaled phosphodiesterase (PDE)4 inhibitors, dual PDE3 and PDE4 inhibitors, p38MAPK (mitogen-activated protein kinase) inhibitors, tyrosine kinase inhibitors and PI (phosphoinositide) 3kinase inhibitors are under development and these compounds may be of benefit in noneosinophilic inflammation. The results of clinical trials of biological agents targeting mediators associated with noneosinophilic inflammation, such as interleukin (IL)-17 and tumor necrosis factor (TNF)-α are disappointing. Greater understanding of the mechanisms of noneosinophilic inflammation in asthma should lead to improved therapies.
Keywords: airway inflammation, asthma, biological agents, biomarkers, cigarette smoking, corticosteroid insensitivity, eosinophils, neutrophils, small molecules
Introduction
Personalized medicine in asthma aims to individualise treatment using noninvasive biomarkers that predict a beneficial response or that identify individuals who are at risk of adverse effects [Agustí et al. 2015]. Several airway inflammatory phenotypes are recognized that help identify a therapeutic response to specific treatments in asthma. For example, eosinophilic airway inflammation, which is usually identified on the basis of sputum or blood eosinophilia, predicts patients with asthma that are likely to obtain a favourable therapeutic response to corticosteroids [Pavord et al. 1999; Little et al. 2000; Green et al. 2002a; Bacci et al. 2006; Berry et al. 2007] and to monoclonal antibodies targeting interleukin (IL)-5 [Pavord et al. 2012; Katz et al. 2014; Thomson, 2014]. Type 2 helper T-cell (Th2)-high subtype of asthma is associated with increased epithelial expression of IL-4, IL-5 and IL-13 [Woodruff et al. 2009; Arron et al. 2013] and is considered to overlap with eosinophilic airway inflammation [Arron et al. 2013]. Evidence from clinical trials suggests that the presence of type-2 eosinophilic inflammation predicts a therapeutic response not only to corticosteroids [Woodruff et al. 2009], but to monoclonal antibodies targeting specific cytokines such as IL-5 [Bel et al. 2014; Ortega et al. 2014] and IL-13 [Corren et al. 2011]. Many patients with asthma have noneosinophilic asthma, sometimes associated with neutrophilic inflammation or have a Th2-low type of inflammation. Compared with type-2 eosinophilic inflammation there are relatively few interventions available for non-type 2 inflammatory subgroups. This article aims to discuss the evidence that noneosinophilic airway inflammation, with or without neutrophilic inflammation, is an appropriate target for therapy in asthma and also aims to assess the results of recent clinical trials of licensed drugs, novel small molecules and biologics agents in the treatment of noneosinophilic asthma.
Is noneosinophilic airway inflammation an appropriate target for therapy in asthma?
A number of factors need to be considered when attempting to answer the question of whether noneosinophilic inflammation is an appropriate target for treatment in asthma including the criteria used to define neutrophilic and eosinophilic inflammation, the stability of noneosinophilic inflammation over time, the prevalence of noneosinophilic inflammation, the strength of evidence for the involvement of noneosinophilic inflammation in clinical features of asthma and the cause(s) of noneosinophilic airway inflammation.
Definition of eosinophilic and neutrophilic airway inflammation
Noneosinophilic airway inflammation is a term used to describe a subtype of asthma associated with normal numbers of sputum eosinophils. The noneosinophilic phenotype is subdivided into neutrophilic inflammation, when neutrophil numbers are raised above a defined cut-off level or paucigranulocytic inflammation, when both eosinophil and neutrophil numbers are normal. In addition, some individuals have a mixed type of inflammation, when there is sputum neutrophilia and eosinophilia. Cut-off levels used to define sputum eosinophilia most commonly used are ⩾2% [Mcgrath et al. 2012; Hastie et al. 2013], >2% [Peters et al. 2014] or ⩾3% [Schleich et al. 2013; Zhang et al. 2014; Wagener et al. 2015]. A ⩾3% cut-off is reported to be the most precise value to identify eosinophilic airway inflammation [Simpson et al. 2010]. Sputum eosinophil counts are associated with bronchial tissue eosinophil numbers suggesting that they provide a good indicator of airway eosinophilic pathology [Arron et al. 2014]. The cut-off for a raised sputum neutrophil count is not clearly established with a wide range of values reported in the literature: >40% [Nair et al. 2012; Moore et al. 2014), ⩾50% [Chaudhuri et al. 2014], >61% [Simpson et al. 2009], >65% [Nair et al. 2015] and ⩾76% [Schleich et al. 2013]. The most appropriate cut-off value that identifies individuals in whom neutrophils are activated and contributing to the pathogenic processes in asthma is not certain. In addition, sputum neutrophils do not correlate with bronchial tissue numbers bringing into doubt their predictive value for identifying neutrophil-induced airway pathology [Arron et al. 2014]. In addition to the presence of noneosinophilic inflammation, Haldar and Pavord proposed that the criteria for a diagnosis of noneosinophilic asthma should include objective evidence of airflow obstruction or airway hyperreactivity, a raised asthma control questionnaire (ACQ) score (>1.5) and the absence of a significant smoking history, fixed airflow obstruction or associated bronchiectasis [Haldar and Pavord, 2007]. In the current article, the criteria for noneosinophilic asthma include the presence of noneosinophilic inflammation as defined above plus objective evidence of asthma, but the review also includes data from patients with both normal and raised ACQ scores, who have a significant smoking history or who have fixed airflow obstruction.
Stability of sputum cell counts
Published data on the long term stability of sputum neutrophil and eosinophil counts is conflicting. Some studies report stable sputum cell counts in patients with mild to severe asthma follow-up over 6 months [Berry et al. 2007], 12 months [Green et al. 2002a], 2 years [Jayaram et al. 2006] and 5 years [Simpson et al. 2006; Van Veen et al. 2009]. In contrast, sputum inflammatory cell phenotype changed in 48.6% of patients with severe asthma over 1 year among patients recruited to the BIOmarkers in Severe Chronic AIRway Disease (BIOAIR) study [Kupczyk et al. 2014a]. Similar variability in sputum cell counts has been reported by others [Hancox et al. 2012] and in one study a stable inflammatory phenotype was found in only one-third of patients [Al-Samri et al. 2010]. Transient sputum eosinophilia is reported in up to 40% patients with noneosinophilic inflammation [Bacci et al. 2012; McGrath et al. 2012]. The potential for the lack of stability in noneosinophilic inflammation over time needs to be accounted for in intervention studies targeting sputum inflammatory cell biomarkers.
Prevalence of noneosinophilic airway inflammation
The different cut-off values used to define elevated sputum cell counts, particularly sputum neutrophils, may explain the variation in prevalence figures for noneosinophilic inflammation between studies. Nevertheless, overall up to 50% of adults and adolescents with stable mild to severe asthma, and in some studies higher proportions, have noneosinophilic inflammation [Gibson et al. 2001; Green et al. 2002b; Simpson et al. 2006; Wang et al. 2011; McGrath et al. 2012; Schleich et al. 2013; Moore et al. 2014; Brooks et al. 2016 ]. For example, a review of sputum cytology data from 995 patients with mild to moderate asthma enrolled in clinical trials undertaken by the Asthma Clinical Research Network (ACRN) reported that noneosinophilic inflammation (sputum eosinophils <2%) was present in 64% of patients not taking inhaled corticosteroid and 83% of patients taking inhaled corticosteroids. In a subgroup of patients followed up for 6 months, 47% of the inhaled corticosteroid-free patients and 72% of those taking inhaled corticosteroids had persistent noneosinophilic inflammation [McGrath et al. 2012]. In a cluster analysis performed on 423 patients recruited to the Severe Asthma Research Program (SARP) cohort, four asthma inflammatory subphenotypes were identified (cut-off values used to define sputum eosinophilia ⩾2% and sputum neutrophilia >40%) [Moore et al. 2014]. Two groups had mild-to-moderate allergic asthma with minimal or eosinophil-predominant sputum inflammation whereas the other two subphenotypes had moderate-to-severe asthma with neutrophil-predominant or mixed granulocytic inflammation [Moore et al. 2014]. A study in a small group of adults with stable asthma found 51.7% of subjects had a paucigranulocytic phenotype, 27.6% neutrophilic inflammation and 17.2% eosinophilic inflammation [Wang et al. 2011].
Involvement of neutrophilic and noneosinophilic airway inflammation in asthma
Evidence for the involvement of noneosinophilic inflammation in asthma is based mainly on studies examining the association between sputum inflammatory phenotypes and clinical outcomes in asthma, including current symptom control, exacerbations, airflow obstruction and therapeutic response to corticosteroids. Further evidence is provided by reports of local activation of neutrophils and systemic inflammation in neutrophilic asthma.
Current symptom control
The severity of current symptoms is in general similar or slightly lower in noneosinophilic or neutrophilic subgroups of asthma compared to eosinophilic subgroups [Cowan et al. 2010; Hastie et al. 2010; Wood et al. 2012; Schleich et al. 2013, 2014; Baines et al. 2014; Newby et al. 2014].
Exacerbations
Sputum neutrophilia is found in up to 80% of exacerbations in adults with asthma [Turner et al. 1995; Fahy et al. 1995; Lamblin et al. 1998; Green et al. 2002a; Jayaram et al. 2006; Maneechotesuwan et al. 2007; Wang et al. 2011], although the predominant sputum cell type can alter during successive exacerbations [D’Silva et al. 2007]. Sputum eosinophilia is a better predictor of future exacerbations than sputum neutrophilia [Jatakanon et al. 2000; Leuppi et al. 2001; Kupczyk et al. 2014b; Schleich et al. 2014]. For example, a cluster analysis performed on patients recruited to the BIOAIR study identified two clusters with raised sputum eosinophil counts that accounted for 83% of patients who had two or more severe exacerbations during follow-up for 1 year [Kupczyk et al. 2014b]. One of these clusters had a mixed inflammatory profile with raised sputum neutrophils (43% percent of patients). A further cluster had a raised neutrophil count and a normal (cut-off values used to define sputum eosinophilia ⩾2% and sputum neutrophilia >40%) eosinophil count (11% of patients) and a noneosinophilic paucigranulocytic inflammation was found in only 6% of cases. Patients with severe asthma associated with eosinophil inflammation have more intubations than noneosinophilic patients [Wenzel et al. 1999].
Airflow obstruction
Sputum neutrophilia is associated with reduced lung function and based on this finding it has been speculated that airway neutrophils may contribute to the development of persistent airflow obstruction in asthma [Little et al. 2002; Shaw et al. 2007]. Against this hypothesis, a recent cluster analysis of lung function decline and sputum eosinophil count performed in 97 patients with severe asthma identified a noneosinophilic group in whom the decline in forced expiratory volume in 1 second (FEV1) was −14 ml per year compared to an eosinophilic group with highly variable eosinophil counts that had a greater rate of decline in FEV1 of −41 ml per year [Newby et al. 2014]. These findings suggest that eosinophilic inflammation, particularly when there is high variability in eosinophil count, is a greater risk factor for the development of persistent airflow obstruction than noneosinophilic inflammation. Bronchodilator reversibility and airway hyperresponsiveness are similar in eosinophilic and noneosinophilic asthma [Berry et al. 2007; McGrath et al. 2012], although one study noted greater airway hyperresponsiveness in persistent or intermittent eosinophilic groups [McGrath et al. 2012].
Impaired response to inhaled corticosteroids
Noneosinophilic inflammation is associated with an impaired therapeutic response to inhaled corticosteroids [Pavord et al. 1999; Green et al. 2002a; Bacci et al. 2006; Berry et al. 2007; Thomson et al. 2009; McGrath et al. 2012], although the lack of efficacy may not be complete. Several clinical studies performed in small numbers of patients with noneosinophilic asthma suggest that this group may obtain some benefit from inhaled corticosteroids although less than that found in eosinophilic patients [Godon et al. 2002; Cowan et al. 2010; Lemière et al. 2011]. Intermittent eosinophilia might be a factor accounting for corticosteroid sensitivity in some of these patients [Bacci et al. 2012; McGrath et al. 2012; Suárez-Cuartín et al. 2015].
Evidence for neutrophil activation in asthma
There is evidence to suggest that the innate immune system is activated in chronic asthma. Firstly, sputum IL-8 and neutrophil elastase concentrations and innate immune receptors –Toll-like receptors (TLR)2, TLR4 and CD14 – as well as pro-inflammatory IL-8 and IL-1β gene expression levels are increased in neutrophilic asthma compared to non-neutrophilic asthma [Simpson et al. 2007; Wood et al. 2012]. Secondly, neutrophil activation, as measured by sputum myeloperoxidase (MPO) levels, is positively associated with sputum neutrophil numbers in asthma [Little et al. 2002]. Thirdly, specific sputum gene expression signatures are reported to discriminate eosinophilic asthma from noneosinophilic asthma as well as to predict a beneficial response to inhaled corticosteroids [Baines et al. 2014]. In this study, noneosinophilic asthma was identified by increased sputum cell expression of IL-1β, alkaline phosphatase, tissuenonspecific isozyme (ALPL) and CXCR2, whereas eosinophilic asthma was characterized by increased expression of Charcot–Leyden crystal protein or galectin-10 (CLC), carboxypeptidase A3 (CPA3) and deoxyribonuclease I-like 3 (DNASE1L3). The NLRP3 inflammasome is upregulated in neutrophilic asthma and may increase the production of IL-1β [Simpson et al. 2014b; Kim et al. 2015a]. Anti-inflammatory responses may be impaired in noneosinophilic asthma based on reduced sputum galectin-3 concentrations, which increases uptake of apoptotic neutrophils and reduced IL-1RA/IL-1β ratio, and which might increase pro-inflammatory actions of IL-1β [Gao et al. 2015]. In addition, soluble receptor for advanced glycation end-products (RAGE), which is a pattern-recognition receptor is deficient in bronchoalveolar lavage (BAL) samples in neutrophilic asthma [Sukkar et al. 2012]. The T-cell granzyme B pathway, which is thought to mediate apoptosis of epithelial cells, might be defective in noneosinophilic asthma, based on the finding of a higher ratio of the expression of granzyme B to its inhibitor in T cells in this group compared to eosinophilic asthma [Simpson et al. 2014a].
Systemic inflammation is increased in patients with neutrophilic airway inflammation. The proportion of patients with elevated CRP, IL-6 and neutrophil elastase concentrations is higher in neutrophilic asthma compared to a non-neutrophilic group [Baines et al. 2011; Wood et al. 2012]. Neutrophilic inflammation is associated with increased α-defensin and neutrophil protease gene expression in blood [Baines et al. 2011]. In noneosinophilic asthma, blood neutrophils released significantly higher levels of IL-8 at rest [Baines et al. 2010]. In a small study, gene expression markers of systemic inflammation were associated with higher basal metabolic index (BMI), greater history of cigarette smoking, lower forced vital capacity (FVC)% predicted, and increased sputum neutrophils [Fu et al. 2013].
Potential inflammatory processes leading to noneosinophilic airway inflammation
Several inflammatory processes could lead to noneosinophilic inflammation and airway damage in asthma although the exact immunological mechanisms are unclear (Figure 1) [Trejo Bittar et al. 2015]. Uncertainty in the clinical relevance of experimental animal models of noneosinophilic inflammation has hampered progress in understanding the involvement of neutrophils and noneosinophilic inflammation in the pathogenesis of asthma. Stimuli such as viruses, cigarette smoke and pollutants could induce the release of chemoattractants including IL-8 to recruit neutrophils to the airways. In experimental asthma models, the release of IL-17A and IL-17F from activated Th17 cells stimulates the synthesis of neutrophil chemoattractants including CXCL1 and IL-8 from the airway epithelium. [Newcomb and Peebles, 2013]. Interferon gamma (IFN-γ) may also be involved in the pathogenesis of severe asthma associated with neutrophilic and eosinophilic inflammation, possibly in part through the release of IFN-γ from Th1 cells [Raundhal et al. 2015]. Data from patients with severe asthma and an experimental murine asthma model implicate high IFN-γ immune responses and low secretory leukocyte protease inhibitor expression (SLPI) in airway epithelial cells with airway hyperresponsiveness [Raundhal et al. 2015]. Neutrophils are a potential source of oxygen free radicals and enzymes and their ability to activate other airway cell types [Futosi et al. 2013]. Neutrophils in asthma are implicated in causing mucus gland hyperplasia and hypersecretion, airway hyperreactivity and remodelling as well as corticosteroid insensitivity. Interestingly, Th1 and Th17 cells may induce airway hyperreactivity or remodelling independently of neutrophil activation.
Figure 1.
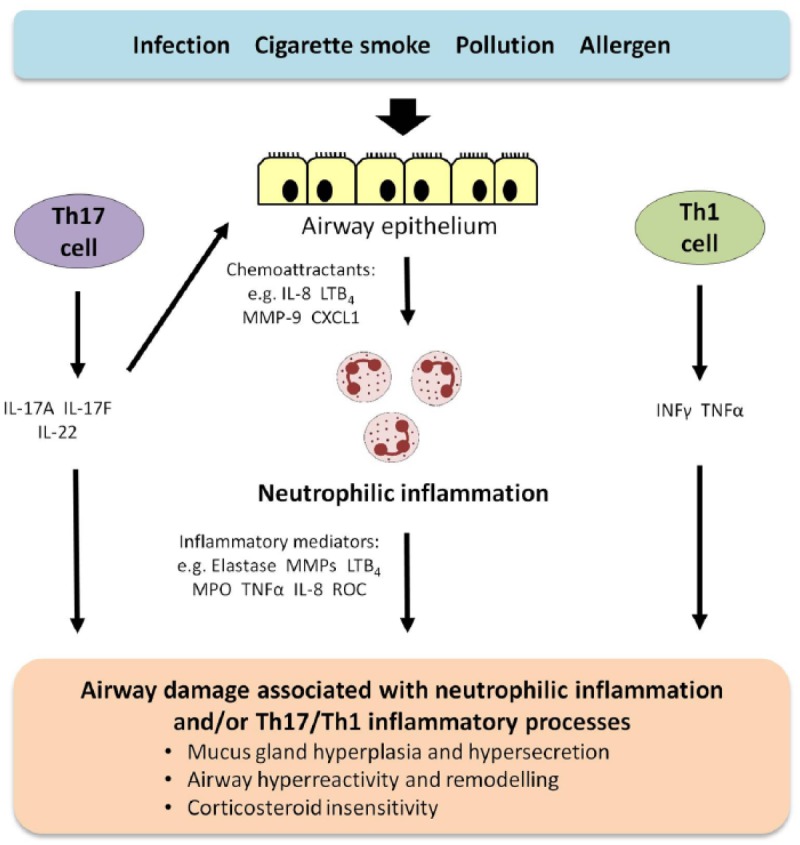
Schematic diagram of potential pathways leading to noneosinophilic inflammation and airway damage in severe asthma.
Several inflammatory pathways could potentially lead to noneosinophilic inflammation and airway damage in asthma although the exact mechanisms are unclear. Possible pathways are briefly summarized in the schematic diagram. Stimuli such as viruses, cigarette smoke and pollutants could induce the release of chemoattractants including IL-8 to recruit neutrophils to the airways. The release of IL-17A and IL-17F from activated Th17 cells could stimulate the synthesis of neutrophil chemoattractants, such as CXCL1 and IL-8 from the airway epithelium. IFN-γ may also be involved in noneosinophilic asthma, possibly in part through its release from Th1 cells. Inflammatory mediators released by neutrophils are implicated in causing mucus gland hyperplasia and hypersecretion, airway hyperreactivity and remodelling as well as corticosteroid insensitivity in asthma. Th1 and Th17 cells may induce airway hyperreactivity or remodelling independently of neutrophil activation.
Abbreviations: CXCL1, chemokine (C-X-C motif) ligand; IFN, interferon; IL, interleukin; LT, leukotriene; MMP, matrix metalloproteinase; MPO, myeloperoxidase; ROC, reactive oxygen species; TNF-α, tumor necrosis factor α.
Limited information has been published on the immunopathological characteristics of noneosinophilic inflammation in asthma compared with other inflammatory airway phenotypes including Th2-low inflammation, Th17-high inflammation or a combination of Th2/Th17 profiles. Bronchial biopsy studies of patients with noneosinophilic asthma or with Th2-low inflammation, report reduced submucosal eosinophil numbers and normal subepithelial basement membrane thickness in both groups [Wenzel et al. 1999; Berry et al. 2007; Woodruff et al. 2009]. In contrast, bronchial eosinophil numbers and subepithelial basement membrane thickness are both increased in eosinophilic asthma and in Th2-high asthma [Wenzel et al. 1999; Berry et al. 2007; Woodruff et al. 2009]. Mast cell numbers are increased in eosinophilic asthma [Wenzel et al. 1999] and Th2-high asthma [Dougherty et al. 2010], whereas mast cell numbers are normal in the submucosa of patients with severe noneosinophilic asthma [Wenzel et al. 1999] and in the epithelium of nonsmokers with Th2-low asthma [Dougherty et al. 2010]. Mast cell numbers in airway smooth muscle are increased in both noneosinophilic and eosinophilic asthma [Berry et al. 2007]. Bronchial biopsy neutrophil numbers are increased by a similar degree in noneosinophilic severe asthma and eosinophilic severe asthma [Wenzel et al. 1999]. Neutrophil numbers in Th2-low asthma have not been reported. A lower proportion of subjects with noneosinophilic asthma are atopic compared to eosinophilic asthma (18% versus 66%) [Berry et al. 2007] and (58% versus 83%) [Gibson et al. 2001]. Severe asthma associated with neutrophilia has significantly higher sputum levels of Th17-related cytokines (CXCL1, CXCL10, Chemokine (C-C motif) ligand (CCL)2, IL-6 and IL-8) compared with severe asthmatics with other inflammatory phenotypes [Manni et al. 2014]. The proportion of Th17 lymphocytes and the ratio of Th17 to regulatory T cells (Treg) in the peripheral blood is greater in patients with noneosinophilic asthma taking inhaled corticosteroids compared to an eosinophilic asthma group [Furukawa et al. 2015]. Approximately one-third of patients with severe eosinophilic asthma have a Th17-high signature that is associated with a Th2-low gene expression profile [Choy et al. 2015]. The number of patients with noneosinophilic severe asthma in this study was not sufficient to determine their Th17 profile [Choy et al. 2015]. Taken together, these findings suggest that noneosinophilic inflammation and Th2-low inflammation in nonsmokers with asthma share some similar immunopathological features, including normal eosinophil numbers, submucosal mast cell numbers and subepithelial basement membrane thickness. There is a need for further studies to establish the similarities and differences in endotypes of noneosinophilic, Th2-low and Th17-high inflammation to help identify subgroups of patients for targeted therapies.
Factors accounting for neutrophilic airway inflammation in asthma
Several factors either alone or in combination could explain raised sputum neutrophil counts in asthma (Table 1). Corticosteroids inhibit apoptosis of neutrophils [Cox, 1995] and their use in asthma may contribute to sputum neutrophilia [Saffar et al. 2011]. In addition, Th2-targeted therapies, including oral corticosteroids may contribute to the development of Th17-high neutrophilic inflammation [Choy et al. 2015; Shum, 2015]. In support of corticosteroids causing neutrophilia in asthma, inhaled corticosteroid withdrawal from patients with moderate asthma resulted in only one patient with neutrophilic inflammation although the reintroduction of inhaled fluticasone for 4 weeks resulted in a raised neutrophil count in only 5% of patients [Cowan et al. 2010]. In one study of patients with severe oral corticosteroid-dependent asthma associated with increased sputum neutrophil number, markers of neutrophil activation including oxidative burst and surface granular receptor expression were similar to patients with mild asthma [Nair et al. 2015]. In contrast, corticosteroid use was not associated with sputum neutrophilia in the SARP cohort, suggesting that continuous corticosteroid exposure may not be the only influence on sputum neutrophil numbers in severe asthma [Moore et al. 2014]. Comorbid conditions such bronchiectasis or severe airflow obstructions occurring in association with asthma may result in neutrophilic inflammation. Delayed human neutrophil apoptosis has been reported in severe asthma [Uddin et al. 2010], possibly due to epithelial growth factor-induced release of mediators with neutrophil chemotactic and anti-apoptotic actions from bronchial epithelial cells [Uddin et al. 2010; Uddin et al. 2013]. Macrophage efferocytosis is impaired in noneosinophilic asthma, which may cause airway neutrophilia [Simpson et al. 2013]. Lastly, an altered airway microbiome has been implicated in airway neutrophilia. Airway colonization determined by terminal restriction fragment length polymorphism (T-RFLP) analysis is associated with more severe airway obstruction and longer duration of disease as well as neutrophilic airway inflammation and raised sputum IL-8 levels [Simpson et al. 2013; Green et al. 2014]. Taken together, these findings suggest that the cause of airway neutrophilia in asthma is likely to be complex, possibly due to corticosteroid treatment inducing impaired apoptosis of neutrophils and Th17 mediated neutrophilic inflammation, delayed apoptosis of neutrophils due to epithelial growth factor release and ineffective macrophage efferocytosis of neutrophils, as well as an altered airway microbiome.
Table 1.
Possible factors accounting for neutrophilic airway inflammation in asthma.
| • Corticosteroid treatment causing reduced apoptosis of neutrophils and contributing to Th17-mediated neutrophilic inflammation |
| • Neutrophilia associated with chronic sinopulmonary infection or bronchiectasis |
| • Delayed human neutrophil apoptosis in severe asthma due to epithelial growth factor-induced release of mediators with neutrophil chemotactic and anti-apoptotic actions from bronchial epithelial cells |
| • Impaired macrophage phagocytosis of neutrophils |
| • Neutrophilia associated with an altered airway microbiome |
Clinical phenotypes associated with noneosinophilic inflammation
Noneosinophilic inflammation, either paucigranulocytic or neutrophilic, occurs in a range of clinical phenotypes that account for approximately 50% of never smokers or exsmokers with mild to severe asthma or that have controlled or uncontrolled asthma (Table 2). Noneosinophilic inflammation, with or without neutrophilic inflammation, is commonly found in smokers with asthma [Chalmers et al. 2002; Boulet et al. 2006; Thomson et al. 2013]. A high BMI is associated with noneosinophilic asthma in some people [Haldar et al. 2008], although others have submucosal eosinophilia [Desai et al. 2013]. Approximately two-thirds of cases of occupational asthma due to low molecular weight agents have noneosinophilic inflammation [Anees et al. 2002], which is associated with a poor asthma prognosis [Lemiere et al. 2014]. Nonoccupational-induced asthma that is exacerbated by work exposures is associated with the noneosinophilic phenotype [Lemière et al. 2013]. Additional factors associated with higher neutrophil counts include older age [Brooks et al. 2013], exposure to environmental pollution through living close to car pollution [Wallace et al. 2011], exposure to occupational particulate matter [Simpson et al. 2015] and respiratory infections.
Table 2.
Clinical phenotypes and factors associated with noneosinophilic airway inflammation in asthma.
| Mild to severe asthma in nonsmokers or exsmokers (both controlled and uncontrolled) |
| Smokers with asthma |
| High basal metabolic index (subgroup) |
| Occupational asthma (subgroup) |
| Factors associated with higher neutrophil counts |
| - Older age |
| - Exposure to environmental pollution |
| - Respiratory infections |
Biomarkers that can identify noneosinophilic airway inflammation
Are there biomarkers that can identify patients with noneosinophilic airway inflammation? Blood eosinophil numbers are moderately associated with sputum eosinophils [Schleich et al. 2013; Zhang et al. 2014; Wagener et al. 2015]. Using a cut-off for a blood eosinophil count of >0.22 × 109/l [Schleich et al. 2013], >0.26 × 109/l [Zhang et al. 2014] or ⩾0.27 × 109/l [Wagener et al. 2015] accurately predicts sputum eosinophilia. In contrast, another study reported that blood eosinophils had a poor predictive value of 47% for sputum eosinophilia (⩾3% cut-off) although this was better in severe asthma (71%) [Hastie et al. 2013]. In patients with mild to severe asthma, blood eosinophils were reported to be better than serum periostin and exhaled nitric oxide in identifying sputum eosinophilia [Wagener et al. 2015]. Blood neutrophil number has a weak relationship with sputum neutrophil count [Schleich et al. 2013; Zhang et al. 2014] and has a poor predictive value for sputum neutrophilia (64% or 38% for a cut-off of ⩾40% or ⩾61% respectively) [Hastie et al. 2013]. In one study, exhaled nitric oxide predicted inhaled corticosteroid response for airway hyperreactivity in noneosinophilic asthma (area under the curve 0.81), with an optimum cut-off point of 33 ppb [Cowan et al. 2010].
Which inflammatory phenotype to target?
In summary, noneosinophilic airway inflammation is found in approximately 50% of patients with mild to severe asthma. The proportion of this group with neutrophilic inflammation is less certain because of variable cut-off points used in different studies to define neutrophilia. Current symptoms, rate of exacerbations and rate of decline in lung function are generally less severe in noneosinophilic asthma compared to eosinophilic asthma. Noneosinophilic inflammation is associated with an impaired response to inhaled corticosteroids. There is some evidence that neutrophils are activated in the airways of patients with neutrophilic asthma and that biomarkers of systemic inflammation is increased in this group. Neutrophilia in asthma may be due to corticosteroids, associated chronic pulmonary infection, an altered airway microbiome or delayed neutrophil apoptosis, particularly in severe disease. Noneosinophilic asthma and Th2-low asthma may share some common immunopathological features, but further investigation is required. Due to the lack of effective specific therapies targeting noneosinophilic inflammation including neutrophilic inflammation there is currently no definitive evidence for the involvement of these inflammatory phenotypes in chronic asthma. Additional pathways may account for poor asthma control in patients with noneosinophilic asthma including Th1 inflammation, Th17 inflammation, or a combination of Th2 and Th17 inflammation, as well as corticosteroid insensitivity (Figure 1). Recent work suggests a reciprocal relationship between Th2 and Th17 pathways in severe disease and that corticosteroid treatment may contribute to the emergence of a Th17-high profile [Choy et al. 2015]. Noninflammatory mechanisms may also be important in some individuals including airway hyperreactivity and airway remodelling.
Treatments targeting noneosinophilic airway inflammation
Many patients with asthma continue to have poorly controlled disease despite treatment with currently available therapies. There is an unmet need for novel treatments that will impact favourably on clinical outcomes in patients with noneosinophilic inflammation. Nonpharmacological interventions, ‘off-label’ use of licensed drugs, novel small molecules and biologics agents are being investigated as possible treatments of noneosinophilic inflammation in asthma (Table 3 and Figure 2).
Table 3.
Treatments targeting noneosinophilic airway inflammation in asthma.
| Nonpharmacological interventions |
| Avoidance from exposure to environmental and occupational pollutants |
| Smoking cessation |
| Dietary supplementation with vitamin D3 |
| ‘Off-label’ use of licensed drugs |
| Macrolides |
| Statins |
| Low-dose theophylline |
| Peroxisome proliferator-activated receptor-γ (PPARγ) agonists |
| Novel small molecule drugs |
| Drugs targeting neutrophilic inflammation |
| C-X-C chemokine receptor (CXCR)2 antagonists |
| 5-lipoxygenase-activating protein (FLAP) inhibitors |
| Phosphodiesterase (PDE) inhibitors |
| PDE4 inhibitors |
| Dual PDE3 and PDE4 inhibitors |
| Protein kinase inhibitors |
| p38 mitogen-activated protein kinase (MAPK) inhibitors |
| Narrow spectrum kinase inhibitors |
| Tyrosine kinase inhibitors |
| Phosphoinositide 3 (PI3)-kinase inhibitors |
| PI3K-δ inhibitors |
| Dual PI3Kδ/γ inhibitors |
| Biological agents |
| Interleukin (IL)-17A receptor blockers |
| IL-17A blockers |
| Tumor necrosis factor (TNF)-α receptor blockers |
| IL-1β monoclonal antibody blockers |
| Soluble IL-1 receptor monoclonal antibody blockers |
| IL-6 monoclonal antibody blockers |
Figure 2.

Targets and potential therapies for treating noneosinophilic airway inflammation in asthma.
Noneosinophilic airway inflammation is found in approximately 50% of patients with asthma. The proportion of this group with neutrophilic inflammation is less certain because of variable cut-off points used to define neutrophilia. The higher the cut-off value used to define sputum neutrophilia the greater the proportion of patients that are classified as having paucigranulocytic inflammation. Pathways that may account for poor asthma control in patients with noneosinophilic asthma including neutrophilic inflammation, associated inflammatory phenotypes (Th1-high inflammation, Th17-high inflammation, a combination of Th2 and Th17 inflammation, mast cell-induced inflammation, and other inflammatory mechanisms) as well as corticosteroid insensitivity. Noninflammatory mechanisms such as airway hyperreactivity and airway remodelling may be important in causing symptoms in some individuals. Potential treatments targeting specific pathways are listed in the diagram. Novel small molecules targeting neutrophilic inflammation, such as CXCR2 antagonists reduce neutrophils, but do not improve clinical outcomes. Smoking cessation in asthmatic smokers and removal from exposure to occupational agents reduces neutrophilic inflammation. The results of clinical trials of biological agents targeting mediators associated with noneosinophilic inflammation, such as IL-17 and TNF-α are disappointing. Preliminary studies of ‘off-label’ use of licensed drugs suggest that macrolides show efficacy in nonsmokers with noneosinophilic severe asthma and statins, low-dose theophylline and PPARγ agonists may benefit asthmatic smokers with noneosinophilic inflammation and associated corticosteroid insensitivity. Inhaled PDE4 inhibitors, dual PDE3 and PDE4 inhibitors, p38MAPK inhibitors, tyrosine kinase inhibitors and PI3kinase inhibitors are under development and these compounds may be of benefit in treating noneosinophilic inflammation and corticosteroid insensitivity. Long-acting bronchodilators or bronchial thermoplasty are possible treatment options for symptomatic patients with paucigranulocytic inflammation in whom there is no evidence of activated inflammatory pathways or corticosteroid insensitivity that could be targeted by specific therapies.
Abbreviations: CXCR, C-X-C chemokine receptor; FLAP, 5-lipoxygenase-activating protein; IL, interleukin; PDE, phosphodiesterase; PI3K, phosphoinositide 3-kinase; PPARγ,: peroxisome proliferator-activated receptor-γ.
Nonpharmacological interventions
Avoidance from exposure to environmental and occupational pollutants may reduce neutrophilic inflammation in asthma. In a study of smokers with asthma, of which a subgroup quit smoking for 6 weeks, the proportion of sputum neutrophils reduced, corticosteroid sensitivity improved and the FEV1 increased compared to those who continued to smoke [Chaudhuri et al. 2006]. After cessation of exposure to occupational agents, neutrophilic inflammation reduced in people where their asthma was cured or improved compared to those where there was no improvement [Maghni et al. 2004].
Several clinical trials have examined the effect of dietary supplements of vitamin D in asthma, based on the anti-inflammatory and corticosteroid-enhancing actions of vitamin D [Nanzer et al. 2013; Zhang et al. 2014; Xystrakis et al. 2006]. Two large randomized clinical trials of vitamin D3 supplementation in patients with asthma and vitamin D insufficiency [VIDA and ViDiAs trials), although not selected for specific airway inflammatory cell profiles or corticosteroid insensitivity, reported no improvement in clinical outcomes [Castro et al. 2014; Martineau et al. 2015; Denlinger et al. 2015 ]. Interestingly, vitamin D supplementation reduces eosinophilic inflammation in patients with nonatopic asthma, suggesting that certain inflammatory phenotypes might benefit from vitamin D3 supplementation [De Groot et al. 2015].
‘Off-label’ use of licensed drugs
Several drugs licensed for the treatment of medical conditions other than asthma have been investigated for their efficacy in asthma, including patients with noneosinophilic inflammation. Candidate drugs have been chosen usually because of preclinical evidence of anti-inflammatory effects that might be relevant to treatment of asthma. Some examples are reviewed below.
Macrolides
Macrolides may be of benefit in the treatment of chronic asthma [Reiter et al. 2013], including noneosinophilic asthma [Simpson et al. 2008], although prescribing macrolides as a long-term treatment increases the risk of adverse drug effects and the development of microbial resistance [Cameron et al. 2012]. The mechanism of action of macrolides in the treatment of airway diseases is not known, but could be due to antibacterial or anti-inflammatory actions, which include inhibition of nuclear factor (NF)-κB and other transcription factors as well as reduction in neutrophil migration or function [Culic et al. 2001; Fujitani and Trifilieff, 2003; Simpson et al. 2008; Cameron et al. 2012; Kobayashi et al. 2013]. Macrolides have additional potentially beneficial properties including antiviral actions [Gielen et al. 2010; Schögler et al. 2015] and an ability to restore corticosteroid sensitivity by inhibiting the phosphoinositide 3-kinase (PI3K) pathway and restoring histone deacetylase (HDAC)2 activity [Spahn et al. 2001; Kobayashi et al. 2013; Hao et al. 2015] and by attenuating TNFα and IL-17 immune responses [Essilfie et al. 2015]. Two recent exploratory clinical trials have investigated the effects of macrolides in noneosinophilic asthma. In one trial, smokers with mild to moderate asthma associated with noneosinophilic inflammation were randomized to azithromycin 250 mg per day or placebo [Cameron et al. 2013]. After 12 weeks, treatment with azithromycin was not associated with improvements in morning peak expiratory flow (PEF), ACQ score, asthma quality of life questionnaire (AQLQ) score and methacholine PC20 compared to placebo and did not alter induced sputum differential counts, bacterial load, C. pneumonia, M. pneumoniae seropositivity or upper airway respiratory virus prevalence. In another randomized controlled trial patients with exacerbation-prone severe asthma received low-dose azithromycin or placebo as an add-on treatment to combination therapy of inhaled corticosteroids and long-acting β2 agonists for 6 months [Brusselle et al. 2013]. The rate of severe exacerbations and lower respiratory tract infections requiring treatment with antibiotics was not reduced by azithromycin. In a predefined subgroup with noneosinophilic severe asthma (blood eosinophilia ⩽200/µl) there was a reduction in the rate of primary endpoints in azithromycin-treated patients [Brusselle et al. 2013]. Azithromycin improved AQLQ scores, but did change ACQ scores or lung function. Based on these findings, further clinical trials of macrolides in noneosinophilic severe asthma are indicated. Novel analogues of macrolides have been developed that have enhanced anti-inflammatory properties than current macrolides, such as solithromycin (CEM-101) [Kobayashi et al. 2013a; Kobayashi et al. 2013b] or that lack antibacterial properties, such as the nonantibiotic azithromycin derivative CSY0073 [Balloy et al. 2014].
Statins
Statins have pleiotropic immunomodulatory actions [Greenwood et al. 2006] that may be of value in the treatment of chronic inflammatory diseases [Greenwood et al. 2006; Hothersall et al. 2006; Yeganeh et al. 2014]. In experimental models of allergic asthma [McKay et al. 2004; Zeki et al. 2009] and tobacco-smoke-induced lung inflammation [Lee et al. 2005; Davis et al. 2013] statins reduce inflammatory pathways potentially relevant to the pathogenesis of asthma and smoke-induced airway diseases. Statins might also restore corticosteroid sensitivity in asthma [Samson et al. 2006; Maneechotesuwan et al. 2010]. Taken together, these findings suggest that statin treatment may have anti-inflammatory effects in people with asthma including smokers with asthma. A randomized double-blind parallel group trial undertaken in 71 smokers with mild to moderate asthma associated with noneosinophilic inflammation compared treatment with atorvastatin 40 mg per day with placebo. After 4 weeks of treatment, inhaled beclometasone at 400 μg per day was added to both treatment arms for a further 4 weeks [Braganza et al. 2011]. At 4 weeks, there was an improvement in ACQ and AQLQ scores with atorvastatin, but not in lung function. There was no significant improvement with atorvastatin and inhaled beclometasone compared to inhaled beclometasone alone in clinical outcome measures at 8 weeks. In a follow-up study the effects of atorvastatin alone and in combination with inhaled corticosteroid was investigated on their ability to suppress the concentration of a range of cytokines, chemokines and growth factors in sputum samples collected during the previous clinical trial [Braganza et al. 2011; Thomson et al. 2015]. Sputum mediator concentrations were not reduced by inhaled beclometasone alone. Atorvastatin significantly reduced sputum concentrations of CCL7, IL-12p70, soluble CD40 ligand (sCD40L), fibroblast growth factor (FGF)-2, CCL4, transforming growth factor alpha (TGF-α) and matrix metalloproteinase (MMP)-8 compared with placebo and, when combined with inhaled beclometasone, reduced sputum concentrations of MMP-8, IL-1β, IL-10, MMP-9, sCD40L, FGF-2, IL-7, granulocyte-colony stimulating factor (G-CSF) and CCL7 compared to ICS alone. Improvements in ACQ or AQLQ scores with atorvastatin and inhaled beclometasone were associated with decreases in G-CSF, IL-7, CCL2 and chemokine (C-X-C motif) ligand (CXCL)8. Interestingly, simvastatin suppresses airway IL-17 and upregulated IL-10 in patients with stable chronic obstructive pulmonary disease (COPD) [Maneechotesuwan et al. 2013]. Taken together, these findings suggest that short-term treatment with atorvastatin alone or in combination with inhaled beclometasone reduces several sputum cytokines, chemokines and growth factor concentrations unresponsive to inhaled corticosteroids alone in asthmatic smokers with noneosinophilic inflammation. There is a need for long-term clinical studies examining effect of statins on exacerbations and airway remodelling in chronic noneosinophilic asthma.
Low-dose theophylline
Low dose theophylline has been shown to restore corticosteroid sensitivity in vitro possibly by increasing HDAC-2 activity, which is suppressed in severe asthma and in smokers with asthma and a similar clinical effect might occur in people with severe disease or who are smokers [Barnes, 2009; To et al. 2010]. Theophylline inhibits oxidative stress-dependent PI3K-δ activation and restores corticosteroid sensitivity in peripheral blood mononuclear cells (PBMCs) from patients with COPD [To et al. 2010]. An exploratory clinical trial examined the effects of low dose theophylline added to inhaled beclometasone compared to inhaled beclometasone alone in smokers with asthma associated with noneosinophilic inflammation [Spears et al. 2009a]. The addition of low dose theophylline to inhaled beclometasone, at a dose titrated to provide a ‘subtherapeutic’ concentration, resulted in increased efficacy as measured by lung function and suggested the restoration of corticosteroid sensitivity in those treated with the combination. Clinical trials to date have not investigated the therapeutic effects of adding low-dose theophylline to patients with severe asthma. A fixed combination of ultra-low dose of theophylline with fluticasone proprionate, SKP-2075 (Skyepharma, London, UK), in a dry powder inhaler is under development for the treatment of COPD. This combination would potentially be of benefit in the treatment of severe asthma and smokers with asthma, possibly in those people with noneosinophilic inflammation.
PPARγ agonist
In preclinical studies peroxisome proliferator-activated receptor-γ (PPARγ) agonists exert anti-inflammatory effects potentially relevant to the treatment of inflammatory airway diseases including asthma and COPD [Spears et al. 2006; Belvisi and Hele, 2008; Seidel et al. 2012; Stephen et al. 2013; Bourke et al. 2014; Lakshmi et al. 2014; Lea et al. 2014; Donovan et al. 2015]. For example, PPARγ agonists reduce eosinophilic and neutrophilic lung infiltration in experimental animal models exposed to allergen or tobacco smoke [Bauer et al. 2010; Lea et al. 2014; Zhao et al. 2014; Morissette et al. 2015]. The oral PPARγ agonist rosiglitazone had a modest effect in attenuating the allergen-induced late asthmatic response [Richards et al. 2010]. A further proof-of-concept study reported that rosiglitazone compared with inhaled beclometasone dipropionate resulted in an improvement in lung function and a borderline reduction in sputum IL-8 concentration in smokers with mild to moderate asthma that was associated with noneosinophilic inflammation [Spears et al. 2009b]. The oral PPARγ agonist pioglitazone is not effective in obese asthmatics [Dixon et al. 2015]. Inhaled PPARγ agonist analogues are under development for the treatment of chronic inflammatory airway diseases and potentially might be of benefit in noneosinophilic asthma.
Novel small molecule drugs
Novel small molecule inhibitors have been developed for treating neutrophilic/noneosinophilic asthma including CXCR2 antagonists, 5-lipoxygenase-activating protein (FLAP) inhibitors, PDE4 inhibitors, dual PDE3 and PDE4 inhibitor and various protein kinase inhibitors.
CXCR2 antagonist
CXCR2 receptors are expressed on neutrophils as well as on airway goblet cells, fibroblasts and airway smooth muscle [Chapman et al. 2009]. Ligands for the CXCR2 receptor include the chemokines CXCL8 (IL-8), growth-related protein (Gro)-α, -β, and -γ (CXCL1–3), epithelial-derived neutrophil attractant-78 (ENA-78; CXCL5), granulocyte chemotactic protein-2 (GCP-2; CXCL6) and neutrophil-activating peptide-2 (NAP-2; CXCL7) [Chapman et al. 2009, Campbell et al. 2013]. Activation of CXCR2 receptors result in neutrophil chemotaxis, proteases production, aairway goblet cell hyperplasia, pulmonary blood vessel angiogenesis, collagen deposition and airway smooth muscle contraction and migration [Chapman et al. 2009]. The effects of CXCR2 antagonists have been studied on airway challenges that induce sputum neutrophilia. The CXCR2 antagonist, AZD8309 inhibits lipopolysaccharide (LPS)-induced airway neutrophilic inflammation in healthy volunteers [Leaker et al. 2013] and the CXCR2 antagonist, SB656933 inhibited ex vivo neutrophil activation and ozone-induced airway inflammation in humans [Lazaar et al. 2011]. The CXCR2 antagonist, SCH527123 inhibits ozone-induced neutrophilia in healthy subjects [Holz et al. 2010]. A randomized, placebo-controlled clinical trial of the CXCR2 antagonist SCH527123 administered for 4 weeks to patients with severe asthma and sputum neutrophils > 40% resulted in a reduction of 36.3% in sputum neutrophil percentage, fewer mild exacerbations and a trend towards improvement in ACQ score [Nair et al. 2012]. A clinical trial of the efficacy and safety of a CXCR2 antagonist AZD5069 in severe, uncontrolled persistent asthma reported that the addition of AZD5069 to combination ICS/long acting β2-agonist (LABA) treatment did not improve clinical outcomes despite a dose-dependent reduction in blood neutrophil counts [O’Byrne et al. 2015]. A lack of improvement in clinical outcome despite a reduction in sputum neutrophil counts was reported with the CXCR2 antagonist AZD5069 in bronchiectasis [De Soyza et al. 2015]. A recent trial of the CXCR2 antagonist Navarixin (SCH527123) in COPD led to significant improvements in FEV1 and reduction in sputum neutrophil count, particularly in current smokers with COPD [Rennard et al. 2015]. The CXCR2 antagonist AZD8309 administered for 4 weeks to patients with moderate to severe COPD was well tolerated with no increase in the rate of infections [Kirsten et al. 2015]. A small-molecule oral CXCR2 antagonists Danirixin (GSK1325756) is undergoing a clinical trial in patients with COPD at risk of exacerbations [ClinicalTrials.gov identifier: NCT02130193]. Oral CXCR2 antagonists could potentially cause neutropenia, and the therapeutic index of these compounds requires careful assessment.
FLAP inhibitors
Pro-inflammatory cysteinyl leukotrienes (LTs) are synthesized from arachidonic acid in inflammatory cells by 5-lipoxygenase (LO) and 5-lipoxygenase activating protein (FLAP). FLAP inhibitors such as GSK-2190915 [Evans et al. 2008] prevent the formation of LTB4, which may be of value in the treatment of neutrophilic asthma. GSK2190915 markedly inhibited ex vivo calcium ionophore stimulated blood LTB4 formation and urinary leukotriene E4 (LTE4) formation [Bain et al. 2013]. Pre-treatment with GSK2190915 reduces the early and late phase response to allergen challenge and results in a significant reduction of sputum LTB4 levels [Kent et al. 2013]. Despite suppressing the target mediator LTB4, the FLAP inhibitor GSK2190915 has no short-term effect on sputum cell counts or clinical endpoints in smokers and nonsmokers with asthma associated with neutrophilic inflammation (sputum neutrophilia ⩾ 50% for one sample and >45% for the other), suggesting that LTB4 suppression alone is inadequate in controlling airway neutrophils in asthma [Chaudhuri et al. 2014]. No active clinical trials of 5-lipoxygenase-activating protein (FLAP) inhibitors in asthma are currently registered on ClinicalTrials.gov.
PDE4 inhibitors and dual PDE3 and PDE4 inhibitors
Phosphodiesterase (PDE)4 inhibitors have immunomodulatory effects on inflammatory cells potentially relevant to the treatment of asthma [Lipworth, 2005; Page and Spina, 2012; Kim et al. 2015b]. In an allergen challenge study the oral PDE4 inhibitor roflumilast attenuated the rise in sputum eosinophils and neutrophil numbers after the late asthmatic response [Gauvreau et al. 2011]. High doses of PDE4 inhibitors may be necessary to treat severe asthma, and gastrointestinal side effects limit their use [Lipworth, 2005; Bateman et al. 2006; Bousquet et al. 2006]. The inhaled administration of PDE4 inhibitors may improve the therapeutic index of PDE4 inhibitors [Chapman et al. 2010; Singh et al. 2010; Nials et al. 2011; De Savi et al. 2014; Moretto et al. 2015]. Inhaled PDE4 inhibitors GSK256066 and CHF6001 both inhibit allergen-induced late asthmatic responses [Singh et al. 2010; Dave et al. 2014] and in patients with moderate COPD, GSK256066 inhaled for 4 weeks was well tolerated although there was no inhibitory effect on sputum and blood inflammatory biomarkers [Watz et al. 2013]. The inhaled dual PDE3 and PDE4 inhibitor RPL554 (Verona Pharma, London, UK) has bronchodilator effects and is well tolerated in patients with asthma and COPD [Franciosi et al. 2013]. In healthy subjects, inhaled RPL554 attenuates the neutrophilic response to lipopolysaccharide (LPS) challenge [Franciosi et al. 2013]. RPL554 is under development for the treatment of asthma and COPD [ClinicalTrials.gov identifiers: NCT02427165, NCT02542254].
Protein kinase inhibitors
Protein kinases are involved in cellular signalling of pro-inflammatory cytokines in asthma and the inhibition of these kinases may have a role in the treatment of severe asthma associated with noneosinophilic asthma [Bhavsar et al. 2010; Cohen and Fleischmann, 2010; Hammaker and Firestein, 2010; Chung, 2011; Guntur and Reinero, 2012]. Several p38MAPK inhibitors restore corticosteroid sensitivity in PBMCs from patients with severe asthma [Bhavsar et al. 2010; Mercado et al. 2012] and COPD [Khorasani et al. 2015]. Clinical trials of p38MAPK inhibitors oral losmapimod (GW856553) (GlaxoSmithKline, London, UK), and inhaled AZD7624 are registered for the treatment of COPD [ClinicalTrials.gov identifiers: NCT02299375, NCT02238483], although neither are registered for the treatment of asthma. Interestingly, a post-hoc analysis of a 6-month clinical trial of oral losmapimod (GW856553) in COPD reported a reduction in exacerbations in a subgroup of patients with a blood eosinophil count ⩽2% [Marks-Konczalik et al. 2015], which may suggest a preferentially beneficial effect of p38MAPK inhibitors in noneosinophilic inflammation. Imatinib, a specific c-kit tyrosine kinase inhibitor that attenuates airway hyperresponsiveness, inflammation and remodelling in a murine model of asthma [Berlin and Lukacs, 2005; Rhee et al. 2011] is under development for patients with severe refractory asthma [ClinicalTrials.gov identifier: NCT01097694]. A tyrosine kinase inhibitor masitinib targets c-kit and platelet-derived growth factor (PDGF) receptor improved asthma control in patients with severe corticosteroid-dependent asthma [Humbert et al. 2009] and a further clinical trial is underway in patients with severe asthma treated with oral corticosteroids [ClinicalTrials.gov identifier: NCT01449162]. An alternative therapeutic strategy to silencing c-kit with small interference ribonucleic acid (RNA) has been shown to attenuate inflammation in a murine model of allergic asthma [Wu et al. 2012; Wu et al. 2014]. Clinical trials of protein kinase inhibitors have not been studied in patients with sputum inflammatory subtypes such as noneosinophilic asthma.
PI3kinase inhibitors
Low dose theophylline is thought to act, at least in part, through the inhibition of PI3K [Ito et al. 2007; To et al. 2010]. Preclinical studies suggest that PI3K-δ inhibitors could potentially reverse corticosteroid insensitivity by increasing HDAC2 activity [Marwick et al. 2009; Marwick et al. 2010] and by reversing fungal-induced steroid resistant airway inflammation through modulation of endoplasmic reticulum stress [Lee et al. 2016]. Selective PI3K inhibitors are being developed as novel therapies for the treatment of chronic inflammatory airway diseases. An inhaled PI3Kδ inhibitor GSK2269557 is undergoing several clinical trials in asthma and COPD. PI3K δ and γ isoforms are involved in inflammatory cell recruitment and activation and dual PI3Kδ/γ inhibitors, such as TG100-115 and IPI-145 reduces airway inflammation induced by allergen or cigarette smoke in murine models [Doukas et al. 2009; Winkler et al. 2013] and restored corticosteroid sensitivity in the smoke model [Doukas et al. 2009]. RV1729, a PI3Kδ/γ inhibitor has undergone early stage clinical evaluation in asthma and COPD. SH2-containing inositol-50-phosphatase 1 (SHIP1) is an endogenous inhibitor of the PI3K pathway. A SPIP1 activator AQX-1125 reduced the allergen-induced late asthmatic response with a non-significant trend for a reduction in sputum eosinophils and neutrophils [Leaker et al. 2014]. Further development of AQX-1125 is underway for the treatment of COPD [ClinicalTrials.gov identifier: NCT01954628].
Biological agents
Monoclonal antibody blockers of inflammatory cytokines such as IL-17 and TNF-α that activate receptors on the surface of neutrophils have been investigated as treatments for asthma.
IL-17 blockers
In preclinical studies Th17 cells and IL-17 are implicated in causing neutrophilic inflammation and corticosteroid insensitivity [Shen et al. 2011; Newcomb and Peebles, 2013; Chesné et al. 2014]. IL-17 concentrations and expression are increased in BAL, sputum and bronchial biopsy samples from patients with severe asthma that correlate with sputum neutrophils. Monoclonal inhibitors of IL-17 are in clinical development [Miossec and Kolls, 2012]. Brodalumab is a human monoclonal antibody that binds with high affinity to human IL-17RA, blocking the biologic activity of IL-17A, IL-17F, IL-17A/F heterodimer, and IL-25. A randomized clinical trial of brodalumab in adults with inadequately controlled moderate to severe asthma receiving regular inhaled corticosteroids, but not selected for neutrophilic inflammation, reported no improvement in the primary outcome ACQ score or in lung function and symptom-free days [Busse et al. 2013]. A subgroup with high bronchodilator reversibility demonstrated a borderline improvement in ACQ score. A further clinical trial of brodalumab in inadequately controlled asthma patients with high bronchodilator reversibility was recently terminated due to a lack of observed efficacy in a pre-specified interim analysis [ClinicalTrials.gov identifier: NCT01902290]. The results of a preliminary proof of efficacy study of the IL-17A monoclonal antibody blocker secukinumab (AIN457) in patients with uncontrolled asthma was also recently terminated. The investigators report that further investigations would require changes in study design, the use of different endpoints, a different IL-17 antibody or a different patient population [ClinicalTrials.gov identifier: NCT01478360].
TNF-α blockers
Neutralizing TNFα restores corticosteroid sensitivity in a mouse model of neutrophilic airway inflammation [Dejager et al. 2015]. Several small clinical studies in severe asthma of the soluble TNF-α receptor blocker etanercept reported beneficial effects on clinical outcomes [Howarth et al. 2005; Berry et al. 2006], whereas larger studies with etanercept [Holgate et al. 2011] and the TNF-α receptor blocker golimumab [Wenzel et al. 2009] did not confirm a consistent beneficial clinical effect. When combined with concerns over increased risk of severe infections and malignancies with TNF-α receptor blocker treatment [Wenzel et al. 2009] it is unlikely that this target will be developed further for the treatment of asthma.
Other monoclonal antibodies
Monoclonal antibodies that block IL-1β, for example, canakinumab or block the soluble IL-1 receptor, for example, anakinra [Hernandez et al. 2015] might be of benefit in neutrophilic asthma, although no clinical studies are currently registered. An IL-6 monoclonal antibody blocker toclizumab is licensed for the treatment of rheumatoid arthritis. Toclizumab could potentially be of benefit in neutrophilic asthma although no clinical studies are registered in asthma.
Conclusions and future developments
Noneosinophilic airway inflammation is a term used to describe a subtype of asthma associated with normal numbers of sputum eosinophils. Up to 50% of never smokers or exsmoker patients with stable, mild to severe asthma have noneosinophilic inflammation and this inflammatory phenotype is also found in smokers with asthma, and some patients with a high BMI or occupational asthma. The noneosinophilic phenotype is subdivided into neutrophilic inflammation, when neutrophil numbers are raised above a defined cut-off level or paucigranulocytic inflammation, when both eosinophil and neutrophil numbers are normal. The relative proportion of each subtype is uncertain because of variable cut-off points used to define neutrophilia. The most appropriate value that indicates that neutrophils are activated and contributing to the pathogenic processes in asthma is not certain. The severity of current symptoms are in general similar or slightly better in noneosinophilic or neutrophilic subgroups of asthma compared to eosinophilic subgroups. Sputum eosinophilia is a better predictor of future exacerbations and a greater risk factor for more rapid decline in lung function than sputum neutrophilia. Noneosinophilic inflammation is associated with an impaired therapeutic response to inhaled corticosteroids. Neutrophilic inflammation is associated with activation of the innate immune system in asthma and systemic inflammation. Several mechanisms either alone or in combination could explain raised sputum neutrophil counts in asthma including corticosteroids, associated chronic sinopulmonary infection, delayed human neutrophil apoptosis due to epithelial growth factor, impaired macrophage phagocytosis and an altered airway microbiome. Limited information has been published on the immunopathological characteristics of noneosinophilic inflammation compared with other inflammatory airway phenotypes including Th2-low inflammation, Th17-high inflammation or a combination of Th2/Th17 profiles in asthma. Taken together, the finding suggest that noneosinophilic inflammation and Th2-low inflammation in nonsmokers with asthma share some similar immunopathological features including normal eosinophil numbers, submucosal mast cell numbers and subepithelial basement membrane thickness. Due to the lack of effective specific therapies targeting noneosinophilic inflammation, including neutrophilic inflammation, there is currently no definitive evidence for the involvement of these inflammatory phenotypes in chronic asthma. Additional pathways may account for poor asthma control in patients with noneosinophilic asthma including Th1 inflammation, Th17 inflammation, or a combination of Th2 and Th17 inflammation, as well as corticosteroid insensitivity (Figure 2). The role of corticosteroid treatment in causing neutrophilic and Th17 inflammation in severe asthma requires further investigation. Noninflammatory mechanisms may also be important in some individuals including airway hyperreactivity and airway remodelling.
There is an unmet need for novel treatments that will impact favourably on clinical outcomes in patients with noneosinophilic inflammation. Nonpharmacological interventions, ‘off-label’ use of licensed drugs, novel small molecules and biologics agents are being investigated as possible treatments of noneosinophilic inflammation in asthma. Smoking cessation in smokers with asthma and cessation of exposure to occupational agents are associated with a reduction in neutrophilic inflammation. Preliminary data of studies of ‘off-label’ use of licensed drugs suggest that macrolides show efficacy in nonsmokers with noneosinophilic asthma and statins, low-dose theophylline and PPARγ agonists may be beneficial in asthmatic smokers with noneosinophilic inflammation and corticosteroid insensitivity. Further clinical studies are indicated to confirm these findings and to determine the role of these therapies in the management of severe asthma. Novel small molecules targeting neutrophilic inflammation in asthma such as CXCR2 antagonists reduce neutrophil counts, but do not improve clinical outcomes. A FLAP inhibitor did not reduce neutrophils or improve symptoms. Inhaled PDE4 inhibitors and dual PDE3 and PDE4 inhibitors are potential therapies for neutrophilic asthma and a dual PDE3 and PDE4 inhibitors is under development for the treatment of asthma and COPD. Additional small molecule drugs including p38MAPK inhibitors, tyrosine kinase inhibitors and PI3kinase inhibitors are under development for asthma. The development of biological agents to target noneosinophilic inflammation in asthma has been disappointing to date with the termination of clinical programmes of monoclonal antibodies targeting IL-17 and TNF-α. In the future, the selection of patients with severe asthma and evidence of Th17 high inflammation may be more likely to identify a subpopulation that respond to IL-17 blockers. Long-acting bronchodilators or bronchial thermoplasty are possible treatment options for symptomatic patients with paucigranulocytic inflammation in whom there is no evidence of activated inflammatory pathways or corticosteroid insensitivity that could be targeted by specific therapies.
Greater understanding of the mechanisms of noneosinophilic inflammation in asthma should lead to improved therapies. International collaborative programmes of research investigating pathogenic mechanism of severe asthma have focused mainly on type 2 eosinophilic inflammation. The Unbiased Biomarkers for the Prediction of Respiratory Disease Outcome (U-BOPRED) study [Gaga et al. 2015; Shaw et al. 2015] and the UK Refractory Asthma Stratification Programme (RASP-UK) [Heaney et al. 2015 ] are designed to identify new phenotypes/endotypes and treatment targets and will hopefully identify new approaches for the treatment of patients with noneosinophilic asthma.
Footnotes
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: In the last three years Professor Thomson has participated in advisory boards or received consultancy/lecture fees from Boston Scientific, Genentech, GlaxoSmithKline, Novartis, Respivert, Roche and Takeda and industry-sponsored grant funding to the University of Glasgow from Boston Scientific, Glaxo SmithKline and Novartis for participating in clinical trials.
References
- Agustí A., Antó J., Auffray C., Barbé F., Barreiro E., Dorca J., et al. (2015) Personalized respiratory medicine: exploring the horizon, addressing the issues. Summary of a BRN-AJRCCM Workshop Held in Barcelona on June 12, 2014. Am J Resp Crit Care Med 191: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Samri M., Benedetti A., Préfontaine D., Olivenstein R., Lemière C., Nair P., et al. (2010) Variability of sputum inflammatory cells in asthmatic patients receiving corticosteroid therapy: a prospective study using multiple samples. J Allergy Clin Immunol 125: 1161–1163. [DOI] [PubMed] [Google Scholar]
- Anees W., Huggins V., Pavord I., Robertson A., Burge P. (2002) Occupational asthma due to low molecular weight agents: eosinophilic and noneosinophilic variants. Thorax 57: 231–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arron J., Choy D., Laviolette M., Kelsen S., Hatab A., Leigh R., et al. (2014) Disconnect between sputum neutrophils and other measures of airway inflammation in asthma. Eur Respir J 43: 627–629. [DOI] [PubMed] [Google Scholar]
- Arron J., Scheerens H., Matthews J., David R. (2013) Redefining approaches to asthma: developing targeted biologic therapies. Adv Pharmacol - Immunopharmacol 66: 1–49. [DOI] [PubMed] [Google Scholar]
- Bacci E., Cianchetti S., Bartoli M., Dente F., Di Franco A., Vagaggini B., et al. (2006) Low sputum eosinophils predict the lack of response to beclomethasone in symptomatic asthmatic patients. Chest 129: 565–572. [DOI] [PubMed] [Google Scholar]
- Bacci E., Latorre M., Cianchetti S., Bartoli M., Costa F., Di Franco A., et al. (2012) Transient sputum eosinophilia may occur over time in noneosinophilic asthma and this is not prevented by salmeterol. Respirology 17: 1199–1206. [DOI] [PubMed] [Google Scholar]
- Bain G., King C., Schaab K., Rewolinski M., Norris V., Ambery C., et al. (2013) Pharmacodynamics, pharmacokinetics and safety of GSK2190915, a novel oral anti-inflammatory 5-lipoxygenase-activating protein inhibitor. Br J Clin Pharmacol 75: 779–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines K., Simpson J., Bowden N., Scott R., Gibson P. (2010) Differential gene expression and cytokine production from neutrophils in asthma phenotypes. Eur Respir J 35: 522–531. [DOI] [PubMed] [Google Scholar]
- Baines K., Simpson J., Wood L., Scott R., Fibbens N., Powell H., et al. (2014) Sputum gene expression signature of 6 biomarkers discriminates asthma inflammatory phenotypes. J Allergy Clin Immunol 133: 997–1007. [DOI] [PubMed] [Google Scholar]
- Baines K., Simpson J., Wood L., Scott R., Gibson P. (2011) Systemic upregulation of neutrophil alpha–defensins and serine proteases in neutrophilic asthma. Thorax 66: 942–947. [DOI] [PubMed] [Google Scholar]
- Balloy V., Deveaux A., Lebeaux D., Tabary O., Le Rouzic P., Ghigo J., et al. (2014) Azithromycin analogue CSY0073 attenuates lung inflammation induced by LPS challenge. Br J Pharmacol 171: 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes P. (2009) Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 71: 451–464. [DOI] [PubMed] [Google Scholar]
- Bateman E., Izquierdo J., Harnest U., Hofbauer P., Magyar P., Schmid-Wirlitsch C., et al. (2006) Efficacy and safety of roflumilast in the treatment of asthma. Ann Allergy Asthma Immunol 96: 679–686. [DOI] [PubMed] [Google Scholar]
- Bauer C., Zavitz C., Botelho F., Lambert K., Brown E., Mossman K., et al. (2010) Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection. PLoS ONE 5: e13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bel E., Wenzel S., Thompson P., Prazma C., Keene O., Yancey S., et al. (2014) Oral glucocorticoid-sparing effect of mepolizumab in eosinophilic asthma. N Eng J Med 371: 1189–1197. [DOI] [PubMed] [Google Scholar]
- Belvisi M., Hele D. (2008) Peroxisome proliferator-activated receptors as novel targets in lung disease. Chest 134: 152–157. [DOI] [PubMed] [Google Scholar]
- Berlin A., Lukacs N. (2005) Treatment of cockroach allergen asthma model with imatinib attenuates airway responses. Am J Respir Crit Care Med 171: 35–39. [DOI] [PubMed] [Google Scholar]
- Berry M., Hargadon B., Shelley M., Parker D., Shaw D., Green R., et al. (2006) Inhibition of tumor necrosis factor α for refractory asthma. N Eng J Med 354: 754–758. [DOI] [PubMed] [Google Scholar]
- Berry M., Morgan A., Shaw D., Parker D., Green R., Brightling C., et al. (2007) Pathological features and inhaled corticosteroid response of eosinophilic and noneosinophilic asthma. Thorax 62: 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar P., Khorasani N., Hew M., Johnson M., Chung K. (2010) Effect of P38 Mapk inhibition on corticosteroid suppression of cytokine release in severe asthma. Eur Respir J 35: 750–756. [DOI] [PubMed] [Google Scholar]
- Boulet L., Lemiere C., Archambault F., Carrier G., Descary M., Deschesnes F. (2006) Smoking and asthma: clinical and radiologic features, lung function, and airway inflammation. Chest 129: 661–668. [DOI] [PubMed] [Google Scholar]
- Bourke J., Bai Y., Donovan C., Esposito J., Tan X., Sanderson M. (2014) Novel small airway bronchodilator responses to rosiglitazone in mouse lung slices. Am J Respir Cell Mol Biol 50: 748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet J., Aubier M., Sastre J., Izquierdo J., Adler L., Hofbauer P., et al. (2006) Comparison of roflumilast, an oral anti-inflammatory, with beclomethasone dipropionate in the treatment of persistent asthma. Allergy 61: 72–78. [DOI] [PubMed] [Google Scholar]
- Braganza G., Chaudhuri R., Mcsharry C., Weir C., Donnelly I., Jolly L., et al. (2011) Effects of short-term treatment with atorvastatin in smokers with asthma – a randomized controlled trial. BMC Pulm Med 11: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks C., Gibson P., Douwes J., Van Dalen C., Simpson J. (2013) Relationship between airway neutrophilia and ageing in asthmatics and non-asthmatics. Respirology 18: 857–865. [DOI] [PubMed] [Google Scholar]
- Brooks C., Van Dalen C., Zacharasiewicz A., Simpson J., Harper J., Le Gros G., et al. (2016) Absence of airway inflammation in a large proportion of adolescents with asthma. Respirology December 22. doi: 10.1111/resp.12701. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Brusselle G., Vanderstichele C., Jordens P., Deman R., Slabbynck H., Ringoet V., et al. (2013) Azithromycin for prevention of exacerbations in severe asthma (AZISAST): a multicentre randomised double-blind placebo-controlled trial. Thorax 68: 322–329. [DOI] [PubMed] [Google Scholar]
- Busse W., Holgate S., Kerwin E., Chon Y., Feng J., Lin J., et al. (2013) Randomized, double-blind, placebo-controlled study of brodalumab, a human anti-L-17 receptor monoclonal antibody, in moderate to severe asthma. Am J Respir Crit Care Med 188: 1294–1302. [DOI] [PubMed] [Google Scholar]
- Cameron E., Chaudhuri R., Mair F., Mcsharry C., Greenlaw N., Weir C., et al. (2013) Randomised controlled trial of azithromycin in smokers with asthma. Eur Respir J 42: 1412–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron E., Mcsharry C., Chaudhuri R., Farrow S., Thomson N. (2012) Long-term macrolide treatment of chronic inflammatory airway diseases: risks, benefits and future developments. Clin Exp Allergy 42: 1302–1312. [DOI] [PubMed] [Google Scholar]
- Campbell L., Maxwell P., Waugh D. (2013) Rationale and means to target pro-inflammatory interleukin-8 (CXCL8) signaling in cancer. Pharmaceuticals 6: 929–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro M., King T., Kunselman S., Hünnemeyer A., Lahu G., Templin S., et al. (2014) Effect of vitamin D3 on asthma treatment failures in adults with symptomatic asthma and lower vitamin D levels: the VIDA randomized clinical trial. JAMA 311(20): 2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers G., Macleod K., Little S., Thomson L., Mcsharry C., Thomson N. (2002) Influence of cigarette smoking on inhaled corticosteroid treatment in mild asthma. Thorax 57: 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman R., House A., Richard J., Prelusky D., Lamca J., Wang P., et al. (2010) Pharmacology of a potent and selective inhibitor of PDE4 for inhaled administration. Eur J Pharmacol 643: 274–281. [DOI] [PubMed] [Google Scholar]
- Chapman R., Phillips J., Hipkin R., Curran A., Lundell D., Fine J. (2009) CXCR2 antagonists for the treatment of pulmonary disease. Pharmacol Ther 121: 55–68. [DOI] [PubMed] [Google Scholar]
- Chaudhuri R., Livingston E., Mcmahon A., Lafferty J., Fraser I., Spears M., et al. (2006) Effects of smoking cessation on lung function and airway inflammation in smokers with asthma. Am J Respir Crit Care Med 174: 127–133. [DOI] [PubMed] [Google Scholar]
- Chaudhuri R., Norris V., Kelly K., Zhu C., Ambery C., Lafferty J., et al. (2014) Effects of a flap inhibitor, GSK2190915, in asthmatics with high sputum neutrophils. Pulm Pharmacol Ther 27: 62–69. [DOI] [PubMed] [Google Scholar]
- Chesné J., Braza F., Mahay G., Brouard S., Aronica M., Magnan A. (2014) IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med 190: 1094–1101. [DOI] [PubMed] [Google Scholar]
- Choy D., Hart K., Borthwick L., Shikotra A., Nagarkar D., Siddiqui S., et al. (2015) Th2 and Th17 inflammatory pathways are reciprocally regulated in asthma. Sci Transl Med 7: 301ra129–301ra129. [DOI] [PubMed] [Google Scholar]
- Chung K. (2011) P38 mitogen-activated protein kinase pathways in asthma and COPD. Chest 139: 1470–1479. [DOI] [PubMed] [Google Scholar]
- Cohen S., Fleischmann R. (2010) Kinase inhibitors: a new approach to rheumatoid arthritis treatment. Curr Opin Rheumatol 22: 330–335 [DOI] [PubMed] [Google Scholar]
- Corren J., Lemanske R., Hanania N., Korenblat P., Parsey M., Arron J., et al. (2011) Lebrikizumab treatment in adults with asthma. N Eng J Med 365: 1088–1098. [DOI] [PubMed] [Google Scholar]
- Cowan D., Cowan J., Palmay R., Williamson A., Taylor D. (2010) Effects of steroid therapy on inflammatory cell subtypes in asthma. Thorax 65: 384–390. [DOI] [PubMed] [Google Scholar]
- Cox G. (1995) Glucocorticoid treatment inhibits apoptosis in human neutrophils. separation of survival and activation outcomes. J Immunol 154: 4719–4725. [PubMed] [Google Scholar]
- Culic O., Erakovic V., Parnham M. (2001) Anti-inflammatory effects of macrolide antibiotics. Eur J Pharmacol 429: 209–229. [DOI] [PubMed] [Google Scholar]
- D’Silva L., Cook R., Allen C., Hargreave F., Parameswaran K. (2007) Changing pattern of sputum cell counts during successive exacerbations of airway disease. Respir Med 101: 2217–2220. [DOI] [PubMed] [Google Scholar]
- Dave S., Brian R., Malcom B., Marie A., Sara C., Fabrizia M., et al. (2014) The effect of the inhaled PDE4 inhibitor CHF6001 on allergen-induced inflammation in asthmatic subjects. Am J Resp Crit Care Med: A4175–A4175. [Google Scholar]
- Davis B., Zeki A., Bratt J., Wang L., Filosto S., Walby W., et al. (2013) Simvastatin inhibits smoke-induced airway epithelial injury: implications for COPD therapy. Eur Respir J 42: 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot J., Van Roon E., Storm H., Veeger N., Zwinderman A., Hiemstra P., et al. (2015) Vitamin D reduces eosinophilic airway inflammation in nonatopic asthma. J Allergy Clin Immunol 135: 670–675. [DOI] [PubMed] [Google Scholar]
- De Savi C., Cox R., Warner D., Cook A., Dickinson M., Mcdonough A., et al. (2014) Efficacious inhaled PDE4 inhibitors with low emetic potential and long duration of action for the treatment of COPD. J Med Chem 57: 4661–4676. [DOI] [PubMed] [Google Scholar]
- De Soyza A., Pavord I., Elborn J., Smith D., Wray H., Puu M., et al. (2015) A randomised, placebo-controlled study of the CXCR2 antagonist AZD5069 in bronchiectasis. Eur Respir J 46: 1021–1032. [DOI] [PubMed] [Google Scholar]
- Dejager L., Dendoncker K., Eggermont M., Souffriau J., Van Hauwermeiren F., Willart M., et al. (2015) Neutralizing TNF[alpha] restores glucocorticoid sensitivity in a mouse model of neutrophilic airway inflammation. Mucosal Immunol 8: 1212–1225. [DOI] [PubMed] [Google Scholar]
- Denlinger L., King T., Cardet J., Craig T., Holguin F., Jackson D., et al. (2015) Vitamin D supplementation and the risk of colds in patients with asthma. Am J Resp Crit Care Med November 5. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai D., Newby C., Symon F., Haldar P., Shah S., Gupta S., et al. (2013) Elevated sputum interleukin-5 and submucosal eosinophilia in obese individuals with severe asthma. Am J Respir Crit Care Med 188: 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon A., Subramanian M., Desarno M., Black K., Lane L., Holguin F. (2015) A pilot randomized controlled trial of pioglitazone for the treatment of poorly controlled asthma in obesity. Respir Res 16: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan C., Bailey S., Tran J., Haitsma G., Ibrahim Z., Foster S., et al. (2015) Rosiglitazone elicits in vitro relaxation in airways and precision cut lung slices from a mouse model of chronic allergic airways disease. Am J Physiol Lung Cell Mol Physiol 309: L1219–1228. [DOI] [PubMed] [Google Scholar]
- Dougherty R., Sidhu S., Raman K., Solon M., Solberg O., Caughey G., et al. (2010) Accumulation of intraepithelial mast cells with a unique protease phenotype in Th2-high asthma. J Allergy Clin Immunol 125: 1046–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doukas J., Eide L., Stebbins K., Racanelli-Layton A., Dellamary L., Martin M., et al. (2009) Aerosolized phosphoinositide 3-kinase {gamma}/{delta} inhibitor TG100-115 [3-[2,4-diamino-6-(3-hydroxyphenyl)pteridin-7-yl]phenol] as a therapeutic candidate for asthma and chronic obstructive pulmonary disease. J Pharmacol Exp Ther 328: 758–765. [DOI] [PubMed] [Google Scholar]
- Essilfie A., Horvat J., Kim R., Mayall J., Pinkerton J., Beckett E., et al. (2015) Macrolide therapy suppresses key features of experimental steroid-sensitive and steroid-insensitive asthma. Thorax 70: 458–467. [DOI] [PubMed] [Google Scholar]
- Evans J., Ferguson A., Mosley R., Hutchinson J. (2008) What’s all the flap about?: 5-lipoxygenase-activating protein inhibitors for inflammatory diseases. Trends Pharmacol Sc 29: 72–78. [DOI] [PubMed] [Google Scholar]
- Fahy J., Kim K., Liu J., Boushey H. (1995) Prominent neutrophilic inflammation in sputum from subjects with asthma exacerbation. J Allergy Clin Immunol 95: 843–852. [DOI] [PubMed] [Google Scholar]
- Franciosi L., Diamant Z., Banner K., Zuiker R., Morelli N., Kamerling I., et al. (2013) Efficacy and safety of RPL554, a dual PDE3 and PDE4 inhibitor, in healthy volunteers and in patients with asthma or chronic obstructive pulmonary disease: findings from four clinical trials. Lancet Respir Med 1: 714–727. [DOI] [PubMed] [Google Scholar]
- Fu J., Baines K., Wood L., Gibson P. (2013) Systemic inflammation is associated with differential gene expression and airway neutrophilia in asthma. OMICS 17: 187–199. [DOI] [PubMed] [Google Scholar]
- Fujitani Y., Trifilieff A. (2003) In vivo and in vitro effects of SAR 943, a rapamycin analogue, on airway inflammation and remodeling. Am J Respir Crit Care Med 167: 193–198. [DOI] [PubMed] [Google Scholar]
- Furukawa T., Sakagami T., Koya T., Hasegawa T., Kawakami H., Kimura Y., et al. (2015) Characteristics of eosinophilic and noneosinophilic asthma during treatment with inhaled corticosteroids. J Asthma 52: 417–422. [DOI] [PubMed] [Google Scholar]
- Futosi K., Fodor S., Mócsai A. (2013) Neutrophil cell surface receptors and their intracellular signal transduction pathways. Internat Immunopharmacol 17: 1185–1197. [DOI] [PubMed] [Google Scholar]
- Gaga M., Brand P., Thomson N. (2015) The quest for the grail: multidimensional efforts for understanding and targeting severe asthma. Eur Respir J 46: 1227–1231. [DOI] [PubMed] [Google Scholar]
- Gao P., Gibson P., Baines K., Yang I., Upham J., Reynolds P., et al. (2015) Anti-inflammatory deficiencies in neutrophilic asthma: reduced galectin-3 and IL-1RA/IL-1β. Respir Res 16: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvreau G., Boulet L., Schmid-Wirlitsch C., Cote J., Duong M., Killian K., et al. (2011) Roflumilast attenuates allergen-induced inflammation in mild asthmatic subjects. Respir Res 12: 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson P., Simpson J., Saltos N. (2001) Heterogeneity of airway inflammation in persistent asthma: evidence of neutrophilic inflammation and increased sputum interleukin-8. Chest 119: 1329–1336. [DOI] [PubMed] [Google Scholar]
- Gielen V., Johnston S., Edwards M. (2010) Azithromycin induces anti-viral responses in bronchial epithelial cells. Eur Respir J 36: 646–654. [DOI] [PubMed] [Google Scholar]
- Godon P., Boulet L., Malo J., Cartier A., Lemière C. (2002) Assessment and evaluation of symptomatic steroid-naive asthmatics without sputum eosinophilia and their response to inhaled corticosteroids. Eur Respir J 20: 1364–1369. [DOI] [PubMed] [Google Scholar]
- Green B., Wiriyachaiporn S., Grainge C., Rogers G., Kehagia V., Lau L., et al. (2014) Potentially pathogenic airway bacteria and neutrophilic inflammation in treatment resistant severe asthma. PLoS ONE 9: e100645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green R., Brightling C., Mckenna S., Hargadon B., Parker D., Bradding P., et al. (2002a) Asthma exacerbations and sputum eosinophil counts: a randomised controlled trial. Lancet 360: 1715–1721. [DOI] [PubMed] [Google Scholar]
- Green R., Brightling C., Woltmann G., Parker D., Wardlaw A., Pavord I. (2002b) Analysis of induced sputum in adults with asthma: identification of subgroup with isolated sputum neutrophilia and poor response to inhaled corticosteroids. Thorax 57: 875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood J., Steinman L., Zamvil S. (2006) Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat Rev Immunol 6: 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guntur V., Reinero C. (2012) The potential use of tyrosine kinase inhibitors in severe asthma. Curr Opin Allergy Clin Immunol 12: 68–75. [DOI] [PubMed] [Google Scholar]
- Haldar P., Pavord I. (2007) Noneosinophilic asthma: a distinct clinical and pathologic phenotype.J Allergy Clin Immunol 119: 1043–1052. [DOI] [PubMed] [Google Scholar]
- Haldar P., Pavord I., Shaw D., Berry M., Thomas M., Brightling C., et al. (2008) Cluster analysis and clinical asthma phenotypes. Am J Resp Crit Car Med 178: 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammaker D., Firestein G. (2010) “Go upstream, young man”: lessons learned from the P38 saga. Ann Rheum Dis 69, Suppl 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancox R., Cowan D., Aldridge R., Cowan J., Palmay R., Williamson A., et al. (2012) Asthma phenotypes: consistency of classification using induced sputum. Respirology 17: 461–466. [DOI] [PubMed] [Google Scholar]
- Hao M., Lin J., Shu J., Zhang X., Luo Q., Pan L., et al. (2015) Clarithromycin might attenuate the airway inflammation of smoke-exposed asthmatic mice via affecting HDAC2. J Thorac Dis 7: 1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie A., Moore W., Li H., Rector B., Ortega V., Pascual R., et al. (2013) Biomarker surrogates do not accurately predict sputum eosinophil and neutrophil percentages in asthmatic subjects. J Allergy Clin Immunol 132: 72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie A., Moore W., Meyers D., Vestal P., Li H., Peters S., et al. (2010) Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol 125: 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaney L., Djukanovic R., Woodcock A., Walker S., Matthews J., Pavord I., et al. (2015) Research in progress: Medical Research Council United Kingdom Refractory Asthma Stratification Programme (RASP-UK). Thorax doi: 10.1136/thoraxjnl-2015-207326. [Epub ahead of print]: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez M., Mills K., Almond M., Todoric K., Aleman M., Zhang H., et al. (2015) IL-1 receptor antagonist reduces endotoxin-induced airway inflammation in healthy volunteers. J Allergy Clin Immunol 135: 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate S., Noonan M., Chanez P., Busse W., Dupont L., Pavord I., et al. (2011) Efficacy and safety of etanercept in moderate-to-severe asthma: a randomised, controlled trial. Eur Respir J 37: 1352–1359. [DOI] [PubMed] [Google Scholar]
- Holz O., Khalilieh S., Ludwig-Sengpiel A., Watz H., Stryszak P., Soni P., et al. (2010) SCH527123, a novel CXCR2 antagonist, inhibits ozone-induced neutrophilia in healthy subjects. Eur Respir J 35: 564–570. [DOI] [PubMed] [Google Scholar]
- Hothersall E., Mcsharry C., Thomson N. (2006) Potential therapeutic role for statins in respiratory disease. Thorax 61: 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth P., Babu K., Arshad H., Lau L., Buckley M., Mcconnell W., et al. (2005) Tumour necrosis factor (TNF{Alpha}) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 60: 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M., De Blay F., Garcia G., Prud’homme A., Leroyer C., Magnan A., et al. (2009) Masitinib, a c-kit/PDGF receptor tyrosine kinase inhibitor, improves disease control in severe corticosteroid-dependent asthmatics. Allergy 64: 1194–1201. [DOI] [PubMed] [Google Scholar]
- Ito K., Caramori G., Adcock I.M. (2007) Therapeutic potential of phosphatidylinositol 3-kinase inhibitors in inflammatory respiratory disease.J Pharmacol Exp Ther 321: 1–8. [DOI] [PubMed] [Google Scholar]
- Jatakanon A., Lim S., Pj B. (2000) Changes in sputum eosinophils predict loss of asthma control. Am J Repir Crit Care Med 161: 64–72. [DOI] [PubMed] [Google Scholar]
- Jayaram L., Pizzichini M., Cook R., Boulet L., Lemiere C., Pizzichini E., et al. (2006) Determining asthma treatment by monitoring sputum cell counts: effect on exacerbations. Eur Respir J 27: 483–494. [DOI] [PubMed] [Google Scholar]
- Katz L., Gleich G., Hartley B., Yancey S., Ortega H. (2014) Blood eosinophil count is a useful biomarker to identify patients with severe eosinophilic asthma. Annals Am Thor Soc 11: 531–536. [DOI] [PubMed] [Google Scholar]
- Kent S., Boyce M., Diamant Z., Singh D., O’Connor B., Saggu P., et al. (2013) The 5-lipoxygenase-activating protein inhibitor, GSK2190915, attenuates the early and late responses to inhaled allergen in mild asthma. Clin Exp Allergy 43: 177–186. [DOI] [PubMed] [Google Scholar]
- Khorasani N., Baker J., Johnson M., Chung K., Bhavsar P. (2015) Reversal of corticosteroid insensitivity by P38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int J Chron Obstruct Pulmon Dis 10: 283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R., Pinkerton J., Gibson P., Cooper M., Horvat J., Hansbro P. (2015a) Inflammasomes in COPD and neutrophilic asthma. Thorax 70: 1199–1201. [DOI] [PubMed] [Google Scholar]
- Kim S., Kim J., Park C., Kim T., Lee S., Kim Y., et al. (2015b) Effect of roflumilast on airway remodeling in a murine model of chronic asthma. Clin Exp Allergy doi: 10.1111/cea.12670. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Kirsten A., Förster K., Radeczky E., Linnhoff A., Balint B., Watz H., et al. (2015) The safety and tolerability of oral AZD5069, a selective CXCR2 antagonist, in patients with moderate-to-severe COPD. Pulm Pharm Therap 31: 36–41. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y., Wada H., Rossios C., Takagi D., Charron C., Barnes P., et al. (2013a) A novel macrolide/fluoroketolide, solithromycin (CEM-101), reverses corticosteroid insensitivity via phosphoinositide 3-kinase pathway inhibition. Br J Pharmacol 169: 1024–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y., Wada H., Rossios C., Takagi D., Higaki M., Mikura S., et al. (2013b) A novel macrolide solithromycin exerts superior anti-inflammatory effect via NF-κB inhibition. J Pharmacol Exp Ther 345: 76–84. [DOI] [PubMed] [Google Scholar]
- Kupczyk M., Dahlén B., Sterk P., Nizankowska-Mogilnicka E., Papi A., Bel E., et al. (2014a) Stability of phenotypes defined by physiological variables and biomarkers in adults with asthma. Allergy 69: 1198–1204. [DOI] [PubMed] [Google Scholar]
- Kupczyk M., Ten Brinke A., Sterk P., Bel E., Papi A., Chanez P., et al. (2014b) Frequent exacerbators – a distinct phenotype of severe asthma. Clin Exp Allergy 44: 212–221. [DOI] [PubMed] [Google Scholar]
- Lakshmi S., Reddy A., Zhang Y., Sciurba F., Mallampalli R., Duncan S., et al. (2014) Down-regulated peroxisome proliferator-activated receptor γ (PPARγ) in lung epithelial cells promotes a pparγ agonist-reversible proinflammatory phenotype in chronic obstructive pulmonary disease (COPD). J Biol Chem 289: 6383–6393. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lamblin C., Gosset P., Tillie-Leblond I., Saulnier F., Marquette C., Wallaert B., et al. (1998) Bronchial neutrophilia in patients with noninfectious status asthmaticus. Am J Respir Crit Care Med 157: 394–402. [DOI] [PubMed] [Google Scholar]
- Lazaar A., Sweeney L., Macdonald A., Alexis N., Chen C., Tal-Singer R. (2011) SB-656933, a novel CXCR2 selective antagonist, inhibits ex vivo neutrophil activation and ozone-induced airway inflammation in humans. Br J Clin Pharmacol 72: 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea S., Plumb J., Metcalfe H., Spicer D., Woodman P., Fox J., et al. (2014) The effect of peroxisome proliferator-activated receptor-γ ligands on in vitro and in vivo models of COPD. Eur Respir J 43: 409–420. [DOI] [PubMed] [Google Scholar]
- Leaker B., Barnes P., O’Connor B. (2013) Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir Res 14: 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaker B., Barnes P., O’Connor B., Ali F., Tam P., Neville J., et al. (2014) The effects of the novel SHIP1 activator AQX-1125 on allergen-induced responses in mild-to-moderate asthma. Clin Exp Allergy 44: 1146–1153. [DOI] [PubMed] [Google Scholar]
- Lee K., Jeong J., Kim S., Cho S., Kolliputi N., Ko Y., et al. (2016) Phosphoinositide 3-kinase-δ regulates fungus-induced allergic lung inflammation through endoplasmic reticulum stress. Thorax 71: 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Lee D., Kim E., Choe K., Oh Y., Shim T., et al. (2005) Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med 172: 987–993. [DOI] [PubMed] [Google Scholar]
- Lemière C., Boulet L., Chaboillez S., Forget A., Chiry S., Villeneuve H., et al. (2013) Work-exacerbated asthma and occupational asthma: do they really differ? J Allergy Clin Immunol 131: 704–710. [DOI] [PubMed] [Google Scholar]
- Lemiere C., Chaboillez S., Bohadana A., Blais L., Maghni K. (2014) Noneosinophilic responders with occupational asthma: a phenotype associated with a poor asthma prognosis. J Allergy Clin Immunol 133: 883–885. [DOI] [PubMed] [Google Scholar]
- Lemière C., Tremblay C., Fitzgerald M., Aaron S., Leigh R., Boulet L., et al. (2011) Effects of a short course of inhaled corticosteroids in noneosinophilic asthmatic subjects. Can Respir J 18: 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuppi J., Salome C., Jenkins C., Anderson S., Xuan W., Marks G., et al. (2001) Predictive markers of asthma exacerbation during stepwise dose reduction of inhaled corticosteroids. Am J Respir Crit Care Med 163: 406–412. [DOI] [PubMed] [Google Scholar]
- Lipworth B. (2005) Phosphodiesterase-4 inhibitors for asthma and chronic obstructive pulmonary disease. Lancet 365: 167–175. [DOI] [PubMed] [Google Scholar]
- Little S., Chalmers G., Macleod K., Mcsharry C., Thomson N. (2000) Non-invasive markers of airway inflammation as predictors of oral steroid responsiveness in asthma. Thorax 53: 232–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little S., Macleod K., Chalmers G., Love J., Mcsharry C., Thomson N.C. (2002) Association of forced expiratory volume with disease duration and sputum neutrophils in chronic asthma. Am J Med 112: 446–452. [DOI] [PubMed] [Google Scholar]
- Maghni K., Lemiere C., Ghezzo H., Yuquan W., Malo J. (2004) Airway inflammation after cessation of exposure to agents causing occupational asthma. Am J Respir Crit Care Med 169: 367–372. [DOI] [PubMed] [Google Scholar]
- Maneechotesuwan K., Ekjiratrakul W., Kasetsinsombat K., Wongkajornsilp A., Barnes P. (2010) Statins enhance the anti-inflammatory effects of inhaled corticosteroids in asthmatic patients through increased induction of indoleamine 2, 3-dioxygenase. J Allergy Clin Immunol 126: 754–762. [DOI] [PubMed] [Google Scholar]
- Maneechotesuwan K., Essilfie-Quaye S., Kharitonov S., Adcock I., Barnes P. (2007) Loss of control of asthma following inhaled corticosteroid withdrawal is associated with increased sputum interleukin-8 and neutrophils. Chest 132: 98–105. [DOI] [PubMed] [Google Scholar]
- Maneechotesuwan K., Kasetsinsombat K., Wongkajornsilp A., Barnes P. (2013) Decreased indoleamine 2,3-dioxygenase activity and IL-10/IL-17a ratio in patients with COPD. Thorax 68: 330–337. [DOI] [PubMed] [Google Scholar]
- Manni M., Trudeau J., Scheller E., Mandalapu S., Elloso M., Kolls J., et al. (2014) The complex relationship between inflammation and lung function in severe asthma. Mucosal Immunol 7: 1186–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks-Konczalik J., Costa M., Robertson J., McKie E., Yang S., Pascoe S. (2015) A post-hoc subgroup analysis of data from a six month clinical trial comparing the efficacy and safety of losmapimod in moderate-severe copd patients with ⩽2% and >2% blood eosinophils. Respir Med 109: 860–869. [DOI] [PubMed] [Google Scholar]
- Martineau A., Hanifa Y., Witt K., Barnes N., Hooper R., Patel M., et al. (2015) Double-blind randomised controlled trial of vitamin D3 supplementation for the prevention of acute respiratory infection in older adults and their carers (Vidiflu). Thorax 70: 953–960. [DOI] [PubMed] [Google Scholar]
- Marwick J., Caramori G., Casolari P., Mazzoni F., Kirkham P., Adcock I., et al. (2010) A Role for phosphoinositol 3-kinase delta in the impairment of glucocorticoid responsiveness in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 125: 1146–1153. [DOI] [PubMed] [Google Scholar]
- Marwick J., Caramori G., Stevenson C., Casolari P., Jazrawi E., Barnes P., et al. (2009) Inhibition of Pi3kdelta restores glucocorticoid function in smoking-induced airway inflammation in mice. Am J Respir Crit Care Med 179: 542–548. [DOI] [PubMed] [Google Scholar]
- McGrath K., Icitovic N., Boushey H., Lazarus S., Sutherland E., Chinchilli V., et al. (2012) A large subgroup of mild-to-moderate asthma is persistently noneosinophilic. Am J Respir Crit Care Med 185: 612–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay A., Leung B., Mcinnes I., Thomson N., Liew F. (2004) A novel anti-inflammatory role of simvastatin in a murine model of allergic asthma. J Immunol 172: 2903–2908. [DOI] [PubMed] [Google Scholar]
- Mercado N., Hakim A., Kobayashi Y., Meah S., Usmani O., Chung K., et al. (2012) Restoration of corticosteroid sensitivity by P38 mitogen activated protein kinase inhibition in peripheral blood mononuclear cells from severe asthma. PLoS ONE 7: e41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P., Kolls J. (2012) Targeting Il-17 and Th17 cells in chronic inflammation. Nat Rev Drug Discov 11: 763–776. [DOI] [PubMed] [Google Scholar]
- Moore W., Hastie A., Li X., Li H., Busse W., Jarjour N., et al. (2014) Sputum neutrophil counts are associated with more severe asthma phenotypes using cluster analysis. J Allergy Clin Immunol 133: 1557–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretto N., Caruso P., Bosco R., Marchini G., Pastore F., Armani E., et al. (2015) Chf6001, a novel highly potent and selective phosphodiesterase 4 inhibitor with robust anti-inflammatory activity and suitable for topical pulmonary administration.J Pharmacol Exp Ther 352: 559–567. [DOI] [PubMed] [Google Scholar]
- Morissette M., Shen P., Thayaparan D., Stämpfli M. (2015) Impacts of peroxisome proliferator-activated receptor-γ activation on cigarette smoke-induced exacerbated response to bacteria. Eur Respir J 45: 191–200. [DOI] [PubMed] [Google Scholar]
- Nair P., Aziz-Ur-Rehman A., Radford K. (2015) Therapeutic implications of ‘neutrophilic asthma’. Curr Opin Pulm Med 21: 33–38 [DOI] [PubMed] [Google Scholar]
- Nair P., Gaga M., Zervas E., Alagha K., Hargreave F., O’Byrne P., et al. (2012) Safety and efficacy of a CXCR2 antagonist in patients with severe asthma and sputum neutrophils: a randomized, placebo-controlled clinical trial. Clin Exp Allergy 42: 1097–1103. [DOI] [PubMed] [Google Scholar]
- Nanzer A., Chambers E., Ryanna K., Richards D., Black C., Timms P., et al. (2013) Enhanced production of IL-17a in patients with severe asthma is inhibited by 1î±,25-dihydroxyvitamin D3 in a glucocorticoid-independent fashion. J Allergy Clin Immunol 132: 297–304. [DOI] [PubMed] [Google Scholar]
- Newby C., Agbetile J., Hargadon B., Monteiro W., Green R., Pavord I., et al. (2014) Lung function decline and variable airway inflammatory pattern: longitudinal analysis of severe asthma. J Allergy Clin Immunol 134: 287–294. [DOI] [PubMed] [Google Scholar]
- Newcomb D., Peebles R., Jr (2013) Th17-mediated inflammation in asthma. Curr Opinion Immunol 25: 755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nials A., Tralau-Stewart C., Gascoigne M., Ball D., Ranshaw L., Knowles R. (2011) In vivo characterization of GSK256066, a high-affinity inhaled phosphodiesterase 4 inhibitor. J Pharmacol Exp Ther 337: 137–144. [DOI] [PubMed] [Google Scholar]
- O’Byrne P., Metev H., Puu M., Richter K., Keen C. (2015) Efficacy and safety of a CXCR2 antagonist, AZD5069, in severe, uncontrolled persistent asthma. Am J Resp Crit Care Med 256: A2503–A2503. [DOI] [PubMed] [Google Scholar]
- Ortega H., Liu M., Pavord I., Brusselle G., Fitzgerald J., Chetta A., et al. (2014) Mepolizumab treatment in patients with severe eosinophilic asthma. N Eng J Med 371: 1198–1207. [DOI] [PubMed] [Google Scholar]
- Page C., Spina D. (2012) Selective PDE inhibitors as novel treatments for respiratory diseases. Curr Opin Pharmacol 12: 275–286. [DOI] [PubMed] [Google Scholar]
- Pavord I., Brightling C., Woltmann G., Wardlaw A. (1999) Noneosinophilic corticosteroid unresponsive asthma. Lancet 353: 2213–2214. [DOI] [PubMed] [Google Scholar]
- Pavord I., Korn S., Howarth P., Bleecker E., Buhl R., Keene O., et al. (2012) Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 380: 651–659. [DOI] [PubMed] [Google Scholar]
- Peters M., Mekonnen Z., Yuan S., Bhakta N., Woodruff P., Fahy J. (2014) Measures of gene expression in sputum cells can identify Th2-high and Th2-low subtypes of asthma. J Allergy Clin Immunol 133: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raundhal M., Morse C., Khare A., Oriss T., Milosevic J., Trudeau J., et al. (2015) High IFN-γ and low SLPI mark severe asthma in mice and humans. J Clin Invest 125: 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J., Demirel N., Mendy A., Gasana J., Vieira E., Colin A., et al. (2013) Macrolides for the long-term management of asthma – a meta-analysis of randomized clinical trials. Allergy 68: 1040–1049. [DOI] [PubMed] [Google Scholar]
- Rennard S., Dale D., Donohue J., Kanniess F., Magnussen H., Sutherland E., et al. (2015) CXCR2 antagonist Mk-7123—a phase 2 proof-of-concept trial for chronic obstructive pulmonary disease. Am J Resp Crit Care Med 191: 1001–1011. [DOI] [PubMed] [Google Scholar]
- Rhee C., Kim J., Park C., Kim J., Kang J., Kim S., et al. (2011) Effect of imatinib on airway smooth muscle thickening in a murine model of chronic asthma. Int Arch Allergy Immunol 155: 243–251. [DOI] [PubMed] [Google Scholar]
- Richards D., Bareille P., Lindo E., Quinn D., Farrow S. (2010) Treatment with a peroxisomal proliferator activated receptor gamma agonist has a modest effect in the allergen challenge model in asthma: a randomised controlled trial. Respir Med 104: 668–674. [DOI] [PubMed] [Google Scholar]
- Saffar A., Ashdown H., Gounni A. (2011) The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr Drug Targets 12: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson K., Minoguchi K., Tanaka A., Oda N., Yokoe T., Yamamoto Y., et al. (2006) Inhibitory effects of fluvastatin on cytokine and chemokine production by peripheral blood mononuclear cells in patients with allergic asthma. Clin Exp Allergy 36: 475–482. [DOI] [PubMed] [Google Scholar]
- Schleich F., Chevremont A., Paulus V., Henket M., Manise M., Seidel L., et al. (2014) Importance of concomitant local and systemic eosinophilia in uncontrolled asthma. Eur Respir J 44: 97–108. [DOI] [PubMed] [Google Scholar]
- Schleich F., Manise M., Sele J., Henket M., Seidel L., Louis R. (2013) Distribution of sputum cellular phenotype in a large asthma cohort: predicting factors for eosinophilic vs neutrophilic inflammation. BMC Pulm Med 13: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schögler A., Kopf B., Edwards M., Johnston S., Casaulta C., Kieninger E., et al. (2015) Novel antiviral properties of azithromycin in cystic fibrosis airway epithelial cells. Eur Respir J 45: 428–439. [DOI] [PubMed] [Google Scholar]
- Seidel P., Alkhouri H., Lalor D., Burgess J., Armour C., Hughes J. (2012) Thiazolidinediones inhibit airway smooth muscle release of the chemokine CXCL10: in vitro comparison with current asthma therapies. Respir Res 13: 90–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw D., Berry M., Hargadon B., Mckenna S., Shelley M., Green R., et al. (2007) Association between neutrophilic airway inflammation and airflow limitation in adults with asthma. Chest 132: 1871–1875. [DOI] [PubMed] [Google Scholar]
- Shaw D., Sousa A., Fowler S., Fleming L., Roberts G., Corfield J., et al. (2015) Clinical and inflammatory characteristics of the European U-Biopred Adult Severe Asthma Cohort. Eur Respir J 46: 1308–1321. [DOI] [PubMed] [Google Scholar]
- Shen N., Wang J., Zhao M., Pei F., He B. (2011) Anti-interleukin-17 antibodies attenuate airway inflammation in tobacco-smoke-exposed mice. Inhal Toxicol 23: 212–218. [DOI] [PubMed] [Google Scholar]
- Shum A. (2015) T cell types that take your breath away. Sci Trans Med 7: 301fs333–301fs333. [DOI] [PubMed] [Google Scholar]
- Simpson J., Gibson P., Yang I., Upham J., James A., Reynolds P., et al. (2013) Impaired macrophage phagocytosis in noneosinophilic asthma. Clin Exp Allergy 43: 29–35. [DOI] [PubMed] [Google Scholar]
- Simpson J., Gibson P., Yang I., Upham J., James A., Reynolds P., et al. (2014a) Altered sputum granzyme B and granzyme B/proteinase inhibitor-9 in patients with noneosinophilic asthma. Respirology 19: 280–287. [DOI] [PubMed] [Google Scholar]
- Simpson J., Grissell T., Douwes J., Scott R., Boyle M., Gibson P. (2007) Innate immune activation in neutrophilic asthma and bronchiectasis. Thorax 62: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J., Guest M., Boggess M., Gibson P. (2015) Occupational exposures, smoking and airway inflammation in refractory asthma. BMC Pulm Med 14: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson J., Mcelduff P., Gibson P. (2010) Assessment and reproducibility of noneosinophilic asthma using induced sputum. Respiration 79: 147–151. [DOI] [PubMed] [Google Scholar]
- Simpson J., Phipps S., Baines K., Oreo K., Gunawardhana L., Gibson P. (2014b) Elevated expression of the NLRP3 inflammasome in neutrophilic asthma. Eur Respir J 43: 1067–1076. [DOI] [PubMed] [Google Scholar]
- Simpson J., Powell H., Boyle M., Scott R., Gibson P. (2008) Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am J Respir Crit Care Med 177: 148–155. [DOI] [PubMed] [Google Scholar]
- Simpson J., Scott R., Boyle M., Gibson P. (2006) Inflammatory Subtypes in Asthma: Assessment and Identification Using Induced Sputum. Respirol 11: 54–61. [DOI] [PubMed] [Google Scholar]
- Simpson J.L., Milne D.G., Gibson P.G. (2009) Neutrophilic Asthma Has Different Radiographic Features to COPD and Smokers. Resp Med 103: 881–887. [DOI] [PubMed] [Google Scholar]
- Singh D., Petavy F., Macdonald A., Lazaar A., O’Connor B. (2010) The inhaled phosphodiesterase 4 inhibitor GSK256066 reduces allergen challenge responses in asthma. Resp Res 11: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spahn J., Fost D., Covar R., Martin R., Brown E., Sj S., et al. (2001) Clarithromycin potentiates glucocorticoid responsiveness in patients with asthma: results of a pilot study. Ann Allergy Asthma Immunol 87: 501–505. [DOI] [PubMed] [Google Scholar]
- Spears M., Donnelly I., Jolly L., Brannigan M., Ito K., McSharry C., et al. (2009a) Effect of low-dose theophylline plus beclometasone on lung function in smokers with asthma: a pilot study. Eur Respir J 33: 1010–1017. [DOI] [PubMed] [Google Scholar]
- Spears M., Donnelly I., Jolly L., Brannigan M., Ito K., McSharry C., et al. (2009b) Bronchodilatory effect of the PPAR-[gamma] agonist rosiglitazone in smokers with asthma. Clin Pharmacol Ther 86: 49–53. [DOI] [PubMed] [Google Scholar]
- Spears M., McSharry C., Thomson N.C. (2006) Peroxisome proliferator-activated receptor-gamma agonists as potential anti-inflammatory agents in asthma and chronic obstructive pulmonary disease. Clin Exp Allergy 36: 1494–1504. [DOI] [PubMed] [Google Scholar]
- Stephen J., Delvecchio C., Spitale N., Giesler A., Radford K., Bilan P., et al. (2013) PPAR ligands decrease human airway smooth muscle cell migration and extracellular matrix synthesis. Eur Respir J 41: 425–432. [DOI] [PubMed] [Google Scholar]
- Suárez-Cuartín G., Crespo A., Mateus E., Torrejón M., Giner J., Belda A., et al. (2015) Variability in asthma inflammatory phenotype in induced sputum. Frequency and causes. Arch Bronconeumol May 4 pii: S0300–2896(15)00110–6. doi: 10.1016/j.arbres.2015.03.007. [Epub ahead of print]: [DOI] [PubMed] [Google Scholar]
- Sukkar M., Wood L., Tooze M., Simpson J., Mcdonald V., Gibson P., et al. (2012) Soluble rage is deficient in neutrophilic asthma and COPD. Eur Respir J 39: 721–729. [DOI] [PubMed] [Google Scholar]
- Thomson N. (2014) Novel therapies targeting eosinophilic inflammation in asthma. Clin Exp Allergy 44: 462–468. [DOI] [PubMed] [Google Scholar]
- Thomson N., Charron C., Chaudhuri R., Spears M., Ito K., McSharry C. (2015) Atorvastatin in combination with inhaled beclometasone modulates inflammatory sputum mediators in smokers with asthma. Pulm Pharmacol Ther 31: 1–8. [DOI] [PubMed] [Google Scholar]
- Thomson N., Chaudhuri R., Heaney L., Bucknall C., Niven R., Brightling C., et al. (2013) Clinical outcomes and inflammatory biomarkers in current smokers and exsmokers with severe asthma. J Allergy Clin Immunol 131: 1008–1016. [DOI] [PubMed] [Google Scholar]
- Thomson N., Chaudhuri R., Thomson N., Chaudhuri R. (2009) Asthma in smokers: challenges and opportunities. Curr Opin Pulm Med 15: 39–45. [DOI] [PubMed] [Google Scholar]
- To Y., Ito K., Kizawa Y., Failla M., Ito M., Kusama T., et al. (2010) Targeting phosphoinositide-3-kinase-delta with theophylline reverses corticosteroid insensitivity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 182: 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo Bittar H., Yousem S., Wenzel S. (2015) Pathobiology of severe asthma. Annu Rev Pathol 10: 511–545. [DOI] [PubMed] [Google Scholar]
- Turner M., Hussack P., Sears M., Dolovich J., Hargreave F. (1995) Exacerbations of asthma without sputum eosinophilia. Thorax 50: 1057–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M., Lau L., Seumois G., Vijayanand P., Staples K., Bagmane D., et al. (2013) EGF-induced bronchial epithelial cells drive neutrophil chemotactic and anti-apoptotic activity in asthma. PLoS ONE 8: e72502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M., Nong G., Ward J., Seumois G., Prince L., Wilson S., et al. (2010) Prosurvival activity for airway neutrophils in severe asthma. Thorax 65:: 684–689. [DOI] [PubMed] [Google Scholar]
- Van Veen I., Ten Brinke A., Gauw S., Sterk P., Rabe K., Bel E. (2009) Consistency of sputum eosinophilia in difficult-to-treat asthma: a 5-year follow-up study. J Allergy Clin Immunol 124: 615–617.e612. [DOI] [PubMed] [Google Scholar]
- Wagener A., De Nijs S., Lutter R., Sousa A., Weersink E., Bel E., et al. (2015) External validation of blood eosinophils, feno and serum periostin as surrogates for sputum eosinophils in asthma. Thorax 70: 115–120. [DOI] [PubMed] [Google Scholar]
- Wallace J., D’Silva L., Brannan J., Hargreave F., Kanaroglou P., Nair P. (2011) Association between proximity to major roads and sputum cell counts. Can Respir J 18: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., He X., Baines K., Gunawardhana L., Simpson J., Li F., et al. (2011) Different inflammatory phenotypes in adults and children with acute asthma. Eur Respir J 38: 567–574. [DOI] [PubMed] [Google Scholar]
- Watz H., Mistry S., Lazaar A., Investigators. I. (2013) Safety and tolerability of the inhaled phosphodiesterase 4 inhibitor GSK256066 in moderate COPD. Pulm Pharmacol Ther 26: 588–595. [DOI] [PubMed] [Google Scholar]
- Wenzel S., Barnes P., Bleecker E., Bousquet J., Busse W., Dahlen S., et al. (2009) A randomized, double-blind, placebo-controlled study of tumor necrosis factor-alpha blockade in severe persistent asthma. Am J Respir Crit Care Med 179: 549–558. [DOI] [PubMed] [Google Scholar]
- Wenzel S., Schwartz L., Langmack E., Al E. (1999) Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med 160: 1001–1008. [DOI] [PubMed] [Google Scholar]
- Winkler D., Faia K., Dinitto J., Ali J., White K., Brophy E., et al. (2013) Pi3k-δ and Pi3k-γ inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chem Biol 20: 1364–1374. [DOI] [PubMed] [Google Scholar]
- Wood L., Baines K., Fu J., Scott H., Gibson P. (2012) The neutrophilic inflammatory phenotype is associated with systemic inflammation in asthma. Chest 142: 86–93. [DOI] [PubMed] [Google Scholar]
- Woodruff P., Modrek B., Choy D., Jia G., Abbas A., Ellwanger A., et al. (2009) T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med 180: 388–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Chen H., Li Y., Wang S., Diao X., Liu K. (2014) Intranasal SIRNA targeting c-kit reduces airway inflammation in experimental allergic asthma. Int J Clin Exp Pathol 7: 5505–5514. [PMC free article] [PubMed] [Google Scholar]
- Wu W., Wang T., Dong J., Liao Z., Wen F. (2012) Silencing of c-kit with small interference RNA attenuates inflammation in a murine model of allergic asthma. Int J Mol Med 30: 63–68. [DOI] [PubMed] [Google Scholar]
- Xystrakis E., Kusumakar S., Boswell S., Peek E., Urry Z., Richards D., et al. (2006) Reversing the defective induction of IL-10-secreting regulatory T cells in glucocorticoid-resistant asthma patients. J Clin Invest 116: 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeganeh B., Wiechec E., Ande S., Sharma P., Moghadam A., Post M., et al. (2014) Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol Ther 143: 87–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeki A., Franzi L., Last J., Kenyon N. (2009) Simvastatin inhibits airway hyperreactivity: implications for the mevalonate pathway and beyond. Am J Respir Crit Care Med 180: 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Simpson J., Powell H., Yang I., Upham J., Reynolds P., et al. (2014) Full blood count parameters for the detection of asthma inflammatory phenotypes. Clin Exp Allergy 44: 1137–1145. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Leung D., Goleva E. (2014) Anti-inflammatory and corticosteroid-enhancing actions of vitamin D in monocytes of patients with steroid-resistant and those with steroid-sensitive asthma.J Allergy Clin Immunol 133: 1744–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Huang Y., He J., Li C., Deng W., Ran X., et al. (2014) Rosiglitazone, a peroxisome proliferator-activated receptor-γ agonist, attenuates airway inflammation by inhibiting the proliferation of effector T cells in a murine model of neutrophilic asthma. Immunol Lett 157: 9–15. [DOI] [PubMed] [Google Scholar]