

SUMMARY
Hotspot mutations in splicing factor genes have been recently reported at high frequency in hematological malignancies, suggesting the importance of RNA splicing in cancer. We analyzed whole-exome sequencing data across 33 tumor types in The Cancer Genome Atlas (TCGA), and we identified 119 splicing factor genes with significant non-silent mutation patterns, including mutation over-representation, recurrent loss of function (tumor suppressor-like), or hotspot mutation profile (oncogene-like). Furthermore, RNA sequencing analysis revealed altered splicing events associated with selected splicing factor mutations. In addition, we were able to identify common gene pathway profiles associated with the presence of these mutations. Our analysis suggests that somatic alteration of genes involved in the RNA-splicing process is common in cancer and may represent an underappreciated hallmark of tumorigenesis.
Graphical abstract
In Brief: Seiler et al. report that 119 splicing factor genes carry putative driver mutations over 33 tumor types in TCGA. The most common mutations appear to be mutually exclusive and are associated with lineage-independent altered splicing. Samples with these mutations show deregulation of cell-autonomous pathways and immune infiltration.
INTRODUCTION
Alternative pre-mRNA splicing is a major source of transcript diversity in mammalian cells and is orchestrated by a megadalton complex called the spliceosome (Papasaikas and Valcárcel, 2016). The major U2-type spliceosome constitutes five small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, U4, U5, and U6) and >150 proteins, while the minor U12-type spliceosome contains five snRNPs and an unknown number of proteins, many of which have analogous genes in the U2 spliceosome. In a dynamic process, pre-mRNA non-coding intron sequences are removed at specific splice sites, leaving coding exons that are ligated to form mature mRNA. These introns and exons contain sequences that are recognized by the core splicing machinery and are essential for recruitment and activation of the splicing process. Additionally, there are cis silencer and enhancer sequences that are recognized by accessory factors, e.g., heterogeneous nuclear ribonucleoproteins (hnRNPs) and serine/arginine-rich (SR) proteins, and these factors are responsible for splicing regulation (Wang et al., 2008). Recurrent somatic mutations of the splicing factor genes SF3B1, SRSF2, U2AF1, and ZRSR2 were first discovered through whole-exome sequencing in myelodysplastic syndrome (MDS) (Yoshida et al., 2011), and they were later reported in other hematological malignancies as well as solid tumors (Makishima et al., 2012; Papaemmanuil et al., 2013; Haferlach et al., 2014; Lindsley et al., 2015; Jeromin et al., 2014; Landau et al., 2015; Patnaik et al., 2013). Differential splicing analysis using RNA sequencing data from patient samples and pre-clinical models revealed that these somatic mutations induced transcriptome-wide splicing alterations (Ferreira et al., 2014; DeBoever et al., 2015; Darman et al., 2015; Zhang et al., 2015; Kim et al., 2015; Okeyo-Owuor et al., 2015; Przychodzen et al., 2013; Madan et al., 2015).
The confluence of both DNA and RNA sequencing in TCGA provide a unique opportunity to interrogate splicing deregulation due to somatic mutation across human cancers. Although systematic analyses of mutations, copy number, and gene expression patterns of RNA-binding proteins (RBPs) have recently been reported (Sebestyén et al., 2016; Neelamraju et al., 2018), here we focus on somatic mutations in known splicing factors and alternative splicing events associated with selected mutations across 33 tumor types and more than 10,000 samples. Furthermore, we compare how these mutations affect gene pathways in the affected lineages, and we examine their potential impact on tumorigenesis.
RESULTS
119 Splicing Factor Genes Carry Recurrent Mutations in Hematological Cancers and Solid Tumors
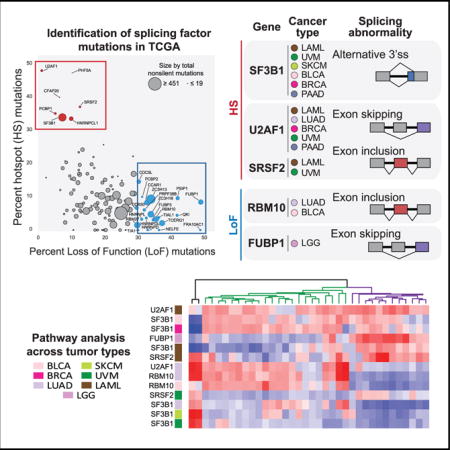
We compiled and curated a catalog of 404 splicing factor genes (Table S1; STAR Methods), and we prioritized genes with likely driver mutations using two complementary approaches (Figure 1A). The first approach, MutSigCV (Lawrence et al., 2013), ranks genes by statistical significance of somatic mutation per cohort adjusted by mutation background of tumor type, gene size, replication time, and gene expression levels. We identified 68 genes as significantly mutated in at least one cohort (q value ≤ 0.1). The second approach, a ratiometric method (Vogelstein et al., 2013), identifies likely oncogenes and tumor suppressors based on the observation that oncogenes are recurrently mutated at the same amino acid position (hotspot, HS), whereas tumor suppressor genes are mutated through loss-of-function (LoF) mutations throughout their length. Using this method, we identified 77 genes as either likely oncogene (OG) or tumor suppressor gene (TSG) using either individual tumor cohorts (72 genes) or a pancan cohort of all samples (5 genes). Similar results were also obtained by a recently published ratio-metric method, 20/20+ (Tokheim et al., 2016) (Figure S1C). Among the 77 genes, 27 were also identified by MutSigCV, while 50 were uniquely identified by this approach only. Finally, ZRSR2 was added as it has been previously identified in hematological tumors as significantly mutated, though it did not meet our driver gene criteria in TCGA. Together, we prioritized 119 genes as likely harboring driver mutations (Table S1).
Figure 1. 119 Splicing Factor Genes Are Mutated across All Tumor Cohorts.
(A) Prioritization of splicing factor genes with likely driver mutations.
(B) Hotspot (HS)- (red) and loss-of-function (LoF)- (blue) mutated genes in the pancan cohort are mapped to spliceosome complexes. The percent non-silent mutation frequency (Table S1) is listed next to each gene.
(C) Genes are plotted as %hotspot or %LoF mutations for non-silent mutations across TCGA (pancan). OG-like genes are colored red and TSG-like genes are colored blue.
(D) Heatmap view of %hotspot (bottom orange panel) or %LoF mutations (top blue panel) of all non-silent mutations per gene in each tumor cohort, sorted by % hotspot mutation high to low and %LoF mutation low to high from left to right. Tumor cohorts are sorted by average mutation counts per sample (right green bar). For comparison, the fraction of samples with non-silent mutations in any of the 119 genes are shown as purple bars on the right. The number of samples with any non-silent mutation in each likely driver gene is given in the top bar chart.
We mapped these 119 genes to known U2 and U12 spliceosome complexes and their associated proteins (Figure 1B; Table S1A). Among components of the U2 spliceosome, we observed that driver mutations primarily impacted proteins involved in the early stages of splicing catalysis, from pre-catalytic (complex A) to the first catalytic step (complex C). Proteins associated with the U2 snRNP were especially well-represented among hotspot mutants, including SF3B1, U2AF1, and PHF5A. In the U12-type spliceosome, prior reports have described ZRSR2 LoF mutations, primarily in MDSand secondary leukemia, that are associated with the retention of U12 spliceosome introns (Yoshida et al., 2011; Papaemmanuil et al., 2013; Haferlach et al., 2014; Lindsley et al., 2015; Madan et al., 2015). Here we identified 3 recurrently mutated genes (SNRNP35, SNRNP48, and ZCRB1) that are also part of the U12 spliceosome. The recurrent hotspot mutations in SNRNP48 and ZCRB1 in acute myeloid leukemia (LAML) indicate that U12-splicing deregulation in hematological malignancies are more prevalent than previously reported.
Globally, the non-silent mutation rate of individual splicing factor genes is low, ranging from 0.16% (PHF5A) to 3.7% (SPEN) (Figure S1A; Table S1B); however, we observed a number of genes with exceptionally high mutation rates in otherwise infrequently mutated tumors (e.g., SF3B1 in uveal melanoma [UVM] and FUBP1 in low-grade glioma [LGG]) (Figure S1A). Segregating LoF and hotspot mutation rates in each gene by tumor type revealed genes with high percentage of HS or LoF mutations across multiple tumor types (Figure 1D), and we found that LoF mutations are much more common than hotspot mutations (Figure 1C). Overall, we observed a significant linear relationship between the number of samples with likely splicing factor driver mutations and the log10 mutation rate per sample in the corresponding cohort (p = 4.02e–11) (Figure S1B). Bladder carcinoma (BLCA), skin cutaneous melanoma (SKCM), and lung adenocarcinoma (LUAD) were most likely to harbor non-silent mutations in any putative driver splicing factor, at more than 60% of patients in each cohort. Of these tumor types, BLCA and UVM had significantly higher rates of splicing factor driver mutations than would be expected by chance (p = 0.01 and 0.03, respectively), suggesting that splicing deregulation is an important hallmark for these tumors.
Due to the importance of splicing factor mutations in cancer, we analyzed the transcriptomic consequences associated with mutations with exceptional frequency in a single cohort and with hotspot (SF3B1, U2AF1, and SRSF2) or LoF mutations (RBM10 and FUBP1) in samples that were not associated with hyper-mutator phenotypes (see the STAR Methods).
SF3B1 Hotspot Mutations Induce Aberrant Splicing
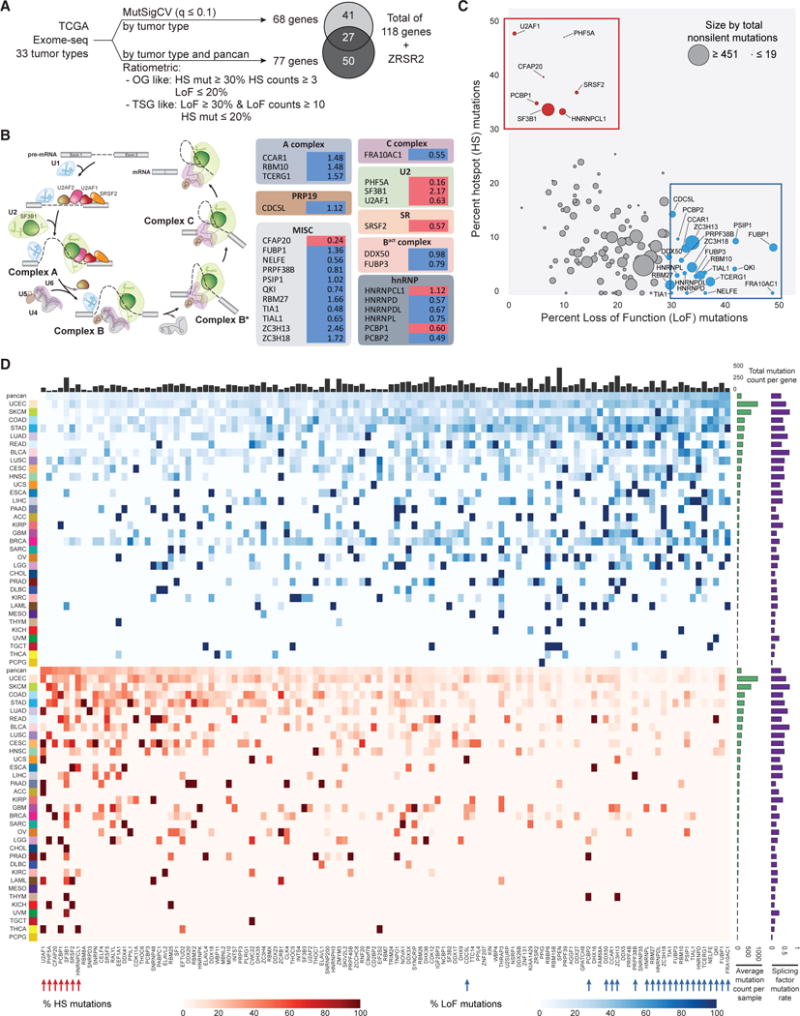
SF3B1 has been reported to be the most frequently mutated splicing factor gene in hematological malignancies and some solid tumors, such as adenoid cystic carcinoma (Martelotto et al., 2015), breast cancer (Cancer Genome Atlas Network, 2012), pancreatic cancer (Biankin et al., 2012), and melanomas (Martin et al., 2013; Cancer Genome Atlas Network, 2015; Hintzsche et al., 2017). It is a member of the U2 complex and, along with SF3B3 and PHF5A, binds to the branch point nucleotide in the pre-catalytic spliceosome (Yan et al., 2016). Here, in a global survey of SF3B1 mutations pan-cancer, we found somatic hotspot mutations that appear to cluster in the C-terminal HEAT repeat domains (HDs) 4–12 (Figures 2A and 2B; Table S2). We previously reported that hotspot mutations in HDs 4-8 display aberrant splicing events enriched with alternative 3′ splice sites (ss), likely as a result of reduced branchpoint fidelity (Darman et al., 2015). Here we also uncovered hotspot mutations in HDs 9-12, including p.L833 (HD 9) in LAML, p.E902 (HD 11) in BLCA, and p.R957 (HD 12) in endometrial cancer (UCEC) samples. These hotspots appeared to be present mainly in these 3 tumor types, resembling previous observations of SF3B1 mutations in position p.R625, which are primarily observed in melanomas.
Figure 2. SF3B1 Hotspot Mutations across Multiple Tumor Types.
(A) SF3B1 somatic mutations in HDs 4-12. Each dot represents a mutant sample colored by tumor cohort.
(B) SF3B1 hotspot mutations are mapped to the structure (PDB: 5GM6). Hotspots in HDs 4-8 are colored purple whereas hotspots in HDs 9-12 are colored gold.
(C) PCA stratifies samples from all solid tumor cohorts with SF3B1 mutations using the expression of alternative 3′ ss associated with SF3B1 hotspot mutations in HDs 4-8. Purple, samples with hotspot mutations in HDs 4-8 (n = 57); gold, samples with p.E902 (n = 7) and p.R957 (n = 5) mutations; green, samples with missense mutations in all other locations (n = 203); gray, WT samples (n = 8,811).
(D) Differential splicing events associated with the BLCA-specific SF3B1 p.E902K mutation (corrected q-value < 0.05). Below each splicing event count, the PSI log2 fold change of each individual event is detailed in a boxplot.
(E) Kernel density estimation plots showing the location of alternative 3′ ss AGs with respect to canonical 3′ ss AGs preferentially used by hotspot mutations in HDs 4-8 (purple) or p.E902K in BLCA (gold).
We observed that overall the occurrence of hotspot mutations in SF3B1 follows a specific periodicity of ~40 amino acids, suggesting a functional role for residues at these positions. Interestingly, the majority of these positions are located at the edge of the HEAT repeat helices of the SF3B1 protein structure (Figure 2B) (Yan et al., 2016; Cretu et al., 2016), suggesting they are important for interactions with RNA or protein or for the conformational flexibility of this super-helical domain. Previously discovered hotspot mutations cluster in HDs 4-8 and near the pre-mRNA-binding region, however, the hotspot mutations in HDs 9-12 are located away from this region, raising the possibility they might induce unique splicing abnormalities.
To test this hypothesis, we used the z-normalized percent spliced-in (PSI) of published alternative 3′ ss associated with SF3B1 hotspot mutations in HDs 4-8 (Darman et al., 2015) to stratify all TCGA solid tumor patient samples using principal-component analysis (PCA) (Figures 2C and S2A). We found that samples with previously identified hotspot mutations in HDs 4-8 were separated from SF3B1 wild-type (WT) samples, as expected. Interestingly, samples with non-hotspot mutations in SF3B1 or mutations in hotspots in HDs 9-12 including those with mutations at position p.E902, were mostly clustered with WT samples, indicating these mutations do not confer the same altered splicing phenotype. We then performed differential splicing analysis using RNA sequencing data directly comparing samples in BLCA with SF3B1 p.E902K (n = 6) to tumors of the same lineage, which were WT with respect to all splicing factor genes (n = 40), resulting in 134 significantly altered junctions (Figure 2D; Table S3). Though splicing alterations as a result of p.E902K also favored alternative 3′ ss, the selected 3′ ss were preferentially located downstream of the 3′ ss used in the WT, while 3′ ss promoted by previously observed hotspots were mostly found upstream (Figure 2E). Similar to 3′ ss promoted by previously identified hotspot mutations, alternative 3′ ss and exon inclusion junctions promoted by SF3B1 p.E902K were also able to stratify solid tumor samples distinctly from samples with other SF3B1 mutations (Figure S2B). The p.R957Q mutation was found to be co-occurring with POLE mutations in UCEC and, thus, in samples with very high mutation rates, reducing the likelihood that this specific SF3B1 mutation is functionally relevant. Other hotspots, such as p.L833, did not have enough samples to allow further functional validation of potential splicing alterations.
U2AF1 and SRSF2 Hotspot Mutations Confer Altered Splicing Based on Sequence Features
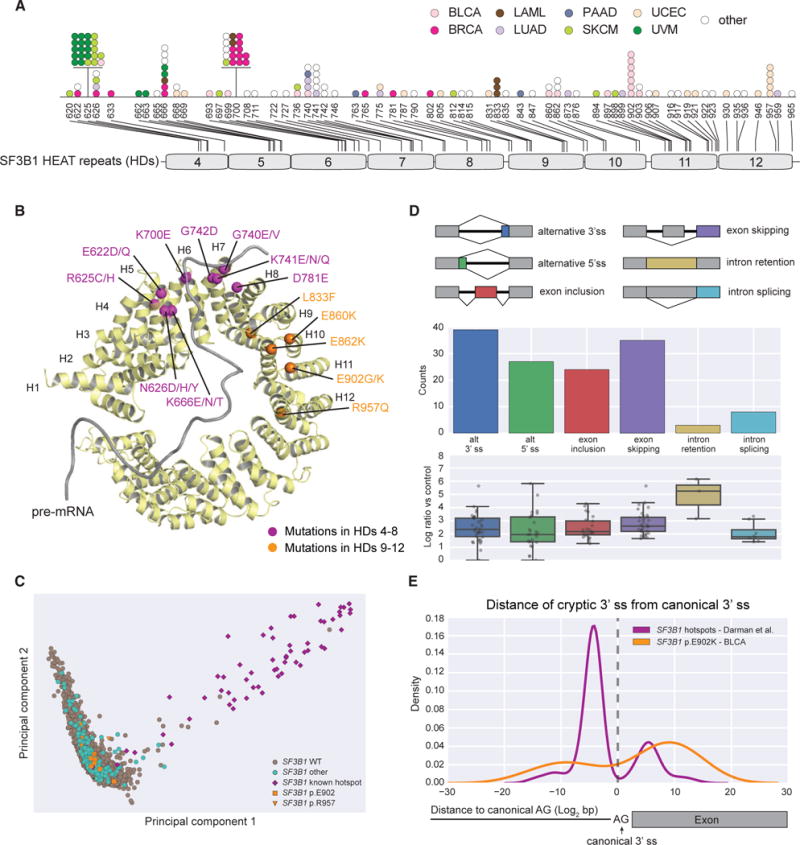
Hotspot mutations of U2AF1 have been reported to alter exon inclusion ratios in both leukemia and lung adenocarcinoma (Przychodzen et al., 2013; Brooks et al., 2014). U2AF1, like SF3B1, is associated with the U2 complex, and it is known to recognize the 3′ dinucleotide AG; and, along with its partner U2AF2 that binds to the 3′ poly-Y tract, it promotes assembly of the pre-catalytic spliceosome (Wu et al., 1999). Hotspot mutations at amino acid positions p.S34 and p.Q157 are common in hematological malignancies (Papaemmanuil et al., 2013; Lindsley et al., 2015) and confer distinct splicing phenotypes (Ilagan et al., 2015), affecting exon inclusion rates based on the nucleotide in the −1 and +1 position relative to the 3′ AG dinucleotide, respectively. In TCGA, p.S34F/Y is the dominant hotspot mutation and is observed in multiple tumor types, most notably LAML, LUAD, and UCEC (Figure 3A; Table S2). In contrast, U2AF1 mutations at p.Q157 are rare and occur in only two samples.
Figure 3. U2AF1 and SRSF2 Mutations in the Pancan Cohort and Differential Splicing Associated with Hotspot Mutations.
(A) U2AF1 somatic mutations mapped to amino acid positions and annotated domains. Each dot represents a single mutant sample colored by tumor cohort.
(B) Differential splicing events associated with U2AF1 p.S34F/Y hotspot mutations in LUAD and LAML (corrected q-value < 0.05). Below each splicing event count, the PSI log2 fold change of each individual event is detailed in a boxplot.
(C) Consensus sequence motifs for exons preferentially used by mutant U2AF1 versus WT U2AF1 across various alternative splicing events.
(D) SRSF2 somatic mutations mapped to amino acid positions and annotated domains. Each bar (in-frame deletion) and dot (other mutations) represents a singlemutant sample colored by tumor cohort.
(E) Differential splicing events associated with SRSF2 in-frame deletions in UVM.
(F) Tetramer (CCNG and GGNG) enrichment analysis comparing cassette exons preferentially included or excluded by SRSF2 mutant samples. Each value is the average tetramer occurrence frequency for all exons in that class. Fold change significance was assessed using Student’s t test.
See also Tables S2 and S3.
To explore the functional impact of the U2AF1 p.S34 hotspot mutations, we focused on LUAD and LAML, comparing mutant samples (n = 15 LUAD and n = 6 LAML) to samples with no known splicing factor gene mutation (n = 87 LUAD and n = 127 LAML). We observed an altered splicing phenotype dominated by alternative 3′ ss and cassette exon events, similar to results obtained by Brooks et al. (2014) (Figure 3B; Table S3). Both exon inclusion and exon skipping events were associated with reduced usage of the 3′ ss trinucleotide TAG, reflecting mutant preference for either C or A in the −1 position. Interestingly, we also observed the same motif selection for alternative 3′ splicing events, which had not been previously reported (Figure 3C).
SRSF2 is an auxiliary splicing factor that has been shown to bind exonic pre-mRNA at specific motifs, where it acts as a splicing enhancer. Both hotspot mutations and in-frame deletions around position p.P95 have been reported, which increase mutant SRSF2 affinity to the nucleotide sequence CCNG relative to the sequence GGNG, resulting in altered exon inclusion rates (Zhang et al., 2015; Kim et al., 2015). We found the majority of SRSF2 somatic mutations in LAML (n = 20) (Figure 3D; Table S2). Interestingly, we identified in-frame deletions in UVM (n = 3), uncovering SRSF2 mutations in this disease. We confirmed that deletions around p.P95 (n = 2) also induced altered exon inclusion and exclusion as compared to WT samples (n = 20) (Figure 3E; Table S3), and we observed that exons with increased inclusion rates displayed an enrichment in CCNG versus GGNG sequence ratios (Figure 3F), consistent with published results in hematological tumors.
RBM10 LoF Mutation Is Associated with Exon Inclusion and a Corresponding Loss of Intron Retention Events in LUAD and BLCA
RBM10 is an RNA-binding protein associated exclusively with splicing repression (Wang et al., 2013), typically acting by binding in the intronic regions both up- and downstream of cassette exons. It is most frequently mutated in LUAD (Cancer Genome Atlas Research Network, 2014) and BLCA (Table S2), and the mutations are mutually exclusive with other splicing factor gene mutations (Figure 4A). RBM10 is located on the X chromosome, and its LoF mutations are the cause of the X-linked recessive disorder TARP syndrome, affecting mainly males (Johnston et al., 2010). We observed that RBM10 LoF mutations in LUAD were also associated with the male gender (p = 0.002, Fisher’s exact test), though this trend was not observed in BLCA, a disease that is found primarily in males. In both diseases, RBM10 LoF mutations resulted in reduced mRNA expression (Figure 4B; p value < 0.0001 in all comparisons, Student’s t test). Differential splicing analyses comparing RBM10 LoF mutant tumors (n = 32) and samples WT for all splicing factor genes (n = 87) identified exon inclusion as the primary alternative splicing event in both LUAD and BLCA (Figure 4C; Figure S3A; Tables S2 and S3). This is consistent with earlier reports correlating the overexpression of RBM10 in HEK293 cells with exon skipping (Wang et al., 2013).
Figure 4. RBM10 LoF Mutations Detected in LUAD and BLCA Induce Global Exon Inclusion Events.
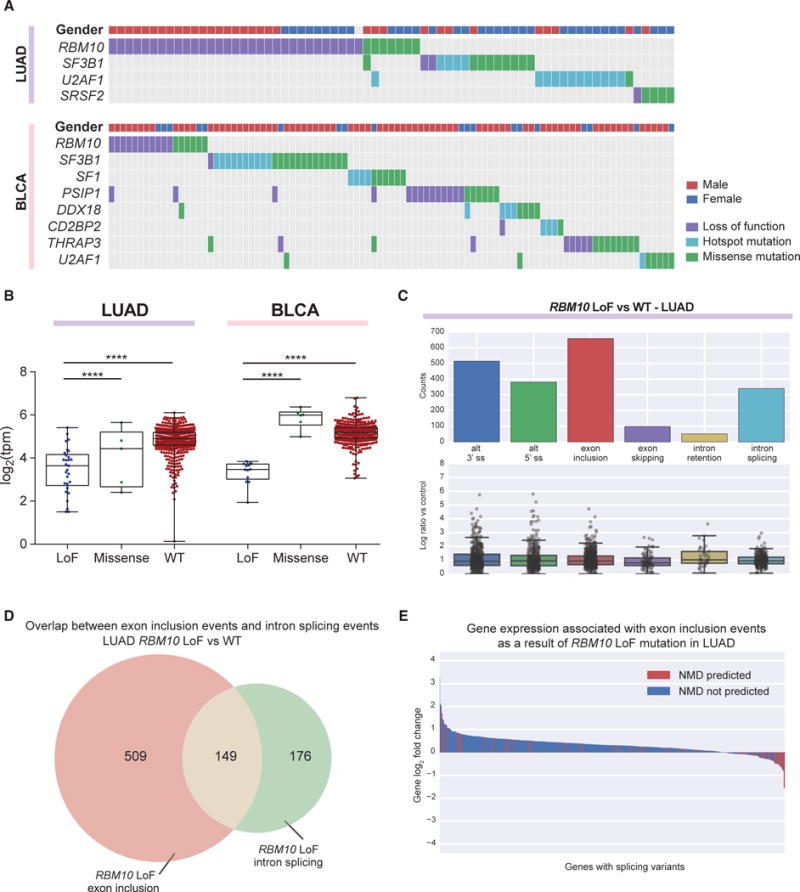
(A) RBM10 LoF mutations are mutually exclusive from other splicing factor gene mutations in LUAD and BLCA.
(B) LoF mutations in RBM10 lead to reduced mRNA expression in both LUAD and BLCA. Each point depicts a sample and the boxplot whiskers depict the complete data range. ****p < 0.0001 in all comparisons, Student’s t test.
(C) Differential splicing events associated with RBM10 LoF mutations in LUAD (corrected q-value < 0.05). Below each splicing event count, the PSI log2 fold change of each individual event is detailed in a boxplot.
(D) Exon inclusion is often concomitant with intron splicing associated with RBM10 LoF mutation in LUAD.
(E) Exons promoted by RBM10 LoF mutation in LUAD may be predicted to contain PTCs (red), leading to reduced gene expression compared to those that do not (blue).
See also Figure S3 and Tables S2 and S3.
We observed a significant overlap in exons included following RBM10 loss in LUAD and exons previously reported to be both excluded upon RBM10 overexpression and included following knockdown (Figure S3B). Interestingly, RBM10 expression has also been shown to correlate with retention of the introns flanking the exons that are skipped due to its activity (Wang et al., 2013; Figure S3C), and we observed the corresponding normal splicing of these introns upon RBM10 loss in LUAD (Figure 4D). The majority of genes with this pattern of altered splicing by RBM10 LoF mutation were upregulated compared to RBM10 WT samples, suggesting that RBM10-mediated cassette exon repression acts as an overall gene regulatory mechanism. We also observed that some RBM10-regulated exons contained a premature termination codon (PTC), which may cause the transcript to be targeted for nonsense-mediated decay (NMD) (Figure 4E). Genes predicted to contain these poison exons were significantly more likely to be downregulated compared to other genes containing RBM10 LoF mutation-induced inclusion events (p = 1.07e–10, Kruskal H test).
FUBP1 LoF Mutation Is Associated with Cassette Exon Events and Gene Downregulation in LGG
FUBP1 (Far upstream element-binding protein 1) was initially described to regulate MYC through binding to its far-upstream element (FUSE), and its overexpression can stimulate MYC expression (Duncan et al., 1994; He et al., 2000). More recently, FUBP1 has been described to bind to AT-rich exons and mediate exon skipping via repression of splicing at the second step reaction (Li et al., 2013). FUBP1 is located at chromosome 1p, and its mutation co-occurs in a subset of glioma samples with 1p deletion. Co-deletion of chromosome 1p and 19q in glioma (Brat et al., 2015), in particular oligodendroglioma, is a common and early event (Jenkins et al., 2006). LoF mutations of FUBP1 in the remaining allele would result in complete loss of FUBP1 in diploid tumor cells. Indeed, we observed significant association of FUBP1 LoF mutation with the oligodendroglioma histology subtype, chromosome 1p deletion, and reduced FUBP1 gene expression in mutant samples compared to WT samples with 1p deletion (Figure 5A).
Figure 5. FUBP1 LoF Mutations in LGG and Associated Differential Splicing.
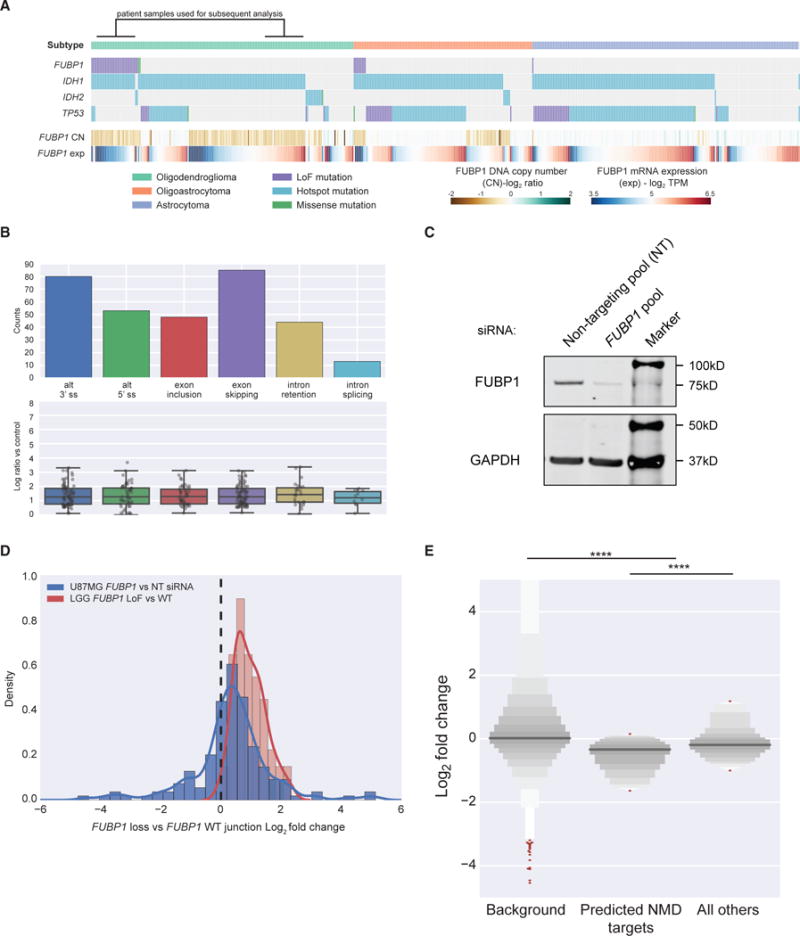
(A) FUBP1 LoF mutations are primarily associated with IDH1 mutation and the oligodendroglioma histology in LGG.
(B) Differential splicing events associated with FUBP1 LoF mutations in LGG (corrected q-value < 0.05). Below each splicing event count, the PSI log2 fold change of each individual event is detailed in a boxplot.
(C) Western blot of FUBP1 protein following transfection of FUBP1 siRNA pool or non-targeting (NT) siRNA pool.
(D) Log2 fold change of splice junctions identified in LGG patient samples (n = 155) in U87MG (blue) compared to LGG patient samples (red).
(E) Letter-value plot showing that genes with alternative splicing events in LGG patient samples (n = 163) are significantly downregulated compared to background (n = 22,982), and genes with splicing changes predicted to result in transcripts targeted by the NMD pathway (n = 79) are significantly downregulated compared to genes not predicted to be targeted (n = 94). The y axis data range has been terminated at −5, +5 for clarity.
See also Figure S4 and Tables S3 and S4.
To investigate the effects of FUBP1 LoF mutations on aberrant splicing and gene expression, we defined our comparison groups to be FUBP1 LoF mutation positive (n = 30) versus WT (n = 31) under IDH1 mutation and chromosome 1p/19q deletion-positive background (Figure 5A; Table S4). Differential splicing analysis identified exon inclusion and exclusion as major alternative splicing events (Figure 5B; Table S3). The FUBP1 RNA expression level and copy number in U87MG, a glioblastoma cell line, are similar to our control LGG patient group, offering an experimental setting to validate our analysis from patient samples. We transfected U87MG cells with a pool of small interfering RNAs (siRNAs) against FUBP1, and we performed RNA sequencing. We first confirmed FUBP1 knockdown at both protein (Figure 5C) and mRNA (67% depletion) levels. Differential splicing analysis showed a similar distribution of aberrant splicing events between transient FUBP1 knockdown in U87MG cells and in FUBP1 LoF patients (Figure S4A; Table S3). Though the overlap of significant splicing events defined by the default q-value threshold of 0.05 was small among events detected in genes that were expressed in both patient samples and U87MG cells (11/155 events), splicing junctions upregulated upon FUPB1 loss in patient samples showed similar, though weaker, upregulation in U87MG (Figure 5D), confirming that the observed splicing changes were modulated by the loss of FUBP1 (p value 4.38e–37, binomial test).
Mechanistically, FUBP1 has been shown to preferentially bind to and inhibit AT-rich exons (Li et al., 2013), and to explore this relationship we calculated the average AT content profiles of the cassette exons and the flanking two exons. Compared to background, we observed significantly higher AT content in all 3 exons of exon-skipping events (p < 0.00015 in all three comparisons, Student’s t test) (Figure S4B), an observation that was recapitulated in FUBP1 siRNA-treated U87MG cells (p < 0.00019 in all three comparisons, Student’s t test) (Figure S4C). Although not statistically significant, we also observed that exons promoted by mutant samples (exon inclusion events) had higher AT content near their 5′ ends compared to exons preferentially included by WT samples, perhaps contributing to this phenotype. Overall, genes with alternative splicing events of any type in patient samples (n = 163) were significantly more likely to be downregulated compared to background (n = 22,982; p = 4.7e–34, Kruskal H test) (Figure 5E), and, among these spliced genes, we observed that those with events predicted to result in a transcript degraded by the NMD pathway were downregulated further (p = 3.0e–4, Kruskal H test).
Given the proposed association between FUBP1 and MYC expression regulation, we also evaluated the potential functional consequences of FUBP1 LoF on MYC expression and downstream MYC signaling. Though we did not see significant reduction in MYC expression, there was a significant downregulation of MYC target genes associated with FUBP1 LoF mutations (Figure S4D; Table S5). We did not observe any MYC target genes to be alternatively spliced, indicating this downregulation was independent of FUBP1 functional splicing alterations. Interestingly, we observed that MYC target gene changes were also correlated in U87MG cells treated with siRNA against FUBP1 (Figure S4E), confirming the independent association between FUBP1 loss and MYC activity.
Driver Mutations in Splicing Factors Affect Cancer Cell-Autonomous Pathways and Immune Infiltration
While extensive studies have characterized the splicing aberrations associated with well-known splicing factor gene mutations, the understanding of how these mutations and splicing changes contribute to selective advantages during tumorigenesis remains unclear. Repeated observations of mutual exclusivity between different splicing factor driver mutations within the same disease (Figure 4A) (Papaemmanuil et al., 2013; Haferlach et al., 2014; Lindsley et al., 2015) suggest either their functional convergence at the pathway level or that cells cannot tolerate more than one splicing factor driver mutation. Hence, we conducted systematic pathway analysis in tumor types harboring driver mutations of the five genes (SF3B1, SRSF2, U2AF1, RBM10, and FUBP1) with confirmed on-target splicing deregulation (Figure 6A).
Figure 6. Pathway Enrichment Analysis Using Curated Gene Sets Indicates that Cancer Pathways Altered by Splicing Factor Mutations Are Lineage Specific.
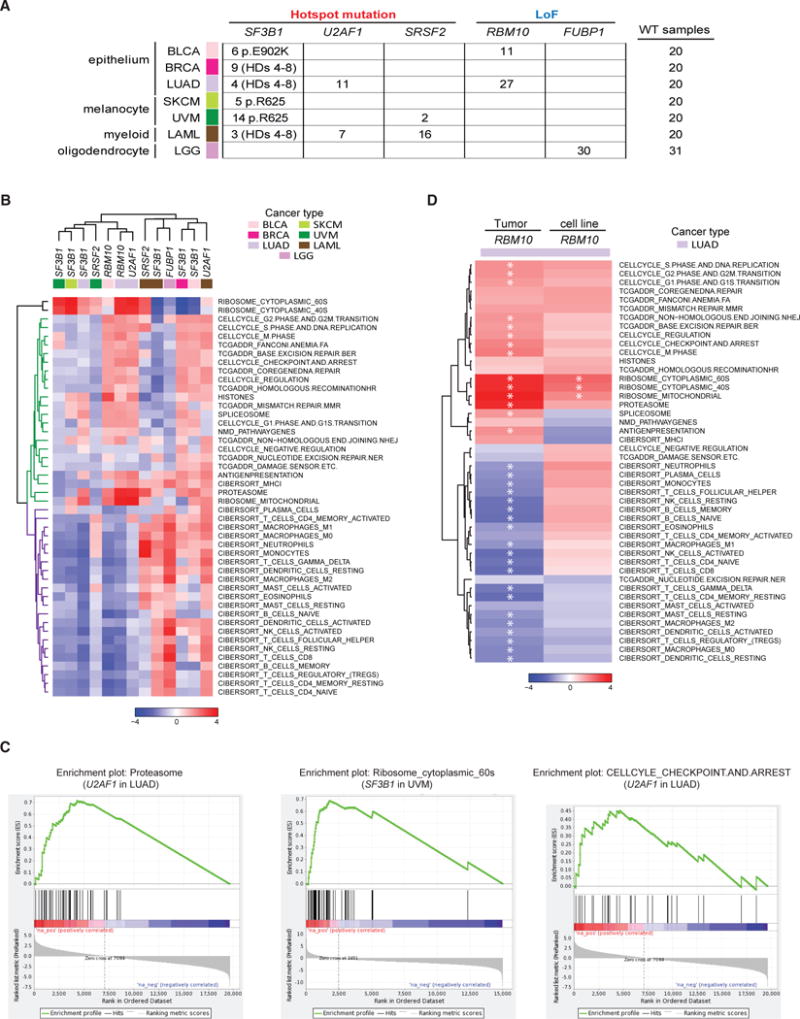
(A) Splicing factor gene mutations and their associated tumor cohorts used in pathway analyses.
(B) Heatmap of gene set enrichment analyses for all comparison groups generated using normalized enrichment scores (NESs) of 46 curated gene sets. Two distinct subclasses of gene sets are cell-autonomous pathways (green) and immune-related signatures (purple).
(C) Representative cancer hallmark gene sets upregulated in splicing factor mutant samples.
(D) Heatmap of NESs comparing patient tumor samples and cell lines, where each column represents the differential pathway modulation of RBM10 LoF mutants (n = 27 TCGA, n = 3 cell lines) versus RBM10 WT (n = 20 TCGA, n = 30 cell lines) of 46 curated gene sets. Significantly modulated gene sets (q value ≤ 0.05) are highlighted with an asterisk.
See also Figure S5 and Tables S4, S5, S6, and S7.
First, we performed gene set enrichment analysis (GSEA) using 50 hallmark gene sets (Subramanian et al., 2005) by comparing all mutant samples of each gene versus their WT control group, which was carefully selected to remove confounding factors of tumor subtype and other splicing factor gene mutations (Table S4). We then clustered all comparison groups and hallmark gene sets using normalized enrichment scores (NESs) (Figure S5A; Table S5). We observed that comparison groups generally clustered by tumor type or similar cell lineage, rather than by specific splicing factor mutations. For example, U2AF1 hotspot mutations in LUAD and RBM10 LoF mutations in epithelial tumors BLCA and LUAD group together, while SF3B1 hotspot mutations in melanomas SKCM and UVM and SRSF2 hotspot mutations in UVM group together. Moreover, certain splicing factor mutations in specific tumor types tended to associate with broad downregulation of cancer hallmark genes, such as SF3B1 hotspot mutations in SKCM and UVM, whereas SF3B1 mutations in BRCA and U2AF1 in LAML were associated with broad upregulation of the same hallmarks. This prompted us to further identify cancer hallmarks commonly regulated by different splicing factor gene mutations in the same tumor type. Within cohorts, hallmark gene sets related to immune response, cell cycle checkpoint and DNA damage response (DDR), and metabolism were associated with splicing factor mutations (Figures S5B–S5E).
Since hallmark gene sets tend to be broadly defined, we also conducted enrichment analysis using a set of custom gene sets containing more specific gene signatures of the hallmark pathways uncovered above. In addition, we included spliceosome, ribosome, proteasome, histone, and NMD pathway genes due to their functional relevance to the splicing process (Table S6). We then re-clustered comparison groups and gene sets using the NESs of these curated gene sets (Figure 6B; Table S7). Strikingly, this analysis revealed that gene sets can be clustered into two large groups: group 1 (colored green in Figure 6B) contains mostly cell-autonomous gene signatures of cell cycle, DDR, and essential cellular machineries; and group 2 (colored purple in Figure 6B) is composed of immune cell signatures. Among cell-autonomous gene sets (Figures 6B and 6C), proteasome genes were upregulated in multiple comparison groups. Ribosomal genes were strongly upregulated in SF3B1 hotspot mutants within SKCM and both SF3B1 and SRSF2 mutants in UVM, three subsets associated with general downregulation of most gene sets. Cell cycle-related gene sets tended to be more consistently upregulated in the splicing factor mutant samples of BLCA and LUAD (Figures 6B and 6C). Among immune cell signatures, we found that certain subgroups, and in particular FUBP1 in LGG, were associated with broad upregulation, suggesting that these conditions harbor an increased immune infiltration. Alternatively, multiple T cell signatures were consistently downregulated in SF3B1 mutants of UVM as well as splicing factor mutant subsets of BLCA and LUAD, suggesting that splicing factor mutations in these tumor types were associated with fewerT cell infiltrates. To test the hypothesis that the low immune cell enrichment scores are most likely due to less immune infiltrates in the tumor microenvironment, we compared lung adenocarcinoma cell lines with RBM10 LoF mutations to the WT (Table S4), and we compared the result with that from LUAD samples (Figure 6D). Three ribosome signatures were significantly upregulated in both comparisons, and other cell-autonomous signatures trended very similarly. However, we observed that most immune cell signatures were only significantly downregulated in patient tumor samples and not in cell lines. Since cancer cell lines are devoid of immune cells, we infer this is most likely due to reduced immune infiltrates in the tumor microenvironment.
DISCUSSION
Using matched DNA and RNA sequencing, we have surveyed 33 tumor types for somatic mutations of over 400 splicing factor genes, and we identified 119 with putative driver mutations. We observed that the most common mutations are mutually exclusive in each cohort, similar to prior hematological surveys (Papaemmanuil et al., 2013; Haferlach et al., 2014; Landau et al., 2015), and furthermore induce altered splicing, which is consistent across tumor lineages. Though splicing factor gene mutations were observed in all tumor types, we found that BLCA and UVM had a significantly higher frequency of putative driver mutations compared to other cohorts. Together, these results suggest that splicing deregulation by somatic mutation in cancer is broader than previously reported.
Curiously, though hotspot mutations in SF3B1 were found in several cohorts, we observed striking lineage specificity for certain amino acid changes. Melanomas of both SKCM and UVM strongly prefer p.R625C/H (21/33 non-silent mutations in HDs 4-12), while BRCA strongly prefer p.K700E (10/18 nonsilent mutations in HDs 4-12), the same most frequently mutated position in hematological malignancies, and p.E902K is only observed in BLCA. Lineage-specific hotspot mutations are likely the outcome of the interplay of several contributing factors, including nucleotide context mutability, gene-specific mutation rate in the tumor type, lineage-specific interacting partner proteins of a protein complex, and the mutational impact on cancer genes and pathways to confer survival advantage in a particular type of cancer. Deconvoluting these complex interactions will be essential to understand the selective pressures underlying these mutations.
How splicing factor gene mutations confer selective advantage to tumor cells is an area of active study. Since splicing factor gene mutations are likely to induce broad transcriptome changes, it is possible these changes can include splicing of oncogenes and tumor suppressors. In hematological malignancies, it has been demonstrated that somatic hotspot mutations in SRSF2 leads to mis-splicing and degradation of EZH2 (Kim et al., 2015), a gene known to be recurrently mutated in those diseases. In another study, SF3B1 mutations in chronic lymphocytic leukemia (CLL) were shown to lead to mis-splicing and the production of a truncated form of ATM, another gene frequently mutated in CLL (Ferreira et al., 2014). In both cases, these splicing factor mutations have been shown to be mutually exclusive with mutations of the aberrantly spliced target gene. In our analysis, we observe previously reported altered splicing of various cancer genes induced by splicing factor gene mutations, including EZH2 in LAML SRSF2 hotspot mutants and NUMB in LUAD RBM10 LoF mutants (Bechara et al., 2013). We also find additional unreported cancer gene alterations. For example, SF3B1 hotspot mutations in BRCA are associated with mis-splicing of CDH1, a gene with frequent LoF mutations in invasive lobular breast cancer (Desmedt et al., 2016). In another example, both RBM10 LoF and U2AF1 hotspot mutants in LUAD are associated with TSC2 mis-splicing, a tumor suppressor of the mTOR pathway (Krymskaya, 2003).
Given the multitude of genes impacted by mis-splicing due to splicing factor gene mutations, the downstream functional impact is unlikely to be solely due to the altered splicing of a single cancer gene. Instead, splicing factor mutations may cause a transcriptome-wide deregulation of normal splicing (spliceosome sickness), which induces broad transcriptional programs beneficial to the tumor. Overall, we observed that different splicing factor genes in the same tumor types are much more likely to be associated with deregulation of the same cancer pathways. These results support the idea that the observed mutual exclusivity of putative driver mutations within a tumor type might be due to functional redundancy, though we cannot rule out that co-occurrence of these mutations may be lethal. Previous functional studies of splicing factor mutations in SF3B1 and U2AF1 using non-hematological tumor cell lines (Zhou et al., 2015; Fei et al., 2016) indicated that the mutant allele is not essential for cell survival and does not provide a proliferation advantage in vitro. Our pathway analysis suggests that, in certain solid tumors, splicing factor mutations are associated with reduced immune infiltration and, therefore, may provide selective advantage to cancer cells through immune evasion. Unlike SF3B1 and U2AF1, RBM10 has been reported to regulate splicing of apoptosis and notch pathway genes, and functional studies of cancer cell lines in vitro and in vivo show that LoF mutations lead to enhanced colony formation or accelerated tumor growth (Bechara et al., 2013; Hernández et al., 2016; Zhao et al., 2017). Our analysis comparing RBM10 LoF mutations in tumor samples and in cancer cell lines complements the existing studies, and it proposes that loss of this splicing factor has an immunosuppressive role in addition to its cell-autonomous growth-promoting role.
Cancer-specific splicing changes are increasingly recognized to contribute to tumorigenesis via various mechanisms. Multiple oncogenes and tumor suppressors have been reported to express cancer-specific or treatment-resistant splice variants (Zhang and Manley, 2013). In another survey of the extent of somatic single-nucleotide variants (SNVs) altering splicing, a large number of SNVs are found to cause intron retention in tumor suppressors and loss of function through NMD or truncated proteins (Jung et al., 2015). Alternatively, splicing factors can act as proto-oncogene or tumor suppressors when their expression is altered in cancer (Anczukow et al., 2015; Jiang et al., 2016). The spectrum of splicing factor gene mutations that occur in multiple tumor types highlights somatic mutation as an important mechanism of splicing deregulation in cancer, the scope of which we are just starting to uncover. Collectively, these observations suggest deregulated RNA splicing as a hallmark of cancer. More functional studies are clearly needed to understand the impact of RNA-splicing changes and splicing factor mutations and, most importantly, their contribution to cancer development.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lihua Yu (Lihua_Yu@h3biomedicine.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
U87MG (male, glioblastoma) cells were obtained from ATCC (ATCC HTB-14) and cultured in ATCC-formulated Eagle’s Minimum Essential Medium (30-2003) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2 and 95% humidity. Cell authentication was conducted at IDEXX BioResearch using STR DNA profiling and found to be 100% matching markers listed in the ATCC database for U87MG cells, with no species contamination.
METHODS DETAILS
Compilation of splicing factor genes
We collected 1512 spliceosome and splicing related genes from three sources: 1)244 spliceosome proteins reported in (Hegele et al., 2012) from a comprehensive yeast two hybrid study using spliceosome components as bait, 2) 254 splicing factors and splicing related proteins annotated in (Barbosa-Morais et al., 2006) Table S6, and 3) 1100 genes from SpliceosomeDB (Cvitkovic and Jurica, 2013). The latter two are curated component lists derived from other publications. All gene identifiers are standardized into HUGO symbol and EntrezID. We prioritized the final list of 404 splicing factor genes (Table S1) by including all genes from sources 1) and 2), and genes from 3) if they are annotated in as “complex-SpliceosomeDB” or “class/family-SpliceosomeDB” excluding “common MS contaminants,” or if they belong to the same protein families from any genes above. The reason we used this conservative approach to prioritize genes in 3) is that some genes, though identified by mass spectrometry experiments in certain spliceosomes, have undefined functions and orthologs across species and hence could simply be contaminants in sample preparations found associated with human spliceosomes.
Detection of somatic mutation and identification of splicing factor genes with driver mutations
Somatic mutation data was provided by TCGA MC3 working group (see Key Resources Table). We considered a sample “splicing factor WT” (and therefore appropriate for use in differential splicing or gene expression contexts) if there were no non-silent mutations in any known splicing factor genes.
MutSigCV analytical results were downloaded from Broad TCGA Firehose dashboard on September 2016 (https://confluence.broadinstitute.org/display/GDAC/Dashboard-Analyses). MutSig2CV3.1 results were used when available. A q-value cut-off of 0.1 was used to define significantly mutated genes in each cohort. We excluded PAAD cohorts from this analysis as samples in this cohort typically had extremely low non-silent mutation counts.
For the ratiometric method, we defined mutational hotspots (HS) as missense or in-frame deletion mutations at the same protein position > = 3 pan-TCGA. Loss of function (LoF) mutations were defined as any of the following mutation classifications (Frame_Shift_Del, Frame_Shift_Ins, Nonsense, Splice_Site). We then calculated %HS or %LoF as the total number of hotspot or LoF mutation-positive samples divided by total number of non-silent mutations per gene per cohort. The “pancan” cohort encompasses all samples in TCGA. We used empirical cut-offs to define genes as HS or LoF type, specifically:
If %HS > = 30% and %LoF ≤ 20% and HS counts > = 3, a gene is called “hotspot” in that cohort, and if %LoF > = 30% and %HS ≤ 20% and LoF mutation counts > = 10, a gene is called “LoF” type in that cohort.
An extended ratiometric method published by Tokheim et al. (Tokheim et al., 2016) called “20/20+” was used as an additional evaluator of putative driver splicing factors. This method uses a random forest-based method trained on known cancer driver genes to identify cohort-level cutoffs appropriate for this identification. For each cohort (as well as the “pancan” cohort), the pre-trained random forest classifier provided by Tokheim et al. was used to assign Benjamini-Hochberg corrected q-values to each gene with q < 0.1 used as a cutoff for significance. These results are given in Table S1. All genes were plotted using oncogene score and tumor suppressor gene score provided by 20/20+, with significant genes labeled and colored based on the larger of the two scores (i.e., red genes have higher oncogene score than tumor suppressor score, whereas blue genes the opposite) (Figure S1C).
Detection of additional samples with hotspot mutations of SF3B1, U2AF1, and SRSF2
Following read alignment by STAR allowing multimapping reads of RNaseq files, samples were interrogated for functional hotspot mutations in known driver splicing genes SF3B1, U2AF1, and SRSF2. For SF3B1, amino acids p.E622, p.Y623, p.R625, p.N626, p.H662, p.T663, p.K666, p.K700, p.V701, p.I704, p.G740, p.K741, p.G742, and p.D781 (Darman et al., 2015, Obeng et al., 2016) were used. For U2AF1, amino acids p.S34, p.R156, and p.Q157 were used (Papaemmanuil et al., 2013; Lindsley et al., 2015). For SRSF2, mutations and deletions in/near amino acid p.P95 were used (Zhang et al., 2015, Kim et al., 2015). Samtools (Li et al., 2009) mpileup was used for genotyping, and only uniquely mapped reads were allowed. A minimum total read coverage of 10 was imposed for the codon encoding amino acid changes in these genes as well as a minimum read coverage of 4 supporting the change. Mutations with allele frequency < 5% were ignored. We also performed visual inspection using Integrative Genomic Viewer (IGV, Robinson et al., 2011) and indel mis-calls were manually corrected.
Prioritization of genes for differential splicing and pathway analysis
We prioritized two groups of genes for in-depth differential splicing and pathway analysis. Group 1 includes SF3B1, SRSF2 and U2AF1. Driver mutations of these genes are well reported with high frequency in hematological tumors and their associated splicing changes are well studied. The goal is to understand how similar or potentially different their somatic mutations and their associated splicing changes are pan-cancer. Group 2 includes other genes with exceptional high mutation frequency and compelling hotspot or LoF mutation patterns. RBM10 and FUBP1 are the top 2 splicing factor genes by frequency of mutation, both with a strong LoF mutation pattern.
Principal component analysis of mutant and wild-type splicing factor samples
Junction counts for all TCGA samples were obtained from Omicsoft® OncoLand® 2016 Q2 release and converted to PSI. SF3B1 mutation information was obtained from TCGA pan-cancer MC3 data and validated using RNA-Seq data. Alternative 3′ splice sites promoted by SF3B1 mutant (HD4-8) activity were obtained from Darman et al., 2015 (Darman et al., 2015). Alternative 3′ splice sites and exon inclusion events promoted by SF3B1 p.E902K versus splicing factor WT samples in BLCA (Table S3) are used to stratify patient samples in Figure S2B.
Letter-value plot
Letter-value plots (Hofmann et al., 2017) are an extension of the standard boxplot for large-scale data. The seaborn python package (see Key Resources Table) was used with the depth parameter “proportion,” where 0.007 is assumed the fraction of samples which are outliers in a given cohort. Letter-value boxes (percentiles of the data, which start at 50% and decrease by half each iteration) are drawn until this fraction is reached. Boxes are colored based on the density of points within, where darker colors indicate higher density.
Pathway analysis
Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was performed using cancer hallmarks and the curated gene sets. Default parameters were chosen except the minimum gene set size was set to 5. The gene expression for each cohort was defined as the mean Log2 transcripts per million (TPM) (i.e., log2(1+tpm)). The R package limma (Ritchie et al., 2015) was used for differential gene expression analysis after filtering out low-expressed genes (maximum TPM < 3), and gene lists ranked by moderated t-statistic values were used as input for GSEA.
Clustering analysis of normalized enrichment score (NES) was done using the R (R Development Core Team, 2011) package using heatmap software from (https://raw.githubusercontent.com/obigriffith/biostar-tutorials/master/Heatmaps/heatmap.3.R)
Curation of gene sets (Table S6)
The following custom gene sets for enrichment analysis:
Cell cycle gene sets were obtained from QIAGEN human cell cycle PCR array Cat. No. PAHS-020Z (http://www.sabiosciences.com/rt_pcr_product/HTML/PAHS-020Z.html).
DNA damage response/repair (DDR) gene sets were shared with us by TCGA PanCanAtlas DDR analysis working group.
Immune gene sets were obtained from the publication by Newman et al., 2015.
Proteasome gene set was obtained from HUGO Gene Nomenclature Committee (HGNC) under gene family proteasome (http://www.genenames.org/cgi-bin/genefamilies/set/690) and Kyoto Encyclopedia of Genes and Genomes (KEGG) proteasome (http://www.genome.jp/kegg/pathway/hsa/hsa03050.html).
Ribosome gene sets were obtained from Ribosomal Protein Gene Database (RPG) (http://ribosome.med.miyazaki-u.ac.jp/) and KEGG ribosome (http://www.genome.jp/kegg/pathway/hsa/hsa03010.html).
Spliceosome is in Table S1.
Nonsense mediated decay (NMD) gene set was curated based on two publications (Nicholson et al., 2010; Kervestin and Jacobson, 2012).
Histone gene list were obtained from HUGO Gene Nomenclature Committee Histone gene family (http://www.genenames.org/cgi-bin/genefamilies/set/864)
Antigen presentation gene set was from Reactome: (http://software.broadinstitute.org/gsea/msigdb/cards/REACTOME_ANTIGEN_PRESENTATION_FOLDING_ASSEMBLY_AND_PEPTIDE_LOADING_OF_CLASS_I_MHC.html). It captures the key elements, while excluding things that are redundant from other customer gene lists (e.g., proteasome).
FUBP1 Knockdown in U87MG and RNA Sequencing
U87MG cells were obtained from ATCC (ATCC HTB-14) and cultured in ATCC-formulated Eagle’s Minimum Essential Medium (30-2003) supplemented with 10% FBS. ON-TARGETplus Non-targeting siRNA Pool (D-001810-10-05) and ON-TARGETplus Human FUBP1 siRNA SMARTpool (L-011548-00-0005) were obtained from Dharmacon. To knockdown FUBP1,250,000 U87MG cells were seeded per well in six-well plates. On the second day, either the non-targeting siRNA pool or the human FUBP1 siRNA pool was transfected into U87MG cells in quadruplicates using Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific), according to the manufacturer’s manual. The final concentration of the siRNA pool was 50 nM in each well; 3 days after transfection, medium was refreshed. At 5 days post-transfection, one well of either non-targeting siRNA pool- or FUBP1 siRNA pool-transfected cells was harvested in radio immunoprecipitation assay (RIPA) buffer supplemented with proteasome complete protease inhibitor cocktail and PhosStop phosphatase inhibitor cocktail (Roche Life Science) for western blot analysis to examine the knockdown efficiency. Specially, equal amounts of protein lysates were loaded onto 4%-12% NuPAGE Bis-Tris gels (Thermo Fisher Scientific) before being transferred onto Nitrocellulose membrane using the iBlot2 dry blotting system (Thermo Fisher Scientific). The membrane was blocked with LI-COR buffer and then incubated with rabbit polyclonal anti-FUBP1 antibody (Abcam ab181111) and monoclonal anti-GAPDH antibody (Sigma G8795) overnight in a cold room. On the second day, the membrane was washed three times with tris-buffered saline Tween 20 (TBST) and incubated with LI-COR IRdye secondary antibodies before TBST wash, and it was scanned and quantified using LI-COR Odyssey imaging system. For RNA extraction, the remaining three wells for each transfection were harvested with RNeasy lysis buffer, and total RNAs were extracted using RNeasy column kit (QIAGEN), following the manufacturer’s protocol. Extracted total RNAs were analyzed on Agilent Tapestation to ensure RNA quality before being submitted to Beijing Genomic Institute (BGI) for polyA+ RNA sequencing (RNA-seq) library preparation and sequenced on Illumina Hiseq 4000.
Differential Splicing Analysis and NMD Prediction
Differential splicing analysis was performed similar to previously described methodology (Darman et al., 2015). In brief, raw sequence reads were extracted from BAM files made available through TCGA, then aligned using STAR using two-pass alignment (Dobin et al., 2013) to human reference genome GRCh37/hg19. Junction PSI was calculated for all sets of junctions that shared a single common ss as the number of raw reads supporting that junction divided by the total number of reads in all junctions sharing that ss. We accounted for intron retention in PSI calculations by counting reads that completely overlapped a 6-nt window around the ss (3 nt within the intron and 3 nt within the exon) as intron retention reads. Read count for each junction was pooled in the FUBP1 siRNA versus non-targeting siRNA cell line comparison to increase statistical power. Each PSI measurement was converted to log odds via the formula log(p/(1-p)) before being compared using either a moderated t test (Ritchie et al., 2015) (patient samples) or binomial test (cell lines) between cohorts. False discovery rate (FDR)-corrected q-values < 0.05 for junctions promoted by the case or mutant cohort (alternative junction) were considered significant. To be reported as a splicing event, at least one junction promoted by the control, orWT case (canonical junction[s]), that shared an ss with the alternative junction was also required to have an FDR-corrected q-value < 0.2, and these are reported in Table S3. For intron retention events, both 5′ and 3′ exon-intron boundaries were required to be significant, and a minimum median threshold for mean intron read coverage over all samples in that cohort was set at 0.1 in order to reduce false positives. NMD prediction was performed for each splicing event by first identifying all RefSeq transcripts that contained an intron that shares an ss with the mutation-promoting junction and then determining the novel peptide sequence that resulted from altering that transcript to contain the splicing event (Darman et al., 2015). Events were predicted to be NMD-targeted if all affected transcripts contained a stop codon > 50 nt from the final exon-exon junction.
QUANTIFICATION AND STATISTICAL ANALYSIS
The details of each statistical test are contained within the Results, including the total number of samples (n) in each case and control condition, as well as the test used. Unless otherwise specified, p values less than 0.05 were considered significant. Multiple testing correction was performed where applicable using the Benjamini-Hochberg FDR correction, and q-values less than 0.05 were considered significant unless otherwise specified.
Differential Gene Expression
Gene differential expression was performed using the limma package following quantification using Kallisto (Bray et al., 2016).
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA sequencing data from U87MG cells reported in this paper is GEO: GSE100530. All other data used are available from the Genomic Data Commons (https://portal.gdc.cancer.gov/).
Supplementary Material
Document S1. Figures S1–S5
Table S1. 404 Spliceosome Genes, Annotation of Membership of the Final 119 Gene List, and Mutation Frequency of the 119 Genes across All Cohorts Including Pancan, Related to Figure 1
Table S2. Samples with SF3B1, U2AF1, SRSF2, and RBM10 Mutations, Including Samples Identified Using RNA-Seq in LAML, Related to Figures 2, 3, and 4
Table S3. Differentially Spliced Junctions Detected in SF3B1 p.E902K Mutation in BLCA, U2AF1 Hotspot Mutation in LUAD and LAML, SRSF2 Hotspot Mutation in UVM, RBM10 LoF in LUAD and BLCA, FUBP1 LoF in LGG, and FUBP1 siRNA KD in U87MG Cells, Related to Figures 2, 3, 4, and 5
Table S4. All Samples Used for Differential Gene Expression and GSEA, Related to Figures 5 (FUBP1) and 6
Table S5. GSEA Output (NES, FDR, and NOM.p) of All Comparison Groups Using MSigDB C1: Hallmark Gene Sets, Related to Figure 6
Table S6. Curated Gene Sets, Related to Figure 6
Table S7. GSEA Output (NES, FDR, and NOM.p) of All Comparison Groups Using Curated Gene Sets, Related to Figure 6
KEY RESOURCES TABLE.
Highlights.
119 splicing factor genes carry putative driver mutations in one or more cancer types
BLCA and UVM carry more driver splicing factor mutations than expected by chance
Common splicing factor mutations associated with lineage-independent altered splicing
Mutations are associated with deregulation of cell-autonomous pathways and immune infiltration
Acknowledgments
We thank all H3 Biomedicine employees for their support in this project. We would also like to thank Collin Tokheim for his help and insight regarding the use of 20/20+. The results shown here are in whole or part based upon data generated by The Cancer Genome Atlas (TCGA) Research Network (https://cancergenome.nih.gov). The members of The Cancer Genome Atlas Research Network for this project are listed in the Supplemental Information. TCGA Research Network is supported by the following NIH grants: U54 HG003273 (Richard A. Gibbs), U54 HG003067 (Stacey Gabriel; Eric S. Lander [contact]), U54 HG003079 (Richard K. Wilson), U24 CA143799 (Terence Paul Speed; Paul T. Spellman [contact]), U24 CA143835 (Ilya Shmulevich), U24 CA143840 (Marc Ladanyi; Chris Sander [contact]), U24 CA143843 (Richard A. Gibbs; David AndrewWheeler[contact]), U24 CA143845 (LyndaChin [contact]; Gad Getz), U24 CA143848 (David N. Hayes; Charles M. Perou [contact]), U24 CA143858 (Joshua Stuart; Christopher Benz; David H. Haussler [contact]), U24 CA143866 (Marco Antonio Marra), U24 CA143867 (Stacey Gabriel; Matthew L. Meyerson [contact]), U24 CA143882 (Stephen B. Baylin; Peter W. Laird [contact]), U24 CA143883 (Gordon B. Mills; John N. Weinstein [contact]; W. K. Alfred Yung), U24 CA144025 (Raju S. Kucherlapati), and P30 CA016672 (Gordon B. Mills).
DECLARATION OF INTERESTS
Michael Seiler, Peter G. Smith, Ping Zhu, Silvia Buonamici, and Lihua Yu are employees of H3 Biomedicine, Inc. Parts of this work are the subject of a patent application: WO2017040526 titled “Splice variants associated with neomorphic sf3b1 mutants.” Shouyoung Peng, Anant A. Agrawal, James Palacino, and Teng Teng are employees of H3 Biomedicine, Inc. Andrew D. Cherniack, Ashton C. Berger, and Galen F. Gao receive research support from Bayer Pharmaceuticals. Gordon B. Mills serves on the External Scientific Review Board of Astrazeneca. Anil Sood is on the Scientific Advisory Board for Kiyatec and is a shareholder in BioPath. Jonathan S. Serody receives funding from Merck, Inc. Kyle R. Covington is an employee of Castle Biosciences, Inc. Preethi H. Gunaratne is founder, CSO, and shareholder of NextmiRNA Therapeutics. Christina Yau is a part-time employee/consultant at NantOmics. Franz X. Schaub is an employee and shareholder of SEngine Precision Medicine, Inc. Carla Grandori is an employee, founder, and shareholder of SEngine Precision Medicine, Inc. Robert N. Eisenman is a member of the Scientific Advisory Boards and shareholder of Shenogen Pharma and Kronos Bio. Daniel J. Weisenberger is a consultant for Zymo Research Corporation. Joshua M. Stuart is the founder of Five3 Genomics and shareholder of NantOmics. Marc T. Goodman receives research support from Merck, Inc. Andrew J. Gentles is a consultant for Cibermed. Charles M. Perou is an equity stock holder, consultant, and Board of Directors member of BioClassifier and GeneCentric Diagnostics and is also listed as an inventor on patent applications on the Breast PAM50 and Lung Cancer Subtyping assays. Matthew Meyerson receives research support from Bayer Pharmaceuticals; is an equity holder in, consultant for, and Scientific Advisory Board chair for OrigiMed; and is an inventor of a patent for EGFR mutation diagnosis in lung cancer, licensed to LabCorp. Eduard Porta-Pardo is an inventor of a patent for domainXplorer. Han Liang is a shareholder and scientific advisor of Precision Scientific and Eagle Nebula. Da Yang is an inventor on a pending patent application describing the use of antisense oligonucleotides against specific lncRNA sequence as diagnostic and therapeutic tools. Yonghong Xiao was an employee and shareholder of TESARO, Inc. Bin Feng is an employee and shareholder of TESARO, Inc. Carter Van Waes received research funding for the study of IAP inhibitor ASTX660 through a Cooperative Agreement between NIDCD, NIH, and Astex Pharmaceuticals. Raunaq Malhotra is an employee and shareholder of Seven Bridges, Inc. Peter W. Laird serves on the Scientific Advisory Board for AnchorDx. Joel Tepper is a consultant at EMD Serono. Kenneth Wang serves on the Advisory Board for Boston Scientific, Microtech, and Olympus. Andrea Califano is a founder, shareholder, and advisory board member of DarwinHealth, Inc. and a shareholder and advisory board member of Tempus, Inc. Toni K. Choueiri serves as needed on advisory boards for Bristol-Myers Squibb, Merck, and Roche. Lawrence Kwong receives research support from Array BioPharma. Sharon E. Plon is a member of the Scientific Advisory Board for Baylor Genetics Laboratory. Beth Y. Karlan serves on the Advisory Board of Invitae.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and seven tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.088.
AUTHOR CONTRIBUTIONS
Conceptualization, L.Y.; Methodology, L.Y., M.S., S.P., and T.T.; Software, M. S. and S.P.; Investigation, M.S., S.P., L.Y., T.T., A.A.A., and J.P.; Visualization, M.S., S.P., A.A.A., and S.B.; Writing – Original Draft, M.S., S.P., A.A.A., J.P., T.T., P.Z., S.B., and L.Y.; Writing – Review & Editing, M.S., S.P., A.A.A., P.Z., P.G.S., S.B., and L.Y.; Resources, TCGA Research Network; Funding Acquisition, TCGA Research Network; Project Administration, TCGA Research Network; Supervision, S.B. and L.Y.
References
- Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. SRSF1-regulated alternative splicing in breast cancer. Mol Cell. 2015;60:105–117. doi: 10.1016/j.molcel.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Carmo-Fonseca M, Aparício S. Systematic genome-wide annotation of spliceosomal proteins reveals differential gene family expansion. Genome Res. 2006;16:66–77. doi: 10.1101/gr.3936206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara EG, Sebestyén E, Bernardis I, Eyras E, Valcarcel J. RBM5, 6, and 10 differentially regulate NUMB alternative splicing to control cancer cell proliferation. Mol Cell. 2013;52:720–733. doi: 10.1016/j.molcel.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J, et al. Australian Pancreatic Cancer Genome Initiative Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA, Rheinbay E, Miller CR, Vitucci M, Morozova O, et al. Cancer Genome Atlas Research Network Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–2498. doi: 10.1056/NEJMoa1402121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. A pan-cancer analysis oftranscriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS ONE. 2014;9:e87361. doi: 10.1371/journal.pone.0087361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretu C, Schmitzová J, Ponce-Salvatierra A, Dybkov O, De Laurentiis EI, Sharma K, Will CL, Urlaub H, Lührmann R, Pena V. Molecular architecture of SF3b and structural consequences of its cancer-related mutations. Mol Cell. 2016;64:307–319. doi: 10.1016/j.molcel.2016.08.036. [DOI] [PubMed] [Google Scholar]
- Cvitkovic I, Jurica MS. Spliceosome database: a tool for tracking components of the spliceosome. Nucleic Acids Res. 2013;41:D132–D141. doi: 10.1093/nar/gks999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep. 2015;13:1033–1045. doi: 10.1016/j.celrep.2015.09.053. [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol. 2015;11:e1004105. doi: 10.1371/journal.pcbi.1004105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmedt C, Zoppoli G, Gundem G, Pruneri G, Larsimont D, Fornili M, Fumagalli D, Brown D, Rothe F, Vincent D, et al. Genomic characterization of primary invasive lobular breast cancer. J Clin Oncol. 2016;34:1872–1881. doi: 10.1200/JCO.2015.64.0334. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, Levens D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8:465–480. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, Varmus H. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet. 2016;12:e1006384. doi: 10.1371/journal.pgen.1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira PG, Jares P, Rico D, Gómez-López G, Martínez-Trillos A, Villamor N, Ecker S, González-Pérez A, Knowles DG, Monlong J, et al. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014;24:212–226. doi: 10.1101/gr.152132.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferlach T, Nagata Y, Grossmann V, Okuno Y, Bacher U, Nagae G, Schnittger S, Sanada M, Kon A, Alpermann T, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Liu J, Collins I, Sanford S, O’Connell B, Benham CJ, Levens D. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 2000;19:1034–1044. doi: 10.1093/emboj/19.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegele A, Kamburov A, Grossmann A, Sourlis C, Wowro S, Weimann M, Will CL, Pena V, Lührmann R, Stelzl U. Dynamic protein-protein interaction wiring of the human spliceosome. Mol Cell. 2012;45:567–580. doi: 10.1016/j.molcel.2011.12.034. [DOI] [PubMed] [Google Scholar]
- Hernández J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13:466–472. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintzsche JD, Gorden NT, Amato CM, Kim J, Wuensch KE, Robinson SE, Applegate AJ, Couts KL, Medina TM, Wells KR, et al. Whole-exome sequencing identifies recurrent SF3B1 R625 mutation and comutation of NF1 and KIT in mucosal melanoma. Melanoma Res. 2017;27:189–199. doi: 10.1097/CMR.0000000000000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann H, Wickham H, Kafadar K. Letter-Value Plots: Boxplots for Large Data. J Comput Graph Stat. 2017;26:467–477. [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 2015;25:14–26. doi: 10.1101/gr.181016.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, Flynn H, Passe S, Felten S, Brown PD, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res. 2006;66:9852–9861. doi: 10.1158/0008-5472.CAN-06-1796. [DOI] [PubMed] [Google Scholar]
- Jeromin S, Weissmann S, Haferlach C, Dicker F, Bayer K, Grossmann V, Alpermann T, Roller A, Kohlmann A, Haferlach T, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28:108–117. doi: 10.1038/leu.2013.263. [DOI] [PubMed] [Google Scholar]
- Jiang L, Huang J, Higgs BW, Hu Z, Xiao Z, Yao X, Conley S, Zhong H, Liu Z, Brohawn P, et al. Genomic landscape survey identifies SRSF1 as a key oncodriver in small cell lung cancer. PLoS Genet. 2016;12:e1005895. doi: 10.1371/journal.pgen.1005895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JJ, Teer JK, Cherukuri PF, Hansen NF, Loftus SK, Chong K, Mullikin JC, Biesecker LG, NIH Intramural Sequencing Center (NISC) Massively parallel sequencing of exons on the X chromosome identifies RBM10 as the gene that causes a syndromic form of cleft palate. Am J Hum Genet. 2010;86:743–748. doi: 10.1016/j.ajhg.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet. 2015;47:1242–1248. doi: 10.1038/ng.3414. [DOI] [PubMed] [Google Scholar]
- Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nat Rev Mol Cell Biol. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell. 2015;27:617–630. doi: 10.1016/j.ccell.2015.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krymskaya VP. Tumour suppressors hamartin and tuberin: intracellular signalling. Cell Signal. 2003;15:729–739. doi: 10.1016/s0898-6568(03)00040-8. [DOI] [PubMed] [Google Scholar]
- Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, Kluth S, Bozic I, Lawrence M, Bottcher S, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–530. doi: 10.1038/nature15395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang Z, Zhou X, Cheng Y, Xie Z, Manley JL, Feng Y. Far upstream element-binding protein 1 and RNA secondary structure both mediate second-step splicing repression. Proc Natl Acad Sci USA. 2013;110:E2687–E2695. doi: 10.1073/pnas.1310607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, Pigneux A, Wetzler M, Stuart RK, Erba HP, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125:1367–1376. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodys-plastic syndrome. Nat Commun. 2015;6:6042. doi: 10.1038/ncomms7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Visconte V, Sakaguchi H, Jankowska AM, Abu Kar S, Jerez A, Przychodzen B, Bupathi M, Guinta K, Afable MG, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119:3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, Piscuoglio S, Wen HC, Lim RS, Shen R, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–189. doi: 10.1002/path.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Maßhöfer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelamraju Y, Gonzalez-Perez A, Bhat-Nakshatri P, Nakshatri H, Janga SC. Mutational landscape of RNA-binding proteins in human cancers. RNA Biol. 2018;15:115–129. doi: 10.1080/15476286.2017.1391436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration ofcell subsets from tissue expression profiles. Nat Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Mühlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci. 2010;67:677–700. doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell. 2016;30:404–417. doi: 10.1016/j.ccell.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia. 2015;29:909–917. doi: 10.1038/leu.2014.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P, Yoon CJ, Ellis P, Wedge DC, Pellagatti A, et al. Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium Clinical and biological implications of driver mutations in myelodys-plastic syndromes. Blood. 2013;122:3616–3627. doi: 10.1182/blood-2013-08-518886. quiz 3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papasaikas P, Valcárcel J. The spliceosome: The ultimate RNA chaperone and sculptor. Trends Biochem Sci. 2016;41:33–45. doi: 10.1016/j.tibs.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Patnaik MM, Lasho TL, Finke CM, Hanson CA, Hodnefield JM, Knudson RA, Ketterling RP, Pardanani A, Tefferi A. Spliceosome mutations involving SRSF2, SF3B1, and U2AF35 in chronic myelomonocytic leukemia: prevalence, clinical correlates, and prognostic relevance. Am J Hematol. 2013;88:201–206. doi: 10.1002/ajh.23373. [DOI] [PubMed] [Google Scholar]
- Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122:999–1006. doi: 10.1182/blood-2013-01-480970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2011. [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebestyén E, Singh B, Miñana B, Pagès A, Mateo F, Pujana MA, Valcarcel J, Eyras E. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016;26:732–744. doi: 10.1101/gr.199935.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokheim CJ, Papadopoulos N, Kinzler KW, Vogelstein B, Karchin R. Evaluating the evaluation of cancer driver genes. Proc Natl Acad Sci USA. 2016;113:14330–14335. doi: 10.1073/pnas.1616440113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Gogol-Döring A, Hu H, Fröhler S, Ma Y, Jens M, Maaskola J, Murakawa Y, Quedenau C, Landthaler M, et al. Integrative analysis revealed the molecular mechanism underlying RBM10-mediated splicing regulation. EMBO Mol Med. 2013;5:1431–1442. doi: 10.1002/emmm.201302663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Romfo CM, Nilsen TW, Green MR. Functional recognition of the 3′ splice site AG by the splicing factor U2AF35. Nature. 1999;402:832–835. doi: 10.1038/45590. [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science. 2016;353:904–911. doi: 10.1126/science.aag0291. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- Zhang J, Manley JL. Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 2013;3:1228–1237. doi: 10.1158/2159-8290.CD-13-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA. 2015;112:E4726–E4734. doi: 10.1073/pnas.1514105112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun Y, Huang Y, Song F, Huang Z, Bao Y, Zuo J, Saffen D, Shao Z, Liu W, Wang Y. Functional analysis reveals that RBM10 mutations contribute to lung adenocarcinoma pathogenesis by deregulating splicing. Sci Rep. 2017;7:40488. doi: 10.1038/srep40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Derti A, Ruddy D, Rakiec D, Kao I, Lira M, Gibaja V, Chan H, Yang Y, Min J, et al. A chemical genetics approach for the functional assessment of novel cancer genes. Cancer Res. 2015;75:1949–1958. doi: 10.1158/0008-5472.CAN-14-2930. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1–S5
Table S1. 404 Spliceosome Genes, Annotation of Membership of the Final 119 Gene List, and Mutation Frequency of the 119 Genes across All Cohorts Including Pancan, Related to Figure 1
Table S2. Samples with SF3B1, U2AF1, SRSF2, and RBM10 Mutations, Including Samples Identified Using RNA-Seq in LAML, Related to Figures 2, 3, and 4
Table S3. Differentially Spliced Junctions Detected in SF3B1 p.E902K Mutation in BLCA, U2AF1 Hotspot Mutation in LUAD and LAML, SRSF2 Hotspot Mutation in UVM, RBM10 LoF in LUAD and BLCA, FUBP1 LoF in LGG, and FUBP1 siRNA KD in U87MG Cells, Related to Figures 2, 3, 4, and 5
Table S4. All Samples Used for Differential Gene Expression and GSEA, Related to Figures 5 (FUBP1) and 6
Table S5. GSEA Output (NES, FDR, and NOM.p) of All Comparison Groups Using MSigDB C1: Hallmark Gene Sets, Related to Figure 6
Table S6. Curated Gene Sets, Related to Figure 6
Table S7. GSEA Output (NES, FDR, and NOM.p) of All Comparison Groups Using Curated Gene Sets, Related to Figure 6