

Abstract
The properties of phospholipase C (PLC) partially purified from Catharanthus roseus transformed roots were analyzed using substrate lipids dispersed in phospholipid vesicles, phospholipid-detergent mixed micelles, and phospholipid monolayers spread at an air-water interface. Using [33P]phosphatidylinositol 4,5-bisphosphate (PIP2) of high specific radioactivity, PLC activity was monitored directly by measuring the loss of radioactivity from monolayers as a result of the release of inositol phosphate and its subsequent dissolution on quenching in the subphase. PLC activity was markedly affected by the surface pressure of the monolayer, with reduced activity at extremes of initial pressure. The optimum surface pressure for PIP2 hydrolysis was 20 mN/m. Depletion of PLC from solution by incubation with sucrose-loaded PIP2 vesicles followed by ultracentrifugation demonstrated stable attachment of PLC to the vesicles. A mixed micellar system was established to assay PLC activity using deoxycholate. Kinetic analyses were performed to determine whether PLC activity was dependent on both bulk PIP2 and PIP2 surface concentrations in the micelles. The interfacial Michaelis constant was calculated to be 0.0518 mol fraction, and the equilibrium dissociation constant of PLC for the lipid was 45.5 μm. These findings will add to our understanding of the mechanisms of regulation of plant PLC.
Ca2+ is the principal second messenger in plants. One of the mechanisms that regulates the levels of this messenger involves the enzyme PLC. PLC catalyzes the hydrolysis of PIP2 to generate two second messengers: IP3 and 1,2-diacylglycerol. PLC is a family of isoenzymes that has been classified into three groups: β, γ, and δ (Rhee and Bae, 1997). In plants several studies have reported the biochemical presence of this enzyme (McMurray and Irvine, 1988; Tate et al., 1989; Melin et al., 1992; Pical et al., 1992; Yotsushima et al., 1992, 1993; Huang et al., 1995; De Los Santos-Briones et al., 1997). Different genes for PLC have also been cloned (Hirayama et al., 1995, 1997; Shi et al., 1995; Yamamoto et al., 1995; Kopka et al., 1998), all of them resembling the δ type. However, the molecular basis for activation of plant PLC isoforms is not clear, nor is it completely understood how PLCs interact with their membrane substrates.
Data concerning the manner in which phospholipases interact with lipid substrates are accumulating, and show general similarities but also some remarkable differences between the different isoforms. A variety of assay procedures have been used to measure lipase activity. Most involve presentation of enzymes with pre-aggregated lipids, including phospholipid vesicles, phospholipid-detergent mixed micelles, pre-immobilized and cross-linked lipids, and phospholipid monolayers. The activity of all PLC isoforms studied to date is affected by the surface pressure of monolayer substrates (Rebecchi et al., 1992; James et al., 1994). PLCs act on substrates that form a lipid-water interface, an arrangement that complicates kinetic analyses of the enzyme catalytic activity. To our knowledge, no investigation has been reported thus far on the kinetics of plant PLCs using different assays. Such studies would provide information if PLC binds the substrate in a noncatalytic manner as a prerequisite to anchoring into the membrane to start catalysis, or if the PLC activity from plant sources is affected when it is measured in monolayers. We now report kinetic analyses, vesicle-binding studies, and monolayer assays using a semipurified enzyme from C. roseus transformed roots.
MATERIALS AND METHODS
Tissue Culture
Hairy root line J1 of Cantharanthus roseus was obtained by infection of leaves with Agrobacterium rhizogenes (Ciau-Uitz et al., 1994) and maintained in B5 medium (Gamborg et al., 1968) supplemented with 30 g/L Suc. The pH was adjusted to 5.7 prior to autoclaving of the medium with 0.1 m KOH/HCl. One hundred milliliters of medium was placed in a 250-mL Erlenmeyer flask and autoclaved for 20 min at 15 p.s.i. Flasks were inoculated with 0.5 g (fresh mass) of hairy roots. Roots were subcultured every 14 d. Cultures were grown in darkness at 25°C on a rotary shaker at 100 rpm.
Preparation of Tissue and Cell Extracts
Roots were quickly frozen with liquid nitrogen and homogenized with a polytron in buffer A (1 g of tissue in 2.5 mL of 50 mm NaCl, 1 mm EGTA, 50 mm Tris-HCl, pH 7.4, 250 mm Suc, 10% [v/v] glycerol, 1 mm PMSF, 10 mm sodium pyrophosphate, 0.2 mm orthovanadate, and 1 mm β-mercaptoethanol). Extracts were passed through gauze, and tissue debris were removed by centrifugation at 14,000g for 30 min at 4°C. The supernatant was further centrifuged at 100,000g for 45 min. The supernatants (protein: 3.5–5.0 mg/mL) was recovered as the soluble fraction. The pellet was resuspended in the same buffer A (protein: 0.5–1.2 mg/mL), and was used as a crude membrane fraction. All steps during the extraction were performed at 4°C. The cell extracts were quickly frozen in liquid nitrogen and stored at −75°C. Protein concentrations of samples were measured with bicinchoninic acid protein assay reagent using BSA as a standard (Smith et al., 1985).
Partial Purification of C. roseus PLC
A method has been developed for the rapid partial purification of C. roseus membrane-associated PLC, and has been used on at least six different occasions with similar results. Twenty grams of fresh roots from the 6th d of culture were extracted as mentioned above. The microsomal crude fraction was resuspended in buffer A in the presence of 2 m KCl. This suspension was sonicated for 3 min. After this, the suspension was centrifuged at 100,000g for 45 min at 4°C. The clarified solution was desalted in a Sephadex G-25 column (Pharmacia) pre-equilibrated with buffer B (20 mm Tris, pH 7.4, 1 mm EDTA, 1 mm DTT, 0.6 mm PMSF, and 2 μg/mL leupeptine) at a flow rate of 2 mL/min. The fractions with PLC activity were applied to a heparin-Sepharose CL-6B column (Pharmacia; 1.6 × 17 cm) pre-equilibrated in buffer C (20 mm K2HPO4, pH 7.3, 1 mm EDTA, 1 mm DTT, 0.6 mm PMSF, and 2 μg/mL leupeptine). The column was developed with a 250-mL linear gradient of 0 to 1.5 m KCl in buffer C and 5-mL aliquots were collected. PLC activity was eluted at 0.6 m KCl. PLC was stored in 20% (v/v) glycerol at −70°C.
PLC Assay
The hydrolysis of [3H]PIP2 was measured as described by Hernández-Sotomayor and Carpenter (1993) and De Los Santos-Briones et al. (1997) in a reaction mixture (50 μL) that contained 35 mm NaH2PO4, pH 6.8, 70 mm KCl, 0.8 mm EGTA, 0.8 mm CaCl2 (final Ca2+ concentration, 25 μm), 200 μm PIP2 (approximately 20,000 cpm), and 0.08% deoxycholate. The reaction was stopped with 100 μL of 1% (w/v) BSA and 250 μL of 10% (w/v) TCA. Precipitates were removed by centrifugation (13,000g for 10 min) and the supernatants were collected for quantification of [3H]IP3 released by liquid scintillation counting using Aquasol (DuPont-New England Nuclear, Boston).
Monolayer Assays
Monolayer assays were performed in a monolayer trough (16-mL subphase volume) supplied by Nima Technology (Coventry, UK). Monolayer surface pressure was measured continuously using filter paper Wihelmy plates suspended from an electronic microbalance. Surface radioactivity was monitored continuously using a remote detector (model FC-006, Bioscan, Washington, DC) suspended 0.5 cm above the monolayer, coupled to computer software (LabChrom v2.10, Lab Logic, Sheffield, UK) that recorded the data. The subphase buffer was composed of 35 mm NaH2PO4 (pH 6.8), 70 mm KCl, 0.8 mm EGTA, and 0.8 mm CaCl2 (final Ca2+ concentration, 25 μm, when added). This buffer was stirred with a Teflon-coated stirrer, and composite lipid monolayers (70% [mol] phosphatidylcoline:27% PS:3% [33P]PIP2) were spread at the surface. An aliquot of 0.7 mL of the subphase was replaced with 0.7 mL of enzyme preparation (20–30 μg of protein) only after a stable monolayer with a constant pressure had formed. After 5 min, Ca2+ ions were added to the subphase to start catalysis and the reaction was either monitored continuously (33P-labeled monolayer) or 0.5-mL aliquots were taken at different times (3H-labeled lipids).
Preparation of [33P]PIP2
[33P]PIP2 was prepared using partially purified PIP kinase (James et al., 1994). PIP (200 μm) was sonicated in PIP kinase buffer (200 mm Hepes, pH 7.4, 40 mm MgCl2, 4 mm EGTA, 4 mm DTT, and 400 mm NaCl) and [33P]ATP (specific radioactivity > 1,000 Ci/mmol) with no unlabeled ATP was added to this substrate suspension. The reaction was initiated with the PIP-kinase preparation and incubated at 37°C for 20 min. The reaction was terminated with 750 μL of chloroform:methanol:concentrated HCl (40:80:1, v/v). Radiolabeled [33P]PIP2 (specific activity > 1,000 Ci/mmol) was purified by HPLC using an amino-cyano analytical column pre-equilibrated with chloroform:methanol:water (20:9:1, v/v).
Vesicle Binding
Large unilamellar vesicles were produced by extrusion through 100-nm polycarbonate membranes using a phospholipid extruder (Lipex Biomembranes, Vancouver) according to the manufacturer's instructions. Fifteen milligrams of lipid (PC:PS:PIP2 [70:27:3 by molarity]) was dried to a film, resuspended by vortexing in 10 mm Hepes, pH 7.4, 200 mm Suc, 3.4 mm EDTA, and 20 mm KCl, and treated with repeated freeze-thawing in a liquid nitrogen-40°C water bath. Lipids were extruded with at least 10 passes through the polycarbonate filters, and large unilamellar vesicles were stored at 4°C. For PLC-binding studies, vesicles were diluted to 100 μm with respect to PIP2 in buffer D (10 mm Hepes, pH 7.4, 3.4 mm EDTA, and 150 mm NaCl), and used as a stock for all lower concentrations required in the binding studies.
Binding was performed in 100 μL of buffer D with 10 μg of C. roseus PLC per tube. Ca2+ was omitted to eliminate PLC-catalyzed PIP2 hydrolysis. Enzyme was incubated with vesicles for 10 min on ice followed by ultracentrifugation at 60,000 rpm for 30 min at 4°C (TLA rotor and TL100 centrifuge, Beckman), and PLC activity remaining in the supernatant was assayed against 100 μm PIP2 as described above.
Data Presentation
All experiments were repeated at least three times using extracts prepared on separate occasions, and all gave similar results. Each figure contains data from a single, representative experiment assayed in duplicate and the errors varied by less than 10%.
Analysis of Kinetic Data
The kinetic analysis of the PLC activity from C. roseus was based on the conditions previously described for secretory PLA2 (phospholipase A2; Hendrickson and Dennis, 1984). The binding of the interface was dependent on the concentration of the substrate (bulk PIP2 concentration), while the binding into the interface was dependent on the substrate mole fraction (case II). All the data were adjusted to the Hill equation (Eq. 1) using the GraFit program (Erithacus Software, Middlesex, UK).
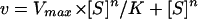 |
1 |
Case I
For the determination of the Ks (binding to the interface), PLC activity was measured increasing concentrations of PIP2 (bulk concentration) with a constant mole fraction of 0.052. To achieve this the concentrations of PIP2 and deoxycholate were varied proportionally, keeping constant the mole fraction.
Equation 1 was reduced to the equation of Henri Michaelis-Menten (Eq. 2) in which the values of Ks were determined.
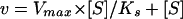 |
2 |
Case II
To determine the Km, PLC activity was assayed keeping the concentration (100 μm) constant and increasing the PIP2 mole fraction by varying the concentrations of deoxycholate. A double-reciprocal Lineweaver-Burk approach was used. The interfacial Km was determined with the intercept to the ordinate ([−1/Km]n).
Case III
To determine if there are several binding sites due to interactions with the lipid interface and the subsequent binding of the substrate to the interface, PLC activity was assayed using a fixed concentration of deoxycholate (1.92 mm) and increasing concentrations of PIP2.
Materials
[3H]PIP2 and Aquasol were purchased from DuPont-New England Nuclear, [33P]ATP was purchased from Amersham, and unlabeled PIP2 was purified from Folch (fraction I) extract of brain lipid (Sigma) by a neomycin affinity column as described in Waldo et al. (1994). The bicinchoninic acid protein assay reagent was supplied by Pierce, and B5 medium PC, PS, and neomycin were obtained from Sigma.
RESULTS
Monolayer Surface Pressure and PLC Activity
To determine the molecular interaction of PLC with lipid interfaces and the reciprocal influences that they have on each other, PLC activity as a function of time was measured in monolayer assays (Fig. 1A). The rate of substrate hydrolysis was followed for 20 min and PIP2 hydrolysis was extended in monolayer assays. A typical time course for PLC-catalyzed hydrolysis of PC-PS-PIP2 monolayers at an initial pressure of 20 mN/m and loss of radioactivity from monolayers due to PIP2 hydrolysis is shown in Figure 1A. Hydrolysis of PIP2-containing monolayers was absolutely dependent on the Ca2+ concentration of the subphase and all data presented were obtained using 25 μm Ca2+ (final concentration), which sustained the maximum rate of PIP2 hydrolysis. Variations of the initial surface pressure of the monolayer resulted in markedly different rates of loss of radioactivity into the subphase. PIP2 hydrolysis in monolayers was transformed into the percentage of PIP2 hydrolyzed per unit time and expressed against the initial surface pressure of the monolayer (Fig. 1B). The percentage of PIP2 hydrolyzed per unit of time was calculated by measuring radioactivities remaining in the monolayer and present in the subphase after 20 min. In the early portion of the pressure-activity curve, PLC activity increased as surface pressure increased (Fig. 1B). A peak in PLC activity was seen at 20 mN/m but as pressure increased beyond this point, PLC activity was markedly reduced.
Figure 1.

Surface pressure-PLC activity relationship for PIP2-containing monolayers. A, Monolayers formed at an initial surface pressure of 20 mN/m and 20 μg of partially purified PLC from C. roseus transformed roots. Ca2+ (25 μm free Ca2+) was added to the subphase after 5 min. Results show a smooth trace, representative of five experiments performed at that pressure. B, PLC activity determined against monolayers formed at increasing initial surface pressures. Reaction time was 20 min after the addition of Ca2+ and data are expressed as percentage of lipid hydrolyzed per minute. Data are means of at least five experiments for each pressure and varied by less than 5%.
PLC-Lipid Binding Effect
Lipid-metabolizing enzymes are considered to bind to and sometimes penetrate lipid interfaces, with subsequent further substrate binding within the interface as component parts of their catalytic mechanism. Therefore, the relationship between bulk substrate concentration and PLC binding was investigated using a noncatalytic vesicle binding assay as described previously for PLC δ (Rebecchi et al., 1992). PLC was incubated with Suc-loaded large unilamellar vesicles with a phospholipid composition of PC:PS:PIP2 (70:27:3 by molarity), and the activity of PLC in the supernatant was determined by assay after ultracentrifugation as described in Methods. When incubated with the vesicles composed of PC-PS-PIP2, PLC was depleted from the supernatant and bound to the large unilamellar vesicle pellet in a manner that was dependent on the total vesicle PIP2 concentration (Fig. 2).
Figure 2.

Binding of C. roseus PLC to Suc-loaded vesicles. Ten micrograms of PLC was incubated with a range of concentrations of PIP2-containing vesicles (PIP2:PC:PS, 3:70:27 mol/mol), followed by ultracentrifugation as described in Methods. PLC activity remaining in the supernatant was assayed against PIP2 and compared with vesicle-free controls in which the activity was 10 pmol/min.
PLC Activity and Dependency on Bulk and Surface Concentrations of PIP2
PIP2 hydrolysis by C. roseus PLC was analyzed according to the method of Hendrickson and Dennis (1984), as has been proposed for animal-PLC δ and γ (Wahl et al., 1992; Cifuentes et al., 1993; Rebecchi et al., 1993). The activity was examined using the three-case kinetic analysis described in detail in “Materials and Methods,” in which bulk and surface PIP2 concentrations were varied independently and concurrently as established for phospholipase A (Hendrickson and Dennis, 1984). Enzyme activity was measured using PIP2-deoxycholate mixed micelles as substrate. Under the conditions used, deoxycholate behaved as a neutral diluent for PIP2. To investigate whether PLC activity involves multiple binding events (a previous different binding site from the catalytic site) due to interactions with the bulk lipid interface and subsequent substrate binding within the interface, the bulk concentration and mole fraction of PIP2 were increased simultaneously and the IP3 production was measured with assays using a single concentration of deoxycholate and increasing concentrations of PIP2. The relationship between PLC activity and bulk and surface concentrations of substrate is presented in Figure 3. Interesting, it did not follow a sigmoidal relationship as described for mammalian PLC β (James et al., 1995). In our model, the behavior of the enzyme was Michaelian, giving a Hill coefficient of 0.968, and thus indicating a single binding site.
Figure 3.

PIP2 hydrolysis by PLC from C. roseus as a function of both bulk concentration and the mole fraction of substrate. The concentration of deoxycholate was held constant at 1.92 mm, and the bulk PIP2 concentration was increased up to 400 μm. Assays were as stated in Methods. Data are representative of four experiments with similar results.
The Ks for interface binding by PLC was determined by assaying PIP2 hydrolysis as a function of bulk concentration at a fixed mole fraction of PIP2 (0.052). This was achieved by varying both bulk PIP2 and deoxycholate concentrations proportionately, while maintaining the mole fraction constant. PLC showed a hyperbolic relationship with PIP2 bulk concentration (Fig. 4).
Figure 4.

PLC activity toward PIP2-deoxycholate mixed micelles as a function of bulk PIP2 concentration. The PIP2 mole fraction was held constant 0.052 and bulk PIP2 plus deoxycholate concentrations varied proportionally. Assays were performed as described in Methods. Data are representative of four experiments.
PLC catalytic activity was also measured at a constant bulk concentration of PIP2 and varied mole fraction. This was achieved by varying the concentration of deoxycholate alone. Experiments were performed at a bulk PIP2 concentration of 100 μm. The relationship between enzyme velocity and substrate mole fraction appeared sigmoidal between 0 and 0.08 (Fig. 5). The constants calculated from this kinetic analysis are: Ks, 45.5 μm, interfacial Km, 0.05, and Vmax, 137.2 pmol/min.
Figure 5.

PLC activity toward PIP2-deoxycholate mixed micelles as a function of PIP2 surface concentration. Bulk PIP2 concentration was held constant at 100 μm with various deoxycholate concentrations to vary PIP2 mole fraction. Other indications are as in Figures 3 and 4.
DISCUSSION
We previously reported that PLC activity is present in C. roseus transformed roots (De Los Santos-Briones et al., 1997). We report here a kinetic study of the partially purified, membrane-associated enzyme using different approaches. Few reports regarding the biochemical characterization of plant PLC are available (Tate et al., 1989; Melin et al., 1992; Pical et al., 1992; Yotsushima et al., 1992, 1993; Hirayama et al., 1995; Huang et al., 1995; Shi et al., 1995; Kopka et al., 1998). To our knowledge, this is the first report in which different assays were used to characterize this enzyme in plants. It is not known if plant PLCs follow a similar mechanism of activation as that reported for PLC from other sources (Hendrickson and Dennis, 1984; Rebecchi et al., 1992, 1993; Wahl et al., 1992; James et al., 1994, 1995, 1997). Most of the PLC enzymatic activity from plant sources is found in the cytosol, but since the substrate of the enzyme is membrane associated, there is the possibility that the cytosolic enzyme under certain physiological circumstances may suffer a redistribution from the cytosol to the membrane.
One of the most characteristic and intriguing features of lipolytic enzymes is their activation by interfaces. To explain the pathways of lipolysis, several investigators have proposed a reversible enzyme adsorption to, or penetration into, the interfaces (Hendrickson and Dennis, 1984; Rebecchi et al., 1993; James et al., 1997). However, such a mechanism has not been demonstrated for plant PLCs. To address this question, we describe the activity of PLC using lipid substrates dispersed in phospholipid vesicles, phospholipid-detergent mixed micelles, and phospholipid monolayers spread at an air-water interface.
We used 33P-labeled substrates to measure PLC activity directly, which showed that the rate and extent of PIP2 hydrolysis in a PC-PS composite monolayer are surface pressure dependent. Although increasing phospholipid mass in the monolayer increased surface pressure, this was not accompanied by a simple increase in enzyme activity throughout the pressure range investigated (Fig. 1B). The reduction in PLC activity, as initial monolayer pressures were increased above the optimum pressure (20 mN/m,; Fig. 1B), was presumably a result of a decrease in the ability of the enzyme to bind the substrate, since this phenomenon has to be very specific and is probably regulated by the PIP2 concentration or another unknown mechanism. However, there were no pressure-induced changes in the rate of catalysis during the course of the experiments (Fig. 1A), which indicates that it was the initial surface pressure that was crucial in determining the subsequent penetration of PLC into the monolayers. It also indicates that the changes in activity were not due to changes in the PIP2 composition of the monolayer after PLC started catalysis. The lower rate of catalysis of lipids at a lower initial surface pressure, which would be expected to permit relatively easy penetration of PLC, may be due to enzyme denaturation by unfolding at the monolayer.
PLC activity may be affected by the composition of the subphase in these assays. The subphase buffer was design to be a simplified intracellular-type buffer solution composed of KCl and NaH2PO4. The resultant free Ca2+ concentration was determined in part by the ionic strength and the ionic composition of the solution, which was characterized in previous experiments in which the Ca2+ requirements of the enzyme were studied (De Los Santos-Briones et al., 1997). The pressure-activity relationship (low activity at pressure below 20 mN/m, reaching a maximum at 20 mN/m, and decreasing above 20 mN/m) for PLC activity against PIP2-containing monolayers presented here (see Fig. 1B) contrasts with that previously reported for PLC δ (Rebecchi et al., 1992, 1993).
Our results (Fig. 1B) resemble those of PLC β (James et al., 1995, 1997), which is surprising since all of the genes cloned to date for plant PLC are of the δ type. For the δ isoform (Rebecchi et al., 1992), it was shown that PLC activity decreased linearly with increasing monolayer surface pressure, with maximum activity being observed at the lowest pressures investigated (15 mN/m). The basis for the differences between PLC from C. roseus and PLC δ activity in monolayers is not clear, but it establishes the phenomenon that different isoforms are affected differently by the quality of the interfaces with which they interact. The data presented here with plant PLC, as well as data from other studies (Rebecchi et al., 1992, 1993; James et al., 1995, 1997), clearly show that PIP2 hydrolysis in monolayers is surface pressure dependent, which is consistent with some element of penetration of lipid interfaces by this family of enzymes.
Our results, together with previous studies using monolayer substrates in which PLC activity was inhibited as the surface pressure increased, suggest that PLCs must penetrate lipid aggregates in order to bind and hydrolyze their substrates. Such a model may seem unnecessary given that the phosphodiester bond in PIP2 is likely to be exposed in the aqueous environment at the surface of the membrane. We propose that this mode of action may facilitate catalysis by restricting diffusion of PLCs into the two-dimensional membrane. Indeed, this may be why a dual substrate mechanism is apparently conserved among a wide range of lipid-metabolizing enzymes.
The data regarding the vesicle binding assay imply association of plant PLC with membrane interfaces through the substrate, PIP2 (Fig. 2). For efficient PLC-catalyzed production of second messengers, PLC may bind to membrane interfaces in a PIP2-specific noncatalytic manner, and subsequent bindings or rearrangements occur within the interface that may help to form a stable anchorage of PLC at the membrane surface. This mechanism may also lead to a series of catalysis reactions whereby PLC could hydrolyze multiple PIP2 molecules before detaching from the interface. The binding of a PIP2 molecule to at least one site in PLC other than the active site is inherent in the above proposal. When PLC from C. roseus was incubated with Suc-loaded PC-PS vesicles lacking PIP2, no measurable binding of the enzyme to the vesicles was exhibited (data not shown). These data strongly support a multisubstrate mechanism in which binding at the interface is a specific process requiring the presence of lipid substrate.
Another unexpected result is shown in Figure 3, in which the kinetic data were analyzed to see if there were multiple binding sites onto the interface and a subsequent binding to the lipid substrate inside the interface. Surprisingly, the curve was not sigmoidal as reported for most mammalian PLCs (Wahl et al., 1992; Rebecchi et al., 1993; James et al., 1995); instead, the curve followed a Michaelis-Menten curve with a Hill coefficient close to 1, probably due to a single binding site. This suggests that plants are regulated in a completely different way from animals. The pleckstrin homology domain, which is found in a broad array of signaling proteins (including all animal PLC isoenzymes), has been suggested to be a sequence that associates proteins with membranes to function (Musacchio et al., 1993), based on evidence for the interaction of PLC δ-pleckstrin homology domains with PIP2. In animal PLC δ enzyme, the amino-terminal region containing the pleckstrin homology domain was necessary for binding to phospholipid vesicles containing PIP2 (Cifuentes et al., 1993). These results suggest that PIP2 might be important for localizing proteins containing pleckstrin homology domains at the membrane surface. However, the pleckstrin homology domain has not to our knowledge been reported in the gene products cloned to date for plant PLC. Perhaps plant PLC first has to bind noncatalytically to PIP2 at the same site where catalysis occurs.
PLCs, which are involved in signal transduction responses to cellular stimuli, are members of a diverse family of enzymes whose mode of interaction with lipid substrates is complex and only partly defined. In summary, we have described the establishment of a controlled monolayer system for studying the family of PLCs, which will permit further investigations into different aspects of the interaction of plant PLCs with their substrates and their regulation. In this study, we have examined the kinetic characteristics of PIP2 hydrolysis by partially purified plant PLC in the absence of their physiological activators. Although it has been proposed that Ca2+ may act as a regulator for plant PLCs, this has never been demonstrated. The data presented here help to establish a basic understanding of how this enzyme behaves toward lipid-water interfaces from which physiologically relevant mechanisms of regulation may eventually be discernible.
ACKNOWLEDGMENTS
We thank Dr. Peter Downes (Biochemistry Department, Dundee University, Scotland) for the facilities provided in his laboratory for the monolayer assays, as well as his generous assistance with reagents, and Dr. Brian Maust (Unidad de Biotecnología, Centro de Investigación Científica de Yucatán) for revision of the English version of the manuscript.
Abbreviations:
- IP3
inositol 1,4,5-trisphosphate
- PC
dioleoylphosphatidylcholine
- PIP
phosphatidylinositol monophosphate
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLC
phospholipase C
- PS
phosphatidylserine
Footnotes
This work was supported by grants from Consejo Nacional de Ciencia y Tecnológia (no. 4119P–N9609) and International Foundation for Science (no. C/2236–2), the interchange program between the Royal Society of London and the Scientific Research Academy of México, and a Consejo Nacional de Ciencia y Tecnología fellowship to C.D.L.S.-B. (no. 88202).
LITERATURE CITED
- Ciau-Uitz R, Miranda-Ham ML, Coello-Coello J, Chi B, Pacheco LM, Loyola-Vargas VM. Indole alkaloid production by transformed and non-transformed root cultures of Catharanthus roseus. In Vitro Cell Dev Biol. 1994;30:84–88. [Google Scholar]
- Cifuentes ME, Honkanen L, Rebecchi MJ (1993) Proteolytic fragments of phosphoinositide-specific phospholipase C-delta. 1. Catalytic and membrane binding properties. J Biol Chem 268: 11586–11593 [PubMed]
- De Los Santos-Briones C, Muñoz-Sánchez JA, Chín-Vera J, Loyola-Vargas VM, Hernández-Sotomayor SMT. Phosphatidylinositol 4,5-bisphosphate-phospholipase C activity during the growing phase of Catharanthus roseus transformed roots. J Plant Physiol. 1997;150:707–713. [Google Scholar]
- Gamborg OL, Miller RA, Ojima K. Nutrient requirements of suspension cultures of soybean root cells. Exp Cell Res. 1968;50:151–158. doi: 10.1016/0014-4827(68)90403-5. [DOI] [PubMed] [Google Scholar]
- Hendrickson HS, Dennis EA. Kinetic analysis of the dual phospholipid model for phospholipase A2 action. J Biol Chem. 1984;259:5734–5739. [PubMed] [Google Scholar]
- Hernández-Sotomayor SMT, Carpenter G. Non-catalytic activation of phospholipase C-γ1 in vitro by epidermal growth factor receptor. Biochem J. 1993;293:507–511. doi: 10.1042/bj2930507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirayama T, Mitsukawa N, Shibata D, Shinozaki K. At-PLC2, a gene encoding phosphoinositide-specific phospholipase C, is constitutively expressed in vegetative and floral tissues in Arabidopsis thaliana. Plant Mol Biol. 1997;34:175–180. doi: 10.1023/a:1005885230896. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Ohto C, Mizoguchi T, Shinosaki K. A gene encoding a phosphatidylinositol-specific phospholipase C is induced by dehydration and salt stress in Arabidopsis thaliana. Proc Natl Acad Sci USA. 1995;92:3903–3907. doi: 10.1073/pnas.92.9.3903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C-H, Tate BF, Drain RC, Coté GG. Multiple phosphoinositide-specific phospholipases C in oat roots: characterization and partial purification. Plant J. 1995;8:257–267. [Google Scholar]
- James SR, Demel RA, Downes CP. Interfacial hydrolysis of phosphatidylinositol 4-phosphate and phosphatidylinositol 4,5-bisphosphate by turkey erythrocyte phospholipase C. Biochem J. 1994;298:499–506. doi: 10.1042/bj2980499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SR, Paterson A, Harden TK, Demel RA, Downes CP. Dependence of the activity of phospholipase Cβ on surface pressure and surface composition in phospholipid monolayers and its implications for their regulation. Biochemistry. 1997;36:848–855. doi: 10.1021/bi962108q. [DOI] [PubMed] [Google Scholar]
- James SR, Paterson A, Harden TK, Downes CP. Kinetic analysis of phospholipase Cβ isoforms using phospholipid-detergent mixed micelles. J Biol Chem. 1995;270:11872–11881. doi: 10.1074/jbc.270.20.11872. [DOI] [PubMed] [Google Scholar]
- Kopka J, Pical C, Gray JE, Müller-Röber B. Molecular and enzymatic characterization of three phosphoinositide-specific phospholipase C isoforms from potato. Plant Physiol. 1998;116:239–250. doi: 10.1104/pp.116.1.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray WC, Irvine RF. Phosphatidylinositol 4,5-bisphosphate phosphodiesterase in higher plants. Biochem J. 1988;249:877–881. doi: 10.1042/bj2490877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melin P-M, Pical C, Jergil B, Sommarin M. Polyphosphoinositide phospholipase C in wheat root plasma membranes: partial purification and characterization. Biochim Biophys Acta. 1992;1123:163–169. doi: 10.1016/0005-2760(92)90107-7. [DOI] [PubMed] [Google Scholar]
- Musacchio A, Gibson T, Rice P, Thompson J, Saraste M. The PH domain: a common piece in the structural patchwork of signaling proteins. Trends Biochem Sci. 1993;18:343–348. doi: 10.1016/0968-0004(93)90071-t. [DOI] [PubMed] [Google Scholar]
- Pical C, Sandelius AS, Melin P-M, Sommarin M. Polyphosphoinositide phospholipase C in plasma membranes of wheat (Triticum aestivum L.): localization of active site and activation by Ca2+ and Mg2+ Plant Physiol. 1992;100:1296–1303. doi: 10.1104/pp.100.3.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebecchi M, Boguslavsky V, Boguslavsky L, McLaughlin S. Phosphoinositide-specific phospholipase C-δ1: effect of monolayer surface pressure and electrostatic surface potentials on activity. Biochemistry. 1992;31:12748–12753. doi: 10.1021/bi00166a006. [DOI] [PubMed] [Google Scholar]
- Rebecchi M, Eberhard R, Delaney T, Ali A, Bittman R. Hydrolysis of short acyl chain inositol lipids by phospholipase C-δ1. J Biol Chem. 1993;268:1735–1741. [PubMed] [Google Scholar]
- Rhee SG, Bae YS. Regulation of phosphoinositide-specific phospholipase C isozymes. J Biol Chem. 1997;272:15045–15048. doi: 10.1074/jbc.272.24.15045. [DOI] [PubMed] [Google Scholar]
- Shi J, Gonzales RA, Bhattacharyya MK. Characterization of a plasma membrane-associated phosphoinositide-specific phospholipase C from soybean. Plant J. 1995;8:381–390. doi: 10.1046/j.1365-313x.1995.08030381.x. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn R, Hermanson GT, Mallia AK, Gartner FM, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Tate BF, Schaller GE, Sussman MR, Crain RC (1989) Characterization of a polyphosphoinositide phospholipase C from the plasma membrane of Avena sativa. Plant Physiol 91: 1275–1279 [DOI] [PMC free article] [PubMed]
- Wahl MI, Jones GA, Nishibe S, Rhee SG, Carpenter G. Growth factor stimulation of phospholipase C-γ1 activity: comparative properties of control and activated enzymes. J Biol Chem. 1992;267:10447–10456. [PubMed] [Google Scholar]
- Waldo GL, Morris AJ, Harden TK. Purification of G-protein-regulated phospholipase C from turkey erythrocytes. Methods Enzymol. 1994;238:195–201. doi: 10.1016/0076-6879(94)38017-1. [DOI] [PubMed] [Google Scholar]
- Yamamoto YT, Conkling MA, Sussex IM, Irish VF. An Arabidopsis cDNA related to animal phosphoinositide-specific phospholipase C genes. Plant Physiol. 1995;107:1029–1030. doi: 10.1104/pp.107.3.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsushima K, Mitsui T, Takaoka T, Hayakawa T, Igaue I. Purification and characterization of membrane-bound inositol phospholipid-specific phospholipase C from suspension-cultured rice (Oryza sativa L.) cells: identification of a regulatory factor. Plant Physiol. 1993;102:165–172. doi: 10.1104/pp.102.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsushima K, Nakamura K, Mitsui T, Igaue I. Purification and characterization of phosphatidylinositol-specific phospholipase C in suspension-cultured cells of rice (Oryza sativa L.) Biosci Biotechnol Biochem. 1992;56:1247–1251. [Google Scholar]