

Abstract
Aims
Heart failure (HF) is an impending complication to myocardial infarction. We hypothesized that the degree of complement activation reflects severity of HF following acute myocardial infarction.
Methods and results
The LEAF trial (LEvosimendan in Acute heart Failure following myocardial infarction) evaluating 61 patients developing HF within 48 h after percutaneous coronary intervention‐treated ST‐elevation myocardial infarction herein underwent a post hoc analysis. Blood samples were drawn from inclusion to Day 5 and at 42 day follow‐up, and biomarkers were measured with enzyme immunoassays. Regional myocardial contractility was measured by echocardiography as wall motion score index (WMSI). The cardiogenic shock group (n = 9) was compared with the non‐shock group (n = 52). Controls (n = 44) were age‐matched and sex‐matched healthy individuals. C4bc, C3bc, C3bBbP, and sC5b‐9 were elevated in patients at inclusion compared with controls (P < 0.01). The shock group had higher levels compared with the non‐shock group for all activation products except C3bBbP (P < 0.05). At Day 42, all products were higher in the shock group (P < 0.05). In the shock group, sC5b‐9 correlated significantly with WMSI at baseline (r = 0.68; P = 0.045) and at Day 42 (r = 0.84; P = 0.036). Peak sC5b‐9 level correlated strongly with WMSI at Day 42 (r = 0.98; P = 0.005). Circulating endothelial cell activation markers sICAM‐1 and sVCAM‐1 were higher in the shock group during the acute phase (P < 0.01), and their peak levels correlated with sC5b‐9 peak level in the whole HF population (r = 0.32; P = 0.014 and r = 0.30; P = 0.022, respectively).
Conclusions
Complement activation discriminated cardiogenic shock from non‐shock in acute ST‐elevation myocardial infarction complicated by HF and correlated with regional contractility and endothelial cell activation, suggesting a pathogenic role of complement in this condition.
Keywords: Complement activation, Inflammation, Myocardial infarction, Acute heart failure, Cardiogenic shock, Wall motion score index
Introduction
The current therapeutic strategy with rapid restoration of blood flow to the ischaemic myocardium by percutaneous coronary intervention (PCI) has markedly reduced the short‐term and long‐term morbidity and mortality in acute ST‐elevation myocardial infarction (STEMI).1, 2 However, acute heart failure (HF) and cardiogenic shock are still important clinical complications of STEMI and remains the leading cause of death in patients with acute myocardial infarction (MI).3, 4, 5 Cardiogenic shock is defined as a state of mismatch between oxygen delivery and oxygen demand caused by critical tissue hypoperfusion due to reduced cardiac output, and the diagnosis is based on haemodynamic (e.g. hypotension), clinical (e.g. cold extremities), and biochemical (e.g. increased lactate) criteria.6
Acute coronary syndromes and MI are associated with inflammation,7 and activation of the innate immune system such as Toll‐like receptors and the complement system are implicated in mediating both adaptive (e.g. tissue repair) and maladaptive (e.g. cardiomyocyte necrosis and apoptosis) responses.8, 9, 10 Cardiogenic shock following MI would exaggerate the inflammatory responses by tissue hypoperfusion and potentially induce a vicious circle.11 Current management of cardiogenic shock involves strategies to increase cardiac output and antithrombotic treatment but do not target the inflammatory response per se.3
The complement system, for long appreciated only as a first line of defence against microbes, is today acclaimed for immune surveillance by much broader means. Damage‐associated molecular patterns can trigger complement activation through three characterized pathways: the classical, the lectin, and the alternative pathway. They all merge at the central complement component C3 and continue into a common terminal pathway with cleavage of C5 and formation of the terminal C5b‐9 complement complex, which, when inserted into membranes as the membrane attack complex, can lyse bacteria and activate host cells. The soluble form of C5b‐9 (sC5b‐9) is a fluid‐phase marker indicating that the terminal pathway has been activated to its very end.12
Whereas a balanced activation of the complement system is regarded as beneficial for the host, an overwhelming activation could promote sustained inflammation and tissue damage, as seen during MI and the following ischaemia/reperfusion injury,13 but its relation to acute HF development following MI is not clear. However, complement is activated in patients with chronic HF, regardless of aetiology, potentially associated with unfavourable outcome,14, 15, 16 and recent studies have highlighted the activation of the lectin pathway as central in ischaemic heart disease and chronic HF.17, 18
The present study is a post hoc study of the LEAF (LEvosimendan in Acute heart Failure following myocardial infarction) trial,19 an interventional study on patients developing HF within 48 h following PCI‐treated STEMI. We hypothesized that enhanced complement activation could be a hallmark of acute HF in this patient group and may discriminate between HF with or without cardiogenic shock.
Materials and methods
Study design and population
The patient population and study design in the LEAF trial have previously been described in detail.19 Briefly, 61 patients with PCI‐treated STEMI who (i) had successful opening of the occluded coronary artery, (ii) had decreased wall motion in at least 3 of 16 segments of the left ventricle evaluated by echocardiography, and (iii) developed clinical signs of HF within 48 h (range: 14–33 h) following PCI were randomized to treatment with the calcium sensitizer levosimendan or placebo.19 HF was defined as dyspnoea at rest and the presence of at least one of the following symptoms: pulmonary oedema, signs of pulmonary congestion on X‐ray, need for continuous positive airway pressure or mechanical ventilation, or need for intravenous diuretics due to symptoms of congestion or persistent oliguria (urine output <0.5 mL/kg/h) after volume therapy. Criteria for subgrouping patients into cardiogenic shock included both of the following: (i) systolic blood pressure < 90 mmHg after 60 min of volume therapy or systolic blood pressure 90–100 mmHg despite vasoactive support and (ii) signs of organ hypoperfusion such as cold and clammy extremities, oliguria, or reduced consciousness. Exclusion criteria were septic shock, acute respiratory distress syndrome, creatinine > 450 μmol/L, severe hepatic failure, age < 20 years, heart rate > 120 b.p.m., pregnancy, significant mechanical outflow obstruction, haemoglobin < 8 g/dL, or allergy to the study medication or any of its components.
In the present study, the STEMI patients who developed cardiogenic shock (n = 9) were compared with patients with HF without any signs of cardiogenic shock (n = 52) in order to investigate differences in complement activation between severe and less severe degree of HF. For comparison, blood samples were obtained from 44 age‐matched and sex‐matched healthy controls. Importantly, to ensure that treatment with levosimendan did not affect the degree of complement activation, we compared the two treatment groups with respect to sC5b‐9 over the whole study period. There was no significant difference between the groups (P = 0.72), and they were thereafter handled as one population.
Blood sampling protocol
Blood samples were collected from patients at the time of inclusion (Day 0), that is at time of HF diagnosis (median 24 h following PCI) and at Days 1, 2, 5 (acute phase of the disease), and 42 following inclusion (follow‐up sample) as previously described.19 Briefly, blood samples were collected in ethylendiaminetetraacetic acid (EDTA), citrate, and serum vacutainer tubes (BD, Plymouth, UK). EDTA and citrated plasma samples were stored on crushed ice immediately after sampling and centrifuged within 30 min at 3000 g for 20 min at 4°C to obtained platelet‐poor plasma. Blood for serum preparation was allowed to clot for 60 min in room temperature and thereafter centrifuged at 2500 g for 10 min for isolation of serum. All samples were stored at −80°C until analysed and thawed only once.
Assays for complement activation markers
The complement activation products C4bc (classical and lectin pathway), C3bc (common pathway), C3bBbP (alternative pathway), and sC5b‐9 (terminal pathway) were measured in EDTA‐plasma samples from patients and controls by in‐house enzyme‐linked immunosorbent assays. All assays are based on either monoclonal antibodies detecting activation‐specific neoepitopes (C4bc, C3bc, and C5b‐9) or pairs of antibodies detecting complexes formed between single components upon activation (C3bBbP) as previously described in detail.20 The level of the respective marker was related to the International Complement Standard #2, defined to contain 1000 complement arbitrary units per millilitre.20
Lectin pathway recognition molecules
Plasma concentrations of mannose‐binding lectin (MBL), ficolin‐1 (FCN1), ficolin‐2 (FCN2), and ficolin‐3 (FCN3) were determined by sandwich enzyme‐linked immunosorbent assays using specific in‐house produced monoclonal antibodies as previously described.21, 22, 23, 24
Markers of endothelial activation
Levels of soluble intercellular adhesion molecule‐1 (sICAM‐1) and soluble vascular cell adhesion molecule‐1 (sVCAM‐1) of the current material have previously been analysed in serum and published.25 In the present study, we extended the data analyses by comparing these markers between patients with and without cardiogenic shock, to explore whether they corresponded with the degree of HF and whether there were any correlations between these markers and markers of complement activation.
Echocardiography
Left ventricular function was measured as wall motion score index (WMSI) by echocardiography as previously described.19 A 16‐segment model was used where a normally contracting or hyperkinetic segment was given a score of 1, a hypokinetic segment scored 2, akinesia gave a score of 3, and a dyskinetic segment scored 4 points. WMSI was calculated by dividing the sum of scores by the number of segments scored. All examinations were performed by two experienced echocardiographers on Days 0, 1, and 42, and the analyses were performed by one observer. An ultrasonic device system (Vivid i or Vivid 7, GE Vingmed Ultrasound, Horten, Norway) was used for the examinations, and the analyses were performed with dedicated software (Echopac GE Vingmed Ultrasound).
Infectious complications
In order to test whether infectious complications contributed to activation of the complement system, levels of activation makers were compared in patients with documented or suspected infection, based on positive culture testing, X‐rays, and clinical evaluation (n = 14), to patients without infection (n = 38). This comparison was only performed in the non‐shock group because the cardiogenic shock group did not include enough patients to ensure statistical testing. Statistical tests for correlation between complement activation and biochemical markers of infection [C‐reactive protein, white blood cell (WBC) count, or interleukin (IL)‐6] were also performed.
Data presentation and statistics
In addition to the patient cohort, a control group comprising 44 age‐matched and sex‐matched healthy individuals was included. The patient cohort was divided into two groups: one group consisting of patients who developed HF without any signs of cardiogenic shock, the non‐shock group (n = 52), and one group consisting of patients who developed cardiogenic shock, referred to as the shock group (n = 9).19 Differences between these two groups during the first 5 days after inclusion (Days 0–5) were analysed with linear mixed model analyses. Differences between the two groups were tested with t‐test or alternatively with the Mann–Whitney U‐test when data were not normally distributed. To compare categorical data between groups, the χ2 test or Fisher's exact test was used. Differences between more than two groups were tested with Kruskal–Wallis test using Dunn's post hoc test. Bonferroni correction was used to correct for multiple testing. Correlation analyses were measured by the Spearman correlation test. All results are given as mean and standard error of the mean. A P value of <0.05 was considered statistically significant. IBM SPSS Statistics version 21 (Armonk, NY) was used for analysis, while GraphPad Prism version 6 (San Diego, CA) was used for data presentation.
Ethics
The study was approved by The Regional Ethics Committee South‐Eastern Norway Regional Health Authority, and the study was conducted in accordance with the principles of the Declaration of Helsinki (http://clinicaltrials.gov http://clinicaltrials.gov/show/NCT00324766). All patients provided written informed consent. If a patient was unable to give informed consent, relatives were informed, and a written consent was acquired from the patient as soon as possible.
Results
Complement activation
Sixty‐one patients were included in the study, and those who developed cardiogenic shock (n = 9) were compared with patients with HF without any signs of cardiogenic shock (n = 52) (Table 1). At the time of inclusion, C4bc, reflecting classical and lectin pathway activation, C3bc, reflecting C3 activation, C3bBbP, reflecting activation of the alternative pathway, and sC5b‐9, reflecting the terminal pathway activation, were significantly elevated in the patient cohort (n = 61) compared with the healthy controls (n = 44) (P < 0.05 for all; Figure 1 A–D). Patients developing shock had significantly higher levels of C4bc, C3bc, and sC5b‐9 in the acute phase of the disease (Days 0–5), compared with patients without shock (P < 0.05 for all; Figure 1 A, B, and D). Even at Day 42, there was an enhanced complement activation reflected by higher levels of all four activation markers (C4bc, C3bc, C3bBbP, and sC5b‐9) in the shock group compared with the non‐shock group (P < 0.05 for all; Figure 1 E–H).
Table 1.
Baseline characteristics of 61 patients with ST‐elevation myocardial infarction developing acute heart failure with or without cardiogenic shock
| Shock | Non‐shock | P value | |
|---|---|---|---|
| Total number (female/male) | 9 (3/6) | 52 (15/37) | 0.89 |
| Age (years, mean, and range) | 57 (49–68) | 66 (56–74) | 0.08 |
| TnTa (ng/L) | 14 640 (7580–20 925) | 12 279 (7811–16 607) | 0.43 |
| Creatininea (μmol/L) | 81 (52–150) | 82 (69–95) | 0.91 |
| eGFRa (mL/min/m2) | 60 (33–60) | 60 (60–60) | 0.24 |
| NT‐proBNPa (pmol/L) | 315 (202–721) | 463 (266–840) | 0.52 |
| C‐reactive proteina (mg/L) | 40 (24–100) | 57 (35–97) | 0.42 |
| WBC count (×109/L)a | 11 (8.9–17) | 12 (10–15) | 0.49 |
| IL‐6a (pg/mL) | 29 (19–40) | 27 (21–33) | 0.54 |
| Previous hypertension, n (%) | 5 (56) | 16 (31) | 0.15 |
| Previous dyslipidemia, n (%) | 1 (11) | 12 (23) | 0.42 |
| Current smoking, n (%) | 6 (67) | 16 (30) | <0.05 |
| Previous diabetes mellitus, n (%) | 1 (11) | 5 (10) | 0.89 |
| Previous statin treatment, n (%) | 2 (22) | 13 (25) | 0.86 |
| Previous myocardial infarction, n (%) | 3 (33) | 8 (15) | 0.20 |
| Multi‐vessel disease, n (%) | 5 (56) | 26 (44) | 0.76 |
| Atrial fibrillationa, n (%) | 1 (9) | 1 (2) | 0.16 |
| Systolic blood pressurea, mmHg | 85 (72–94) | 106 (96–117) | <0.001 |
| Diastolic blood pressurea, mmHg | 55 (48–58) | 67 (60–72) | <0.001 |
| Hours from symptom start to PCI | 3 (2–8) | 3 (2–6) | 0.80 |
| Hours from PCI to baseline | 17 (10–23) | 23 (14–32) | 0.07 |
| LVEFa, % | 44 (34–49) | 41 (38–47) | 0.88 |
| Antimicrobial treatment, n (%) | 8 (89) | 14 (27) | <0.001 |
| Mortality within 6 months, n (%) | 3 (33) | 2 (4) | <0.05 |
GFR, glomerular filtration rate; IL‐6, interleukin 6; LVEF, left ventricular ejection fraction; NT‐proBNP, N terminal pro brain natriuretic peptide; PCI, percutaneous coronary intervention; TnT, troponin T; WBC, white blood cell.
Data are given as median (25th and 75th percentile) or number (%).
At the time of inclusion, that is median 24 h following PCI.
Figure 1.
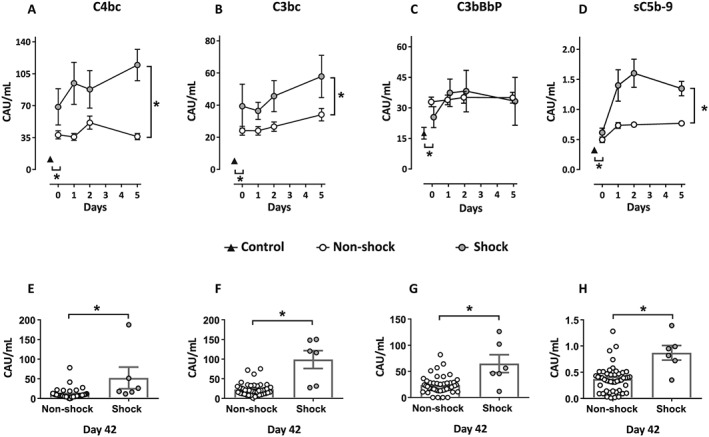
Complement activation products during the first 5 days of the disease and at Day 42 after inclusion. Figures in the upper panel (A–D) show values at inclusion and throughout the acute phase of the disease (Days 0–5). (A) Plasma levels for C4bc (classical and lectin pathway activation), (B) C3bc (common activation of all initial pathways), (C) C3bBbP (alternative pathway activation), and (D) sC5b‐9 (terminal pathway activation) are shown for patients with cardiogenic shock (n = 9, grey circles), patients with heart failure without cardiogenic shock (n = 52, open circles), and healthy controls (n = 44, black triangles). Statistical differences between the shock group and the non‐shock group of patients from inclusion (Day 0) to Day 5 (the acute phase of the disease) are indicated with brackets and *(P < 0.05) at the right‐hand side of the graph. Statistical difference between patients and controls at the time of inclusion are indicated with *(P < 0.05). Figures at the lower panel (E–H) show plasma levels at Day 42 for (E) C4bc, (F) C3bc, (G) C3bBbP, and (H) sC5b‐9 for patients with cardiogenic shock (n = 7, grey columns) and patients without cardiogenic shock (n = 45, white columns). Data are given as mean ± standard error of the mean. CAU, complement arbitrary units.
Lectin pathway recognition molecules
The level of FCN2 was at the time of inclusion lower among patients vs. controls (P < 0.05), whereas no significant differences were observed for MBL, FCN1, or FCN3 (Figure 2 A–D). During the acute phase of the disease (Days 0–5), FCN2 increased significantly (P < 0.05) in the patient cohort as a whole, but there were no significant group differences between those with and without cardiogenic shock (Figure 2 C). At Day 42, however, the shock group had a significantly higher level of FCN2 compared with the non‐shock group (P < 0.05; Figure 2 G). No significant group differences were found for MBL, FCN1, or FCN3 (Figure 2 E, F, and H). Furthermore, there was no correlation between C4bc and MBL or the ficolins.
Figure 2.
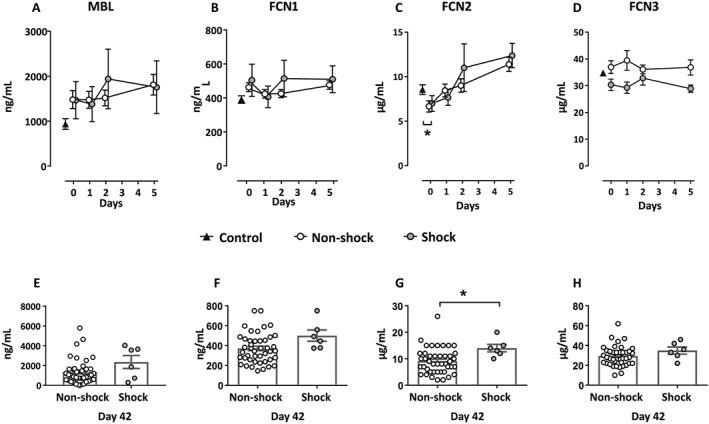
Lectin pathway proteins levels during the first 5 days of the disease and at Day 42. Figures at the upper panel (A–D) show plasma levels for the (A) mannose‐binding lectin (MBL), (B) Ficolin‐1 (FCN1), (C) Ficolin‐2 (FCN2), and (D) Ficolin‐3 (FCN3) for patients from inclusion (Day 0) to Day 5 (the acute phase of the disease). Figures in the lower panel (E–H) show plasma levels for the (E) MBL, (F) FCN1, (G) FCN2, and (H) FCN3 at Day 42. The figures are shown with the same patient populations and details as described in the Figure 1 legend. *P < 0.05.
Markers of endothelial activation
We have previously published data on endothelial activation in these patients.25 When now analysing their relation to cardiogenic shock, we found that sICAM‐1 and sVCAM‐1 were significantly higher in the shock group compared with the non‐shock group during the acute phase of the disease (Days 0–5) (P < 0.01 for both; Figure 3 A,B) with no significant differences at Day 42, (Figure 3 C,D).
Figure 3.
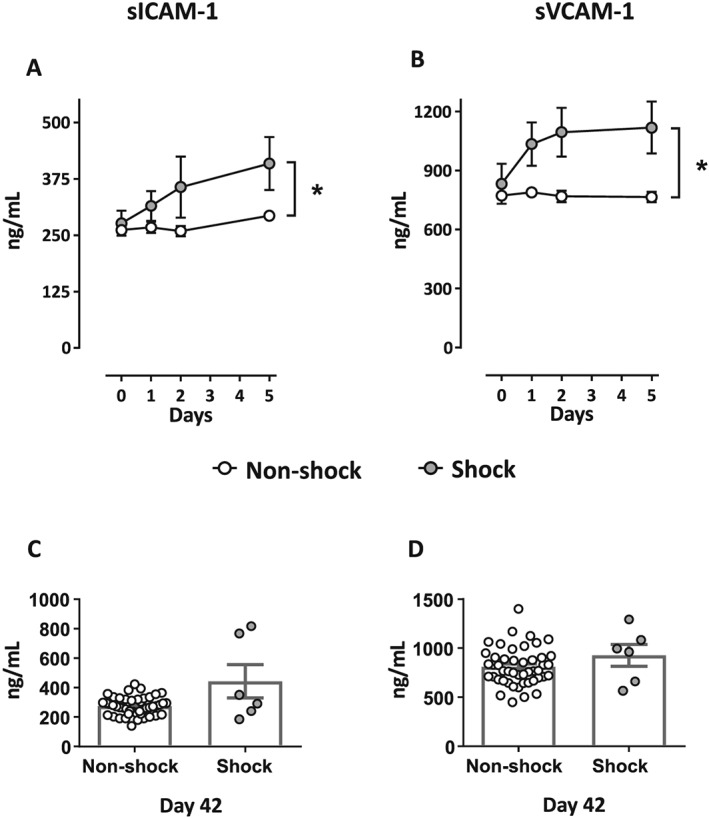
Serum levels of the endothelial cell activation markers sICAM‐1 and sVCAM‐1 during the first 5 days of the disease and at Day 42. Figures in the upper panel show serum levels of the (A) soluble intercellular adhesion molecule 1 (sICAM‐1) and the (B) soluble vascular adhesion molecule 1 (sVCAM‐1) for patients from inclusion (Day 0) to Day 5 (the acute phase of the disease). Figures in the lower panel show serum levels of (C) sICAM‐1 and (D) sVCAM‐1 at the control measurement at Day 42. The figures are shown with the same patient populations and details as described in the Figure 1 legend. * P < 0.05.
Correlation between complement activation and regional myocardial contractility
In the shock group, there was a significant correlation between complement activation as measured by sC5b‐9 and WMSI at the day of inclusion (Day 0) (r = 0.678, P = 0.045) and at Day 42 (r = 0.841, P = 0.036; Table 2). At these two time points, both blood sampling and WMSI were performed, and thus, direct correlation tests could be performed. sC5b‐9 reached its highest level at Day 2, where WMSI was not performed. Interestingly, this peak sC5b‐9 level correlated significantly with WMSI at Day 42 in the shock group (r = 0.975, P = 0.005; Table 2). Weaker or no correlations were found for the other complement activation products: WMSI Day 1 correlated with C3bBbP measured at Days 2 (r = 0.943, P = 0.005) and 42 (r = 0.829, P = 0.042), and WMSI measured at Day 0 correlated with C4bc measured at Day 0 (r = 0.703, P = 0.035, data not shown). No correlations were found between WMSI and C3bc. In the non‐shock group, the only significant correlation was found between C4bc measured at Day 1 and WMSI at Day 0 (data not shown).
Table 2.
Correlation between left ventricular regional contractility measured as wall motion score index and complement activation measured by sC5b‐9 in patients with cardiogenic shock (n = 9) following percutaneous coronary intervention‐treated ST‐elevation myocardial infarction
| sC5b‐9 | sC5b‐9 | sC5b‐9 | ||
|---|---|---|---|---|
| Day 0 | Day 2 | Day 42 | ||
| WMSI Day 0 | r | 0.678 | 0.206 | 0.522 |
| P | 0.045 | 0.696 | 0.288 | |
| WMSI Day 1 | r | 0.311 | 0.812 | 0.488 |
| P | 0.415 | 0.050 | 0.329 | |
| WMSI Day 42 | r | 0.551 | 0.975 | 0.841 |
| P | 0.257 | 0.005 | 0.036 | |
WMSI, wall motion score index. Statistical significance is shown in bold.
Correlation between complement activation and the markers of endothelial activation
There was a significant correlation between complement activation in the whole patient group (n = 61) at Day 2, when sC5b‐9 peaked, and peak level of sVCAM‐1 (r = 0.296, P = 0.022) and sICAM‐1 (r = 0.317, P = 0.014), whereas no correlation was found when the shock and non‐shock groups were analysed separately.
Complement activation following infection
There was no significant difference in complement activation, measured as sC5b‐9, between patients with infection (n = 14) and patients without infection (n = 38) in the non‐shock group during the acute phase of the disease (Days 0–5) (P = 0.44). There was no difference between the non‐shock and the shock group with respect to levels of C‐reactive protein, WBC count, or IL‐6 (Table 1). We found no association between peak values of sC5b‐9 and markers of infection (C‐reactive protein, WBC count, or IL‐6, all measured as peak values) (Table S1 ).
Discussion
In the present study, we found increased complement activation in patients who developed acute HF following PCI‐treated STEMI compared with healthy controls. Notably, the degree of complement activation discriminated those patients developing cardiogenic shock from those in the non‐shock group. The increased activation persisted even 6 weeks after STEMI in the shock group. In these patients, there was also a strong correlation between complement activation and regional contractility measured as WMSI both at inclusion and at 6 weeks. Although complement activation has been shown to be involved in the progress of HF, this is, to the best of our knowledge, the first study to document that the degree of complement activation is directly related to the disease severity and impaired myocardial function in patients developing acute HF following STEMI.
The patient population in this study was characterized by large MIs determined by high levels of troponins and clinical and echocardiographic findings.19 In the present study, we show that the complement activation products C4bc, C3bc, C3bBbP, and sC5b‐9, representing complement activation from initiation to terminal activation, were increased at the time when the patients were diagnosed with HF (14–33 h following PCI treatment), compared with healthy individuals. Furthermore, there was stronger and more persistent complement activation in the most severely affected patients. This persistent activation indicates that complement might play an important role in the pathophysiological process of HF. In fact, the peak level of sC5b‐9 during the acute phase correlated significantly with WMSI after 6 weeks, suggesting that complement‐mediated mechanisms could promote myocardial damage with subsequent development of severe HF following STEMI.
Because of its amplification loop, the alternative pathway can contribute substantially to complement activation from the level of C3 and further downstream the activation cascade.26, 27 The lack of difference between the two patient groups with respect to the activation product C3bBbP is therefore somewhat surprising. The amplification loop is, however, under strict control by regulatory proteins like factor H, and complement activation triggered presuming via the lectin pathway with a tight regulatory control of the alternative pathway in both groups in the early phase of disease may be a reasonable explanation for this finding. The regulatory balance may then have changed after the initial phase, explaining the significant difference in C3bBbP in the two groups at Day 42.
Several clinical and experimental studies have previously demonstrated increased complement activation in cardiovascular disease and HF.14, 15, 28, 29, 30, 31, 32 Particularly, the lectin pathway has been linked to complement‐mediated myocardial injury and HF,18, 33, 34, 35 and lectin pathway recognition molecules were therefore thoroughly investigated in the present study. MBL and the ficolins are circulating recognition molecules binding to molecular structures on damaged host cells further activating the mannose‐binding serine proteases, MASP1 and MASP2.36 MBL is also an acute phase reactant,37 and altered levels of FCN1–3 are reported in various pathological conditions, either due to consumption or changed expression.38 The lower level of FCN2 in the patient population at inclusion compared with healthy controls is in line with a previous observation seen in STEMI patients35 and is suggested to reflect consumption in the early phase of the disease. From the inclusion level, FCN2 increased significantly during the first 5 days of the disease and was at Day 42 significantly higher in the shock group. The other recognition molecules, MBL, FCN1, and FCN3, did not differ significantly from the healthy controls at inclusion. FCN1 was higher than the controls, although not significant, and showed no change during the course. The reason for the different patterns for FCN1 and FCN2 is uncertain but might be related to their different profiles for release and consumption, which makes it difficult to compare these two. FCN1 is synthesized by peripheral leukocytes. Upon cell activation, secretion of FCN1 increases, but the majority is tethered to the cell membrane of the activated cell.39 This can explain the small, however non‐significant, early increase of FCN1 in patients. FCN2 is synthesized in the liver as a soluble protein.40 Increased secretion of FCN2 is, in relation to FCN1, delayed, which enables a consumption profile early after MI. Further on, FCN1 and FCN2 are highly homologous, but FCN2 has four carbohydrate‐binding domains, whereas FCN1 has only one.41 FCN2 might therefore bind its ligand more tightly as compared with FCN1, but without knowing the exact target, this remains speculative.
C4bc reflects both classical and lectin pathway activation. Although classical pathway activation cannot be excluded, our findings of increased C4bc is in accordance with lectin pathway activation during the acute phase,33, 42 although the role of the lectin pathway in post‐MI HF is still elusive.
Microbial infections are well‐known activators of the complement system.43 We therefore compared complement activation in patients with or without signs of infection. The non‐shock group contained a sufficient amount of patients treated for infections, documented or suspected, to enable statistical analysis regarding infectious complications and complement activation. Notably, there was no difference in complement activation in patients with or without infection in this group. Antibiotics were given mainly because of suspected aspiration, and septic patients were excluded from the trial. Furthermore, there were no correlations between peak levels of sC5b‐9, C‐reactive protein, IL‐6, or WBC count. Thus, there is no evidence that the increased complement activation is caused by infections but rather by the cardiogenic shock per se.
The patients in the shock group were characterized by significantly increased levels of the soluble adhesion molecules sVCAM‐1 and sICAM‐1 as compared with the non‐shock group reflecting enhanced endothelial cell activation in those with the most severe HF. Activated endothelial cells have been shown to secrete complement components and to express adhesion molecules ICAM‐1 and VCAM‐1 in response to sC5b‐9 and are also targets for complement activation products.44 Herein, we also found a significant correlation between sC5b‐9 and the adhesion molecules in the whole HF group, further suggesting crosstalk between endothelial cells and terminal complement activation in patients with acute, severe HF following MI. With a positive correlation of sustained complement activation and development of cardiogenic shock, the critical question arises whether complement activation solely is the result of hypoperfusion caused by cardiogenic shock, or whether it also contributes to exacerbation of shock and, in extension, if these patients would benefit from complement inhibition. Increased systemic complement activation has previously been shown in patients with chronic HF consistent with tissue hypoperfusion, acidosis, and endothelial cell damage.14 Neoantigens exposed in ischaemic tissue are linked to recognition by natural IgM and subsequent lectin pathway activation,45 which would support sustained complement activation. If complement significantly aggravates the shock syndrome, there would be fear for a vicious circle. By being part of the innate immune system, complement is instantly activated upon ‘danger’ and has the potential for initiating a broad range of inflammatory responses. Specific complement inhibition may therefore be suitable in patients where attenuation of inflammation is desired, including patients with post‐MI HF and particularly those with cardiogenic shock. Various clinical trials targeting different parts of the inflammatory response have failed to reach significance with regard to their primary endpoints.46 However, in the COMplement inhibition in Myocardial infarction treated with Angioplasty trial,47 where complement inhibition with the C5‐inhibitor pexelizumab was given as a bolus dose and with continuous infusion for 20 h following MI, a significant reduction in 90 day mortality was seen. The incidence of cardiogenic shock was reduced with 45%, however, non‐significantly. The Assessment of Pexelizumab in Acute Myocardial Infarction trial did not show any effect of pexelizumab,48 but there is a remaining question whether C5 was appropriately inhibited.49 In order to rule out if complement inhibition would be beneficial in patients with acute severe HF and cardiogenic shock due to MI, more clinical trials are needed.
The current study is of explorative character, however, on a well‐defined cohort with close follow‐up and careful plasma preparation, which is critical for accurate complement analysis. The low numbers of patients in the group of cardiogenic shock as well as the lack of blood samples before PCI are limitations of the present study. The major differences found between the groups, with statistical significance for all complement activation products and endothelial cell markers, however, increase the impact of the data because the risk of type I error can be regarded as small.
The patients included in this study represent a group of patients often excluded from clinical trials due to the severity of the disease. However, our results, consistently demonstrating an increased and persistent complement activation correlating to disease severity and endothelial cell activation, indicating that patients with advanced HF complicating large MI, may particularly benefit from therapy targeting complement activation. Our findings add new understanding to the inflammatory profile in patients with acute severe HF, which can pave the way for new prognostic markers and targets for therapy.
Conflict of interest
None declared.
Funding
This study was financially supported by The Research Council of Norway, The Norwegian Council on Cardiovascular Disease, The Odd Fellow Foundation, the European Community's Seventh Framework Programme under grant agreement no. 602699 (DIREKT), the Novo Nordisk Research Foundation, The Danish Research Foundation of Independent Research, and The Svend Andersen Research Foundation and Rigshospitalet (Copenhagen, Denmark).
Supporting information
Table S1. Spearman correlation analysis and linear regression analyses between peak values of sC5b‐9 and various relevant variables.
Orrem, H. L. , Nilsson, P. H. , Pischke, S. E. , Grindheim, G. , Garred, P. , Seljeflot, I. , Husebye, T. , Aukrust, P. , Yndestad, A. , Andersen, G. Ø. , Barratt‐Due, A. , and Mollnes, T. E. (2018) Acute heart failure following myocardial infarction: complement activation correlates with the severity of heart failure in patients developing cardiogenic shock. ESC Heart Failure, 5: 292–301. doi: 10.1002/ehf2.12266.
References
- 1. Fach A, Bunger S, Zabrocki R, Schmucker J, Conradi P, Garstka D, Fiehn E, Hambrecht R, Wienbergen H. Comparison of outcomes of patients with ST‐segment elevation myocardial infarction treated by primary percutaneous coronary intervention analyzed by age groups (<75, 75 to 85, and >85 years); (Results from the Bremen STEMI Registry). Am J Cardiol 2015; 116: 1802–1809. [DOI] [PubMed] [Google Scholar]
- 2. Weir R, McMurray J. Epidemiology of heart failure and left ventricular dysfunction after acute myocardial infarction. Curr Heart Fail Rep 2006; 3: 175–180. [DOI] [PubMed] [Google Scholar]
- 3. Thiele H, Ohman EM, Desch S, Eitel I, de Waha S. Management of cardiogenic shock. Eur Heart J 2015; 36: 1223–1230. [DOI] [PubMed] [Google Scholar]
- 4. Kolte D, Khera S, Aronow WS, Mujib M, Palaniswamy C, Sule S, Jain D, Gotsis W, Ahmed A, Frishman WH, Fonarow GC. Trends in incidence, management, and outcomes of cardiogenic shock complicating ST‐elevation myocardial infarction in the United States. J Am Heart Assoc 2014; 3: e000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Diepen S, Katz JN, Albert NM, Henry TD, Jacobs AK, Kapur NK, Kilic A, Menon V, Ohman EM, Sweitzer NK, Thiele H, Washam JB, Cohen MG. Contemporary management of cardiogenic shock: a scientific statement from the American Heart Association. Circulation 2017; 136: e232–e268. [DOI] [PubMed] [Google Scholar]
- 6. Reynolds HR, Hochman JS. Cardiogenic shock: current concepts and improving outcomes. Circulation 2008; 117: 686–697. [DOI] [PubMed] [Google Scholar]
- 7. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol 2014; 11: 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matzinger P. The danger model: a renewed sense of self. Science 2002; 296: 301–305. [DOI] [PubMed] [Google Scholar]
- 9. Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell 1997; 91: 295–298. [DOI] [PubMed] [Google Scholar]
- 10. Fang L, Moore XL, Dart AM, Wang LM. Systemic inflammatory response following acute myocardial infarction. J Geriatr Cardiol 2015; 12: 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shpektor A. Cardiogenic shock: the role of inflammation. Acute Card Care 2010; 12: 115–118. [DOI] [PubMed] [Google Scholar]
- 12. Mollnes TE, Jokiranta TS, Truedsson L, Nilsson B, Rodriguez de Cordoba S, Kirschfink M. Complement analysis in the 21st century. Mol Immunol 2007; 44: 3838–3849. [DOI] [PubMed] [Google Scholar]
- 13. Banz Y, Rieben R. Role of complement and perspectives for intervention in ischemia‐reperfusion damage. Ann Med 2012; 44: 205–217. [DOI] [PubMed] [Google Scholar]
- 14. Aukrust P, Gullestad L, Lappegard KT, Ueland T, Aass H, Wikeby L, Simonsen S, Froland SS, Mollnes TE. Complement activation in patients with congestive heart failure: effect of high‐dose intravenous immunoglobulin treatment. Circulation 2001; 104: 1494–1500. [DOI] [PubMed] [Google Scholar]
- 15. Clark DJ, Cleman MW, Pfau SE, Rollins SA, Ramahi TM, Mayer C, Caulin‐Glaser T, Daher E, Kosiborod M, Bell L, Setaro JF. Serum complement activation in congestive heart failure. Am Heart J 2001; 141: 684–690. [DOI] [PubMed] [Google Scholar]
- 16. Gombos T, Forhecz Z, Pozsonyi Z, Szeplaki G, Kunde J, Fust G, Janoskuti L, Karadi I, Prohaszka Z. Complement anaphylatoxin C3a as a novel independent prognostic marker in heart failure. Clin Res Cardiol 2012; 101: 607–615. [DOI] [PubMed] [Google Scholar]
- 17. Timmers L, Pasterkamp G, de Hoog VC, Arslan F, Appelman Y, de Kleijn DP. The innate immune response in reperfused myocardium. Cardiovasc Res 2012; 94: 276–283. [DOI] [PubMed] [Google Scholar]
- 18. Prohaszka Z, Munthe‐Fog L, Ueland T, Gombos T, Yndestad A, Forhecz Z, Skjoedt MO, Pozsonyi Z, Gustavsen A, Janoskuti L, Karadi I, Gullestad L, Dahl CP, Askevold ET, Fust G, Aukrust P, Mollnes TE, Garred P. Association of ficolin‐3 with severity and outcome of chronic heart failure. PLoS One 2013; 8: e60976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Husebye T, Eritsland J, Muller C, Sandvik L, Arnesen H, Seljeflot I, Mangschau A, Bjornerheim R, Andersen GO. Levosimendan in acute heart failure following primary percutaneous coronary intervention‐treated acute ST‐elevation myocardial infarction. Results from the LEAF trial: a randomized, placebo‐controlled study. Eur J Heart Fail 2013; 15: 565–572. [DOI] [PubMed] [Google Scholar]
- 20. Bergseth G, Ludviksen JK, Kirschfink M, Giclas PC, Nilsson B, Mollnes TE. An international serum standard for application in assays to detect human complement activation products. Mol Immunol 2013; 56: 232–239. [DOI] [PubMed] [Google Scholar]
- 21. Bastrup‐Birk S, Skjoedt MO, Munthe‐Fog L, Strom JJ, Ma YJ, Garred P. Pentraxin‐3 serum levels are associated with disease severity and mortality in patients with systemic inflammatory response syndrome. PLoS One 2013; 8: e73119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Munthe‐Fog L, Hummelshoj T, Hansen BE, Koch C, Madsen HO, Skjodt K, Garred P. The impact of FCN2 polymorphisms and haplotypes on the Ficolin‐2 serum levels. Scand J Immunol 2007; 65: 383–392. [DOI] [PubMed] [Google Scholar]
- 23. Munthe‐Fog L, Hummelshoj T, Honore C, Moller ME, Skjoedt MO, Palsgaard I, Borregaard N, Madsen HO, Garred P. Variation in FCN1 affects biosynthesis of Ficolin‐1 and is associated with outcome of systemic inflammation. Genes Immun 2012; 13: 515–522. [DOI] [PubMed] [Google Scholar]
- 24. Munthe‐Fog L, Hummelshoj T, Ma YJ, Hansen BE, Koch C, Madsen HO, Skjodt K, Garred P. Characterization of a polymorphism in the coding sequence of FCN3 resulting in a Ficolin‐3 (Hakata antigen) deficiency state. Mol Immunol 2008; 45: 2660–2666. [DOI] [PubMed] [Google Scholar]
- 25. Husebye T, Eritsland J, Arnesen H, Bjornerheim R, Mangschau A, Seljeflot I, Andersen GO. Association of interleukin 8 and myocardial recovery in patients with ST‐elevation myocardial infarction complicated by acute heart failure. PLoS One 2014; 9: e112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harboe M, Garred P, Karlstrom E, Lindstad JK, Stahl GL, Mollnes TE. The down‐stream effects of mannan‐induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol 2009; 47: 373–380. [DOI] [PubMed] [Google Scholar]
- 27. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol 2004; 138: 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gorsuch WB, Chrysanthou E, Schwaeble WJ, Stahl GL. The complement system in ischemia‐reperfusion injuries. Immunobiology 2012; 217: 1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hill JH, Ward PA. The phlogistic role of C3 leukotactic fragments in myocardial infarcts of rats. J Exp Med 1971; 133: 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orn S, Manhenke C, Ueland T, Damas JK, Mollnes TE, Edvardsen T, Aukrust P, Dickstein K. C‐reactive protein, infarct size, microvascular obstruction, and left‐ventricular remodelling following acute myocardial infarction. Eur Heart J 2009; 30: 1180–1186. [DOI] [PubMed] [Google Scholar]
- 31. Vakeva AP, Agah A, Rollins SA, Matis LA, Li L, Stahl GL. Myocardial infarction and apoptosis after myocardial ischemia and reperfusion: role of the terminal complement components and inhibition by anti‐C5 therapy. Circulation 1998; 97: 2259–2267. [DOI] [PubMed] [Google Scholar]
- 32. Van Der Pals J, Koul S, Andersson P, Gotberg M, Ubachs J, Kanski M, Arheden H, Olivecrona G, Larsson B, Erlinge D. Inhibition of c5a related neutrophil activation by adc‐1004 reduces myocardial infarct in a porcine ischemia‐reperfusion model. Eur Heart J 2010; 31: 979–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Busche MN, Pavlov V, Takahashi K, Stahl GL. Myocardial ischemia and reperfusion injury is dependent on both IgM and mannose‐binding lectin. Am J Physiol Heart Circ Physiol 2009; 297: H1853–H1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Trendelenburg M, Theroux P, Stebbins A, Granger C, Armstrong P, Pfisterer M. Influence of functional deficiency of complement mannose‐binding lectin on outcome of patients with acute ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention. Eur Heart J 2010; 31: 1181–1187. [DOI] [PubMed] [Google Scholar]
- 35. Schoos MM, Munthe‐Fog L, Skjoedt MO, Ripa RS, Lonborg J, Kastrup J, Kelbaek H, Clemmensen P, Garred P. Association between lectin complement pathway initiators, C‐reactive protein and left ventricular remodeling in myocardial infarction‐a magnetic resonance study. Mol Immunol 2013; 54: 408–414. [DOI] [PubMed] [Google Scholar]
- 36. Garred P, Honoré C, Ma YJ, Munthe‐Fog L, Hummelshøj T. MBL2, FCN1, FCN2 and FCN3—the genes behind the initiation of the lectin pathway of complement. Mol Immunol 2009; 46: 2737–2744. [DOI] [PubMed] [Google Scholar]
- 37. Dean MM, Minchinton RM, Heatley S, Eisen DP. Mannose binding lectin acute phase activity in patients with severe infection. J Clin Immunol 2005; 25: 346–352. [DOI] [PubMed] [Google Scholar]
- 38. Endo Y, Matsushita M, Fujita T. New insights into the role of ficolins in the lectin pathway of innate immunity. Int Rev Cell Mol Biol 2015; 316: 49–110. [DOI] [PubMed] [Google Scholar]
- 39. Rorvig S, Honore C, Larsson LI, Ohlsson S, Pedersen CC, Jacobsen LC, Cowland JB, Garred P, Borregaard N. Ficolin‐1 is present in a highly mobilizable subset of human neutrophil granules and associates with the cell surface after stimulation with fMLP. J Leukoc Biol 2009; 86: 1439–1449. [DOI] [PubMed] [Google Scholar]
- 40. Garred P, Genster N, Pilely K, Bayarri‐Olmos R, Rosbjerg A, Ma YJ, Skjoedt MO. A journey through the lectin pathway of complement‐MBL and beyond. Immunol Rev 2016; 274: 74–97. [DOI] [PubMed] [Google Scholar]
- 41. Garlatti V, Belloy N, Martin L, Lacroix M, Matsushita M, Endo Y, Fujita T, Fontecilla‐Camps JC, Arlaud GJ, Thielens NM, Gaboriaud C. Structural insights into the innate immune recognition specificities of L‐ and H‐ficolins. EMBO J 2007; 26: 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwaeble WJ, Lynch NJ, Clark JE, Marber M, Samani NJ, Ali YM, Dudler T, Parent B, Lhotta K, Wallis R, Farrar CA, Sacks S, Lee H, Zhang M, Iwaki D, Takahashi M, Fujita T, Tedford CE, Stover CM. Targeting of mannan‐binding lectin‐associated serine protease‐2 confers protection from myocardial and gastrointestinal ischemia/reperfusion injury. Proc Natl Acad Sci U S A 2011; 108: 7523–7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res 2010; 20: 34–50. [DOI] [PubMed] [Google Scholar]
- 44. Fischetti F, Tedesco F. Cross‐talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 2006; 39: 417–428. [DOI] [PubMed] [Google Scholar]
- 45. Zhang M, Takahashi K, Alicot EM, Vorup‐Jensen T, Kessler B, Thiel S, Jensenius JC, Ezekowitz RA, Moore FD, Carroll MC. Activation of the lectin pathway by natural IgM in a model of ischemia/reperfusion injury. J Immunol 2006; 177: 4727–4734. [DOI] [PubMed] [Google Scholar]
- 46. Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 2012; 122: 23–35. [DOI] [PubMed] [Google Scholar]
- 47. Granger CB, Mahaffey KW, Weaver WD, Theroux P, Hochman JS, Filloon TG, Rollins S, Todaro TG, Nicolau JC, Ruzyllo W, Armstrong PW. Pexelizumab, an anti‐C5 complement antibody, as adjunctive therapy to primary percutaneous coronary intervention in acute myocardial infarction: the COMplement inhibition in Myocardial infarction treated with Angioplasty (COMMA) trial. Circulation 2003; 108: 1184–1190. [DOI] [PubMed] [Google Scholar]
- 48. Armstrong PW, Granger CB, Adams PX, Hamm C, Holmes D Jr, O'Neill WW, Todaro TG, Vahanian A, Van de Werf F. Pexelizumab for acute ST‐elevation myocardial infarction in patients undergoing primary percutaneous coronary intervention: a randomized controlled trial. JAMA 2007; 297: 43–51. [DOI] [PubMed] [Google Scholar]
- 49. Martel C, Granger CB, Ghitescu M, Stebbins A, Fortier A, Armstrong PW, Bonnefoy A, Theroux P. Pexelizumab fails to inhibit assembly of the terminal complement complex in patients with ST‐elevation myocardial infarction undergoing primary percutaneous coronary intervention. Insight from a substudy of the Assessment of Pexelizumab in Acute Myocardial Infarction (APEX‐AMI) trial. Am Heart J 2012; 164: 43–51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Spearman correlation analysis and linear regression analyses between peak values of sC5b‐9 and various relevant variables.